

Abstract
Castleman disease (CD) describes a group of three rare and poorly understood lymphoproliferative disorders that have heterogeneous clinical symptoms and common lymph node histopathological features. Unicentric CD (UCD) involves a single region of enlarged nodes. Multicentric CD (MCD) involves multiple regions of enlarged lymph nodes, constitutional symptoms, and organ dysfunction due to a cytokine storm often including interleukin 6. MCD is further divided into Human Herpes Virus-8 (HHV-8)-associated MCD, which occurs in immunocompromised individuals, and HHV-8-negative/idiopathic MCD (iMCD). Recently, iMCD has been further sub-divided into patients with TAFRO syndrome, which involves thrombocytopenia (T), anasarca (A), fevers (F), reticulin myelofibrosis (R), organomegaly (O), and normal or only slightly elevated immunoglobulin levels, and those who do not have TAFRO syndrome. Non-TAFRO iMCD patients typically have thrombocytosis, less severe fluid accumulation, and hypergammaglobulinemia. iMCD patients with TAFRO syndrome may have a worse prognosis, but more research is needed.
KEY WORDS: TAFRO, Castleman disease, lymphoproliferative disorders, interleukin-6, cytokines, cytokine storm
INTRODUCTION
Castleman disease (CD) is a rare and still inadequately described and understood disease that could be fatal if not properly treated. CD was named after Benjamin Castleman, who was the first to describe the characteristic histopathological findings of angiofollicular lymph node hyperplasia in a localized region of lymph nodes in the 1950s [1,2]. The disease was subsequently observed to also occur in more than one lymph node region leading to the first major classification of the disease into unicentric CD (UCD) and multicentric CD (MCD) [3]. UCD is in most cases treated successfully with surgery [4,5]. MCD was strongly associated with the HIV epidemic and Human Herpesvirus (HHV-8) infection was found to cause MCD in immunocompromised individuals [6-10]. Approximately one-half of MCD cases are HHV-8-positive and the other half are HIV-negative/HHV-8-negative and idiopathic. This has led to the classification of MCD into HHV-8-associated MCD and HHV-8-negative, idiopathic MCD (iMCD) [11,12].
There is limited epidemiological data regarding CD mostly due to historical lack of a unique international classification of disease (ICD) code for this disease. This is changing due to valiant efforts of patients, clinicians and researchers assembled through the Castleman Disease Collaborative Network (CDCN), which has built a global community for targeted and patient-centered collaborative research [13]. The estimated incidence of all forms of CD is 6500-7700 individuals every year in the USA or about 2.2 per 100,000 [14]. Three year disease-free survival rate derived from a systematic literature review from 2011 was 45.7% among 84 HIV negative, HHV-8 unknown MCD cases, and a 2012 series of 60 HHV-8 unknown MCD cases showed a 10-year overall survival rate of around 40% [12,15,16]. The CDCN is currently building a CD registry, which will provide more reliable data on epidemiology, clinical characteristics, and the outcome of this disease.
UCD, HHV-8-associated MCD, and iMCD all share common lymph node histopathological features, which can be separated into hyaline vascular (HV), plasma cell (PC), mixed, and plasmablastic. Plasmablastic is connected exclusively with HHV-8-associated MCD [17]. HV involves atrophic germinal centers, hypervascularization, including sclerotic blood vessels, and follicular dendritic cell (FDC) prominence as well as FDC dysplasia. There is controversy surrounding HV type since many hematopathologists consider this type to occur only in UCD, since sclerotic vessels and FDC dysplasia are not often seen in MCD. More recently, “hypervascular” (HyperV) has been used to describe this histopathological subtype (Fajgenbaum et al., in press) [18]. PC involves hyperplastic germinal centers, hypervascularization, sheet-like plasmacytosis, and occasional atrophic germinal centers. Mixed involves features of both HV and PC. The value of these histopathological subtypes to inform patient care is limited and somewhat controversial.
The etiology of iMCD is unknown, but the symptoms are a result of a cytokine storm, which often includes interleukin 6 (IL-6) and other pro-inflammatory cytokines [12]. The etiology of the cytokine storm in iMCD may be viral, autoinflammatory, autoimmune, or neoplastic [9,10]. iMCD can occur at any age, but is most common in 30-40 year olds and is slightly more frequent in men than in women [19,20]. iMCD symptoms can range from mild lymphadenopathy to more severe cases involving intense inflammation, hepatosplenomegaly, capillary leak syndrome with anasarca, pleural effusion and ascites, organ failure, and death [12,19]. Skin involvement in the form of cherry hemangioma is often noticed [21]. Patients of Asian descent often display large violaceous skin lesions and interstitial pneumonitis, while Polynesian patients demonstrate a mild clinical course despite significant biochemical derangements [12,22-24].
iMCD can be further divided clinically into patients with TAFRO syndrome and those without TAFRO syndrome. TAFRO syndrome patients have thrombocytopenia (T), anasarca (A), fevers (F), reticulin myelofibrosis (R), organomegaly (O), and normal or only slightly elevated immunoglobulin levels. Patients with these clinical symptoms were first described as having “TAFRO syndrome” in 2010 by Takai et al. [25], but iMCD patients with TAFRO symptoms have been observed and reported for decades. Although the vast majority of reported cases of so-called “TAFRO patients” are from Japan, a Caucasian case was reported in Europe [26] and 2 cases from the United States were included in a predominantly Japanese analysis of 25 cases in 2016 [27]. There is no doubt that TAFRO cases are present in other populations, however more recognition and reports are of great importance for elucidation of this clinical syndrome. iMCD patients without TAFRO syndrome typically have thrombocytosis, less severe anasarca, and hypergammaglobulinemia (Figure 1). This group of patients has been described as having the idiopathic plasmacytic lymphadenopathy with polyclonal hyperimmunoglobulinemia or IPL-type [28]. The enlarged lymph nodes in IPL/non-TAFRO patients are generally more enlarged than the TAFRO cases. TAFRO cases are typically more acute in presentation.
FIGURE 1.
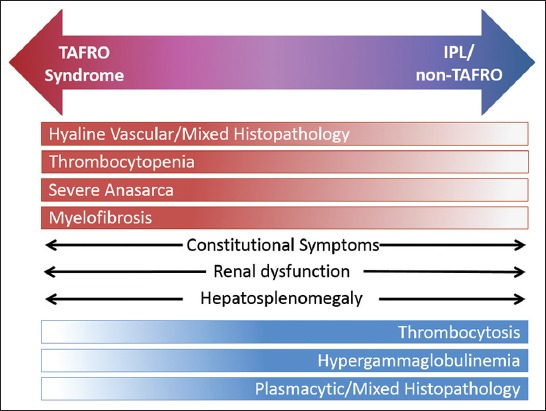
Clinical spectrum of iMCD. iMCD includes a broad spectrum of clinical and laboratory abnormalities. iMCD patients tend to experience either TAFRO syndrome abnormalities (top) or IPL/non-TAFRO abnormalities (bottom). Constitutional symptoms, renal dysfunction, and hepatosplenomegaly do not tend to segment to TAFRO or IPL. iMCD: Idiopathic multicentric Castleman disease; TAFRO: Thrombocytopenia, anasarca, fevers, reticulin myelofibrosis, organomegaly; IPL: Idiopathic plasmacytic lymphadenopathy.
A major issue in the clinical approach to TAFRO syndrome was raised recently by Dr. Frits van Rhee (personal communication). It is not still clear if TAFRO syndrome simply represents the most “extreme clinical presentation” of iMCD or if TAFRO syndrome is an overlapping entity with iMCD with distinct pathophysiological, laboratory, and clinical characteristics. These issues are also raised by two papers focused on clinical, pathological and diagnostic criteria of TAFRO syndrome [27,29]. In their paper published in the American Journal of Hematology in 2016, Iwaki et al. [27] characterized the disease as TAFRO-iMCD, a distinct entity within the larger clinical area of iMCD. Although TAFRO syndrome is characterized by a more aggressive clinical course, corticosteroid refractoriness, thrombocytopenia, higher frequency of anasarca, elevated levels of alkaline phosphatase and especially normal levels of gammaglobulins, in the authors’ opinion it has enough similar characteristics, particularly histological to keep it within the larger iMCD umbrella (Figure 2). Therefore, the authors recommend further subclassification of iMCD into TAFRO-iMCD and iMCD-NOS (not otherwise specified). Masaki et al. [29] take a slightly different approach to diagnosis and classification of TAFRO syndrome. In their diagnostic criteria, histological features consistent with CD in lymph nodes is not a necessary part of the diagnosis, as only 2 out of 4 minor categories are necessary for the diagnosis in addition to 3 major categories (anasarca, thrombocytopenia, and systemic inflammation). Two histological categories (CD-like features on lymph node biopsy and reticulin myelofibrosis or increased number of megakaryocytes in bone marrow) could be absent if mild organomegaly and progressive renal insufficiency are present. Therefore, patients without histological characteristics of MCD could be assigned a diagnosis of the TAFRO syndrome based on their clinical presentation. Case reports from TAFRO patients seem to suggest that IL-6 may be less important and vascular endothelial growth factor (VEGF) more important in those cases, but further research is necessary. However, this distinction could be artificial considering the number of elevated cytokines in the process of a cytokine storm. At the end, Masaki et al. [29] consider TAFRO and iMCD as overlapping syndromes, since some of the TAFRO patients do not fulfill the criteria of iMCD.
FIGURE 2.
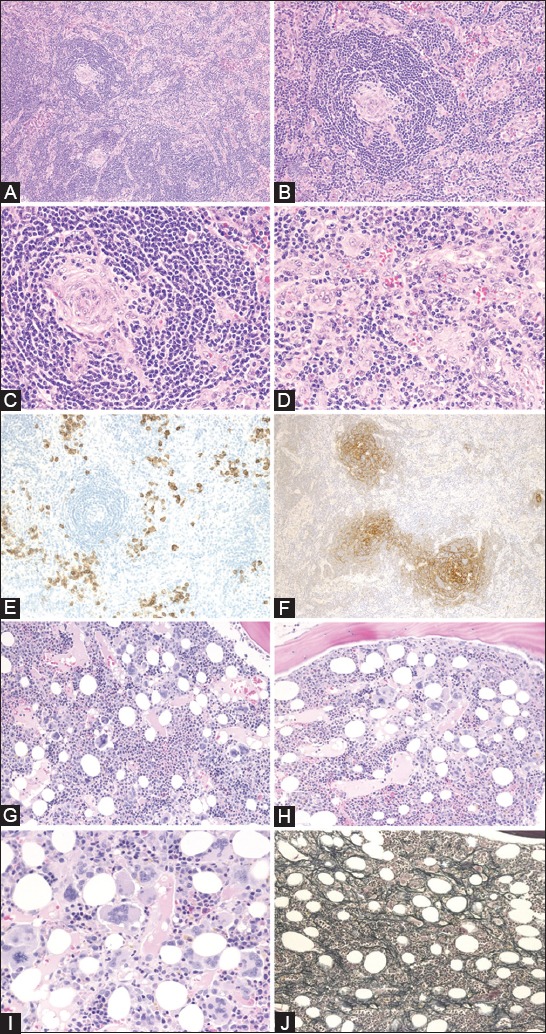
Histological findings of TAFRO-iMCD lymph nodes (A-F) and bone marrow (G-J). (A) The biopsy of a mildly enlarged lymph node shows atrophic germinal centers and intact sinuses (H&E staining); (B) Marked proliferation of high endothelial venules were observed in the germinal centers and interfollicular zone; (C) Atrophic germinal centers with endothelial cells demonstrating enlarged nuclear proliferation without prominent penetrating hyalinized blood vessels (H&E staining); (D) The interfollicular zone is expanded and there is proliferation of highly dense endothelial venules with enlarged nuclei (H&E staining); (E) There are small numbers of CD138-positive plasma cells in the interfollicular zone (CD138 staining); (F) CD21 immunostaining shows expanded or disrupted patterns of follicular dendritic cell networks (CD21 immunostaining); (G) Fifty-nine percent (13/22) of cases show hypercellular marrow (H&E staining); (H) Megakaryocytes tended to be hyperplastic (H&E staining); (I) Megakaryocytes were slightly atypical, with multiple and widely separated nuclei. Marked plasmacytosis was not observed (H&E staining); (J) Silver stain shows very loose network of reticulin fibers (Silver staining). iMCD: Idiopathic multicentric Castleman disease; TAFRO: Thrombocytopenia, anasarca, fevers, reticulin myelofibrosis, organomegaly; H&E: Hematoxylin and eosin. Reprinted from Iwaki et al. [27] with permission.
In conclusion, the etiology, pathology, and overall strategy for the optimal treatment of the TAFRO syndrome remain unclear. TAFRO syndrome could have a unique pathogenesis that is different from the pathogenesis of other cases with iMCD. Therefore, it is possible that different therapeutic approaches will be necessary in the treatment of TAFRO patients as opposed to iMCD patients. Deep ‘omics’ analyses could potentially provide us with new therapeutic targets in the near future. There are lot of unknowns about this new clinical entity, however there is very strong resolution among patients and the research community to disentangle this Gordian Knot.
DECLARATION OF INTERESTS
G.S., I.M., and M.B.S. declare no conflict of interests. D.C.F. has received research funding and served on an advisory board for Janssen Pharmaceuticals.
REFERENCES
- 1.Castleman B, Towne VW. Case records of the Massachusetts General Hospital: Case No. 40231. N Engl J Med. 1954;250(23):1001–5. doi: 10.1056/NEJM195406102502308. https://doi.org/10.1056/NEJM195406102502308. [DOI] [PubMed] [Google Scholar]
- 2.Castleman B, Iverson L, Menendez VP. Localized mediastinal lymph node hyperplasia resembling thymoma. Cancer. 1956;9(4):822–30. doi: 10.1002/1097-0142(195607/08)9:4<822::aid-cncr2820090430>3.0.co;2-4. https://doi.org/10.1002/1097-0142(195607/08)9: 4<822: AID-CNCR2820090430>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 3.Gaba AR, Stein RS, Sweet DL, Varikojis D. Multicentric giant lymph node hyperplasia. Am J Clin Pathol. 1978;69(1):86–90. doi: 10.1093/ajcp/69.1.86. https://doi.org/10.1093/ajcp/69.1.86. [DOI] [PubMed] [Google Scholar]
- 4.Bowne WB, Lewis JJ, Filippa DA, Niesvizky R, Brooks AD, Burt ME, et al. The management of unicentric and multicentric Castleman’s disease: A report of 16 cases and a review of the literature. Cancer. 1999;85(3):706–17. doi: 10.1002/(sici)1097-0142(19990201)85:3<706::aid-cncr21>3.0.co;2-7. https://doi.org/10.1002/(SICI)1097-0142(19990201)85: 3<706: AID-CNCR21>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- 5.Waterston A, Bower M. Fifty years of multicentric Castleman’s disease. Acta Oncol. 2004;43(8):698–704. doi: 10.1080/02841860410002752. https://doi.org/10.1080/02841860410002752. [DOI] [PubMed] [Google Scholar]
- 6.Soulier J, Grollet L, Oksenhendler E, Cacoub P, Cazals-Hatem D, Babinet P, et al. Kaposi’s sarcoma-associated herpesvirus-like DNA sequences in multicentric Castleman’s disease. Blood. 1995;86(4):1276–80. [PubMed] [Google Scholar]
- 7.Chang Y, Cesarman E, Pessin MS, Lee F, Culpepper J, Knowles DM, et al. Identification of herpesvirus-like DNA sequences in AIDS-associated Kaposi’s sarcoma. Science. 1994;266(5192):1865–9. doi: 10.1126/science.7997879. https://doi.org/10.1126/science.7997879. [DOI] [PubMed] [Google Scholar]
- 8.Moore PS, Boshoff C, Weiss RA, Chang Y. Molecular mimicry of human cytokine and cytokine response pathway genes by KSHV. Science. 1996;274(5293):1739–44. doi: 10.1126/science.274.5293.1739. https://doi.org/10.1126/science.274.5293.1739. [DOI] [PubMed] [Google Scholar]
- 9.Suda T, Katano H, Delsoi G, Kakiuchi C, Nakamura T, Shiota M, et al. HHV-8 infection status of AIDS-related and AIDS-associated multicentric Castleman’s disease. Pathol Int. 2001;51(9):671–9. doi: 10.1046/j.1440-1827.2001.01266.x. https://doi.org/10.1046/j.1440-1827.2001.01266.x. [DOI] [PubMed] [Google Scholar]
- 10.Dossier A, Meignin V, Fieshi C, Boutboul D, Oksenhendler E, Galicier L. Human herpesvirus 8-related Castleman disease in the absence of HIV infection. Clin Infect Dis. 2013;56(6):833–42. doi: 10.1093/cid/cis1009. https://doi.org/10.1093/cid/cis1009. [DOI] [PubMed] [Google Scholar]
- 11.El-Osta HE, Kurzrock R. Castleman’s disease: From basic mechanisms to molecular therapeutics. Oncologist. 2011;16(4):497–511. doi: 10.1634/theoncologist.2010-0212. https://doi.org/10.1634/theoncologist.2010-0212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Faigenbaum DC, van Rhee F, Nabel CS. HHV-8-negative, idiopathic multicentric Castleman disease: Novel insights into biology, pathogenesis, and therapy. Blood. 2014;123(19):2924–33. doi: 10.1182/blood-2013-12-545087. https://doi.org/10.1182/blood-2013-12-545087. [DOI] [PubMed] [Google Scholar]
- 13.Fajgenbaum DC, Ruth JR, Kelleher D, Rubenstein AH. The collaborative network approach: A new framework to accelerate Castleman’s disease and other rare disease research. Lancet Haematol. 2016;3(4):e150–2. doi: 10.1016/S2352-3026(16)00007-7. https://doi.org/10.1016/S2352-3026(16)00007-7. [DOI] [PubMed] [Google Scholar]
- 14.Munshi N, Mehra M, van de Velde H, Desai A, Potluri R, Vermeulen J. Use of a claims database to characterize and estimate the incidence rate for Castleman disease. Leuk Lymphoma. 2015;56(5):1252–60. doi: 10.3109/10428194.2014.953145. https://doi.org/10.3109/10428194.2014.953145. [DOI] [PubMed] [Google Scholar]
- 15.Talat N, Schulte KM. Castleman’s disease: Systemic analysis of 416 patients from the literature. Oncologist. 2011;16(9):1316–24. doi: 10.1634/theoncologist.2011-0075. https://doi.org/10.1634/theonclogist.2011-0075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Peterson BA, Frizzera G. Multicentric Castleman’s disease. Semin Oncol. 1993;20(6):636–47. [PubMed] [Google Scholar]
- 17.Liu AY, Nabel CS, Finkelman BS, Ruth JR, Kurzrock R, van Rhee F, et al. Idiopathic multicentric Castleman’s disease: A systematic literature review. Lancet Haematol. 2016;3(4):e163–75. doi: 10.1016/S2352-3026(16)00006-5. https://doi.org/10.1016/S2352-3026(16)00006-5. [DOI] [PubMed] [Google Scholar]
- 18.Fajgenbaum DC, Uldrick TS, Bagg A, Frank D, Wu D, Srkalovic G, et al. International evidence-based consensus diagnostic criteria for HHV-8-negative/idiopathic multicentric Castleman disease. Blood 2017 (in press) doi: 10.1182/blood-2016-10-746933. https://doi.org/10.1182/blood-2016-10-746933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dispenzieri A, Armitage JO, Loe MJ, Geyer SM, Allred J, Camoriano JK, et al. The clinical spectrum of Castleman’s disease. Am J Hematol. 2012;87(11):997–1002. doi: 10.1002/ajh.23291. https://doi.org/10.1002/ajh.23291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Waterston A, Bower M. Fifty years of multicentric Castleman’s disease. Acta Oncol. 2004;43(8):698–704. doi: 10.1080/02841860410002752. https://doi.org/10.1080/02841860410002752. [DOI] [PubMed] [Google Scholar]
- 21.Fajgenbaum D, Rosenbach M, van Rhee F, Nasir A, Reutter J. Eruptive cherry hemangiomatosis associated with multicentric Castleman disease: A case report and diagnostic clue. JAMA Dermatol. 2013;149(2):204–8. doi: 10.1001/jamadermatol.2013.1552. https://doi.org/10.1001/jamadermatol.2013.1552. [DOI] [PubMed] [Google Scholar]
- 22.Nishmoto N, Kanakura Y, Aozasa K, Johkoh T, Nakamura M, Nakano S, et al. Humanized anti-interleukin-6 receptor antibody treatment of multicentric Castleman disease. Blood. 2005;106(8):2627–32. doi: 10.1182/blood-2004-12-4602. https://doi.org/10.1182/blood-2004-12-4602. [DOI] [PubMed] [Google Scholar]
- 23.Ahmed B, Tschen JA, Cohen PR, Zaki MH, Rady PL, Tyring SK, et al. Cutaneous castleman’s disease responds to anti interleukin-6 treatment. Mo Cancer Ther. 2007;6(9):2386–90. doi: 10.1158/1535-7163.MCT-07-0256. https://doi.org/10.1158/1535-7163.mct-07-0256. [DOI] [PubMed] [Google Scholar]
- 24.Zhai S, Simpson D. Polynesian variant of idiopathic multicentric Castleman disease [abstract] Blood. 2013;122(21):Abstract 5127. [Google Scholar]
- 25.Takai K, Nikkuni K, Shibuya H, Hashidate H. Thrombocytopenia with mild bone marrow fibrosis accompanied by fever, pleural effusion, ascites and hepatosplenomegaly. [Article in Japanese] Rinsho Ketsueki. 2010;51(5):320–5. http://doi.org/10.11406/rinketsu.51.320. [PubMed] [Google Scholar]
- 26.Abdo LA, Morin CP, Collarino RP, Cabane JP, Gatfosse MA. First European case of TAFRO syndrome associated with Sjogren disease. Am J Intern Med. 2014;2(6):102–5. https://doi.org/10.11648/j.ajim.20140206.12. [Google Scholar]
- 27.Iwaki N, Fajgenbaum DC, Nabel CS, Gion Y, Kondo E, Kawano M, et al. Clinicopathologic analysis of TAFRO syndrome demonstrates a distinct subtype of HHV-8-negative multicentric Castleman disease. Am J of Hematol. 2016;91(2):220–6. doi: 10.1002/ajh.24242. https://doi.org/10.1002/ajh.24242. [DOI] [PubMed] [Google Scholar]
- 28.Kawabata H, Takai K, Kojima M, Nakamura N, Aoki S, Nakamura S, et al. Castleman-Kojima disease (TAFRO syndrome): A novel systemic inflammatory disease characterized by a constellation of symptoms, namely, thrombocytopenia, ascites (anasarca), microcytic anemia, myelofibrosis, renal dysfunction, and organomegaly: A status report and summary of Fukushima (6 June 2012) and Nagoya meetings (22 September 2012) J Clin Exp Hematop. 2013;53(1):57–61. doi: 10.3960/jslrt.53.57. https://doi.org/10.3960/jslrt.53.57. [DOI] [PubMed] [Google Scholar]
- 29.Masaki Y, Kawabata H, Takai K, Kojima M, Tsukamoto N, Ishigaki Y, et al. Proposed diagnostic criteria, disease severity classification and treatment strategy for TAFRO syndrome. Int J Hematol. 2016;103(6):686–92. doi: 10.1007/s12185-016-1979-1. https://doi.org/10.1007/s12185-016-1979-1. [DOI] [PubMed] [Google Scholar]