

Abstract
Purpose
Pan-class I/II histone deacetylase (HDAC) inhibitors are effective treatments for select lymphomas. Isoform-selective HDAC inhibitors are emerging as potentially more targeted agents. ACY-1215 (ricolinostat) is a first in class selective HDAC6 inhibitor. To better understand the discrete function of HDAC6 and its role in lymphoma, we developed a lymphoma cell line resistant to ACY-1215.
Experimental Design
The diffuse large B-cell lymphoma cell line OCI-Ly10 was exposed to increasing concentrations of ACY-1215 over an extended period of time, leading to the development of a resistant cell line. Gene expression profiling (GEP) was performed to investigate differentially expressed genes. Combination studies of ACY-1215 and ibrutinib were performed in cell lines, primary human lymphoma tissue and a xenograft mouse model.
Results
Systematic incremental increases in drug exposure led to the development of distinct resistant cell lines with IC50 values 10-20 fold greater than that for parental lines. GEP revealed up-regulation of MAPK10, HELIOS, HDAC9 and FYN, as well as down-regulation of SH3BP5 and LCK. Gene set enrichment analysis (GSEA) revealed modulation of the BTK pathway. Ibrutinib was found to be synergistic with ACY-1215 in cell lines as well as in 3 primary patient samples of lymphoma. In vivo confirmation of anti-tumor synergy was demonstrated with a xenograft of DLBCL.
Conclusion
The development of this ACY-1215 resistant cell line has provided valuable insights into the mechanistic role of HDAC6 in lymphoma and offered a novel method to identify rational synergistic drug combinations. Translation of these findings to the clinic is underway.
Introduction
Pan-class I/II histone deacetylase (HDAC) inhibitors have proven to be successful agents for the treatment of lymphoma, though their clinical application has been restricted predominantly to the T-cell lymphomas [1-5]. While the precise role of individual HDAC isoforms in lymphoma has remained an area of active research, isoform selective HDAC inhibitors have begun to emerge. The first example of this is ACY-1215 (ricolinostat), an isoform selective HDAC6 inhibitor. HDAC6 belongs to the class 2b family of HDACs and differs from other HDACs in that it resides predominantly in the cytoplasm. It is known to play a role in protein homeostasis and the unfolded protein response (UPR) [6, 7]. HDAC6 inhibition has demonstrated activity in preclinical models of lymphoma and multiple myeloma and is presently being studied in clinical studies both as a single agent and in combination. Although very well tolerated clinically, activity as a single agent has been limited and combination strategies have proven more efficacious thus far. Combinations of ACY-1215 with lenalidomide, pomalidomide and bortezomib are presently in clinical study for patients with multiple myeloma [8-11].
In an effort to gain insights into the role of HDAC6 in lymphoma and to identify novel pathways that may be synergistic with ACY-1215, a lymphoma cell line was developed to be resistant to the HDAC6 selective inhibitor ACY-1215. Drug resistance can be defined as intrinsic or acquired. Intrinsic drug resistance is often difficult to demonstrate in tissue culture, and is defined as cells that harbor preexisting conditions which render them unresponsive to a particular drug or drug combination. Acquired drug resistance typically emerges in stages, and at least theoretically is attributed to the emergence of pathways that bypass the inhibition posed by a particular drug. We reasoned that if the emergence of compensatory pathways could mitigate sensitivity to exposure of a specific drug, then such pathways could represent logical targets for rational drug : drug combinations, preempting acquired drug resistance, at least in that specific context This paradigm, if validated, could create a logic informing the development of ‘rational’ upfront combinations that could improve the efficacy of new drug combinations.
We employed a strategy of gradual drug acclimation to identify a resistant cell line in order to try and capture emerging compensatory pathways of resistance to ACY-1215. We conducted gene expression profiling (GEP) of the resistant line which was compared to the parental line. The GEP data revealed modulation of the B-cell receptor (BCR) pathway, including down-regulation of the negative regulator of BTK (SH3BP5), increased FYN and IKZF2. These observations led to systematic evaluation of the combination of ACY-1215 with ibrutinib, a first in class BTK inhibitor, which demonstrated strong synergy. This synergy was demonstrated across a large panel of cell lines, including ones representing DLBCL and mantle cell lymphoma (MCL), and primary human samples including chronic lymphocytic lymphoma (CLL), lymphoplasmacytic lymphoma, and marginal zone lymphoma (MZL). In addition, an in vivo murine xenograft model of DLBCL (OCI-LY10) confirmed the therapeutic benefits of the combination over the individual drugs. Interestingly, the UPR has been shown to be linked to the BCR pathway through IRE-1 and XBP-1 [12, 13] in models of CLL. Work conducted in the present manuscript further substantiates this link as a potential mechanism in models of DLBCL, MCL and MZL. We believe these findings add credence to the idea that understanding mechanisms of acquired drug resistance could lead to the development of rational drug : drug combinations with novel agents now emerging in the clinic. The findings of these series of experiments lay the foundation for clinical studies of ACY-1215 and ibrutinib in B-cell lymphoid malignancies.
Materials and Methods
Drugs and reagents
ACY-1215 was provided by Acetylon Pharmaceuticals, Inc. (Boston, MA). Bortezomib, ibrutinib, romidepsin, verapamil and vorinostat were obtained from Selleck Chemicals (Houston, TX). All drugs were diluted in DMSO. Deuterated internal standard ibrutinib-d5 was purchased from TLC Pharmaceutical Standards Ltd (Ontario, Canada). All solvents and other chemicals for sample extraction were LCMS grade.
Cell lines and culture
Hbl-1, OCI-Ly10, and Riva are activated B-cell (ABC) DLBCL cell lines; OCI-Ly1, OCI-Ly7, Su-DHL-6 are germinal center (GC) DLBCL cell lines; JVM2, MAVER, Hbl-2, Jeko-1, and Rec-1 are mantle cell lymphoma (MCL) cell lines; HH and H9 are T-cell lymphoma (TCL) cell lines. Su-DHL-6, HBL-1, Riva, HBL2, Jeko-1, Rec-1, MAVER, HH, and H9 were obtained from ATCC. OCI-Ly1, OCI-Ly7, and OCI-Ly10 were obtained from DSMZ. Su-DHL-6, Hbl-1, Hbl-2, HH, H9, Jeko-1, Jvm-2, MAVER, and Rec-1 were grown in RPMI (10% FBS). OCI-Ly7, OCI-Ly10 and Riva were grown in IMDM (10% FBS). All cell lines were authenticated and screened for mycoplasma using the ATCC/Promega STR Authentication Testing Kit and Lonza MycoAlert for mycoplasma testing. Primary patient lymphoma samples were collected on an approved IRB protocol. Peripheral blood was collected and peripheral blood mononuclear cells were extracted by Ficoll-Paque density gradient media and centrifugation.
Cell viability assays
Cells (3×105 cells/well) were incubated with 1:100 dilution of drug (listed above) alone or in combination. Cell viability was assessed using the CellTiter-Glo Luminescent Cell Viability Assay (Promega Corporation, Madison, W, USA) and confirmed by Vi-Cell Series Cell Viability Analyzer [14, 15]. The ACY-1215 resistant cell line was derived from the OCI-Ly10 cell line. OCI-Ly10 was exposed to increasing concentrations of ACY-1215 over time. Systematic incremental increases in drug concentration exposure led to the development of distinct cell lines with IC50 values 10-20 fold greater than that for parental lines.
BH3 Profiling
BH3 profiling was performed as previously described [16]. Briefly, whole cells were permeabilized using the detergent digitonin which selectively permeabilizes the cell membrane, but not the mitochondrial membrane. BH3 peptides (synthesized by the Tufts University Peptide Synthesis Core and Genscript) were administered to the cells in prescribed concentrations. Depolarization of the mitochondrial membrane potential was assessed by staining the cells with JC-1 dye in 384 well, black, flat bottom, non-treated plates. Following JC-1 exposure, the JC-1 emission was read on a GloMax (Promega) plate reader once every 5 minutes for 180 minutes to generate a kinetic trace of mitochondrial membrane potential over time.
Flow Cytometry/JC-1
FACS Calibur System was used to acquire the fluorescence signals (≥1×105 events/sample); data was analyzed using Flowjo 8.8.6. Cells (3×105/mL) were quantitated for apoptosis using Alex Fluor 488/Annexin V (Dead cell apoptosis kit Invitrogen #V13240) or for the determination of the transmembrane mitochondrial membrane potential (Δψm) via staining with JC-11.3 ug/mL JC-1 dye (Invitrogen, Carlsbad, CA) as previously described [14].
Western blotting
Western blotting (WB) was performed as previously described [14]. Antibodies used were as follows: anti-acetylated lysine, anti-GRP78/BiP, anti-HDAC6, anti-HDAC9, anti-PERK and p-PERK, anti-p-eif2α, anti-IRE1α, anti-FYN, anti-eif2α, anti-p-eif2α, anti-BTK, anti-p-BTK, anti-PCL-ɣ2, anti-p-PCL-ɣ2, anti-CARD11, anti-ATF4, anti-AKT, anti-p-AKT, anti-CHOP, anti-Helios, anti-β-actin, anti-BIM (C23C5), anti-Bcl2 (Cell Signaling Technology); anti-MAPK10, anti-p-IRE1α and anti-SH3BP5 (Abcam); anti-XBP-1 (Santa Cruz). Densitometry analysis was performed on scanned images using ImageJ software (NIH).
Gene Expression Profiling
RNA was extracted from cells using the RNeasy minikit (Qiagen). RNA was quantified and assessed for integrity via Bioanalyzer 2100 (Agilent) for an RNA Integrity Number greater than 8 for RNA sequencing by the Columbia Genome Center. mRNA was enriched from total RNA samples using poly-A pull down and libraries were prepared using the TruSeq RNA prep kit (Illumina). Sample derived libraries were then sequenced using the HiSeq2500 platform (Illumina).
Base calling was performed using Real Time Analysis (Illumina) and bcl2fastq (v 1.8.4) for converting BCL to fastq format. Reads were then mapped to a reference genome (Human:NCBI/build 37.2) using TopHat (v2.0.4). Relative abundance was estimated using Cufflinks (version 2.0.2). Differentially expressed genes were tested for using the R package DESeq2.
Gene set enrichment analysis was performed using a pre-ranked tool from the Broad Institute's GSEA software [17]. The metric of -log10 of the p value*sign of the log fold change was used to rank the genes. Pathway analysis on this pre-ranked list of genes was then performed using Gene Ontology and Lymphoid biology gene sets from the Staudt lab [18].
Semi-Quantitative PCR
Semi-quantitative Multiplex RT-PCR was performed as previously described [19]. Reverse transcription (RT) was performed in a 20 μL reaction system with a total of 2 mg of RNase free DNase treated RNA (Omniscript RT kit, Qiagen) and oligo-d(T). Multiplex PCR reactions were run in 30 cycles. Primers (Fisher Scientific) were designed to span multiple exons of the target genes. Primer sequences can be found in Table 1. Images were analyzed using the ImageJ software (NIH) and band densities were normalized to GAPDH.
Table 1. Primers Used for semi-quantitative RT-PCR.
| Gene | Forward | Reverse |
|---|---|---|
| MAPK10 | 5′-CGGACTCCGAGCACAATAAA-3′ | 5′-GGAGCAGGTAGATGCAAGATAG-3′ |
| SH3BP5 | 5′-ATGAGGGTAGGAGGGACTTATAG-3′ | 5′-CGTCACCACCTCGTCTTTATC-3′ |
| FYN | 5′-GGATTGGCCCGATTGATAGA-3′ | 5′-GGTTTCACTCTCGCGGATAAG-3′ |
| HDAC9 | 5′-GAGGCAAGAACAGGAAGTAGAG-3′ | 5′-GGATGGAGATGTTCCACTAAGG-3′ |
| IKZF2 | 5′-CACCCAACCACTCCCAAATA-3′ | 5′-CTGAGCCATATCCACCTCTTTC-3′ |
| HDAC6 | 5′-AGTGGCCGCATTATCCTTATC-3′ | 5′-GAGGGTCCTTCTCTGTCTTCTA-3′ |
| LCK | 5′-CTACGGGACATTCACCATCAA-3′ | 5′-CTCTCTGTGGTCTCAGGAAATG-3′ |
| GAPDH | 5′- -3′ | 5′- -3′ |
In Vivo Studies
Animals were housed and maintained in accordance with an IUCAC-approved protocol. OCI-Ly10 1×107 (either resistant or parental) in 50% Matrigel (BD Biosciences) were subcutaneously injected into the flanks of 5-7-week-old beige/SCID mice (Taconic Farms, INC, NY). Treatment was initiated when tumor volume measured 80 mm3. Tumors volume was assessed using the 2 largest perpendicular axes (l=length; w=width) and calculated using the formula v=0.5 (l2 × w). Mice were divided into 4 cohorts of 8-10 mice per cohort as follows: (1) untreated control; (2) ACY-1215: 50 mg/kg days 1-5, 8-12, 15-19; (3) ibrutinib: 3 mg/kg days 1-20; (4) ACY-1215 plus ibrutinib. Drugs were diluted in sterile dextrose 5% in water and were administered via the intraperitoneal (IP) route [20]. Mice were assessed for weight loss and tumor volume 3×/week. Animals were sacrificed when the tumor volume exceeded 2000 mm3 or after sustained loss of >10% body weight in accordance with institutional guidelines.
Pharmacokinetic/Pharmacodynamic in vivo studies
Mice were studied for pharmacokinetic and pharmacodynamic effects of ACY-1215 and ibrutinib. Approximately 250 uL of blood was collected by sub-mandibular vein bleed at 0.5, 1, and 2 hours after treatment. Mice were sacrificed at 4, 6, and 8 hours after treatment; blood and tumor tissue were collected for measurement of drug concentration and WB analysis.
LC-MS/MS analysis for quantitation of ACY-1215 and ibrutinib
ACY1215 and ibrutinib were measured in mouse serum and tumor tissues using ultra performance Liquid Chromatography-tandem Mass Spectrometry (LC-MSMS) after liquid-liquid extraction using deuterated ibrutinib as internal standard. Serum samples (100 μL) or aqueous tissue homogenate containing 10 mg wet tissue disrupted by a Tissue Tearer homogenizer were spiked with deuterated internal standard at a level of 50 ng/mL. This was mixed with 100 μL of acetonitrile, vortexed and incubated for 10 minutes followed by addition of 750 μL of methyl tert-butyl ether. The mixture was vortexed for 10 minutes, centrifuged and the organic layer was transferred to a LCMS vial, evaporated under nitrogen stream and re-suspended in 50% methanol. Calibration standards and QC samples for both the compounds were prepared spanning a range of 0.5 ng/mL to 1000 ng/mL and extracted same as the samples.
LC-MSMS analysis was performed on a platform comprising Agilent 1290 Infinity UHPLC integrated to Agilent 6410 triple quad mass spectrometer controlled by MassHunter v 3.1 (Agilent Technologies, Santa Clara, CA). Chromatographic separation was performed on an Agilent Poroshell C18 column (50×2.1 mm, 2.7 u, 100A) maintained at 40°C. The flow rate was maintained at 500μL/min. The initial flow conditions were 50% solvent A (water containing 0.1% formic acid) and 50% solvent B (Methanol with 0.1% formic acid). Solvent B was raised to 70% over 1.75 min and to 95% by 1.95 min, held until 3.5 min and back to initial conditions by 4 min with a total run time of 6 min. The retention time for ACY-1215 and ibrutinib was 1.34 and 2.08 min respectively. The mass spectrometer was operated under multiple reaction monitoring (MRM) mode with positive electrospray ionization. For MRM, following transitions were utilized for quantitation: ACY1215 434.2>274.0; ibrutinib 441.2>304.1 and ibrutinib-d5 446.2>309.1. Mass spectrometer was operated using the following parameters: gas temperature, 300°C; gas flow, 13 L/minute; nebulizer, 30 psi; capillary 3 Kv; desolvation gas flow, 500 L/h; cone gas flow, 50 L/h and collision energy, 30 v.
Lower limit of quantification (LLOQ), defined as the lowest concentration with an accuracy and precision of <20% was determined to be 0.5 ng/mL for ACY1215 and 2.5 ng/mL for ibrutinib in serum. In tumor samples the LLOQ was determined to be 1ng/mL for both ACY1215 and 2.5ng/mL for ibrutinib. The intra-assay accuracy and precision for ACY1215 was 96.1% and 1.32% respectively while for ibrutinib, the intra-assay accuracy was 99.6% with a precision of 1.72%. The assay showed an inter-assay precision for ACY1215 0.90% and for ibrutinib 1.52%.
Statistical analysis
For determination of the inhibitory concentration of 50% of cells (IC50) and synergy, all experiments were run in triplicate and repeated at least twice. IC50 was calculated with Calcusyn software (Biosoft, Cambridge, United Kingdom). Relative risk ratio (RRR) was used as a model for establishing synergy between 2 drugs [21]. RRR is based on calculating the ratio between the actual value and expected value (EV). In the case of 2 cytotoxic compounds, EV is calculated by the formula: EV = (NA × NB)/100, where NA represents the percentage of viable cells treated with drug A and NB represents the percentage of viable cells treated with drug B. RRR<1 represents a synergistic effect; values equal to 1 indicate the mean additive effect; and values>1 represent an antagonistic effect. Flow cytometry assays were performed in duplicate, repeated at least twice, and reported as the mean with associated standard deviations.
In vivo statistical analysis was performed using Prism GraphPad's Two-Way ANOVA Analysis. Overall survival (OS) was estimated using the Kaplan-Meier method, and presented as the mean OS with 95% confidence intervals. Area under the serum concentration time curve (AUC) and half-life were determined non-compartmentally using Phoenix Winnonlin software version 6.3 (Certara, St. Louis, MO). All drug concentrations are represented as the mean with the standard deviation where applicable.
Results
The development of an ACY-1215 resistant cell line is stable and not dependent on efflux pumps
The diffuse large B-cell lymphoma cell line OCI-Ly10 was exposed to increasing concentrations of ACY-1215 over the course of 1 year. Systematic incremental increases in drug exposure led to the development of a distinct cell line with an IC50 value 10-20 fold greater than that of the parental line. The resistant R10-OCI-LY10 (R10) exhibited an IC50 of 10 uM as compared to parental IC50 of 0.9 uM after 48 hours of exposure (Figure 1a). Resistance was maintained after repeated passages for ≥1 month in the absence of drug and was not overcome by inhibition of efflux pumps as determined via verapamil co-exposure (Figure 1a)[22]. Interestingly the resistant line was cross resistant to vorinostat which strongly inhibits HDAC6 (R10 IC50=not reached vs P IC50=0.6 uM). Treatment of the resistant line with bortezomib demonstrated intermediate activity underscoring the notion that although their molecular targets are vastly different (i.e. proteasome vs HDAC6), both drug mechanisms converge on the processing of misfolded proteins (R10 IC50=8 nM vs P IC50=3.3 nM. In contrast, the resistant line was sensitive to romidepsin which is known to predominantly inhibit HDAC1, 2 and 3 with minimal activity against HDAC6 (R10 IC50=3 uM vs P IC50=2.25 uM) (Figure 1b)[23].
Figure 1. Development and Characterization of Selective HDAC6 Inhibitor Resistant Cell Line.
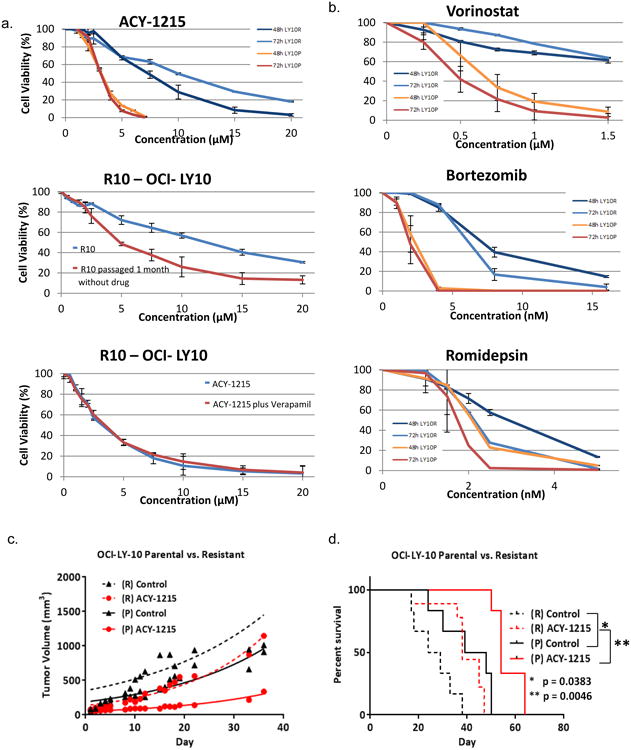
The DLBCL cell line, OCI-LY10 was exposed to increasing concentrations of ACY-1215 over time. (A) Concentration : effect relationships were established for resistant and parental Ly10 at 48 and 72 hours following exposure to ACY-1215. 48 hour concentration : effect relationship of increasing concentrations of ACY-1215 in the resistant line after immediate exposure and after a wash-out period of 1 month. Concentration : effect relationship was determined with ACY-1215 alone or in combination with verapamil 20 uM to inhibit efflux pumps. (B) Concentration : effect relationships were established for resistant and parental Ly10 at 48 and 72 hours following exposure to, vorinostat, bortezomib, and romidepsin. (C) SCID-beige mice were injected with LY10 107 in their flanks and treated with ACY-1215 50 mg/kg days 1-5, 8-12, 15-19 via the intraperitoneal route. (D) Kaplan-Meier Curve was calculated for the resistant control mice as compared to the resistant ACY-1215 mice, parental control and treatment cohorts
Both the resistant and parental LY10 cell lines were xenografted into the flanks of SCID beige mice at 107 cells. Mice were divided into 4 cohorts, resistant (R) control, parental (P) control, (R) ACY-1215, (P) ACY-1215. Mice were treated with ACY-1215 50 mg/kg days 1-5, 8-12, 15-19 by intraperitoneal route. The tumor volume doubling time was calculated for each cohort using GraphPad Prism software. The resistant cohorts exhibited accelerated tumor growth and decreased survival demonstrating that the resistant line maintains a highly aggressive phenotype over time in vivo. Treating the resistant mouse with ACY-1215 had no impact on the tumor doubling time as compared to the untreated resistant control mice (6.857 versus 7.004 days). In addition, the rate of growth was greater in the resistant control mouse cohort as compared to the parental control cohort (7.004 versus 8.039 days to doubling). The volumetric doubling time of (P) ACY-1215 treated tumors was more than twice as long compared to those of the (R) line treated with ACY-1215 (15.62 versus 6.857 days) (Figure 1c). This translated into a shorter survival for both the resistant cohorts as compared to parental cohorts. Median survival in days was as follows for each cohort: resistant control (26.5) < resistant ACY-1215 (38) < parental control (43.5) < parental ACY-1215 (54) (Figure 1d).
The IRE-1/XBP-1 pathway is upregulated in cells resistant to ACY-1215
In order to evaluate for intrinsic drug resistance, basal levels of Bcl2 and Bim were correlated with the IC50 of a panel of lymphoma cell lines. It has been demonstrated by others that decreased levels of Bim and increased levels of Bcl2 correlate with drug resistance [24, 25]. The Bcl2 :Bim ratio correlated to sensitivity to ACY-1215 and cell lines with relatively high levels of Bcl2 and low levels of Bim were relatively resistant to ACY-1215 (Figure 2a). Next we investigated whether resistance to apoptosis in the resistant cell line was due to alterations in the BCL2 family proteins, which are known to play critical roles in regulating apoptosis. Mitochondrial membrane depolarization was evaluated following treatment with ACY-1215 in parental and resistant cells to in an effort to study changes following acquired drug resistance. Compared to the parental line the resistant line did not depolarize the mitochondrial membrane following treatment with ACY-1215 2.5 μM, confirming resistance to induction of apoptosis (Figure 2b). BH3 profiling is a functional assay which informs the cellular dependence on anti-apoptotic proteins for evasion of cell death [26]. There was no difference in the BH3 profiles of the resistant and parental cell lines (Figure 2c). Taken together, these findings conclude that functional alterations in the Bcl2 family of proteins do not contribute to resistance to ACY-1215.
Figure 2. The IRE-1/XBP-1 pathway is upregulated in cells resistant to ACY-1215.
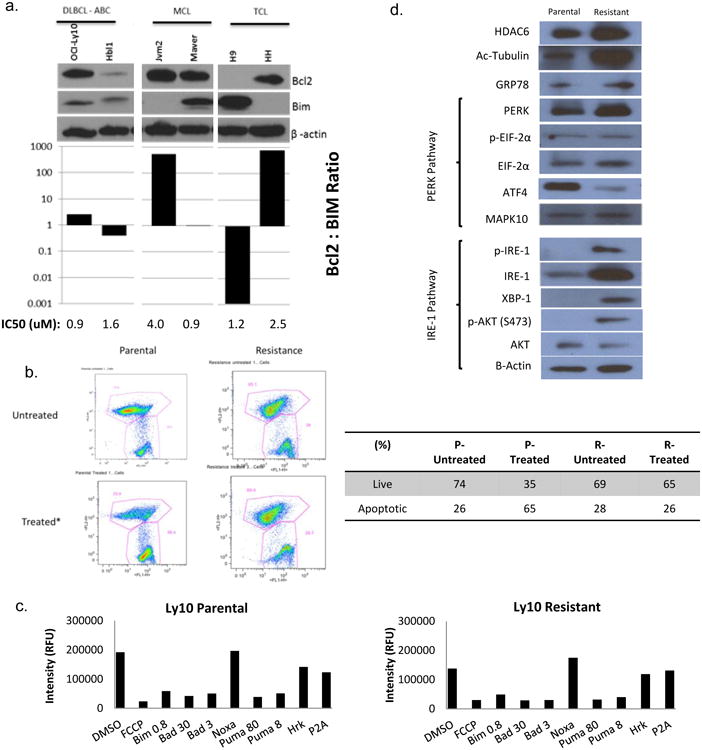
, (A) Western blot analysis of a panel of lymphoma cell lines for Bcl2 and Bim was compared to the IC50 in these cell lines. (B) Mitochondrial membrane potential was measured following 48 hour exposure of cells to ACY-1215 2.5 uM via flow cytometry. (C) BH3 profiling was performed on the parental and resistant cell lines. (D) Baseline characteristics of the resistant line was compared to the parental line with respect to the unfolded protein response (UPR).
Given the known influence of ACY-1215 on the unfolded protein response (UPR)[6, 7], evaluation of PERK and IRE-1 pathways were evaluated by immunoblot (Figure 2d). The resistant line displayed relatively stable GRP78, the master regulator of the UPR. The GRP78- PERK pathway also remained relatively stable. The IRE-1 pathway however was upregulated with concomitant activation of XBP-1 and AKT as compared to the parental line perhaps driving accelerated growth in these ACY-1215 resistant cells. This corresponds to previously published preclinical studies of CLL. Kriss, et al. demonstrated that the Eμ-TCL1 mouse model was found to have up-regulated levels of the ER stress response pathway, in particular IRE-1 and XBP-1 [12]. In the Eμ-TCL1 model, the tonic IRE-1/XBP-1 pathway led to a constitutively active B-cell receptor pathway. Additionally, in an XBP-1KD/Eμ-TCL1 mouse model, mice with knocked-down XBP-1 had significantly slower progression to B-cell leukemia than XBP-1WT/Eμ-TCL1 mice [13]. Together, these findings suggest a functional link between the IRE-1/XBP-1 and B-cell receptor pathways.
Gene expression profiling reveals distinct modulation of the B-cell receptor pathway in the resistant cell line as compared to the parental cell line
Gene expression profiling was performed on both the resistant and parental cell lines. The parental and resistant cell lines had distinct gene expression signatures as represented by principal component analysis and as can be visualized on the heat map (Figure 3a). The gene expression data were analyzed by GSEA and gene cluster analysis. There were 1363 genes from resistant line 2-log fold up-regulated as compared to the parental and 1825 genes 2-log fold down-regulated as compared to the parental (p < 0.05). GSEA revealed increased expression of pathways known to drive lymphomagenesis of activated B-cell (ABC) lymphoma such as the BTK pathway (Figure 3b). In addition to changes in the UPR, differentially expressed genes in the resistant line included up-regulation of MAPK10, HELIOS, HDAC9 and FYN, as well as down-regulation of SH3BP5 (a negative regulator of BTK) and LCK. The change in expression was confirmed by PCR (Figure 3c) and western blot analysis (Figure 3d). Given the up-regulation of FYN, a tyrosine kinase in the B-cell receptor pathway, and the down-regulation of SH3BP5 a negative regulator of the Bruton's Tyrosine Kinase, as well as findings generated by GSEA, the resistant line was treated with ibrutinib. Cell viability of the resistant and parental lines were evaluated over time. Ibrutinib 2 uM was able to overcome resistance with a 70% viability in the resistant line versus 64% viability in the parental line at 72 hours (Figure 3e). This data reveals that cells exposed to ACY-1215, a selective HDAC6 inhibitor, rely heavily on the B-cell receptor pathway for expansion. It also suggests that treatment with the combination of ACY-1215 and ibrutinib may lead to a synergistic interaction.
Figure 3. Resistant cell line has differentially expressed gene profile as compared to parental line and identifies that the BCR pathway is upregulated in resistant cells.
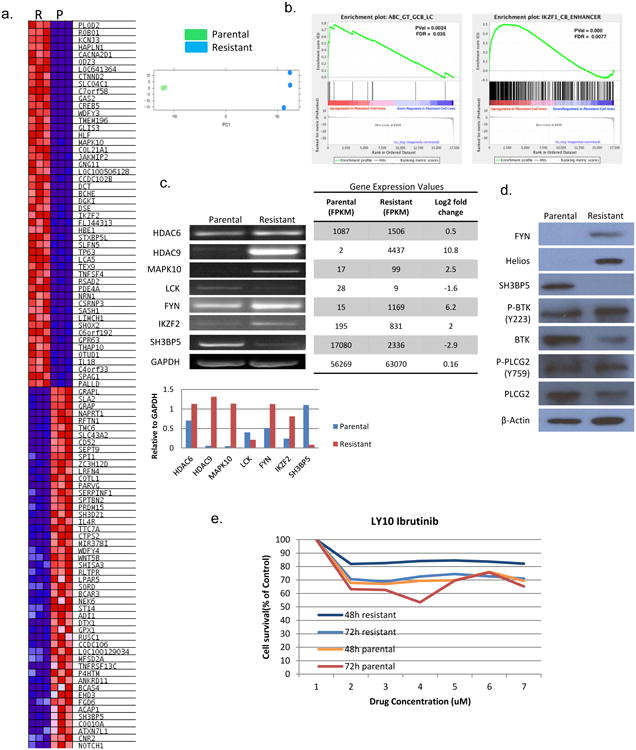
(A) Resistant (R) cells and parental cells (P) were evaluated by RNA Seq for gene expression. The two cells lines were distinct as demonstrated by principle component analysis. The heat map represents the top 100 genes with significant overall 2-log fold change between the resistant and parental lines. (B) Gene set enrichment analysis (GSEA) was performed to compare enrichment of pathways in resistant verses parental lines. (C) Differentially expressed genes of interest were confirmed via PCR for the resistant line as compared to the parental line. (D) Protein expression of genes of interest was confirmed with immunoblot analysis. (E) Concentration : effect relationships of ibrutinib in resistant and parental lines was evaluated at 48 and 72 hours.
ACY-1215 plus Ibrutinib is highly synergistic in lymphoma cell lines and primary human lymphoma samples
Based on the findings generated from the resistant cell line, a panel of lymphoma cell lines were treated with ACY-1215, ibrutinib, or the combination and viability and synergy were measured over 24, 48, and 72 hours (Figure 4a). Cell lines included the GCB-DLBCL OCI-Ly7, the ABC-DLBCL cell lines HBL1, OCI-Ly10, RIVA, mantle cell lymphoma (MCL) lines HBL2, JEKO1, REC1, and the T-cell lymphoma line H9. As expected, synergy was most pronounced in the ABC-DLBCL and MCL cell lines with synergy coefficients as low as 0.10 (Ly10) and 0.15 (JEKO1) respectively (where RRR<1 connotes synergy). Both of these disease entities are known to be driven by tonic signaling of the B-cell receptor (BCR) pathway and are highly sensitive to BTK inhibitors. There was little-to-no synergy in GCB-DLBCL and T-cell lymphoma respectively as these cell lines do not rely on the BCR pathway for growth. The heat maps represent the viability or synergy of a panel of cell lines following treatment with ACY-1215, ibrutinib or the combination at 24, 48, and 72 hours. Red boxes indicate lower viability and higher synergy whereas blue boxes represent high viability and no synergy. Synergy was calculated by the relative risk ration (RRR) where a RRR < 1 connotes synergy.
Figure 4. Ibrutinib plus ACY-1215 is synergistic in cell lines and primary human lymphoma samples.
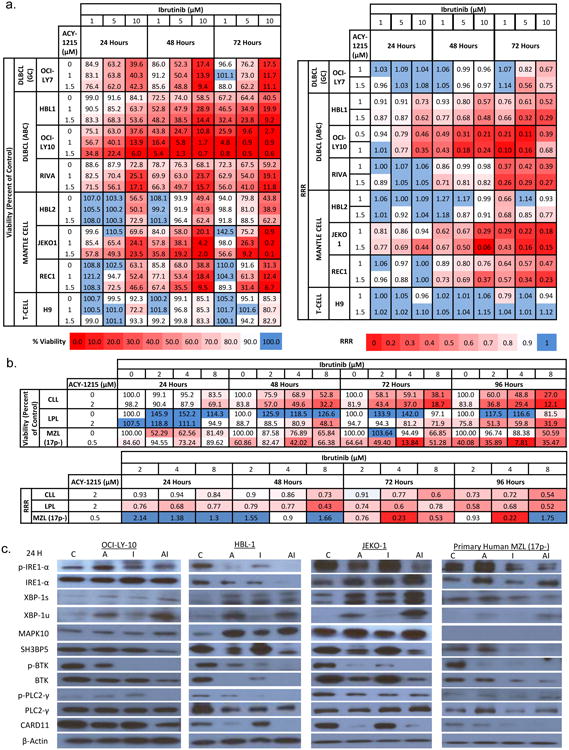
(A). Heat map represents the viability of a panel of cell lines following treatment with ACY-1215, ibrutinib or the combination at 24, 48, and 72 hours. Red boxes indicate lower viability. Synergy was calculated by the relative risk ration (RRR). RRR < 1 connotes synergy and is represented by red boxes. (B) Primary human lymphoma samples, chronic lymphocytic leukemia (CLL), lymphoplasmacytic lymphoma (LPL), and 17p deleted marginal zone lymphoma (MZL), were treated with ACY-1215, ibrutinib or the combination over 24 to 96 hours. Viability was measured and synergy was calculated by RRR and represented in the heat maps. (C) The IRE-1 and BTK pathways were evaluated following treatment with the combination of ibrutinib and ACY-1215 by immunoblot analysis.
To confirm the clinical relevance of this observation, three primary patient samples of leukemic phase lymphoma were collected under an IRB approved protocol. The patient samples spanned 3 subtypes of lymphoma including chronic lymphocytic lymphoma (CLL), lymphoplasmacytic B-cell lymphoma (LPL), and marginal zone lymphoma (MZL) harboring a 17p deletion. These three diseases are known for their sensitivity to BTK inhibitors. Cells were treated with increasing concentrations of ibrutinib in combination with ACY-1215 and viability and synergy were measured after 24, 48, 72, and 96 hours. All three of the lymphoma subtypes achieved marked synergy with RRR of 0.54, 0.43, and 0.22 respectively (Figure 4b).
The molecular effects of the combination treatment was evaluated in three lymphoma cell lines, 2 ABC-DLBCL (LY-10 and HBL-1) and one MCL line (JEKO1) as well as the primary human MZL 17p- patient sample by immunoblot (Figure 4c). The combination led to a decrease in p-IRE1- α which has been shown to interplay with the BTK pathway [12, 13]. This corresponded to a decrease p-BTK, total BTK and its down-stream targets such as p-PLC2-γ and CARD11. There was decreased expression of SH3BP5, the negative regulator of the BTK in all cell lines. This is expected as its function is to inhibit auto-phosphorylation of BTK and the cells treated with the combination exhibited complete inhibition of p-BTK [27]. These findings show that targeting the IRE1 pathway of the UPR and the BTK pathway with a selective HDAC6 inhibitor and ibrutinib are synergistic at both the cytotoxic and biologic level.
The combination of ACY-1215 and ibrutinib led to marked tumor growth delay and prolonged overall survival in a xenograft model of lymphoma
The parental LY10 cell line was xenografted into the flanks of SCID beige mice at 107 cells. Mice were divided into 4 cohorts, control, ACY-1215, ibrutinib, and the combination. Mice were treated with ACY-1215 50 mg/kg days 1-5, 8-12, 15-19, and ibrutinib 3 mg/kg once daily on days 1-20. Both drugs were given by intraperitoneal route (Figure 5a). The combination of ACY-1215 and ibrutinib was well tolerated with weight loss observed in both the ACY-1215 and combination cohorts that returned to baseline by day 20 (Figure 5b). This finding is similar to that seen in prior mouse studies of single agent and combinations with ACY-1215 [6, 7]. Mice demonstrated significant tumor growth delay following treatment with the combination as compared to either ACY-1215, ibrutinib, or untreated mice (p=0.0003, p=0.0006, and p<0.0001 respectively) (Figure 5c). Mice treated with the combination had a prolonged overall survival compared to the control (Figure 5d). Of the mice that completed one cycle of therapy, the mean survival in days was as follows: combination (66.5) > ibrutinib (57.6) > ACY-1215 (56.6) > control (40.1) > resistant control (26.5).
Figure 5. The combination of ACY-1215 and ibrutinib leads to statistically significant tumor growth delay compared to single agent treatment in a xenograft mouse model of lymphoma.
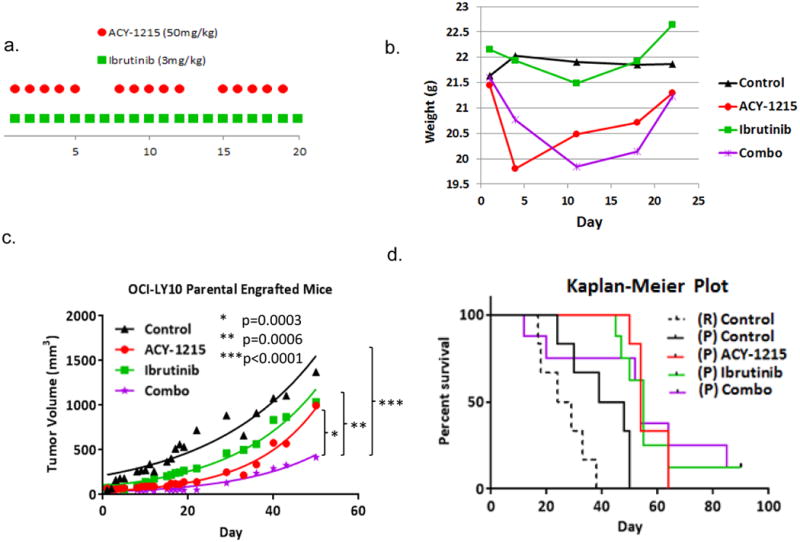
(A) SCID-beige mice were injected with LY10 107 in their flanks and treated with ACY-1215 50 mg/kg days 1-5, 8-12, 15-19, ibrutinib 3 mg/kg days 1-20, or the combination via the intraperitoneal route. (B) Mice were weighed every 3-4 days as a measurement for toxicity. (C) Tumor volume was measure over time as a function of treatment group (D) Kaplan-Meier Curve was calculated for the resistant control mice as compared to the parental control and treatment cohorts.
Pharmacokinetic and pharmacodynamic effects of ACY-1215 in combination with ibrutinib
The concentration of ACY-1215 and ibrutinib was measured at sequential time points in serum and tumor tissue after a single intraperitoneal injection of ACY-1215 (50mg/kg) or ibrutinib (3mg/kg) or a combination of both. The concentrations achieved for both drugs are similar or greater than the concentrations found to induce cytotoxicity in the in vitro experiments. In addition, these concentrations also recapitulate what has been described in human pharmacokinetic studies. There was no difference in the concentrations of either drug in resistant or parental mice therefore these data were pooled for analysis (figure 6a, b).
Figure 6. Pharmacokinetic and pharmacodynamics effects of ACY-1215 in combination with ibrutinib in mice.
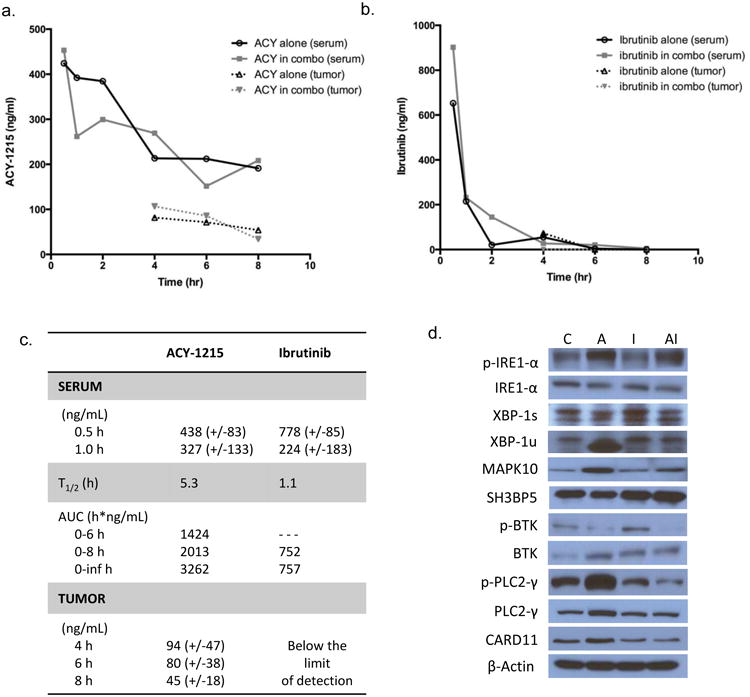
Serum and tumor tissue was collected from mice at sequential time points and analyzed for concentration of ACY-1215 and ibrutinib by LC-MS/MS. Mice were treated with ACY-1215 at 50 mg/kg alone and ACY-1215 50 mg/kg with ibrutinib 3 mg/kg. Drug concentrations are represented as mean values. (A) Graphical representation of ACY-1215 concentration over time. Mice received one dose of ACY-1215 50 mg/kg or ACY-1215 50 mg/kg plus ibrutinib 3 mg/kg for analysis of serum concentration of ACY-1215. For analysis of drug concentration in tumor tissue, ACY-1215 was administered at 50 mg/kg with or without ibrutinib. (B) Graphical representation of ibrutinib concentration over time analyzed in serum and tumor tissue. Mice received one dose of ibrutinib 3 mg/kg via i.p. route. (C) Summary of pharmacokinetic data for ACY-1215 and ibrutinib. (D) Immunoblot analysis of the IRE1 pathway of the UPR and the BTK pathway from whole cell lysates of mouse tumor tissue treated with ACY-1215, ibrutinib or the combination. Mice were treated with a single i.p. injection and analyzed at 6 hours.
The average ACY-1215 serum concentration reached at 0.5 and 1 hour after administration was 438 (+- 85) and 327 (+- 133) ng/mL, respectively. The average serum concentration of ACY-1215 at 6 and 8 hours was 182 (+-80) and 200 (+-89) ng/mL, resulting in an average half-life of 5.3 hours and an average AUC0-6h, AUC0-8h and AUC0-inf of 1425, 2013 and 3262 h*ng/mL, respectively for both single administration of ACY-1215 and in combination with ibrutinib in both parental and resistant groups. There was no substantial difference in serum pharmacokinetics of ACY-1215 between groups and treatments, which was also observed for tumor concentrations of the drug. Mean (+SD) tumor concentrations for single administration of ACY1215 and the combination with ibrutinib in the parental and resistant group at 4, 6 and 8 hours after injection were 94 (+-47), 80 (+-38) and 45 (+-18) ng/mL, respectively (figure 6C).
The serum pharmacokinetics of ibrutinib exhibited no substantial differences when the drug was administered alone or in combination with ACY-1215. The average ibrutinib serum concentration reached at 0.5 and 1 hour after administration was 778 (+- 85) and 224 (+- 183) ng/mL, respectively. The average serum concentration of ibrutinib at 6 and 8 hours was 13 (+-22) and 6 (+-6) ng/mL, resulting in an average half-life of 1.1 hours and an average AUC0-8h and AUC0-inf of 752, and 757 h*ng/mL, respectively for both single administration of ibrutinib and in combination with ACY-1215 in both parental and resistant groups. Ibrutinib tumor concentrations were below the lowest limit of quantification in most samples (figure 6b).
Immunoblot analysis of mouse tumor tissue was evaluated for modulation of the IRE1 and BTK pathways following 6 hours of exposure to ACY-1215, ibrutinib, or the combination. Following the short, single exposure to the drugs there was reduction of p-BTK and corresponding reduction of the downstream targets p-PLC2-γ and CARD11 (Figure 6d) which is most pronounced in the combination. These results confirm those demonstrated in the cell lines treated with the combination.
Discussion
The development of this ACY-1215 resistant cell line has proven to be a powerful tool to identify rational drug partners. As treatment strategies for malignancies evolve, targeted approaches have become exceedingly specific. Although new highly selective drugs have efficacy against singular targets, what collateral effects these new agents have on other pathways is not immediately apparent. Utilizing gene expression profiling and gene set enrichment analysis we identified that the BTK pathway circumnavigated resistance to tonic HDAC6 inhibition.
The resistant line displayed up-regulation of the IRE1 pathway of the UPR as compared to the parental line. This is in keeping with observations made in models of CLL where inhibition of IRE1 led to impaired growth through XBP-1 mediated down-regulation of the BTK pathway. In Eμ-TCL1 mice knocked down for XBP-1 mice displayed decreased signaling through the B-cell receptor pathway as revealed by decreased p-BTK. This corresponded to slower progression to leukemia relative to age matched controls [13]. By inhibiting IRE1, similar findings were replicated establishing a link between the unfolded protein response and the B-cell receptor pathway. Recent publications have established other links with HDACs and signaling of the BTK pathway. HDAC inhibition has been shown to restore the expression of BTK-targeting miRNA, leading to decreased signaling of the BTK pathway and ultimately apoptosis [28]. Combinations of panobinostat or abexinostat with ibrutinib were synergistic and overcame BTK mutations known to induce ibrutinib resistance. In addition, HDAC6 has been linked to the de-acetylation and activation of MYD88, a toll-like receptor kinase known to interplay with the BTK pathway. HDAC6 inhibition with tubacin or vorinostat acetylated and abrogated the effects of MYD88 leading to cell cycle arrest and cell death [29]. These three examples point to the interconnectedness of HDAC modulation and BTK signaling.
Our findings reveal that tonic inhibition of HDAC6 leads to up-regulation of the IRE1 pathway of the UPR. This in turn was associated with up-regulation of the B-cell receptor pathway as evidenced by RNA sequencing, GSEA and immunoblot analysis. Confirmation of the interplay between the UPR and the BCR pathway were further established by demonstrating synergy between ACY-1215, a selective HDAC6 inhibitor, and ibrutinib across a panel of cell lines (ABC-DLBCL and MCL) known to be sensitive to BTK inhibitors. Furthermore this synergy was also established in three primary human lymphoma samples of subtypes also known to be sensitive to BTK inhibitors: CLL, LPL and MZL. The combination was highly synergistic in a rapidly growing 17p- deleted MZL demonstrating a novel approach to these challenging diseases.
The combination of ACY-1215 and ibrutinib led to marked inhibition of p-IRE1 and p-BTK compared to either of the drugs alone and this corresponded to inhibition of down-stream targets of the BCR such as p-PLC-γ and CARD11. These findings were observed in cell lines, primary human lymphoma samples and xenograft models of lymphoma. In addition, the combination was well-tolerated in mice. Ly10 xenografted mice treated with the combination had statistically significant tumor growth delay compared to those treated with either drug alone.
Although it is well established that ibrutinib is effective for patients with 17p- CLL and MCL, clinical experience has informed us that these responses though durable often are not permanent [30, 31]. It has been observed that patients relapsing off of ibrutinib have highly aggressive and often explosive disease. Our findings demonstrate that combining ibrutinib with ACY-1215 has potent synergy and that this combination is effective at inhibiting the most aggressive subtypes of lymphoma.
The gene expression data derived from the resistant cell-line also revealed other potential druggable targets. FYN, a Src kinase which plays a crucial role in signaling in the T-cell and B-cell receptor pathways, was also found to be upregulated in the resistant cell line [32, 33]. This kinase is known to be inhibited by dasatinib and combination therapy of dasatinib plus ACY-1215 is being explored. Helios was also upregulated in the resistant line. Helios is a member of the Ikaros family of DNA-binding protiens and is found to be mainly expressed in precursor cells of T-cell lineage. Little is known about Helios' effects in B lymphocytes but ectopic overexpression has led to lymphomagenesis in transgenic mice [34]. Most of Helios is found to be bound to Ikaros, the effects of which are unknown. In models of myeloma, Ikaros in known to be selectively ubiquitinated and degraded by lenalidomide via cereblon ubiquitin ligase [35]. Studies evaluating the combination of lenalidomide and ACY-1215 are currently underway in myeloma and how these findings may be translated to B-cell lymphoma are presently under study. Finally, HDAC9 was found to be upregulated in the resistant cell line. HDAC9 is a member of the class IIa family and resides in both the nucleus and cytoplasm. Although the function of HDAC9 is not well studied, it is likely that it's targets may overlap with HDAC6. Upregulation of HDAC9 may have been induced in resistant line as a collateral effect to overcome tonic HDAC6 inhibition. Pan-class HDAC inhibition with romidepsin was able to overcome this effect as the resistant line was sensitive to treatment with romidepsin.
Developing a drug resistant cell line is an under-utilized strategy to identify rational and synergistic drug: drug combinations. This strategy could be applied broadly and may proactively identify pathways co-opted by malignant cells upon inhibition by highly selective targeted agents. The findings of this manuscript identified the synergistic interaction of ACY-1215, a selective HDAC6 inhibitor, and ibrutinib the first in class BTK inhibitor via this technique. Translation of these findings to the clinic are now underway.
Supplementary Material
Translational Relevance.
To better understand the function of HDAC6 and its role in lymphoma, we developed lymphoma cell lines resistant to the selective HDAC6 inhibitor ACY-1215. ACY-1215 has demonstrated activity in preclinical models of multiple myeloma and lymphoma and is currently being studied clinically for both of these diseases. Gene expression profiling was performed on the resistant line and compared to the parental line which pointed to modulation of the BTK pathway revealing a potential drug partner. Synergy was confirmed with the combination of ACY-1215 and ibrutinib in lymphoma cell lines, primary human lymphoma tissue and a mouse model of lymphoma. The development and characterization of this ACY-1215-resistant cell line has proven to be a valuable tool to advance our understanding of the functional role of HDAC6 in lymphoma. This strategy is a practical method to identify potential rational and synergistic drug : drug combinations. Translation of these findings in lymphoma are now underway.
Acknowledgments
We would like to kindly thank Anthony Letai, M.D., PhD. for providing technical support for BH3 profiling functional assays.
Funding: This work was supported by the Amos Medical Faculty Development Program of the American Society of Hematology and Robert Wood Johnson Foundations and the Columbia University Provost Award for Junior Faculty, research funding from Acetylon Pharmaceuticals, Inc (J.E.A.), the American Society of Hematology HONORS Award Program (P.M.J.) and the National Center for Advancing Translational Sciences, National Institutes of Health, (UL1 TR00004) (S.C.) and (R01 CA194547) (O.E.). We would also like to acknowledge the Lymphoma Research Fund of Columbia University for its generous support.
Footnotes
Author Contributions: Contribution: J.E.A., S.A.P., M.G.L., K.Z., P.M.J., O.A.O. designed the study, analyzed and interpreted the data, J.E.A. wrote the paper, J.E.A., S.A.P., X.S., L.S., R.N., S.C. contributed to the in vivo experiments, J.E.A., S.A.P, P.M.J., M.G.L, K.M.Z., Y.G. contributed with in vitro experiments; J.D., Y.W., A.V, O.E. conducted the statistical analysis.
Disclosure Conflicts of Interest: J.E.A. received investigational drug and research funding from Acetylon Pharmaceuticals, Inc.
J.E.A. and O.A.O. have provided consultancy for Acetylon Pharmaceuticals, Inc
Uncategorized References
- 1.Duvic M, et al. Phase 2 trial of oral vorinostat (suberoylanilide hydroxamic acid, SAHA) for refractory cutaneous T-cell lymphoma (CTCL) Blood. 2007;109(1):31–9. doi: 10.1182/blood-2006-06-025999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Piekarz RL, et al. Phase 2 trial of romidepsin in patients with peripheral T-cell lymphoma. Blood. 2011;117(22):5827–34. doi: 10.1182/blood-2010-10-312603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.O'Connor OA, et al. Belinostat in Patients With Relapsed or Refractory Peripheral T-Cell Lymphoma: Results of the Pivotal Phase II BELIEF (CLN-19) Study. J Clin Oncol. 2015;33(23):2492–9. doi: 10.1200/JCO.2014.59.2782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ning ZQ, et al. Chidamide (CS055/HBI-8000): a new histone deacetylase inhibitor of the benzamide class with antitumor activity and the ability to enhance immune cell-mediated tumor cell cytotoxicity. Cancer Chemotherapy and Pharmacology. 2012;69(4):901–909. doi: 10.1007/s00280-011-1766-x. [DOI] [PubMed] [Google Scholar]
- 5.San-Miguel JF, et al. Panobinostat plus bortezomib and dexamethasone versus placebo plus bortezomib and dexamethasone in patients with relapsed or relapsed and refractory multiple myeloma: a multicentre, randomised, double-blind phase 3 trial. The Lancet Oncology. 2014;15(11):1195–1206. doi: 10.1016/S1470-2045(14)70440-1. [DOI] [PubMed] [Google Scholar]
- 6.Amengual JE, et al. Dual targeting of protein degradation pathways with the selective HDAC6 inhibitor ACY-1215 and bortezomib is synergistic in lymphoma. Clinical Cancer Research. 2015;21(20):4663–4675. doi: 10.1158/1078-0432.CCR-14-3068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Santo L, et al. Preclinical activity, pharmacodynamic, and pharmacokinetic properties of a selective HDAC6 inhibitor, ACY-1215, in combination with bortezomib in multiple myeloma. Blood. 2012;119(11):2579–2589. doi: 10.1182/blood-2011-10-387365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yee AJ, et al. Ricolinostat (ACY-1215), a selective HDAC6 inhibitor, in combination with lenalidomide and dexamethasone: results of a phase 1b trial in relapsed and relapsed refractory multiple myeloma. Blood. 2014;124(21):4772–4772. [Google Scholar]
- 9.Niesvizky R, et al. ACY-241, a Novel, HDAC6 Selective Inhibitor: Synergy with Immunomodulatory (IMiD®) Drugs in Multiple Myeloma (MM) Cells and Early Clinical Results (ACE-MM-200 Study) Blood. 2015;126(23):3040–3040. [Google Scholar]
- 10.Vogl DT, et al. Phase 1B results of ricolinostat (ACY-1215) combination therapy with bortezomib and dexamethasone in patients with relapsed or relapsed and refractory multiple myeloma (MM) Blood. 2014;124(21):4764–4764. [Google Scholar]
- 11.Raje NS, et al. Ricolinostat (ACY-1215), the First Selective HDAC6 Inhibitor, Combines Safely with Pomalidomide and Dexamethasone and Shows Promosing Early Results in Relapsed-and-Refractory Myeloma (ACE-MM-102 Study) Blood. 2015;126(23):4228–4228. [Google Scholar]
- 12.Kriss CL, et al. Overexpression of TCL1 activates the endoplasmic reticulum stress response: a novel mechanism of leukemic progression in mice. Blood. 2012;120(5):1027–1038. doi: 10.1182/blood-2011-11-394346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tang CHA, et al. Inhibition of ER stress–associated IRE-1/XBP-1 pathway reduces leukemic cell survival. The Journal of clinical investigation. 2014;124(6):2585–2598. doi: 10.1172/JCI73448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Paoluzzi L, et al. Targeting Bcl-2 family members with the BH3 mimetic AT-101 markedly enhances the therapeutic effects of chemotherapeutic agents in in vitro and in vivo models of B-cell lymphoma. Blood. 2008;111(11):5350–5358. doi: 10.1182/blood-2007-12-129833. [DOI] [PubMed] [Google Scholar]
- 15.Marchi E, et al. Pralatrexate is synergistic with the proteasome inhibitor bortezomib in in vitro and in vivo models of T-cell lymphoid malignancies. Clin Cancer Res. 2010;16(14):3648–58. doi: 10.1158/1078-0432.CCR-10-0671. [DOI] [PubMed] [Google Scholar]
- 16.Ryan J, Letai A. BH3 profiling in whole cells by fluorimeter or FACS. Methods. 2013;61(2):156–164. doi: 10.1016/j.ymeth.2013.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Subramanian A, et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proceedings of the National Academy of Sciences. 2005;102(43):15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shaffer AL, et al. A library of gene expression signatures to illuminate normal and pathological lymphoid biology. Immunological Reviews. 2006;210(1):67–85. doi: 10.1111/j.0105-2896.2006.00373.x. [DOI] [PubMed] [Google Scholar]
- 19.Kalac M, et al. HDAC inhibitors and decitabine are highly synergistic and associated with unique gene-expression and epigenetic profiles in models of DLBCL. Blood. 2011;118(20):5506–5516. doi: 10.1182/blood-2011-02-336891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yang Y, et al. Exploiting Synthetic Lethality for the Therapy of ABC Diffuse Large B Cell Lymphoma. Cancer Cell. 2012;21(6):723–737. doi: 10.1016/j.ccr.2012.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhao L, Wientjes MG, Au JL. Evaluation of combination chemotherapy integration of nonlinear regression, curve shift, isobologram, and combination index analyses. Clinical Cancer Research. 2004;10(23):7994–8004. doi: 10.1158/1078-0432.CCR-04-1087. [DOI] [PubMed] [Google Scholar]
- 22.Gottesman MM, Fojo T, Bates SE. Multidrug resistance in cancer: role of ATP-dependent transporters. Nat Rev Cancer. 2002;2(1):48–58. doi: 10.1038/nrc706. [DOI] [PubMed] [Google Scholar]
- 23.Bradner JE, et al. Chemical phylogenetics of histone deacetylases. Nat Chem Biol. 2010;6(3):238–243. doi: 10.1038/nchembio.313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen S, et al. A Bim-targeting strategy overcomes adaptive bortezomib resistance in myeloma through a novel link between autophagy and apoptosis. Blood. 2014;124(17):2687–97. doi: 10.1182/blood-2014-03-564534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Thomas A, et al. Drug-induced apoptosis in B-cell chronic lymphocytic leukemia: relationship between p53 gene mutation and bcl-2/bax proteins in drug resistance. Oncogene. 1996;12(5):1055–62. [PubMed] [Google Scholar]
- 26.Moore VDG, Letai A. BH3 profiling–measuring integrated function of the mitochondrial apoptotic pathway to predict cell fate decisions. Cancer letters. 2013;332(2):202–205. doi: 10.1016/j.canlet.2011.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yamadori T, et al. Bruton's tyrosine kinase activity is negatively regulated by Sab, the Btk-SH3 domain-binding protein. Proceedings of the National Academy of Sciences of the United States of America. 1999;96(11):6341–6346. doi: 10.1073/pnas.96.11.6341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bottoni A, et al. Targeting BTK through microRNA in chronic lymphocytic leukemia. Blood. 2016 doi: 10.1182/blood-2016-07-727750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.New M, et al. TLR adaptor protein MYD88 mediates sensitivity to HDAC inhibitors via a cytokine-dependent mechanism. Cancer Res. 2016 doi: 10.1158/0008-5472.CAN-16-0504. [DOI] [PubMed] [Google Scholar]
- 30.Woyach JA, et al. Resistance Mechanisms for the Bruton's Tyrosine Kinase Inhibitor Ibrutinib. New England Journal of Medicine. 2014;370(24):2286–2294. doi: 10.1056/NEJMoa1400029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Landau D, et al. Clonal evolution in patients with chronic lymphocytic leukemia (CLL) developing resistance to BTK inhibition. Blood. 2013;122(21):866–866. doi: 10.1038/ncomms11589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gauld SB, Cambier JC. Src-family kinases in B-cell development and signaling. Oncogene. 2004;23(48):8001–6. doi: 10.1038/sj.onc.1208075. [DOI] [PubMed] [Google Scholar]
- 33.Palomero T, et al. Recurrent mutations in epigenetic regulators, RHOA and FYN kinase in peripheral T cell lymphomas. Nat Genet. 2014;46(2):166–70. doi: 10.1038/ng.2873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dovat S, et al. Transgenic expression of Helios in B lineage cells alters B cell properties and promotes lymphomagenesis. J Immunol. 2005;175(6):3508–15. doi: 10.4049/jimmunol.175.6.3508. [DOI] [PubMed] [Google Scholar]
- 35.Kronke J, et al. Lenalidomide causes selective degradation of IKZF1 and IKZF3 in multiple myeloma cells. Science. 2014;343(6168):301–5. doi: 10.1126/science.1244851. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.