

Abstract
Purpose
Human papillomavirus (HPV) 16 plays an etiologic role in a growing subset of HNSCCs, where viral expression of the E6 and E7 oncoproteins are necessary for tumor growth and maintenance. Although patients with HPV(+) tumors have a more favorable prognosis, there are currently no HPV-selective therapies. Recent studies identified differential receptor tyrosine kinase (RTK) profiles in HPV(+) vs. HPV(−) tumors. One such RTK, HER3, is overexpressed and interacts with phosphoinositide 3-kinase (PI3K) in HPV(+) tumors. Therefore, we investigated the role of HPV oncoproteins in regulating HER3-mediated signaling, and determined whether HER3 could be a molecular target in HPV(+) HNSCCs.
Experimental Design
HER3 was investigated as a molecular target in HPV(+) HNSCC using established cell lines, patient-derived xenografts (PDX), and human tumor specimens. A mechanistic link between HPV and HER3 was examined by augmenting E6 and E7 expression levels in HNSCC cell lines. The dependency of HPV(+) and HPV(−) HNSCC models on HER3 was evaluated with anti-HER3 siRNAs and the clinical stage anti-HER3 monoclonal antibody KTN3379.
Results
HER3 was overexpressed in HPV(+) HNSCCs, where it was associated with worse overall survival in patients with pharyngeal cancer. Further investigation indicated that E6 and E7 regulated HER3 protein expression and downstream PI3K pathway signaling. Targeting HER3 with siRNAs or KTN3379 significantly inhibited the growth of HPV(+) cell lines and PDXs.
Conclusions
This study uncovers a direct relationship between HPV infection and HER3 in HNSCC, and provides rationale for the clinical evaluation of targeted HER3 therapy for the treatment of HPV(+) patients.
Keywords: Human papillomavirus (HPV), HER3, Head and Neck Squamous Cell Carcinoma (HNSCC)
INTRODUCTION
Human papillomavirus (HPV) is a double stranded DNA virus that infects squamous and cutaneous epithelia lining the anogenital and the upper aerodigestive tracts. HPV causes virtually all cervical cancers and has now been shown to play an etiologic role in a growing subset of head and neck squamous cell carcinomas (HNSCCs) (1). Approximately 75% of tumors that arise in the oropharynx are associated with HPV infection (2–4). It is projected that the total number of oropharynx cancers will be greater than the number of cervical cancers by 2020 in the USA alone, highlighting the epidemic state of this disease (5–7). The oncogenic potential of HPV is attributed to the expression of the viral oncoproteins E6 and E7, which promote the degradation of the tumor suppressors p53 and retinoblastoma protein (pRb), respectively. Viral oncogene expression influences tumor formation, growth, and maintenance, mediating the unique biology of HPV-associated cancers (4, 8, 9).
HPV(+) HNSCC patients are typically younger in age, male, non-smokers/drinkers, and present with poorly differentiated lymph node positive tumors (3, 10). Although HPV(+) patients respond more favorably to chemoradiation and have a better prognosis, approximately one quarter of HPV(+) HNSCCs are lethal (3, 11–14). Furthermore, there are currently no HPV-selective therapies (15). Thus, innovative approaches targeting viral oncogene-driven mechanisms may reduce morbidities associated with standard chemoradiation and improve outcomes for this patient population.
The HER family of receptor tyrosine kinases is comprised of four receptors, (EGFR/HER1, ErbB2/HER2, ErbB3/HER3, and ErbB4/HER4) and is one of the most well studied receptor families in all of oncology. The epidermal growth factor receptor (EGFR) was established as a molecular target in HNSCC over a decade ago (16), resulting in approval by the Food and Drug Administration (FDA) of the anti-EGFR monoclonal antibody, cetuximab, for the treatment of HNSCC (17–19). In an effort to deintensify standard-of-care chemoradiation, several clinical studies have evaluated the efficacy of cetuximab in HPV(+) HNSCC patients. The results to date have been inconclusive, although some studies indicate that HPV(+) patients experience less benefit from EGFR inhibitors compared with HPV(−) disease (20, 21). To better understand these findings, several studies have investigated HER family expression signatures in large cohorts of HPV(+) and HPV(−) tumors. The results indicate an inverse correlation between EGFR expression and HPV positivity (22–24), while the HER family receptors, HER2 and HER3, were more highly expressed in HPV(+) tumors (25–27). HER3 is a unique HER family member, as it has limited kinase activity and can directly bind and activate phosphoinositol-3-kinase (PI3K) (28, 29). We recently reported that HER3 was overexpressed and highly bound to PI3K in HPV(+) tumors using a proximity-based immunohistochemical (IHC) assay (25). Collectively, these findings suggest that HPV(+) and HPV(−) tumors have distinct HER family expression signatures, which may inform future targeted therapies for each patient population.
In the present study, we investigated a possible mechanistic link between HPV and HER3 to determine whether HER3 could be a promising molecular target in HPV(+) tumors. Our results support previous studies examining HER3 expression in HPV(+) patients, where HER3 was overexpressed in HPV(+) cell lines, PDX models, and pharyngeal tumor specimens. Moreover, high HER3 expression was a poor prognostic factor in a pharyngeal cancer patient cohort. Further analyses indicated that HPV oncoproteins, E6 and E7, regulated HER3 protein abundance and downstream PI3K/AKT signaling, uncovering a direct relationship between HPV infection and HER3. HPV(+) HNSCC preclinical models were dependent on HER3 for cellular proliferation and tumor growth, where targeting HER3 with an anti-HER3 monoclonal antibody reduced the growth of HPV(+) tumors. Collectively, these data indicate that HER3 is a potential molecular target in HPV(+) tumors, and provide rationale for the clinical evaluation HER3 targeted therapy in HPV(+) HNSCC patients.
MATERIALS AND METHODS
Cell Lines
The HPV(+) HNSCC cell lines, UM-SCC47 (SCC47), 93-VU-147T, UPCI-SCC90 (SCC90), UD-SCC2 (SCC2), and UM-SCC104 (SCC104) and the HPV(−) cell line UM-SCC22B were kindly provided from Dr. Randal Kimple (University of Wisconsin-Madison). The HPV(+) HNSCC cell line UPCI-SCC152 was purchased from American Type Culture Collection (ATCC). All HPV(+) cell lines were maintained in Dulbecco’s Modified Eagle’s Medium ((DMEM), Mediatech Inc.) with 10% FBS (Gemini Bio-Products) and 1% penicillin and streptomycin (Life Technologies) supplemented with 1% non-essential amino acids (Gibco). The HPV(−) cell lines, FaDu, SCC9, and Detroit562 were purchased from ATCC. PE/CA-PJ34 and PE/CA-PJ49 cells were purchased from Sigma-Aldrich. CAL33 was kindly provided from Dr. Gerard Milano (University of Nice, Nice, France). UM-SCC4 (SCC4), UM-SCC1 (SCC1), and TU138 were kindly provided from Thomas E. Carey (University of Michigan). TU146 was kindly provided from Jeffrey Meyers (MD Anderson Cancer Center). UT-SCC45 (SCC45) was kindly provided by Reidar Grenman (University of Turku, Finland). Normal oral keratinocytes-spontaneously immortalized (NOKSI) were kindly provided by Dr. Nevan Krogan (University of California, San Francisco). All cell lines were authenticated by the indicated sources or confirmed via short-tandem repeat testing (Genetica DNA Laboratories). All HPV(−) cell lines were maintained in DMEM with 10% FBS and 1% penicillin and streptomycin. NOKSI cells were maintained in Keratinocyte SFM and supplemented with defined keratinocyte-SFM growth supplement (Gibco).
Antibodies and compounds
All antibodies used are indicated below: Cell Signaling Technology: HER3-XP, p-HER3-Y1197, HER2, p-AKT-S473, AKT, p-rpS6-S235/236, rpS6, PI3K-p110a, p53, p-MAPK-T202/204, MAPK, p-SFK-Y419, and GAPDH. Millipore: EGFR. Abcam: β-Actin and β-Tubulin. Euromedex: E6 (2E-3F8). Invitrogen: E7. Santa Cruz Biotechnology Inc: E7, horseradish peroxidase (HRP)–conjugated goat–anti-rabbit IgG, and goat–anti-mouse IgG. A combination of both E7 antibodies (1:1) was used for immunoblotting. KTN3379 and the control IgG1 antibody KTN0062C, were kindly provided by Kolltan Pharmaceuticals under a Materials Transfer Agreement.
Plasmids, transfection, and siRNA technology
pLXSN16-E6E7 was a gift from Dr. Denise Galloway (Addgene plasmid # 52394)(30). pDONR223-ErbB3 was a kind gift from Drs. William Hahn & David Root (Addgene plasmid #23874)(31) and subsequently cloned into pLX302 using the gateway system; pLX302 was a gift from David Root (Addgene plasmid # 25896) (32). HER3 shRNA (shHER3) or non-targeting shRNA (shNT) was cloned into pSIREN-RetroQ vector (Clontech) using the BamHI and EcoRI restriction sites. shHER3: GCGATGCTGAGAACCAATA; shNT: AATTGTACTACACAAAAGTAC. Virus was generated and used to establish 93-VU-147T-HER3 stable cell lines, where individual clones were selected and grown in puromycin (0.5ug/mL). Stable cell lines expressing shHER3, shNT, or pLXSN16-E6E7 were established via transfection using FuGENE HD (Promega) or Lipofectamine LTX (Life Technology); single cell clones were chosen after selection with puromycin (0.5 ug/mL) or neomycin (500 ug/mL).
For siRNA-mediated silencing, cells were transiently transfected with two separate HER3 siRNAs (siHER3-1, used in Figure 4; ON-TARGETplus, SMARTpool #L-003127; GE Dharmacon, or siHER3-2, used in Supplemental Figure 2; Origene HER3 siRNA #6263) or non-targeting siRNA (siNT; ON-TARGETplus Non-targeting Pool, #D-001810; Dharmacon). siRNAs targeting E6 and E7 were purchased from Santa Cruz Biotechnology, sc-156008 and sc-270423. Lipofectamine RNAiMAX was used for all siRNA experiments according to the manufacturer’s instructions (Life Technology).
Figure 4. HPV(+) HNSCC cell lines are dependent on HER3 for cellular proliferation and tumor growth.

(A) HPV(−) and HPV(+) HNSCC cell lines were transfected with siHER3 (30 nM) or non-targeting siRNA (siNT) for 72 hours before performing proliferation assays. Proliferation is plotted as a percentage of growth relative to siNT transfected cells (n= 3–6 replicates in three independent experiments). Whole cell lysates were harvested at the same time point to confirm HER3 knockdown and investigate changes in phospho-AKT and phospho-rpS6 expression. β-actin was used as a loading control. Data points are represented as mean ± s.e.m. **, P <0.01. (B) SCC47 cells stably expressing an shRNA targeting HER3 (shHER3) or a non-targeting control (shNT) were inoculated into the left or right dorsal flank in 4 athymic nude mice (n=4 tumor pairs, 8 tumors total). Tumor volume was monitored for three weeks and plotted at the last time point. Data points are represented as mean ± s.e.m. **, P < 0.01. Whole cell lysates were harvested from each tumor and evaluated for the indicated proteins by immunoblot analysis. (C) pLX302-HER3 was stably overexpressed in the HPV(+) cell line, 93-VU-147T. Two HER3 stable clones (HER3-C1 and HER3-C2) and a vector control were transfected with siE6E7 (30 nM) or siNT for 72 hours before performing proliferation assays (n=3 replicates in three independent experiments). Whole cell lysates were harvested at the same time point to analyze HER3 expression and downstream PI3K pathway signaling. β-actin was used as a loading control. Data points are represented as mean ± s.e.m. **, P<0.01.
Cell proliferation assays
Exponentially growing cells were seeded in six-well or 96-well plates. Following 72-hour treatment with siRNAs or KTN3379, cells were washed with phosphate-buffered saline (PBS) and fixed/stained with 0.5% crystal violet diluted in methanol. Plates were air dried overnight and dye was eluted with 0.1 M sodium citrate (pH 6.0) diluted in 50% ethanol. Eluant from 6-well dishes was transferred to 96-well plates, and the absorbance was measured at 595 nm (A595) to determine cellular proliferation. The percentage of proliferating cells was calculated by comparison of the A595 reading from drug-treated or siRNA transfected cells versus control cells. The anti-proliferative effect of KTN3379 was also demonstrated using MTT assay.
Immunoblot analysis
Whole cell lysates were obtained using lysis buffer containing 1M Tris-HCl, pH 7.6, 0.5M EDTA, 5M NaCl, 100mM Na4P2O7, 0.5M NaF, 50mM Na3VO4, and 1% Triton X-100, supplemented with protease and phosphatase inhibitors as previously described (25, 33). Samples were sonicated and then centrifuged at 14,000 RPM for 10 minutes at 4°C. Protein concentrations were determined by Bradford assay (Bio-Rad Laboratories). Equal amounts of protein were fractionated by SDS-PAGE, transferred to a PVDF membrane (Bio-Rad Laboratories), and incubated with the indicated primary antibodies. Proteins were detected via incubation with HRP-conjugated secondary antibodies and ECL Western Blotting Substrate (Santa Cruz Biotechnology) or SuperSignal West Femto Maximum Sensitivity Chemiluminescent Substrate (Thermo Fisher Scientific).
Immunoprecipitation (IP)
Whole cell lysate were obtained using NP40 lysis buffer (50 mM HEPES, pH 7.4, 150 mM NaCl, 1% NP-40, 0.5% deoxycholic acid, 10% glycerol, 2.5 mM EGTA, 1 mM EDTA, 1 mM DTT supplemented with protease and phosphatase inhibitors). Protein A/G magnetic beads were used for IP following the manufacturer’s instructions (Thermo Fisher Scientific). The anti-HER3 antibody used for IP was KTN3379.
cDNA synthesis and quantitative PCR
Total RNA was isolated using RNeasy kit purchased from Qiagen. 0.5 μg of total RNA was used to make cDNA using the iScript Reverse Transcription Supermix Kit (Bio-Rad Laboratories). qRT-PCR was conducted using the CFX96 Touch Real-Time PCR Detection System (Bio-Rad). All reactions were performed in triplicate and repeated three times. To determine the normalized value, 2ΔΔCt values were compared between E6, E7, HER3, and β-Actin, 18S, or TBP, where the change in crossing threshold (ΔCt) =Ct Target –Ct Control and ΔΔCt = ΔCt (NT or Vector) –ΔCt (siE6E7 or E6E7). The sequences of primer sets used for this analysis are as follows: E6-F: 5′-CTGCAATGTTTCAGGACCCA-3′, E6-R: 5′-TCATGTATAGTTGTTTGCAGCTCTGT-3′; E7-F: 5′-ACCGGACAGAGCCCATTACA-3′, E7-R: 5′-GCCCATTAACAGGTCTTCCAAA-3′, HER3-F: 5′-CAGAGTGATGTCTGGAGCTATG-3′, HER3-R: TCTCTAGCAGGTCTGGTACTT, B-β-Actin-F: 5′-CAGCCATGTACGTTGCTATCCAGG-3′, β-Actin-R: 5′-AGGTCCAGA CGCAGGATGGCATG-3′, 18S-F: AACCCGTTGAACCCCATT, 18S-R: CCATCCAATCGGTAGTAGCG, TBP-F: CCCATGACTCCCATGACC, TBP-R: TTTACAACCAAGATTCACTGTGG. The NRG1 (Hs00247620_m1) primer set was a TaqMan Gene Expression Assay (Thermo Fisher).
Annexin-V Staining
Cells were plated in six-well dishes and transfected with non-targeting siRNA (siNT) or siE6/E7. 72 hours after transfection the cells were harvested with 0.5% trypsin, washed with phosphate-buffered saline (PBS), and re-suspended in 100 μL of binding buffer (BD Biosciences). Cells were stained with 2 μL of FITC-conjugated annexin-V antibody and 2 μL of propidium iodide (FITC Annexin-V apoptosis detection kit, BD Biosciences). The cells were analyzed by flow cytometry (Calibur DxP8). FlowJo Software was used to analyze the data.
Cell line xenografts
Female Hsd:athymic Nude-Foxn1nu mice were injected bilaterally with HPV(+) cell lines (1×106 cells) and tumors were allowed grow to 100 mm3. Three mice were randomized to KTN3379 treatment (10 mg/kg) and three mice to vehicle control (0.9% normal saline) for SCC47 xenografts (n=6 mice, 12 tumors total). Two mice were randomized to KTN3379 treatment and two mice to vehicle control for SCC90 xenografts (n=4 mice, 8 tumors total). Mice were treated by intraperitoneal (IP) injection twice weekly. Tumors were treated for a total of 3 weeks or until visual sign of ulceration occurred, at which point tumors were harvested 3 hours after the last treatment. Tumor volume measurements were determined by digital calipers twice weekly and calculated using the formula (π)/6 × (large diameter) × (small diameter)2.
Patient derived xenografts (PDXs)
HNSCC PDXs were established from patients with newly diagnosed or recurrent HNSCC upon written consent in accordance with IRB approval as described previously (34). NOD/SCID gamma (NSG) female mice were established with HPV(+) PDXs bilaterally. Three mice were randomized to KTN3379 treatment (10 mg/kg) and three mice to vehicle control (0.9% normal saline) (n=6 mice, 12 tumors total).
Pharyngeal cancer patient cohort and tissue microarray
Pharyngeal cancer patients completed written consent in accordance with IRB approval from the University of Pittsburgh (protocol number 991206). Three separate cores from each tumor were spotted on the tissue microarray (2mm size) as previously described (35). HPV in situ hybridization and p16 staining were performed as previously described (3, 25).
Immunohistochemistry and scoring
The pharyngeal cancer patient TMA was stained using the Universal quick kit (Vector Laboratories, Inc., PK-8800) with the anti-HER3-XP antibody (Cell Signaling, 1:50) or no primary antibody as a control. Antibody binding was revealed by addition of 3,3′-diaminobenzidine substrate and counterstained with Mayer’s hematoxylin (Vector Laboratories). Tissues were examined using a Nikon Eclipse microscope by an expert pathologist (R.J). The H-score was calculated by assessing the staining intensity of HER3 for each core (0 to 3+) and multiplying it by the percentage of cells expressing the highest level of HER3 in the tumor section. This categorical scale was used to define low HER3 expressing tumors, scoring below 200, and high-HER3 expressing tumors, scoring 200–300.
Statistical analysis
Student t-tests were used to evaluate differences in cell proliferation and differences in E6, E7, HER3, and NRG1 mRNA expression. Differences were considered statistically significant if *, P < 0.05. To evaluate the sensitivity of cell line xenografts or PDXs to KTN3379, the mean fractional tumor volumes were compared between the vehicle treated and KTN3379 treatment arms at the last time point of the study using two-sample t-tests with equal standard deviations. For clinical parameters, 2×2 contingency tables were analyzed by Fisher’s exact test. Overall survival was determined from date of diagnosis to date of death, with censoring at date of last follow-up for surviving patients. The log-rank test was used for univariate survival analysis. These statistical analyses were carried out using Graphpad Prism v6.0d. Multivariate survival analysis was calculated by Cox proportional hazards regression using R v3.2.2. Proportional hazards assumptions were verified using Schoenfeld residuals. Differences were considered statistically significant if *, P < 0.05.
RESULTS
HER3 is overexpressed in HPV(+) HNSCC preclinical models
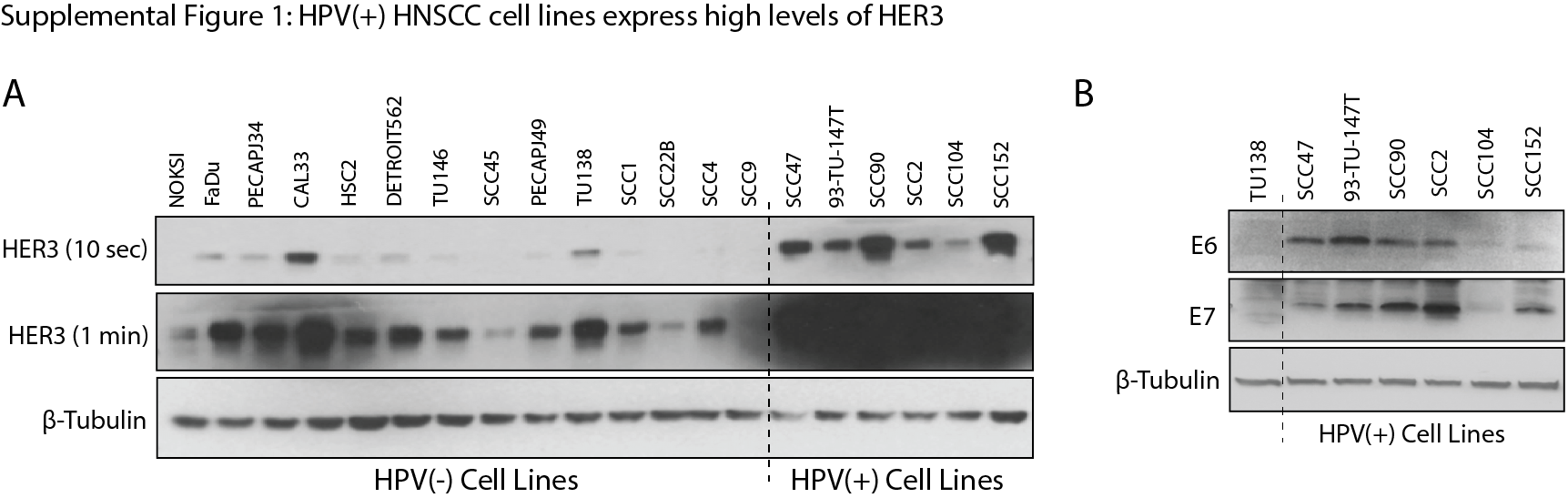
We recently reported that HPV(+) HNSCCs express significantly elevated mRNA and protein levels of the RTK HER3 (25). Further, HER3:PI3K complexes were significantly elevated in HPV(+) tumors (25). To determine if this HER3 expression signature was detected in HNSCC preclinical models, HER3 protein expression was evaluated in a panel of 13 HPV(−) and 6 HPV(+) HNSCC cell lines. All HPV(+) cell lines examined were PIK3CA wild-type. HER3 was found to be overexpressed in all HPV(+) cell lines (UM-SCC47, 93-VU-147T, UPCI-SCC90, UD-SCC2, UM-SCC104, and UPCI-SCC152) compared to the HPV(−) cell lines and one normal oral keratinocyte cell line (NOKSI) (Figure 1A). This observation was consistent in a longer immunoblot exposure, which depicts varied levels of HER3 expression in HPV(−) cell lines (Supplemental Figure 1A). Furthermore, HPV positivity was confirmed in the cell lines by immunoblot analysis for E6 and E7 (Supplemental Figure 1B). To evaluate whether HER3 signals through the PI3K pathway in HPV(+) cell lines, HER3 and PI3K complexes were examined by co-immunoprecipitation (IP) with an anti-HER3 monoclonal antibody in HPV(−) and HPV(+) cell lines (Figure 1B). Similar to our previous findings in HPV(+) tumors (25), HER3:PI3K complexes were elevated in HPV(+) cell lines as compared to HPV(−) cell lines. Furthermore, HER3 expression was evaluated in a panel of 8 HPV(−) and 7 HPV(+) patient derived xenografts (PDXs), and found to be expressed at elevated levels in all HPV(+) PDXs relative to HPV(−) PDXs (Figure 1C). HPV positivity was confirmed in all PDXs by immunoblot analysis for E6 and E7. Collectively, these data indicate that HER3 is overexpressed and highly associated with PI3K in HPV(+) HNSCC preclinical models.
Figure 1. HER3 is overexpressed in HPV(+) HNSCC preclinical models.

(A) Whole cell lysates were harvested from 12 HPV(−) and 6 HPV(+) HNSCC cell lines, and one normal oral epithelial cell line (NOKSI) and evaluated for HER3 expression by immunoblot analysis. β-Tubulin was used as a loading control. (B) 500 μg of whole cell lysates harvested from cell lines in (A) were subjected to immunoprecipitation with anti-HER3 antibody followed by immunoblotting for total HER3 protein and the p110α subunit of PI3K. IgG heavy chain staining was used as a loading control. (C) Whole cell lysates were harvested from 8 HPV(−) PDXs and 7 HPV(+) PDXs and evaluated for HER3, E6, and E7 protein expression by immunoblot analysis. β-Tubulin was used as a loading control. ImageJ software was used to quantitate p110α:HER3 association, and HER3 expression levels in HPV(−) and HPV(+) models. Experiments were performed in triplicate with similar results.
HER3 is a poor prognostic factor in pharyngeal cancer patients
Since HER3 was overexpressed in HPV(+) HNSCC cell lines, PDX models, and human tumors, we next evaluated the prognostic significance of HER3 in a cohort of pharyngeal cancer patients. A tissue microarray (TMA) consisting of 38 pharyngeal tumors was immunohistochemically stained for HER3 using a HER3 monoclonal antibody. This cohort was representative of previously annotated pharyngeal cancer patient cohorts, which are primarily HPV(+), male, and representing a younger demographic (see Supplemental Table 1 for clinical information of this patient cohort). A blinded pathologist evaluated the TMA for HER3 expression, and each tumor was given a H-score on a categorical scale of 0 to 300. Representative tumors expressing low and high HER3 are shown in Figure 2A. While HPV in-situ hybridization (ISH) and p16 expression did not correlate with high HER3 in this cohort of patients, previous work from our laboratory reported a significant correlation between HER3 and p16 using the quantitative VeraTag staining method in a larger cohort of HNSCC patients (25). Overall survival (OS) of this cohort was determined by evaluating the vital status of patients in the low and high HER3 groups. There was a significant association between OS and HER3 score (P=0.006), where median OS was shorter in patients with high HER3 (2.9 years) as compared to patients expressing low HER3 (6.5 years) (Figure 2B and Table 1). The hazard ratio (HR) for OS among patients with high HER3 versus low HER3 score was 2.20 (95% CI, 0.72–6.73), indicating a higher probability of death in patients with high HER3 expression. To further analyze this cohort, overall survival was evaluated using a multivariate Cox proportional hazard model that adjusts for HPV status and high versus low HER3. When controlling for HPV status, high HER3 increased the risk of death by a factor of 1.47 (P = 0.0324). Taken together, these data suggest that HER3 is overexpressed in more aggressive pharyngeal cancers, and is predictive of worse overall survival irrespective of HPV status.
Figure 2. High HER3 expression is associated with worse overall survival in pharyngeal cancer patients.

(A) HER3 is differentially expressed in a cohort of 38 pharyngeal cancer patients. Pathologic IHC quantitation was determined via calculation of a Histo-Score. Representative images of low and high HER3 expressing tumors are shown (10×). (B) High HER3 expression was significantly associated with shorter overall survival (OS) in pharyngeal cancer patients as analyzed by the log-rank test. **, P < 0.01.
Table 1.
Median overall survival (95% confidence interval) for pharyngeal cancer patients with high and low HER3 scores
| HER3 Score | Deaths/Total | Median OS (Years) | H.R. (95% CI) | Log Rank p-value |
|---|---|---|---|---|
| Low | 8/29 | 6.5 | REF | ——— |
| High | 5/9 | 2.9 | 2.20 (0.72, 6.73) | 0.0060 |
Statistically signficant, P<0.01
HPV oncoproteins, E6 and E7, regulate HER3 protein expression and downstream PI3K/AKT signaling
Previous in vitro studies using human primary keratinocytes and human mammary epithelial cells suggest that HPV oncoproteins regulate the expression of several different RTKs, including EGFR, HER2, insulin receptor (IR), and insulin-like growth factor receptor (IGF1R) (36–39). To investigate a functional link between HPV and HER3 in HNSCC, the bicistronic E6/E7 mRNA was targeted in several HPV(+) cell lines using pooled siRNAs targeting both coding sequences. The knockdown of E6/E7 in three HPV(+) cell lines (SCC47, 93-VU-147T, and SCC90) robustly diminished the phosphorylation and total protein expression of HER3 (Figure 3A). Furthermore, the phosphorylation of the HER3 downstream effector AKT and ribosomal protein S6 (rpS6) were decreased following E6/E7 knockdown in all HPV(+) cell lines tested. The knockdown of E6 and E7 was validated by examining mRNA, and by immunoblotting for p53 and E7, respectively (Figure 3A). HER3 mRNA levels were unchanged in cells depleted of E6 and E7, suggesting that these oncoproteins may regulate HER3 via a posttranscriptional mechanism. To verify that the loss of HER3/AKT signaling was not due to a massive apoptotic response induced by ablating E6/E7 expression, annexin-V analysis was conducted in HPV(+) cell lines 72 hours after E6/E7 knockdown (Supplemental Figure 2A). These results indicated that there was only a minor increase (5–11%) in apoptosis in cells depleted of E6/E7 as compared to cells transfected with non-targeting control. Moreover, E6/E7 knockdown had no effect on the phosphorylation of MAPK or Src Family Kinases, demonstrating specificity for the loss in HER3/AKT signaling we observed (Supplemental Figure 2B).
Figure 3. HPV regulates HER3 protein expression and downstream PI3K/AKT signaling.

(A) Whole cell lysates and mRNA were harvested from three HPV(+) HNSCC cell lines 72 hours after transfection with E6/E7 siRNA or a non-targeting (siNT) control. (B) Whole cell lysates and mRNA were harvested from SCC1 and SCC22B clones (E6/E7-C1 and E6/E7-C2) stably overexpressing pLXSN-E6E7 or pLXSN-vector control. β-Tubulin was used as a loading control for immunoblotting in (A) and (B). E6, E7, and HER3 mRNA expression was detected by quantitative RT-PCR and normalized to the expression of each target in siNT-transfected cells or vector controls (n=3 in three independent experiments). The expression of each target gene is shown for only one clone per cell line. 18S expression was used as an endogenous control. Data points are represented as mean ± s.e.m. **, P <0.01.
To confirm a role for E6/E7 in the regulation of HER3, E6 and E7 were stably overexpressed in two HPV(−) cell lines, SCC1 and SCC22B. As compared to vector controls, two independent clones overexpressing E6 and E7 from each cell line had dramatically increased total and phosphorylated HER3 protein levels (Figure 3B). Furthermore, phospho-AKT and phospho-rpS6 expression levels were markedly increased in E6/E7 stable clones, indicating a direct role for E6 and E7 in mediating the activation of the PI3K pathway. While increases in HER3 protein levels were detected in each E6/E7 overexpressing clone, HER3 mRNA levels remained unchanged as compared to vector controls (expression is depicted for E6/E7-C1 for each cell line). These findings further support a role for E6 and E7 in the posttranscriptional regulation of HER3.
HPV(+) HNSCC cell lines are dependent on HER3 for cellular proliferation and tumor growth
Since E6 and E7 regulated HER3 protein expression and PI3K/AKT signaling, we hypothesized that HPV(+) cell lines would be dependent on HER3 for cellular proliferation. To test this hypothesis, proliferation assays were performed 72 hours after transfecting five HPV(−) and four HPV(+) HNSCC cell lines with a pooled siRNA targeting HER3 or a non-targeting siRNA control. Genetic ablation of HER3 resulted in statistically significant inhibition of cellular proliferation (21–55%) in all HPV(+) cell lines tested, whereas the HPV(−) cell lines were relatively unaffected (Figure 4A). Analysis of HPV(−) and HPV(+) cell lines after HER3 knockdown demonstrated that AKT and rpS6 phosphorylation were reduced only in HPV(+) cell lines, indicating that HER3 signals to the PI3K/AKT signaling pathway more prominently in HPV(+) cells. These results were further validated with a second independent siRNA targeting HER3 (Supplemental Figure 3). To evaluate the role of HER3 on tumor growth, HER3 was stably knocked down with shRNA (shHER3) in the HPV(+) cell line, SCC47. An equal number of cells expressing shHER3 were inoculated into the right dorsal flank of athymic nude mice, while cells expressing a non-targeting control (shNT) were inoculated into the left dorsal flank (n=4 tumor pairs, 8 tumors total). After three weeks, shHER3 tumors were significantly smaller compared to shNT tumors (Figure 4B). Tumors were harvested and analyzed for HER3 expression and downstream PI3K signaling by immunoblot analysis. As compared to shNT control tumors, all shHER3 tumors expressed reduced levels of phospho-AKT and phoshpo-rpS6 (Figure 4B). Collectively, these observations indicate that HPV(+) preclinical models are dependent on HER3 for cellular proliferation, tumor growth, and activation of the PI3K/AKT signaling pathway.
To further evaluate the role of HER3 in HPV-mediated cellular proliferation, HER3 was ectopically overexpressed in the HPV(+) cell line, 93-VU-147T. Two clones overexpressing HER3 (clone 1 and clone 2) or a vector control were subsequently transfected with siRNA targeting E6 and E7 (siE6E7) or a non-targeting control (siNT) (Figure 4C). Consistent with previous reports (40), knockdown of E6 and E7 inhibited the proliferation of 93-VU-147T-Vector (93-VU-147T-V) cells by approximately 60%, while cells ectopically overexpressing HER3 displayed a more modest reduction in proliferation (19–25%) compared to siNT cells. Immunoblot analysis of each cell line indicated that more HER3 was retained in the clones as compared to vector control cells post E6 and E7 knockdown. Further, while E6 and E7 knockdown reduced PI3K/AKT signaling in all cell lines, there was more AKT and rpS6 phosphorylation retained in the clones overexpressing HER3. These findings suggest that HER3 contributes to the oncogenic effects of E6 and E7 in HPV(+) cells by activating the PI3K/AKT signaling pathway.
HER3 is a promising molecular target in HPV(+) HNSCC preclinical models
Monoclonal antibodies that inhibit HER3 activation are now in clinical development, however, predictive biomarkers to identify patients who may respond to these agents are lacking. Since HER3 is overexpressed in HPV(+) cell lines, PDXs, and human tumors, we hypothesized that HPV(+) preclinical models would be highly sensitive to anti-HER3 therapy. KTN3379 is a clinically advanced monoclonal antibody that effectively inhibits both ligand dependent and independent activation of HER3 (41, 42). To investigate the efficacy of KTN3379 in vitro, five HPV(−) and four HPV(+) cell lines were treated with 100 nM of KTN3379 for 72 hours prior to performing proliferation assays (Figure 5A). While all four HPV(+) cell lines were significantly growth inhibited upon treatment with KTN3379, only a subset (2 of 5) of the HPV(−) cell lines tested were responsive. Consistent with previous studies (42), KTN3379 prevented the phosphorylation of HER3 in all cell lines tested over a time course of 72 hours. However, evaluation of downstream PI3K/AKT signaling indicated that phospho-AKT and phospho-rpS6 were more substantially inhibited in three HPV(+) cell lines (SCC47, 93-VU-147T, and SCC90) as compared to three HPV(−) cell lines (TU138, PECAPJ49, and SCC1) (Figure 5A). Furthermore, siRNA-mediated knockdown of EGFR and HER2 only moderately prevented the phosphorylation of HER3 in HPV(+) cell lines (Supplemental Figure 4), supporting the necessity of targeting HER3 directly with agents like KTN3379.
Figure 5. HPV(+) HNSCC models are sensitive to therapeutic blockade of HER3 with the anti-HER3 monoclonal antibody, KTN3379.

(A) HPV(−) and HPV(+) HNSCC cell lines were treated with 100 nM KTN3379 for 72 hours before performing proliferation assays. Proliferation is plotted as a percentage of growth relative to vehicle treated cells (n=6 in three-five independent experiments). Data points are represented as mean ± s.e.m. *, P <0.05, **, P <0.01. Whole cell lysates were harvested from three HPV(−) cell lines (PECAPJ49, TU138, and SCC1), and three HPV(+) cell lines (SCC47, 93-VU-147T, and SCC90) over a time course of 72 hours of KTN3379 treatment (100 nM) followed by immunoblotting for the indicated proteins. β-actin was used as a loading control. (B) HPV(+) cell lines (SCC47 and SCC90) were grown as xenografts in athymic nude mice (n=6 tumors per treatment group for SCC47, and n=4 tumors per treatment group for SCC90). The HPV(+) PDX-10 was implanted into the dorsal flanks of NOD/SCIDγ mice (n=6 tumors per treatment group). All xenografts were treated with KTN3379 (10 mg/kg) or vehicle twice weekly for the indicated time period by IP injection. Tumor volumes for all xenograft experiments were calculated as fractions of the average starting volume for each group, and the mean tumor volume ± s.e.m is shown. **, P<0.01.
To further evaluate the efficacy of KTN3379, the HPV(+) cell lines SCC47 and SCC90, and the HPV(+) PDX-10 were established as xenografts in athymic nude or NSG mice (Figure 5B). Once tumors reached approximately 100 mm3, mice were randomized and treated with either KTN3379 (10 mg/kg twice weekly by intraperitoneal injection) or vehicle control. All HPV(+) xenografts treated with KTN3379 demonstrated statistically significant reductions in tumor volumes over several weeks of treatment (Figure 5B). Evaluation of tumor whole cell lysates indicated that KTN3379 treated tumors expressed reduced levels of both phospho- and total HER3, suggesting that KTN3379 may induce the degradation of HER3 in vivo (two representative tumors per treatment group are shown). Consistent with in vitro data, the HPV(+) tumors treated with KTN3379 expressed reduced levels of phospho-AKT and phospho-rpS6. Taken together, these observations indicate that HPV(+) HNSCC preclinical models are highly sensitive to anti-HER3 therapy, and suggest that HER3 may be a promising therapeutic target in HPV-associated HNSCCs.
DISCUSSION
HER3 is emerging as a molecular target in several different cancers, however, biomarkers that predict patient response to HER3 targeted therapy are lacking (43). Several reports examining large cohorts of HNSCC patients have found HER3 expression to be associated with decreased survival, however, these early studies did not include HPV status in their evaluations (44). More recently, one group reported that HER3 and neuregulin 1 (NRG1), the cognate ligand for HER3, are independently associated with decreased overall survival in oropharyngeal cancer patients (45). In the current study, HER3 was overexpressed in HPV(+) HNSCC cell lines, PDX models, and pharyngeal tumors, where high levels of HER3 protein expression was associated with reduced survival of pharyngeal cancer patients. However, examination of a larger cohort of HPV(+) patients with a matched control group is needed to better determine the prognostic value of HER3 in HPV(+) HNSCC.
We found that HPV oncoproteins, E6 and E7, regulated HER3 protein expression in HNSCC cell lines. These data are consistent with previous studies suggesting a role for HPV oncoproteins in the regulation of RTKs. In a study by Spangle et al., overexpression of HPV16-E6 in primary human foreskin keratinocytes resulted in increased EGFR and insulin receptor internalization and prolonged activation upon ligand withdrawal (46). A study in mammary epithelial cells indicated that E6 and E7 cooperate with HER2 by increasing its expression and promoting HER2 dependent growth (36). While two groups have reported that E6 and E7 transcriptionally activate EGFR expression (38, 39), others have observed contrary findings (46). Our data support the posttranscriptional regulation of HER3 by HPV. These data suggest that HER3 mRNA or protein stability are regulated by HPV oncoproteins in HPV(+) HNSCC. A potential mechanism may involve E6/E7 acting to prevent proteasomal degradation of HER3. In preliminary studies we have found that proteasome inhibition abrogates the loss of HER3 abundance upon knockdown of E6/E7 in HPV(+) cells (unpublished data). E6 or E7 may sequester the E3 ubiquitin ligases Nrdp1 or NEDD4, which have been shown to promote ubiquitination and negative regulation of HER3 (47, 48). Furthermore, we suspect that increased HER3 levels leads to increased HER3 dimerization with other HER family members or RTKs resulting in HER3 phosphorylation and downstream signaling.
Interestingly, a recent study in a cervical cancer cell line found a posttranscriptional role for E2 in the regulation of HER3 via sequestration of Nrdp1 (49). This study found that viral episomal forms were necessary for HER3 expression, as viral genome integration has been reported to disrupt the E2 open reading frame (50, 51). The HPV(+) HNSCC cell lines used in our study have integrated viral genomes, where portions of the E2 gene have been reported to be lost in SCC47 cells (52, 53). Thus, whether E2 is fully expressed and functional in HNSCC is unclear. Finally, some reports have suggested a role for the HPV oncoprotein E5 in the regulation of EGFR-containing endosomes. However, the relevance of this observation in HNSCC and the impact on HER receptor expression and/or signaling has not been fully addressed (54, 55).
Previous studies have indicated that phospho-HER3 levels can be predictive of response to anti-HER3 therapy in preclinical models (42, 56). IHC for phosphorylated HER3 is often unreliable in clinical samples, and thus may not be an accurate way to predict response (43). In the current study, HER3 was overexpressed in HPV(+) models, which correlated with response to anti-HER3 therapy in vivo. Unlike other studies in breast and lung cancer models (42, 56), NRG1 mRNA and protein expression did not correlate with response to anti-HER3 therapy in the current HNSCC cell lines tested (Supplemental Figure 5). This may reflect the fact that HPV E6/E7 maintain high levels of HER3 protein expression, and thus HER3 can readily dimerize with other RTKs without an over abundance of ligand present. Collectively, our data suggest that HPV infection may be a biomarker for HER3 activity and response to HER3 targeted therapy in HNSCC, however, examination of tumor lysates from larger clinical cohorts and HPV-selective clinical trials will be necessary to support this hypothesis.
In prior studies we have shown that HER2, HER3, HER2:HER3, and HER3:PI3K protein complexes are elevated in HPV(+) versus HPV(−) HNSCC tumors (25). Furthermore, HPV(+) cell lines were found to be moderately sensitive to the dual EGFR/HER2 tyrosine kinase inhibitor, afatinib (25). Recently, data from the afatinib versus methotrexate LUX-Head and Neck 1 trial indicated that HPV(+) patients exhibited minimal response to afatinib (57), highlighting a discrepancy between preclinical response and patient response to this agent. This discrepancy may be due to several factors, including increased potency of afatinib under in vitro conditions and the lack of patient selection based on the activation status of HER family receptors. While the LUX-Head and Neck 1 trial found that HPV(+) patients exhibited minimal responses to afatinib, the ability of afatinib to effectively inhibit HER3 was not examined. The rational design of future clinical trials will benefit from further preclinical modeling comparing the efficacies of different HER family targeting agents in HPV(+) models.
There are currently several anti-HER3 targeted therapies in clinical trials (clinicaltrials.gov), all of which are monoclonal antibodies. Here, KTN3379 reduced HER3 phosphorylation and total HER3 protein levels in all xenograft models examined. However, KTN3379 only reduced total levels of HER3 in SCC90 cells in vitro. The differential effects of KTN3379 on total HER3 protein levels in vitro is difficult to explain, but it suggests a greater potency of KTN3379 in vivo, which has also been observed in other studies (58). While KTN3379 effectively blocked the phosphorylation of HER3 in both HPV(+) and HPV(−) models in the current study, HPV(+) models appeared to be more responsive to this agent. However, it is noted that KTN3379 has demonstrated antitumor responses in other HPV(−) HNSCC models (58, 59), and thus additional biomarkers will be needed to select for responsive HPV(+) and HPV(−) tumors. Taken together, our findings suggest that HER3 is a promising molecular target in HPV(+) tumors, and provide rationale for the clinical evaluation of anti-HER3 therapies for the treatment of HPV(+) patients.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
STATEMENT OF TRANSLATIONAL RELEVANCE.
Human papillomavirus (HPV) 16 plays an etiologic role in a growing number of head and neck squamous cell carcinomas (HNSCCs) that arise in the oropharynx, where viral expression of the E6 and E7 oncoproteins are necessary for tumor growth and maintenance. While the molecular pathogenesis of HPV-driven HNSCC is distinct from HPV(−) tumors, there are no HPV-selective therapies and standard treatment approaches result in debilitating lifelong toxicities. Thus, understanding viral oncogene driven mechanisms may uncover new molecular targets in HPV(+) tumors. In the current study, the receptor tyrosine kinase HER3 was found to be overexpressed in HPV(+) HNSCC models, and significantly associated with worse overall survival in pharyngeal cancer patients. HPV oncoproteins, E6 and E7, mediated HER3 protein expression and downstream PI3K signaling, uncovering a direct relationship between HPV infection and HER3. Furthermore, HPV(+) preclinical HNSCC models were sensitive to HER3 targeting with siRNAs and the clinical stage anti-HER3 monoclonal antibody, KTN3379. Collectively, these studies provide a rationale for the clinical evaluation of anti-HER3 therapeutics for the treatment of HPV(+) HNSCCs.
Acknowledgments
Financial Support: The project described was supported by the grant CRP-13-308-06-COUN from the American Cancer Society (J.R. Grandis), career development award CDA-2-057-10S, from the Department of Veterans Affairs (U. Duvvuri), grant Z-2/59 from the Interdisciplinary Center for Clinical Research of the University Würzburg (S. Hartmann), postdoctoral fellowship PF-16-087-01-TBG from the American Cancer Society (T.M. Brand), and by NIH grant T32 CA108462 for Molecular and Cellular Mechanisms of Disease (T.M. Brand).
Footnotes
Conflicts of Interest: None
References
- 1.Jemal A, Simard EP, Dorell C, Noone AM, Markowitz LE, Kohler B, Eheman C, Saraiya M, Bandi P, Saslow D, Cronin KA, Watson M, Schiffman M, Henley SJ, Schymura MJ, Anderson RN, Yankey D, Edwards BK. Annual Report to the Nation on the Status of Cancer, 1975–2009, featuring the burden and trends in human papillomavirus(HPV)-associated cancers and HPV vaccination coverage levels. J Natl Cancer Inst. 2013;105(3):175–201. doi: 10.1093/jnci/djs491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kang H, Kiess A, Chung CH. Emerging biomarkers in head and neck cancer in the era of genomics. Nat Rev Clin Oncol. 2015;12(1):11–26. doi: 10.1038/nrclinonc.2014.192. [DOI] [PubMed] [Google Scholar]
- 3.Ang KK, Harris J, Wheeler R, Weber R, Rosenthal DI, Nguyen-Tan PF, Westra WH, Chung CH, Jordan RC, Lu C, Kim H, Axelrod R, Silverman CC, Redmond KP, Gillison ML. Human papillomavirus and survival of patients with oropharyngeal cancer. N Engl J Med. 2010;363(1):24–35. doi: 10.1056/NEJMoa0912217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gillison ML, Koch WM, Capone RB, Spafford M, Westra WH, Wu L, Zahurak ML, Daniel RW, Viglione M, Symer DE, Shah KV, Sidransky D. Evidence for a causal association between human papillomavirus and a subset of head and neck cancers. J Natl Cancer Inst. 2000;92(9):709–20. doi: 10.1093/jnci/92.9.709. [DOI] [PubMed] [Google Scholar]
- 5.Ferlay J, Shin HR, Bray F, Forman D, Mathers C, Parkin DM. Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int J Cancer. 2010;127(12):2893–917. doi: 10.1002/ijc.25516. [DOI] [PubMed] [Google Scholar]
- 6.Chaturvedi AK, Engels EA, Pfeiffer RM, Hernandez BY, Xiao W, Kim E, Jiang B, Goodman MT, Sibug-Saber M, Cozen W, Liu L, Lynch CF, Wentzensen N, Jordan RC, Altekruse S, Anderson WF, Rosenberg PS, Gillison ML. Human papillomavirus and rising oropharyngeal cancer incidence in the United States. J Clin Oncol. 2011;29(32):4294–301. doi: 10.1200/JCO.2011.36.4596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Marur S, D’Souza G, Westra WH, Forastiere AA. HPV-associated head and neck cancer: a virus-related cancer epidemic. Lancet Oncol. 2010;11(8):781–9. doi: 10.1016/S1470-2045(10)70017-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bodily J, Laimins LA. Persistence of human papillomavirus infection: keys to malignant progression. Trends in microbiology. 2011;19(1):33–9. doi: 10.1016/j.tim.2010.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jabbar S, Strati K, Shin MK, Pitot HC, Lambert PF. Human papillomavirus type 16 E6 and E7 oncoproteins act synergistically to cause head and neck cancer in mice. Virology. 2010;407(1):60–7. doi: 10.1016/j.virol.2010.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gillison ML, D’Souza G, Westra W, Sugar E, Xiao W, Begum S, Viscidi R. Distinct risk factor profiles for human papillomavirus type 16-positive and human papillomavirus type 16-negative head and neck cancers. J Natl Cancer Inst. 2008;100(6):407–20. doi: 10.1093/jnci/djn025. [DOI] [PubMed] [Google Scholar]
- 11.Fakhry C, Westra WH, Li S, Cmelak A, Ridge JA, Pinto H, Forastiere A, Gillison ML. Improved survival of patients with human papillomavirus-positive head and neck squamous cell carcinoma in a prospective clinical trial. J Natl Cancer Inst. 2008;100(4):261–9. doi: 10.1093/jnci/djn011. [DOI] [PubMed] [Google Scholar]
- 12.Ragin CC, Taioli E. Survival of squamous cell carcinoma of the head and neck in relation to human papillomavirus infection: review and meta-analysis. Int J Cancer. 2007;121(8):1813–20. doi: 10.1002/ijc.22851. [DOI] [PubMed] [Google Scholar]
- 13.Fakhry C, Zhang Q, Nguyen-Tan PF, Rosenthal D, El-Naggar A, Garden AS, Soulieres D, Trotti A, Avizonis V, Ridge JA, Harris J, Le QT, Gillison M. Human papillomavirus and overall survival after progression of oropharyngeal squamous cell carcinoma. J Clin Oncol. 2014;32(30):3365–73. doi: 10.1200/JCO.2014.55.1937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ang KK, Zhang Q, Rosenthal DI, Nguyen-Tan PF, Sherman EJ, Weber RS, Galvin JM, Bonner JA, Harris J, El-Naggar AK, Gillison ML, Jordan RC, Konski AA, Thorstad WL, Trotti A, Beitler JJ, Garden AS, Spanos WJ, Yom SS, Axelrod RS. Randomized phase III trial of concurrent accelerated radiation plus cisplatin with or without cetuximab for stage III to IV head and neck carcinoma: RTOG 0522. J Clin Oncol. 2014;32(27):2940–50. doi: 10.1200/JCO.2013.53.5633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dimitrios Colevas A. Systemic Therapy for Metastatic or Recurrent Squamous Cell Carcinoma of the Head and Neck. J Natl Compr Canc Netw. 2015;13(5):e37–48. doi: 10.6004/jnccn.2015.0080. [DOI] [PubMed] [Google Scholar]
- 16.Ford AC, Grandis JR. Targeting epidermal growth factor receptor in head and neck cancer. Head Neck. 2003;25(1):67–73. doi: 10.1002/hed.10224. [DOI] [PubMed] [Google Scholar]
- 17.Bonner JA, Harari PM, Giralt J, Azarnia N, Shin DM, Cohen RB, Jones CU, Sur R, Raben D, Jassem J, Ove R, Kies MS, Baselga J, Youssoufian H, Amellal N, Rowinsky EK, Ang KK. Radiotherapy plus cetuximab for squamous-cell carcinoma of the head and neck. N Engl J Med. 2006;354(6):567–78. doi: 10.1056/NEJMoa053422. Epub 2006/02/10. 354/6/567 [pii] [DOI] [PubMed] [Google Scholar]
- 18.Baselga J, Trigo JM, Bourhis J, Tortochaux J, Cortes-Funes H, Hitt R, Gascon P, Amellal N, Harstrick A, Eckardt A. Phase II multicenter study of the antiepidermal growth factor receptor monoclonal antibody cetuximab in combination with platinum-based chemotherapy in patients with platinum-refractory metastatic and/or recurrent squamous cell carcinoma of the head and neck. J Clin Oncol. 2005;23(24):5568–77. doi: 10.1200/JCO.2005.07.119. [DOI] [PubMed] [Google Scholar]
- 19.Vermorken JB, Mesia R, Rivera F, Remenar E, Kawecki A, Rottey S, Erfan J, Zabolotnyy D, Kienzer HR, Cupissol D, Peyrade F, Benasso M, Vynnychenko I, De Raucourt D, Bokemeyer C, Schueler A, Amellal N, Hitt R. Platinum-based chemotherapy plus cetuximab in head and neck cancer. N Engl J Med. 2008;359(11):1116–27. doi: 10.1056/NEJMoa0802656. [DOI] [PubMed] [Google Scholar]
- 20.Vermorken JB, Stohlmacher-Williams J, Davidenko I, Licitra L, Winquist E, Villanueva C, Foa P, Rottey S, Skladowski K, Tahara M, Pai VR, Faivre S, Blajman CR, Forastiere AA, Stein BN, Oliner KS, Pan Z, Bach BA investigators S. Cisplatin and fluorouracil with or without panitumumab in patients with recurrent or metastatic squamous-cell carcinoma of the head and neck (SPECTRUM): an open-label phase 3 randomised trial. Lancet Oncol. 2013;14(8):697–710. doi: 10.1016/S1470-2045(13)70181-5. [DOI] [PubMed] [Google Scholar]
- 21.Machiels JP, Haddad RI, Fayette J, Licitra LF, Tahara M, Vermorken JB, Clement PM, Gauler T, Cupissol D, Grau JJ, Guigay J, Caponigro F, de Castro G, Jr, de Souza Viana L, Keilholz U, Del Campo JM, Cong XJ, Ehrnrooth E, Cohen EE, Lux H investigators N. Afatinib versus methotrexate as second-line treatment in patients with recurrent or metastatic squamous-cell carcinoma of the head and neck progressing on or after platinum-based therapy (LUX-Head & Neck 1): an open-label, randomised phase 3 trial. Lancet Oncol. 2015;16(5):583–94. doi: 10.1016/S1470-2045(15)70124-5. [DOI] [PubMed] [Google Scholar]
- 22.Troy JD, Weissfeld JL, Youk AO, Thomas S, Wang L, Grandis JR. Expression of EGFR, VEGF, and NOTCH1 suggest differences in tumor angiogenesis in HPV-positive and HPV-negative head and neck squamous cell carcinoma. Head Neck Pathol. 2013;7(4):344–55. doi: 10.1007/s12105-013-0447-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fei J, Hong A, Dobbins TA, Jones D, Lee CS, Loo C, Al-Ghamdi M, Harnett GB, Clark J, O’Brien CJ, Rose B. Prognostic significance of vascular endothelial growth factor in squamous cell carcinomas of the tonsil in relation to human papillomavirus status and epidermal growth factor receptor. Ann Surg Oncol. 2009;16(10):2908–17. doi: 10.1245/s10434-009-0579-1. [DOI] [PubMed] [Google Scholar]
- 24.Reimers N, Kasper HU, Weissenborn SJ, Stutzer H, Preuss SF, Hoffmann TK, Speel EJ, Dienes HP, Pfister HJ, Guntinas-Lichius O, Klussmann JP. Combined analysis of HPV-DNA, p16 and EGFR expression to predict prognosis in oropharyngeal cancer. Int J Cancer. 2007;120(8):1731–8. doi: 10.1002/ijc.22355. [DOI] [PubMed] [Google Scholar]
- 25.Pollock N, Wang L, Wallweber G, Gooding WE, Huang W, Chenna A, Winslow J, Sen M, DeGrave K, Li H, Zeng Y, Grandis JR. Increased expression of HER2, HER3, and HER2:HER3 heterodimers in HPV-positive HNSCC using a novel proximity-based assay: Implications for targeted therapies. Clin Cancer Res. 2015 doi: 10.1158/1078-0432.CCR-14-3338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stankiewicz E, Prowse DM, Ng M, Cuzick J, Mesher D, Hiscock F, Lu YJ, Watkin N, Corbishley C, Lam W, Berney DM. Alternative HER/PTEN/Akt pathway activation in HPV positive and negative penile carcinomas. PLoS One. 2011;6(3):e17517. doi: 10.1371/journal.pone.0017517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mazibrada J, Longo L, Vatrano S, Cappia S, Giorcelli J, Pentenero M, Gandolfo S, Volante M, dell’Oste V, Lo Cigno I, Biolatti M, Landolfo S, Papotti M. Differential expression of HER2, STAT3, SOX2, IFI16 and cell cycle markers during HPV-related head and neck carcinogenesis. New Microbiol. 2014;37(2):129–43. [PubMed] [Google Scholar]
- 28.Prigent SA, Gullick WJ. Identification of c-erbB-3 binding sites for phosphatidylinositol 3′-kinase and SHC using an EGF receptor/c-erbB-3 chimera. Embo J. 1994;13(12):2831–41. doi: 10.1002/j.1460-2075.1994.tb06577.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jura N, Shan Y, Cao X, Shaw DE, Kuriyan J. Structural analysis of the catalytically inactive kinase domain of the human EGF receptor 3. Proc Natl Acad Sci U S A. 2009;106(51):21608–13. doi: 10.1073/pnas.0912101106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Halbert CL, Demers GW, Galloway DA. The E7 gene of human papillomavirus type 16 is sufficient for immortalization of human epithelial cells. J Virol. 1991;65(1):473–8. doi: 10.1128/jvi.65.1.473-478.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Johannessen CM, Boehm JS, Kim SY, Thomas SR, Wardwell L, Johnson LA, Emery CM, Stransky N, Cogdill AP, Barretina J, Caponigro G, Hieronymus H, Murray RR, Salehi-Ashtiani K, Hill DE, Vidal M, Zhao JJ, Yang X, Alkan O, Kim S, Harris JL, Wilson CJ, Myer VE, Finan PM, Root DE, Roberts TM, Golub T, Flaherty KT, Dummer R, Weber BL, Sellers WR, Schlegel R, Wargo JA, Hahn WC, Garraway LA. COT drives resistance to RAF inhibition through MAP kinase pathway reactivation. Nature. 2010;468(7326):968–72. doi: 10.1038/nature09627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yang X, Boehm JS, Yang X, Salehi-Ashtiani K, Hao T, Shen Y, Lubonja R, Thomas SR, Alkan O, Bhimdi T, Green TM, Johannessen CM, Silver SJ, Nguyen C, Murray RR, Hieronymus H, Balcha D, Fan C, Lin C, Ghamsari L, Vidal M, Hahn WC, Hill DE, Root DE. A public genome-scale lentiviral expression library of human ORFs. Nat Methods. 2011;8(8):659–61. doi: 10.1038/nmeth.1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kirken RA, Malabarba MG, Xu J, Liu X, Farrar WL, Hennighausen L, Larner AC, Grimley PM, Rui H. Prolactin stimulates serine/tyrosine phosphorylation and formation of heterocomplexes of multiple Stat5 isoforms in Nb2 lymphocytes. J Biol Chem. 1997;272(22):14098–103. doi: 10.1074/jbc.272.22.14098. [DOI] [PubMed] [Google Scholar]
- 34.Li H, Wheeler S, Park Y, Ju Z, Thomas SM, Fichera M, Egloff AM, Lui VW, Duvvuri U, Bauman JE, Mills GB, Grandis JR. Proteomic Characterization of Head and Neck Cancer Patient-Derived Xenografts. Mol Cancer Res. 2016;14(3):278–86. doi: 10.1158/1541-7786.MCR-15-0354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Oakley GJ, Fuhrer K, Seethala RR. Brachyury, SOX-9, and podoplanin, new markers in the skull base chordoma vs chondrosarcoma differential: a tissue microarray-based comparative analysis. Modern Pathology. 2008;21(12):1461–9. doi: 10.1038/modpathol.2008.144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Narisawa-Saito M, Handa K, Yugawa T, Ohno S, Fujita M, Kiyono T. HPV16 E6-mediated stabilization of ErbB2 in neoplastic transformation of human cervical keratinocytes. Oncogene. 2007;26(21):2988–96. doi: 10.1038/sj.onc.1210118. [DOI] [PubMed] [Google Scholar]
- 37.Hu Z, Muller S, Qian G, Xu J, Kim S, Chen Z, Jiang N, Wang D, Zhang H, Saba NF, Shin DM, Chen ZG. Human papillomavirus 16 oncoprotein regulates the translocation of beta-catenin via the activation of epidermal growth factor receptor. Cancer. 2015;121(2):214–25. doi: 10.1002/cncr.29039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Akerman GS, Tolleson WH, Brown KL, Zyzak LL, Mourateva E, Engin TS, Basaraba A, Coker AL, Creek KE, Pirisi L. Human papillomavirus type 16 E6 and E7 cooperate to increase epidermal growth factor receptor (EGFR) mRNA levels, overcoming mechanisms by which excessive EGFR signaling shortens the life span of normal human keratinocytes. Cancer Res. 2001;61(9):3837–43. [PubMed] [Google Scholar]
- 39.Sizemore N, Choo CK, Eckert RL, Rorke EA. Transcriptional regulation of the EGF receptor promoter by HPV16 and retinoic acid in human ectocervical epithelial cells. Exp Cell Res. 1998;244(1):349–56. doi: 10.1006/excr.1998.4179. [DOI] [PubMed] [Google Scholar]
- 40.Rampias T, Sasaki C, Weinberger P, Psyrri A. E6 and e7 gene silencing and transformed phenotype of human papillomavirus 16-positive oropharyngeal cancer cells. J Natl Cancer Inst. 2009;101(6):412–23. doi: 10.1093/jnci/djp017. [DOI] [PubMed] [Google Scholar]
- 41.Bauer T, Infante J, Cannon S, Eder P, LoRusso P, Lavallee T, Gedrich R, Sidor C, Falchook G, Cannon S. A Phase 1, Open-label Study to Evaluate the Safety and Pharmacokinetics of the Anti-ErbB3 Antibody, KTN3379, Alone or in Combination with Targeted Therapies in Patients with Advanced Tumors. J Clin Oncol. 2015;33(suppl) abstr 2598. (ASCO Annual Meeting) [Google Scholar]
- 42.Xiao Z, Carrasco RA, Schifferli K, Kinneer K, Tammali R, Chen H, Rothstein R, Wetzel L, Yang C, Chowdhury P, Tsui P, Steiner P, Jallal B, Herbst R, Hollingsworth RE, Tice DA. A Potent HER3 Monoclonal Antibody That Blocks Both Ligand-Dependent and -Independent Activities: Differential Impacts of PTEN Status on Tumor Response. Mol Cancer Ther. 2016 doi: 10.1158/1535-7163.MCT-15-0555. [DOI] [PubMed] [Google Scholar]
- 43.Campbell MR, Amin D, Moasser MM. HER3 comes of age: new insights into its functions and role in signaling, tumor biology, and cancer therapy. Clin Cancer Res. 2010;16(5):1373–83. doi: 10.1158/1078-0432.CCR-09-1218. Epub 2010/02/25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ocana A, Vera-Badillo F, Seruga B, Templeton A, Pandiella A, Amir E. HER3 Overexpression and Survival in Solid Tumors: A Meta-analysis. Jnci-J Natl Cancer I. 2013;105(4):266–73. doi: 10.1093/Jnci/Djs501. [DOI] [PubMed] [Google Scholar]
- 45.Qian G, Jiang N, Wang D, Newman S, Kim S, Chen Z, Garcia G, MacBeath G, Shin DM, Khuri FR, Chen ZG, Saba NF. Heregulin and HER3 are prognostic biomarkers in oropharyngeal squamous cell carcinoma. Cancer. 2015;121(20):3600–11. doi: 10.1002/cncr.29549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Spangle JM, Munger K. The HPV16 E6 oncoprotein causes prolonged receptor protein tyrosine kinase signaling and enhances internalization of phosphorylated receptor species. PLoS Pathog. 2013;9(3):e1003237. doi: 10.1371/journal.ppat.1003237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Diamonti AJ, Guy PM, Ivanof C, Wong K, Sweeney C, Carraway KL. An RBCC protein implicated in maintenance of steady-state neuregulin receptor levels. Proceedings of the National Academy of Sciences of the United States of America. 2002;99(5):2866–71. doi: 10.1073/pnas.052709799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Huang Z, Choi BK, Mujoo K, Fan X, Fa M, Mukherjee S, Owiti N, Zhang N, An Z. The E3 ubiquitin ligase NEDD4 negatively regulates HER3/ErbB3 level and signaling. Oncogene. 2015;34(9):1105–15. doi: 10.1038/onc.2014.56. [DOI] [PubMed] [Google Scholar]
- 49.Paolini F, Curzio G, Melucci E, Terrenato I, Antoniani B, Carosi M, Mottolese M, Vici P, Mariani L, Venuti A. Human papillomavirus 16 E2 interacts with neuregulin receptor degradation protein 1 affecting ErbB-3 expression in vitro and in clinical samples of cervical lesions. Eur J Cancer. 2016;58:52–61. doi: 10.1016/j.ejca.2016.02.001. [DOI] [PubMed] [Google Scholar]
- 50.Jeon S, Lambert PF. Integration of human papillomavirus type 16 DNA into the human genome leads to increased stability of E6 and E7 mRNAs: implications for cervical carcinogenesis. Proc Natl Acad Sci U S A. 1995;92(5):1654–8. doi: 10.1073/pnas.92.5.1654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Romanczuk H, Howley PM. Disruption of either the E1 or the E2 regulatory gene of human papillomavirus type 16 increases viral immortalization capacity. Proc Natl Acad Sci U S A. 1992;89(7):3159–63. doi: 10.1073/pnas.89.7.3159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Steenbergen RD, Hermsen MA, Walboomers JM, Joenje H, Arwert F, Meijer CJ, Snijders PJ. Integrated human papillomavirus type 16 and loss of heterozygosity at 11q22 and 18q21 in an oral carcinoma and its derivative cell line. Cancer Res. 1995;55(22):5465–71. [PubMed] [Google Scholar]
- 53.Akagi K, Li J, Broutian TR, Padilla-Nash H, Xiao W, Jiang B, Rocco JW, Teknos TN, Kumar B, Wangsa D, He D, Ried T, Symer DE, Gillison ML. Genome-wide analysis of HPV integration in human cancers reveals recurrent, focal genomic instability. Genome Res. 2014;24(2):185–99. doi: 10.1101/gr.164806.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Suprynowicz FA, Krawczyk E, Hebert JD, Sudarshan SR, Simic V, Kamonjoh CM, Schlegel R. The human papillomavirus type 16 E5 oncoprotein inhibits epidermal growth factor trafficking independently of endosome acidification. J Virol. 2010;84(20):10619–29. doi: 10.1128/JVI.00831-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Straight SW, Hinkle PM, Jewers RJ, McCance DJ. The E5 oncoprotein of human papillomavirus type 16 transforms fibroblasts and effects the downregulation of the epidermal growth factor receptor in keratinocytes. J Virol. 1993;67(8):4521–32. doi: 10.1128/jvi.67.8.4521-4532.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Schoeberl B, Faber AC, Li D, Liang MC, Crosby K, Onsum M, Burenkova O, Pace E, Walton Z, Nie L, Fulgham A, Song Y, Nielsen UB, Engelman JA, Wong KK. An ErbB3 antibody, MM-121, is active in cancers with ligand-dependent activation. Cancer Res. 2010;70(6):2485–94. doi: 10.1158/0008-5472.CAN-09-3145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Machiels JPH, Haddad RI, Fayette J, Licitra LF, Tahara M, Vermorken JB, Clement PM, Gauler T, Cupissol D, Grau JJ, Guigay J, Caponigro F, de Castro G, Viana LD, Keilholz U, del Campo JM, Cong XYJL, Ehrnrooth E, Cohen EEW Investigators L-HN. Afatinib versus methotrexate as second-line treatment in patients with recurrent or metastatic squamous-cell carcinoma of the head and neck progressing on or after platinum-based therapy (LUX-Head & Neck 1): an open-label, randomised phase 3 trial. Lancet Oncology. 2015;16(5):583–94. doi: 10.1016/S1470-2045(15)70124-5. [DOI] [PubMed] [Google Scholar]
- 58.Alvarado D, Wallweber J, Seibel S, Chenna A, Ravanera R, Huang W, Stathas D, Theobald P, Lavallee T. Association of ErbB/HER Biomarkers with Antitumor Activity of the anti-ErbB3/HER3 Monoclonal Antibody KTN3379 in SCCHN. American Association for Cancer Resaerch Annual Meeting; 2015. (Abstract Number 1558) [Google Scholar]
- 59.Ligon G, Lilliquist J, Seibe S, Wallweber J, Neumeister V, Rimm D, LaVallee T, Alvarado D. Combination of neuregulin with EGFR activation signatures predict activity of the anti-ErbB3 antibody KTN3379 in SCCHN. American Association for Cancer Resaerch Annual Meeting; 2016. (Abstract Number 1196) [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.