

Abstract
Purpose
Reports of cytomegalovirus (CMV) detection in high-grade gliomas (HGG)/glioblastoma (GBM) have been conflicting. We undertook a comprehensive approach to determine presence or absence of CMV in tissue, plasma and serum of HGG patients.
Experimental Design
In a retrospective arm, 25 fresh frozen tissues from GBM patients were tested for CMV by real-time PCR. Tissue microarrays from 70 HGG patients were tested by immunohistochemistry (IHC) and 20 formalin-fixed paraffin-embedded (FFPE) GBM tissues by IHC and chromogenic in situ hybridization (CISH), targeting CMV-encoded IE1/2 and pp65. In a prospective arm, 18 patients with newly-diagnosed HGG provided tissue and blood samples.
Results
All retrospectively collected tissues were negative for CMV by all methods. In the prospective cohort, 18 patients with newly-diagnosed HGG provided blood samples at the time of diagnosis and during follow-up. Of 38 plasma specimens, CMV DNA was detected in 3 of 18 samples at baseline and 1 of 20 follow-up samples. Serum CMV IgG was positive in 8 of 15 (53%) of patients. Among the FFPE samples tested in the prospective arm, all were negative for CMV by IHC, CISH and PCR.
Conclusions
Utilizing 6 highly-sensitive assays with 3 orthogonal technologies on multiple specimens and specimen types, no evidence for CMV in GBM tissues was found. Our findings call for multicenter blinded analyses of samples collected from different geographical areas with agreed upon study designs and determination of causality or lack thereof of CMV in HGG/GBM for future guidance on the necessity anti-viral and/or CMV-based therapies.
Keywords: Cytomegalovirus, glioblastoma (GBM), anaplastic astrocytoma, immunohistochemistry (IHC), in-situ hybridization
Introduction
The question of whether cytomegalovirus (CMV) plays a role in gliomagenesis and whether antiviral therapy would modify the prognosis and outcome of patients with gliomas, has been intensively debated since the initial report on CMV detection in GBM tissues. While several groups reported on their ability to detect CMV (1–9), others were unable to replicate these finding, raising questions about the assays used and their sensitivities for CMV detection (10–16). The ongoing controversy of CMV detection in gliomas has significant clinical implications since suppression of CMV replication with antiviral therapy could potentially modify the prognosis of these cancers. If CMV plays a role in glioblastomas (GBM), future clinical trials could potentially support changes to its standard of care. In addition to antiviral therapy, the presence of CMV in GBM may suggest that immunotherapies, such as anti-CMV vaccines (e.g., clinicaltrials.gov NCT01109095) might be of value (17–19).
We undertook a comprehensive approach for the detection of CMV in HGG. Using retrospectively-collected HGG tissues, and prospectively collected samples from newly-diagnosed HGG patients who underwent standard therapy with radiation and chemotherapy, we investigated whether CMV was present in tumor cells or in the tumor microenvironment. Three complementary technologies: real-time PCR for CMV DNA (two different genomic targets), chromogenic in situ hybridization for CMV DNA (from two distinct genome regions), and immunohistochemical staining (against two different CMV polypeptides) were used for this analysis.
Materials and Methods
Retrospective specimen collections
Archived HGG samples for CMV detection included fresh frozen tissue (n = 25), formalin-fixed paraffin embedded (FFPE) whole sections (n = 20) and a separate set of FFPE tissues in a microarray (n = 70). Methods used for CMV detection with each sample type are summarized in Table 1.
Table 1.
Overall specimen sources, tests performed and numbers of patients/samples tested.
| Specimen source | Total number of patients (samples) | Real-time PCR | CISH | IHC | IgG avidity index | |
|---|---|---|---|---|---|---|
| Retrospective specimen collections | Fresh frozen tissue | 25 | 25 | |||
| Tissue microarray | 70 | 70 | ||||
| FFPE whole sections | 19 | 19 | 19 | |||
| Prospective study N = 18 patients |
FFPE whole sections | 11 | 8 | 11 | 8 | |
| Plasma | 38 | 18 baseline, 20 follow-up samples | ||||
| Serum | 15 | 15 | ||||
| Totals | 178 | 71 | 30 | 97 | 15 | |
Prospective cohort of newly-diagnosed HGG
Adults with newly diagnosed high-grade gliomas (WHO grade III or IV) who were scheduled to undergo standard chemoradiation followed by adjuvant temozolomide were asked to participate in this IRB-approved research protocol. Formalin-fixed paraffin embedded (FFPE) tumor tissue obtained from the surgery that led to the diagnosis of HGG was used for CMV detection by real-time PCR, chromogenic in-situ hybridization (CISH) and immunohistochemistry (IHC). Serum and plasma samples, collected at baseline prior to the start of chemoradiation, were assayed for CMV IgG avidity and real-time PCR, respectively. Additional plasma samples were collected at follow-up visits for real-time PCR (usually at the time of scheduled MRI scans) for as long as the patient agreed to participate. Beta-actin was tested as a housekeeping gene (quality control) to assure sufficient DNA in the tested samples. Serum IgG avidity index was determined using standard ELISA analysis (Focus Diagnostics, Cypress, CA). Interpretive criteria for the ELISA assay were: An index of ≤ 50 was considered low, 51–59 intermediate, and > 60 high. A total of 11 FFPE whole sections, 38 plasma samples, and 15 serum samples were tested within this prospective cohort.
DNA extraction and real-time PCR
FFPE Tissues
FFPE blocks (prospective cohort) initially underwent pathological review to determine tumor cellularity. Tumors were then macrodissected to remove surrounding normal tissue. DNA was extracted using the DNA FFPE tissue kit (Qiagen, Valencia, CA), following the manufacturer’s instructions.
Fresh Frozen Tissues
Patient samples were obtained as excess tissues not needed for diagnosis from tissues removed from patients undergoing resection and found to have glioblastoma or high grade gliomas by immediate frozen section analyses. Samples were snap frozen and processed later for DNA extraction. DNA was isolated using the Qiagen DNeasy tissue kit (Valencia, CA), following the manufacturer’s instructions.
Plasma
Plasma samples were tested for the presence of CMV DNA by both the quantitative CMV US17 real-time PCR assay and the COBAS AmpliPrep/COBAS TaqMan CMV IVD test. DNA was isolated from plasma samples for CMV US17 real-time PCR using automated DNA extraction on the Biorobot M48 instrument with the Virus Mini protocol and MagAttract Virus Mini Kit (Qiagen, Valencia, CA). The sample input volume was 400 μL, and the elution volume was 100 μL. For the COBAS AmpliPrep/COBAS TaqMan CMV IVD test (Roche Diagnostic Corp., Indianapolis, IN), nucleic acid extraction is the first process performed by the instrumentation.
Real-time PCR
The US17 real-time PCR assay used for fresh frozen tissues (retrospective collection), FFPE tissues and plasma samples (prospective cohort) targets a 151-bp fragment from a highly conserved region of the CMV genome. The primers and probe for US17 were: forward – 5′ GCGTGCTTTTTAGCCTCTGCA-3′, reverse 5′- AAAAGTTTGTGCCCCAACGGTA-3′ and US17 probe FAM – 5′ TGATCGGGCGTTATCGCGTTCT-3′ (20). Quantification standards were prepared by cloning the US17 amplicon in the pCR2.1-TOPO plasmid vector (Invitrogen/ThermoFisher Scientific, Carlsbad, CA). Serial 10-fold dilutions of plasmid from 7.0 to 1.0 log10 copies/reaction were included with each assay and used to establish a standard curve. The measurable range of the assay is 1.0E+02 to 1.0E+08 copies/mL and the limit of detection is 1.0E+02 copies/mL. This assay has been used extensively in our laboratory to detect CMV DNA and measure CMV loads in different body fluids (21). Beta-actin real-time PCR was used to assess efficiency of DNA extraction from FFPE tissues (Applied Biosystems/ThermoFisher Scientific, Foster City, CA).
In addition to US17 real-time PCR, CMV DNA in plasma was quantified using The COBAS AmpliPrep/COBAS TaqMan CMV test which is based on simultaneous amplification, detection, and quantification of both target CMV DNA and internal control quantitative standard. The measurable range for this assay is 1.37E+02 to 9.10E+06 IU/mL, and the limit of detection is 9.1E+01 IU/mL. Results can be converted manually to CMV DNA copies/mL by multiplying the result in IU/mL by 1.1 copies/IU.
Immunohistochemistry (IHC)
Immunostaining of glioblastoma TMA sections was performed with PowerVision kit according to the manufacturer’s protocol (Leica Biosystems). Briefly, the slides were heated at 60°C for 10 min, deparaffinized and hydrated through xylene, graded ethyl alcohols, dH2O, dH2O with 20% Tween 20 (P-7949, Sigma-Aldrich). After antigen retrieval (25 minutes of steaming in citrate buffer using Black and Decker Handy Steamer Plus), sections were treated 5 minutes with Dual Blocking Solution (S2003, Dako) and incubated overnight at room temperature with either mouse monoclonal antibody against CMV pp65 protein (VP-C422, Vector Laboratories, Burlingame, CA; 1:800) or mouse monoclonal antibody against CMV immediate early antigen (MAB810, Millipore, Billerica, MA; 1:8000) followed by secondary anti-mouse IgG-reagent provided in the Powervision+PolyDAB kit (PV6119, Leica Biosystems). Immunostaining was visualized with DAB chromogen (D4293, Sigma-Aldrich) and sections were counterstained with Mayer’s hematoxylin.
Control experiments for CMV protein detection by IHC were carried out using human foreskin fibroblasts passage 12–16 (ATCC, CRL-2088) that were either mock infected (negative control cells) or infected with the CMV Towne strain (ATCC VR-977) (positive control cells) for 72 hours (Figure 1). Cells were grown in Dulbecco’s Modified Eagle Medium (DMEM) containing 10% fetal bovine serum (FBS) (Gibco, Carlsbad, CA) in a 5% CO2 incubator at 37 °C. After infection cells were harvested by trypsinization, washed with PBS and transferred to a microfuge tube that had been prepared with a 2% solidified agarose plug at the bottom. The cells were then centrifuged to form a pellet and fixed in neutral buffered formalin overnight at room temperature by gentle layering of formalin on freshly isolated cell pellets and then and were processed and embedded into paraffin blocks by slicing the tube and placing the sliced tube into a tissue cassette for tissue processing and embedding (22) to simulate FFPE tissue. CMV positive tissues from surgical gastrointestinal mucosal biopsies, that were fixed in formalin and processed in the CLIA clinical lab similarly to the clinical brain FFPE tumor tissues used in this study, were also used as additional positive controls (Figure 1). Prostate tumor and normal tissues (negative for HCMV DNA (23) were used to construct a tissue microarray using 48 tissue cores from 8 patients (2–4 each tumor and normal per patient). These served as additional negative controls for the IHC.
Figure 1. Analytical validation of CISH and IHC in human fibroblast cell lines.
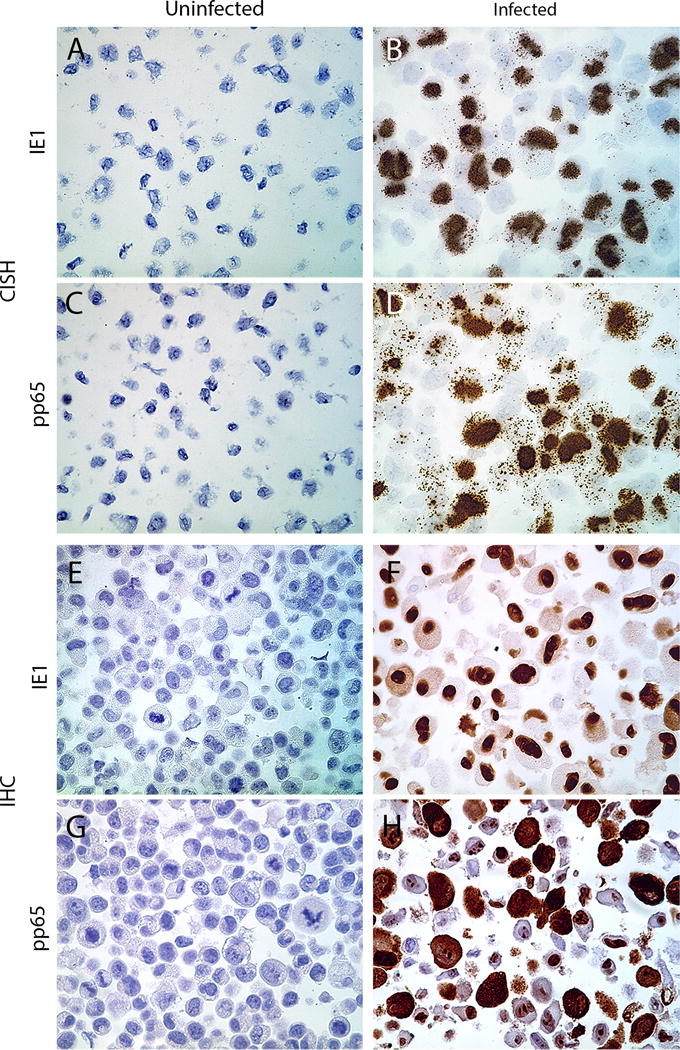
Human foreskin fibroblasts were uninfected or infected with a human CMV Towne for 72 h. Cells were then fixed in neutral buffered formalin overnight and processed into paraffin blocks. CISH assay depicts targeting IE1 DNA in uninfected (A) and CMV-infected (B), and pp65 DNA in uninfected (C) and CMV-infected cells (D). Representative figures for IHC staining of IE1- and pp65 proteins in uninfected (E,G) and CMV-infected (F,H) cells are shown (all images are original magnification of ×400).
In situ hybridization (CISH)
In conjunction with Advanced Cell Diagnostics (ACD) we designed two probe sets that recognize two distinct regions of the CMV genome (ACD 425971 – V-HHPV5-IE1 and ACD 425981 – V-HHPV5-PP65) which are specific for CMV DNA and not RNA. CMV-infected human foreskin fibroblasts prepared into FFPE blocks were used as positive control. As a control for brain tumor tissue DNA integrity for in situ hybridization we designed an ACD probe set that recognizes human telomeric DNA (22 and Q Zheng, C Heaphy, AM Meeker, AM De Marzo, manuscript in process). Mock infected fibroblasts prepared identically into FFPE were used an isogenic negative control to demonstrate specificity of hybridizations. Chromogenic in situ hybridization was carried out according to the manufacturer’s instructions for RNA detection (Advanced Cell Diagnostics (ACD) RNAscope 2.0 Brown Kit) (25). FFPE slides were baked at 60°C for 1 hour, then deparaffinized with exposure to xylene twice, 10 minutes each time, followed by stirring in 100% ethanol twice and air-drying, then rehydration with dH2O for 2 minutes. Pretreatment solution 1 was applied to the slides for 10 minutes at room temperature. The slides were then boiled in pretreatment solution 2, at 100°C for 15 minutes, followed by protease digestion in pretreatment solution 3 for 30 minutes at 40°C to allow target accessibility. ACD target probe (HCMV IE1, ACD 425971 or HCMV pp65, ACD 425981) was applied and the slides were incubated in a HybEZ TM Oven at 40°C for 2 hours. Slides were washed twice with 1× wash buffer for 2 min at room temperature. Signal amplification steps followed: amplification reagents 1, 3 and 5 were incubated for 30 minutes, amplification reagents 2, 4 and 6 for 15 minutes; amplification steps 1, 2 and 4 took place in the oven at 40°C, 5 and 6 at room temperature. Slides were washed with the 1× wash buffer between each amplification step. DAB solution was applied for 10 minutes at room temperature, and slides were washed with dH2O. 50% Gill’s Hematoxylin was applied for 2 minutes for counterstaining, slides were then rinsed in 0.01% ammonia dH2O for 10 seconds. Slides were passed through 100% ethanol, then xylene and coverslipped with Cytoseal mounting medium. CMV-infected and non-infected human foreskin fibroblasts (Figure 1) and CMV positive gastrointestinal (colonic mucosal) tissues (Figure 2) were used as positive controls.
Figure 2. CMV detection by CISH and IHC from CMV-positive human tissues.
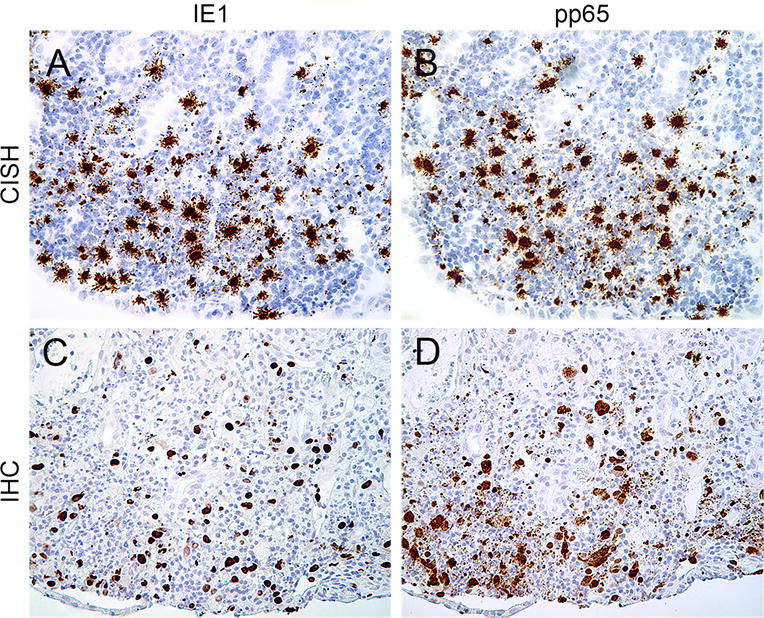
CISH assays targeting IE1 DNA (A), pp65 DNA (B); IHC staining for IE1 (C), pp65 (D) proteins performed on a colonic biopsy with known CMV infection (all images are ×100).
Tissue microarray construction
FFPE glioblastoma tissues were obtained from the pathology archives of Johns Hopkins Hospital for the construction of a TMA (tissue microarray) by the Oncology Tissue Services Laboratory at the Johns Hopkins Medical Institutions. Details of TMA preparation have been described elsewhere (26). Each tumor was spotted 4–8 times for TMA construction with 0.6 mm punches.
Statistical considerations
All outcome data are presented with standard descriptive summaries.
Results
Retrospective tissue dataset analysis
Fresh frozen tissues from 25 patients with GBM were analyzed by a highly sensitive US17 real-time PCR for presence or absence of CMV. All 25 patients had the diagnosis of GBM (WHO grade IV). Median age was 46 (range, 2–79; 56% male, 44% female). All samples were negative for CMV by real-time PCR (Supplemental Table 1). Positive control tissues consisted of DNA isolated from FFPE tissue biopsies with known positive CMV status from both colon and gastric mucosal biopsies.
The presence of CMV DNA was also interrogated by two novel in situ hybridization assays developed in conjunction with ACD, in which each probe set detects a different region of the CMV genome. This approach, which is identical to using ACD RNAScope, except the probes are designed to hybridize to the antisense DNA strand instead of the transcribed sense strand of RNA, has been shown to detect single molecules in single cells (25, 27).
Both probe sets (ACD 425971 – V-HHPV5-IE1 and ACD 425981 – V-HHPV5-PP65) showed strongly positive hybridization signals in the control HCMV infected cells but not in the mock infected cells (Figure 1). The majority of the signals were nuclear, but some signal was also present in the cytoplasm, consistent with the life cycle of CMV. We verified that the assay was specific for DNA by pre-treating slides with either RNAse or DNAse and found that there was no signal reduction with RNAse, but a substantial reduction with DNAse pretreatment (data not shown).
To gain insight into the sensitivity of these assays, we infected the same human fibroblasts used as negative control cells with different multiplicity of infection plaque forming units (2, 0.2, 0.02, 0.002) and harvested the cells after 3 hours of infection before new viral particles were produced. Supplemental Figure 1A shows that both probe sets displayed robust signals in cells infected with MOI of 2, with many less cells with signals using a MOI of 0.2. No signals were detected in cells infected with M0I of 0.02 or 0.002.
Assay performance was also confirmed using known CMV positive human biopsy tissues which were strongly positive for hybridization signals using both probe sets (Figure 2). Using these in situ hybridization assays on a set of 30 cases (11 prospective and 19 retrospective) of high grade glioma FFPE clinical specimens showed that none were positive for either probe (summarized in Supplemental Table 2; representative example in Figure 3)i. As a positive control for DNA integrity for in-situ hybridization using this ACD technology, we used a novel probe set against human telomeric DNA, and as shown in Supplemental Figure 2, there were robust signals within nuclei consistent with telomeric DNA foci (22) in all samples tested.
Figure 3. Lack of CMV detection in human high grade gliomas.
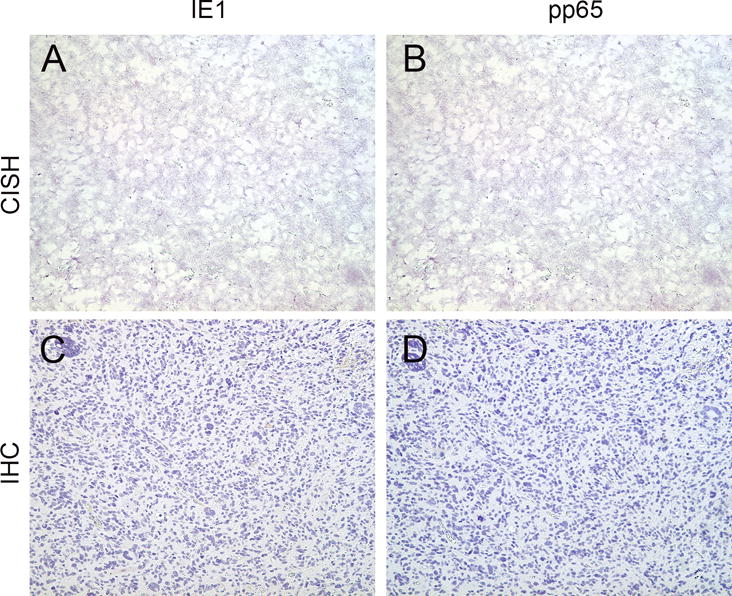
IE1 (A) and pp65 (B) CISH assays performed on FFPE samples of a representative high grade glioma were negative for their corresponding DNA segments. By immunohistochemistry the same sample was negative for IE1 (C) and pp65 (D) proteins as well (all images are ×200).
The presence of CMV proteins in tissue samples was tested by IHC assays, targeting either IE1 or pp65, using the same tissues in which adequate tissue was present (N= 27 of 30). Figures 1 and 2 show that the IHC assays show similar specificity to the in-situ probe sets.
In terms of sensitivity of the IHC assays, there were robust signals in human fibroblasts infected with HCMV MOI of 2, and 0.2, with some positive staining for IE1 protein down to an MOI of 0.02 (Supplemental Figure 1B). While these results do not reveal a precise number of molecules per cell that can be detected, it is clear that even rare cells infected with low levels of HCMV DNA can be detected after 3 hours of infection.
All 27 human brain tumor tissues were negative for CMV IE1 and pp65 protein (Supplemental Table 2). To expand the number of and type of specimens examined, we performed IHC using the same antibodies on samples from a tissue microarray (TMA) of 68 GBM (WHO grade IV) and 2 malignant gliomas (WHO grade III), that included 24 pediatric tissue samples (age <18 years). IHC was negative in all of the TMA samples (Supplemental Table 3; representative example in Figure 3). To determine the tissue integrity for IHC staining we performed IHC against vimentin (28) and the proliferation marker Ki67, which is commonly used in both clinical and research laboratories for IHC. All tissues with enough tumor tissue available showed robust staining for both markers (Supplemental Table 2, and Supplemental Figure 3). We did not perform in situ hybridization on the TMA because samples were significantly older (taken between 1987 and 2009) and we have found that ACD in situ hybridization signals on tissues more than 5 years old are generally markedly diminished (J Baena Del Valle, Q Zheng, AM De Marzo, manuscript in process).
To further assess the specificity of our IHC assay and to determine whether altered conditions could generate false positive signals, we performed another set of experiments in which we altered the dilution of our antibodies for IHC staining. We reasoned that any signals obtained in our negative control fibroblast cells, that are consistently negative for CMV DNA by a highly sensitive PCR assay, are false positive signals. Using the 8B1.2 mouse monoclonal antibody (Millipore #MAB810), our determined optimal antibody dilution was 1:8000. However, when we used a higher concentration of antibody (from e.g. 1:500), there was false positive staining and this was even more marked at a final dilution of 1:50 (Supplemental Figure 4A). Thus, by simply altering the concentration of the primary antibody we can generate false positive signals. Similarly, we obtained false positive staining using the anti-pp65 monoclonal antibody by IHC when we dropped the dilution from our optimal dilution of 1:800 to either 1:50 or 1:10 (Supplemental Figure 4B). In terms of detecting CMV protein in human brain tumor FFPE sections, when we applied the 8B1.2 1E1 antibody at 1:50 to a number of the same brain tumor samples used above, we obtained positive staining (ranging from weak to robust) in all cases that had tumor tissue to evaluate (18 of 20). An example of intermediate levels of staining with the IE1 antibody is shown in (Supplemental Figure 5). These results showing that we can readily obtain positive IHC signals in human brain tumors with both antibodies when using suboptimal antibody concentrations (without changing any other parameters of staining) provide a plausible explanation why some studies using IHC have found positive staining in human high grade gliomas; false positive staining can be readily achieved simply by using high antibody concentrations with antibodies that are otherwise thought to be specific.
Prospective patient cohort
Between September 2012 and August 2013, 18 patients with newly-diagnosed HGG were enrolled in a prospective protocol. These included 10 patients with GBM, 2 with gliosarcoma and 6 with anaplastic astrocytomas. The median age of the participants was 56 years (range, 30–89) and 50% were male. Available tissue samples were analyzed for CMV using real-time PCR, CISH and IHC, using the same protocol as for the retrospective tissue samples (see above). Real-time PCR for CMV was negative in all samples (n=8; 3 tissues were eliminated from the analysis due to a negative β-actin PCR result; for 7 tissues, no sufficient tissue was available for analysis as part of this research study). Analysis by CISH (n=11) and IHC (n=8) was also negative (Table 2). The serum IgG avidity index for CMV was determined at enrollment to assess for recent or past prior infection. Eight of 15 (53%) patients were CMV-seropositive (n=15; index range, 0.55 to 0.82), a prevalence consistent with the lower end of that reported in the general population (20, 21). Of 38 plasma samples that were collected at baseline and during participation in the study, low level viremia was detected by US17 real-time PCR in 3 samples at baseline (Table 2) and only in one follow-up sample. Of the 4 low level positive samples by the CMV US17 assay, 3 were also low level positive by the COBAS AmpliPrep/COBAS TaqMan CMV test. All 4 patients with positive CMV DNA were seropositive at baseline as determined by the serum IgG avidity index.
Table 2.
Prospective CMV analysis of tissue, serum and plasma from 18 patients with newly diagnosed high-grade glioma.
| Patient | Histology | Demographics | Blood | |||
|---|---|---|---|---|---|---|
| Diagnosis | WHO grade | Age at diagnosis | Gender | Real-time PCR* plasma (baseline), copy number/mL | Serum CMV IgG avidity index | |
| 1 | GBM | IV | 56 | M | NEGATIVE | 0.81 |
| 2 | GBM | IV | 76 | M | NEGATIVE | N/A |
| 3 | GBM | IV | 60 | M | NEGATIVE | NEGATIVE |
| 4 | GBM | IV | 70 | F | NEGATIVE | NEGATIVE |
| 5 | GBM | IV | 51 | M | NEGATIVE | 0.55 |
| 6 | GBM | IV | 48 | F | NEGATIVE | NEGATIVE |
| 7 | GBM | IV | 59 | F | NEGATIVE | NEGATIVE |
| 8 | GBM | IV | 46 | F | NEGATIVE | 0.76 |
| 9 | GBM | IV | 68 | F | 1.15E+02 | 0.82 |
| 10 | GBM | IV | 42 | M | NEGATIVE | NEGATIVE |
| 11 | GS | IV | 45 | M | 1.06E+02 | 0.73 |
| 12 | GS | IV | 89 | F | 2.49E+02 | 0.62 |
| 13 | AA | III | 30 | M | NEGATIVE | N/A |
| 14 | AA | III | 42 | M | NEGATIVE | 0.68 |
| 15 | AA | III | 32 | F | NEGATIVE | NEGATIVE |
| 16 | AA | III | 62 | M | NEGATIVE | NEGATIVE |
| 17 | AA | III | 64 | F | NEGATIVE | 0.73 |
| 18 | AA | III | 55 | F | NEGATIVE | N/A |
| Positive controls | N/A | N/A | N/A | N/A | Towne CMV spiked into plasma at 1E3 and 1E5 copies/mL | N/A |
Abbreviations: GBM, glioblastoma; GS, gliosarcoma; AA, anaplastic astrocytoma; N/A, not analyzed/not available.
Both the quantitative US17 real-time PCR assay and the COBAS AmliPrep/COBAS TaqMan IVD test were used for this analysis (results in the table are from testing with US17).
Discussion
Using 3 different highly-sensitive and specific tissue-based detection methods (real-time PCR, CISH, and IHC) and several different specimen procurements, including fresh frozen tissue samples and archived FFPE samples, we did not detect CMV in tissues from patients with HGG. This included patients who were CMV seropositive (53% of patients in the prospective cohort). Our data are consistent with other reports that also did not identify CMV in tissue of patients with these cancers (10–16), but they stand in contrast to several studies by other investigators that reported on detection of CMV in the majority of tissues and cells analyzed (1–9). The key question underlying this controversy is how different investigators and laboratories could come to entirely opposite conclusions when studying the same cancer type and virus. Reporting on absence of CMV in gliomas, especially if several other groups reported high detection rates, raises the question of whether our results may be false negative due to technical problems or lack of sensitivity of our assays. This question has been previously raised in the literature in the context of a report stating that CMV could not be detected with standard pathological and virological diagnostics (12). It was argued that the detection limit of the IHC assay used with standard techniques may have been too low, leading to false negative results (12, 27). Since we used several rigorous and highly sensitive methodological approaches, the probability that CMV was prevalent in the tissue samples and yet could not be detected is very low. The US17 real-time PCR assay is highly-sensitive and can detect CMV DNA down to a level of a few copies per reaction. Recent advances in the ability to perform CISH on FFPE tissues using commercially available probes and staining kits have led to dramatic improvements in the ability to routinely interrogate the presence, localization and relative levels of RNA or DNA species in situ (25, 27, 30). Improvements in assay specificity results from the requirement for simultaneous hybridization of two separate but adjacent probes for signal detection, and assay sensitivity results from amplification of the signals based on both branching DNA hybridizations and enzymatic reactions. The Advanced Cell Diagnostics (ACD) commercial CISH assay can detect a single DNA or RNA molecule in individual cells (25, 27, 31). Moreover, almost half of our prospective population was CMV seronegative, and it would be highly unlikely that relatively high tissue levels of CMV would be present without evidence of prior infection by serum immunological measures such as the one used here. In all, the combination of several techniques applied in this study is appropriately powered to detect low level CMV and poor sensitivity is an unlikely to explanation for our results.
The blood-based analysis of patients in the prospectively studied cohort in this report highlights several key points: Although the number of patients was small, results from the CMV IgG avidity assay were consistent with seropositivity rates in the general population. In other words, the rate of seropositive patients was not increased, which might have supported the hypothesis of a linkage between CMV reactivation and gliomagenesis. Three patients had detectable but relatively low CMV DNA copies at baseline, prior to chemoradiation. Interestingly, however, in these patients CMV DNA was not detected during follow-up testing. Only one patient was found to have viremia during follow-up.
Our study had several limitations. These included the relatively low number of patients that were enrolled in the prospective cohort and in some cases the limited amount of available tissue so that not all tissues could undergo analysis by real-time PCR, IHC and CISH (Table 2).
Our findings do not support an association of CMV with HGG or a role of CMV infection in gliomagenesis. Nonetheless, there are reports suggesting potential activity of antiviral agents, including valganciclovir, in gliomas (32, 33). Based on our findings and findings by others, however, we suspect that this phenomenon is unrelated to CMV, and that if indeed these drugs have clinically relevant anti-tumor activity in these patients, then it is likely that those effects may have another, CMV-independent antineoplastic effect in gliomas.
In contrast to another herpes virus, HHV8, which since its initial report of detection was confirmed to be the cause of AIDS-associated Kaposi’s sarcoma (KS) (34), in the case of CMV, its inconsistent detection in GBM led to the hypothesis that when present it might modulate the behavior of this cancer. However, as we show in our study many patients with GBM are seronegative for CMV, and the virus cannot be detected in tumor tissue in either seropositive or seronegative individuals using three diagnostic modalities with 6 different tests. While our methods are highly sensitive for detection of CMV, it is still possible that there is CMV present in some patients (e.g. less than a few copies per many thousands of tumor cells), but that it is below the level of detectability. However, we submit that in this case there are no known molecular or cellular mechanisms by which such low levels could drive cancer. Further, we provide a plausible explanation regarding the reports of positive IHC staining in that a positive stain can be technically achieved by simply changing the antibody concentrations.
A number of recent studies used bioinformatics approaches to test for the presence of HCMV DNA in unmapped, non-human, next generation sequencing reads, and these studies reported either the complete absence of HCMV, or, the present of sporadic low level HCMV reads homologous to HCMV laboratory expression vector sequences (Supplemental Table 4).
In summary, our findings support prior studies indicating absence of CMV in human glioma tissues. Because of the importance of this topic, and the continued controversy (e.g. a consensus conference proceedings published in 2012 indicated that “there is sufficient evidence to conclude that HCMV sequences and viral gene expression exist in most, if not all, malignant gliomas”) (35), we suggest that an independent study, coordinated by a central neutral laboratory with expertise in human viral-disease related testing and research be performed. Identical proficiency samples could be distributed by the coordinating center to multiple investigators for CMV testing by their individual respective methods. To increase the credibility of the final study results, the study would also be designed to include centrally coded blinded control samples (both positive and PCR-negative) for both solution-based and in-situ based (e.g. in-situ hybridization and IHC) methods. A similar approach was used previously to help settle questions regarding another highly controversial association between a virus and human diseases (e.g. prostate cancer and chronic fatigue syndrome) (36, 37).
Statement of Translational Relevance
Over the past years, there has been significant controversy regarding a potential association between cytomegalovirus (CMV) and glioblastomas (GBM) and other high-grade gliomas (HGG). This is of great importance as clinical trials are being conducted that are based on the hypothesis that CMV is associated with and/or has a causative relationship with HGG/GBM. Despite comprehensive analysis of several different specimen collections, including fresh frozen and formalin-fixed, paraffin-embedded tissue, with 6 highly sensitive assays, we did not detect CMV in any of the samples studied. Our results call for an independent multicenter blinded analysis of samples collected from different geographical areas throughout the world with agreed upon study designs and determination of causality or lack thereof of CMV in HGG for future guidance on anti-viral and/or CMV-based vaccine therapies.
Supplementary Material
Acknowledgments
This work was supported by the Sidney Kimmel Comprehensive Cancer Center core grant, P30CA006973. We thank Ms. Wendy Jachman, the Robert H. Gross Memorial Fund, and the Retired Professional Fire Fighters Cancer Fund, Inc. (RPFFCF) for generous support of this project. We thank Silvia Petrik and Joy Fisher from the Johns Hopkins Brain Cancer Program for their regulatory and data management support.
Financial support: Sidney Kimmel Comprehensive Cancer Center core grant, P30CA006973; Ms. Wendy Jachman; the Robert H. Gross Memorial Fund, and the Retired Professional Fire Fighters Cancer Fund, Inc. (RPFFCF).
Footnotes
Conflict of Interest: The authors have no conflict of interest related to this manuscript.
Occasional weak signals were detected in a small minority of cells for both probes; these cases are considered negative (e.g. signals are considered non-specific background) since similar weak nuclear signals can be seen at times using bacterial probes in human cells.
References
- 1.Cobbs CS, Harkins L, Samanta M, Gillespie GY, Bharara S, King PH, et al. Human cytomegalovirus infection and expression in human malignant glioma. Cancer Res. 2002;62(12):3347–50. [PubMed] [Google Scholar]
- 2.Mitchell DA, Xie W, Schmittling R, Learn C, Friedman A, McLendon RE, et al. Sensitive detection of human cytomegalovirus in tumors and peripheral blood of patients diagnosed with glioblastoma. Neuro Oncol. 2008;10(1):10–8. doi: 10.1215/15228517-2007-035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Scheurer ME, Bondy ML, Aldape KD, Albrecht T, El-Zein R. Detection of human cytomegalovirus in different histological types of gliomas. Acta Neuropathol. 2008;116(1):79–86. doi: 10.1007/s00401-008-0359-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ranganathan P, Clark PA, Kuo JS, Salamat MS, Kalejta RF. Significant association of multiple human cytomegalovirus genomic loci with glioblastoma multiforme samples. J Virol. 2012;86(2):854–64. doi: 10.1128/JVI.06097-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rahbar A, Orrego A, Peredo I, Dzabic M, Wolmer-Solberg N, Straat K, et al. Human cytomegalovirus infection levels in glioblastoma multiforme are of prognostic value for survival. J Clin Virol. 2013;57(1):36–42. doi: 10.1016/j.jcv.2012.12.018. [DOI] [PubMed] [Google Scholar]
- 6.Libard S, Popova SN, Amini RM, Karja V, Pietilainen T, Hamalainen KM, et al. Human cytomegalovirus tegument protein pp65 is detected in all intra- and extra-axial brain tumours independent of the tumour type or grade. PLoS One. 2014;9(9):e108861. doi: 10.1371/journal.pone.0108861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shamran HA, Kadhim HS, Hussain AR, Kareem A, Taub DD, Price RL, et al. Detection of human cytomegalovirus in different histopathological types of glioma in iraqi patients. Biomed Res Int. 2015;2015:642652. doi: 10.1155/2015/642652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bhattacharjee B, Renzette N, Kowalik TF. Genetic analysis of cytomegalovirus in malignant gliomas. J Virol. 2012;86(12):6815. doi: 10.1128/JVI.00015-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stangherlin LM, Castro FLF, Medeiros RSS, Guerra JM, Kimura LM, Shirata NK, et al. Human Cytomegalovirus DNA Quantification and Gene Expression in Gliomas of Different Grades. Plos One. 2016;11(7):e0159604. doi: 10.1371/journal.pone.0159604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lau SK, Chen YY, Chen WG, Diamond DJ, Mamelak AN, Zaia JA, et al. Lack of association of cytomegalovirus with human brain tumors. Mod Pathol. 2005;18(6):838–43. doi: 10.1038/modpathol.3800352. [DOI] [PubMed] [Google Scholar]
- 11.Tang KW, Alaei-Mahabadi B, Samuelsson T, Lindh M, Larsson E. The landscape of viral expression and host gene fusion and adaptation in human cancer. Nat Commun. 2013;4:2513. doi: 10.1038/ncomms3513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Baumgarten P, Michaelis M, Rothweiler F, Starzetz T, Rabenau HF, Berger A, et al. Human cytomegalovirus infection in tumor cells of the nervous system is not detectable with standardized pathologico-virological diagnostics. Neuro Oncol. 2014;16(11):1469–77. doi: 10.1093/neuonc/nou167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yamashita Y, Ito Y, Isomura H, Takemura N, Okamoto A, Motomura K, et al. Lack of presence of the human cytomegalovirus in human glioblastoma. Mod Pathol. 2014;27(7):922–9. doi: 10.1038/modpathol.2013.219. [DOI] [PubMed] [Google Scholar]
- 14.Tang KW, Hellstrand K, Larsson E. Absence of cytomegalovirus in high-coverage DNA sequencing of human glioblastoma multiforme. Int J Cancer. 2015;136(4):977–81. doi: 10.1002/ijc.29042. [DOI] [PubMed] [Google Scholar]
- 15.Strong MJ, Blanchard E, 4th, Lin Z, Morris CA, Baddoo M, Taylor CM, et al. A comprehensive next generation sequencing-based virome assessment in brain tissue suggests no major virus – tumor association. Acta Neuropathol Commun. 2016;4(1):71. doi: 10.1186/s40478-016-0338-z. 016-0338-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lin CT, Leibovitch EC, Almira-Suarez MI, Jacobson S. Human herpesvirus multiplex ddPCR detection in brain tissue from low- and high-grade astrocytoma cases and controls. Infect Agent Cancer. 2016;11:32. doi: 10.1186/s13027-016-0081-x. 016-0081-x. eCollection 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nair SK, De Leon G, Boczkowski D, Schmittling R, Xie W, Staats J, et al. Recognition and killing of autologous, primary glioblastoma tumor cells by human cytomegalovirus pp65-specific cytotoxic T cells. Clin Cancer Res. 2014;20(10):2684–94. doi: 10.1158/1078-0432.CCR-13-3268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mitchell DA, Batich KA, Gunn MD, Huang MN, Sanchez-Perez L, Nair SK, et al. Tetanus toxoid and CCL3 improve dendritic cell vaccines in mice and glioblastoma patients. Nature. 2015;519(7543):366–9. doi: 10.1038/nature14320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lawler SE. Cytomegalovirus and glioblastoma; controversies and opportunities. J Neurooncol. 2015;123(3):465–71. doi: 10.1007/s11060-015-1734-0. [DOI] [PubMed] [Google Scholar]
- 20.Machida U, Kami M, Fukui T, Kazuyama Y, Kinoshita M, Tanaka Y, et al. Real-time automated PCR for early diagnosis and monitoring of cytomegalovirus infection after bone marrow transplantation. J Clin Microbiol. 2000;38(7):2536–42. doi: 10.1128/jcm.38.7.2536-2542.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Barger-Kamate B, Forman M, Sangare CO, Haidara AS, Maiga H, Vaidya D, et al. Effect of artemether-lumefantrine (coartem) on cytomegalovirus urine viral load during and following treatment for malaria in children. J Clin Virol. 2016;77:40–5. doi: 10.1016/j.jcv.2016.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Esopi D, Gambichler B, Fedor H, Southerland M, Hicks J, Zheng Q, et al. Biospecimen Research Network (BRN) Symposium. Washington DC: 2011. A cell line TMA approach for tissue biomarker development and validation. [Google Scholar]
- 23.Sfanos KS, Sauvageot J, Fedor HL, Dick JD, De Marzo AM, Isaacs WB. A molecular analysis of prokaryotic and viral DNA sequences in prostate tissue from patients with prostate cancer indicates the presence of multiple and diverse microorganisms. Prostate. 2008;68(3):306–20. doi: 10.1002/pros.20680. [DOI] [PubMed] [Google Scholar]
- 24.Meeker AK, Gage WR, Hicks JL, Simon I, Coffman JR, Platz EA, et al. Telomere length assessment in human archival tissues: Combined telomere fluorescence in situ hybridization and immunostaining. Am J Pathol. 2002;160(4):1259–68. doi: 10.1016/S0002-9440(10)62553-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang F, Flanagan J, Su N, Wang LC, Bui S, Nielson A, et al. RNAscope: A novel in situ RNA analysis platform for formalin-fixed, paraffin-embedded tissues. J Mol Diagn. 2012;14(1):22–9. doi: 10.1016/j.jmoldx.2011.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fedor HL, De Marzo AM. Practical methods for tissue microarray construction. Methods Mol Med. 2005;103:89–101. doi: 10.1385/1-59259-780-7:089. [DOI] [PubMed] [Google Scholar]
- 27.Deleage C, Wietgrefe SW, Del Prete G, Morcock DR, Hao XP, Piatak M, Jr, et al. Defining HIV and SIV reservoirs in lymphoid tissues. Pathog Immun. 2016;1(1):68–106. doi: 10.20411/pai.v1i1.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Battifora H. Assessment of antigen damage in immunohistochemistry the vimentin internal control. Am J Clin Pathol. 1991;96(5):669–71. doi: 10.1093/ajcp/96.5.669. [DOI] [PubMed] [Google Scholar]
- 29.Cobbs C. Response to “human cytomegalovirus infection in tumor cells of the nervous system is not detectable with standardized pathologico-virological diagnostics”. Neuro Oncol. 2014;16(11):1435–6. doi: 10.1093/neuonc/nou295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kerr DA, Arora KS, Mahadevan KK, Hornick JL, Krane JF, Rivera MN, et al. Performance of a branch chain RNA in situ hybridization assay for the detection of high-risk human papillomavirus in head and neck squamous cell carcinoma. Am J Surg Pathol. 2015;39(12):1643–52. doi: 10.1097/PAS.0000000000000516. [DOI] [PubMed] [Google Scholar]
- 31.Wang H, Wang MX, Su N, Wang LC, Wu X, Bui S, et al. RNAscope for in situ detection of transcriptionally active human papillomavirus in head and neck squamous cell carcinoma. J Vis Exp. 2014;(85) doi: 10.3791/51426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Stragliotto G, Rahbar A, Solberg NW, Lilja A, Taher C, Orrego A, et al. Effects of valganciclovir as an add-on therapy in patients with cytomegalovirus-positive glioblastoma: A randomized, double-blind, hypothesis-generating study. Int J Cancer. 2013;133(5):1204–13. doi: 10.1002/ijc.28111. [DOI] [PubMed] [Google Scholar]
- 33.Soderberg-Naucler C, Rahbar A, Stragliotto G. Survival in patients with glioblastoma receiving valganciclovir. N Engl J Med. 2013;369(10):985–6. doi: 10.1056/NEJMc1302145. [DOI] [PubMed] [Google Scholar]
- 34.Chang Y, Cesarman E, Pessin MS, Lee F, Culpepper J, Knowles DM, et al. Identification of herpesvirus-like DNA sequences in AIDS-associated kaposi’s sarcoma. Science. 1994;266(5192):1865–9. doi: 10.1126/science.7997879. [DOI] [PubMed] [Google Scholar]
- 35.Dziurzynski K, Chang SM, Heimberger AB, Kalejta RF, McGregor Dallas SR, Smit M, et al. Consensus on the role of human cytomegalovirus in glioblastoma. Neuro Oncol. 2012;14(3):246–55. doi: 10.1093/neuonc/nor227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Alter HJ, Mikovits JA, Switzer WM, Ruscetti FW, Lo SC, Klimas N, et al. A multicenter blinded analysis indicates no association between chronic fatigue syndrome/myalgic encephalomyelitis and either xenotropic murine leukemia virus-related virus or polytropic murine leukemia virus. MBio. 2012;3(5) doi: 10.1128/mBio.00266.12. Print 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Simmons G, Glynn SA, Komaroff AL, Mikovits JA, Tobler LH, Hackett J, Jr, et al. Failure to confirm XMRV/MLVs in the blood of patients with chronic fatigue syndrome: A multi-laboratory study. Science. 2011;334(6057):814–7. doi: 10.1126/science.1213841. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.