

Abstract
Enterohemorrhagic Escherichia coli (EHEC) are strains of E. coli that express Shiga toxins (Stx) and cause hemorrhagic colitis. In some cases, disease can progress to hemolytic uremic syndrome, a potentially fatal form of kidney disease. Both enteric and renal disease are associated with the expression of stx genes, which are often carried on lysogenic phage. Toxin is expressed following induction and conversion of the phage to lytic growth. The authors previously used a germ-free mouse model to demonstrate that toxin gene expression is enhanced during growth in vivo and that renal disease is dependent on both prophage induction and expression of Stx2. In the current study, the authors document and quantify necrotizing colitis, examine the progression of enteric and renal disease, and determine the role of Stx2, phage genes, and the type 3 secretion system (T3SS) in bacterial colonization and colitis and systemic disease. By 1 day after inoculation, EHEC-monocolonized mice developed colitis, which decreased in severity thereafter. Systemic disease developed subsequently. Infection with EHEC mutant strains revealed that renal failure and splenic necrosis were absolutely dependent on the expression of Stx2 but that T3SS function and prophage excision were not necessary for systemic disease. In contrast, colitis was only partly dependent on Stx2. This study demonstrates that in germ-free mice, like in human patients, EHEC causes early colitis followed by renal failure and that systemic disease but not colitis is Stx2 dependent.
Keywords: animal models, bacterial infections, Escherichia coli, gastrointestinal diseases, germ-free, mice, pathogenicity, virulence factors
Strains of Escherichia coli that produce Shiga toxins (Stx) include isolates referred to as enterohemorrhagic Escherichia coli (EHEC), because they are associated with severe hemorrhagic colitis in human patients. In some patients, hemorrhagic colitis may progress to hemolytic uremic syndrome (HUS), a form of acute renal failure associated with hemolysis and microthrombosis, often leading to chronic renal failure and occasionally to death. Both HUS and hemorrhagic colitis have been attributed to the production of Stx by EHEC.25 Shiga toxins are macromolecular cytotoxins that are most commonly carried on complete or partial genomes of prophages of the lambdoid family, all of which share a common genetic organization and life cycle.11 Prophage integrate into and replicate with the bacterial chromosome (a process called lysogeny), and infectious phage and Stx are rarely produced. Prophage is activated by a process known as induction, resulting in entry into the lytic cycle of phage growth, excision of the prophage genome, production of infectious phage, and expression of virulence genes encoded in the prophage genome (Stx, in the case of EHEC). Enterohemorrhagic E. coli strains may produce 1 or both of 2 major forms of Stx, Stx1 and Stx2, including several Stx2 subtypes.24 Animal model and clinical studies suggest that Stx2 is the more pathogenic in vivo.8,22,26,42
In addition to Stx, a major virulence determinant of Shiga-toxigenic E. coli is believed to be the locus of enterocyte effacement (LEE). The LEE is a pathogenicity island that is present on most EHEC isolates and that is similar to the LEE of enteropathogenic E. coli. In both strains, the island encodes a type 3 secretion system (T3SS) by means of which bacterially encoded effectors are translocated into host cells. Among these effectors are products that promote formation of adherence pedestals, thought to contribute to disease progression.10
Study of the pathogenesis of Stx-mediated hemorrhagic colitis, and to a lesser extent HUS, has been hindered by the fact that few animal species other than humans are susceptible to Stx-mediated disease (reviewed in Ritchie31). Cattle and other ruminants, considered the principal source of EHEC in human outbreaks, do not develop disease due to EHEC.27 Laboratory animal models have varying responses to infection. Rabbits become infected by disease strains and in some cases develop bloody diarrhea and renal disease.28 Conventional rodents are resistant to colonization by EHEC and do not develop disease,7,32 but EHEC does colonize germ-free mice8 and mice whose microbiota has been altered by anti-biotic treatment.40,41
Enterohemorrhagic E. coli infection of germ-free or streptomycin-treated mice has been commonly used to model HUS in humans.8,29,40,41 Reports vary widely, however, concerning the outcome of mouse infections. Most publications report development of acute renal failure associated with acute renal tubular necrosis,8,40,41 but reports of other manifestations of disease in mice are inconsistent.12,37 Hemorrhagic colitis has not been described in mice. We previously showed that in germ-free mice, acute renal tubular necrosis as well as clinical disease are dependent on both Stx2 production and prophage induction.8,39 We also showed that although the rate of prophage induction is low and stable during in vitro growth, intestinal colonization results in markedly enhanced prophage induction and increased luminal Stx production.39
In our previously published studies, we investigated the role of Stx2 in bacterial colonization and acute renal failure in mice, but we did not examine colonic disease or bacterial virulence factors other than Stx2. We showed that in germ-free mice, EHEC colonizes the cecum and colon and, to a lesser extent, the ileum and that in the ileum and cecum, the bacteria are sometimes found closely associated with the epithelium. The goal of this study was to document and quantify necrotizing colitis in EHEC-infected mice, document progression of disease, and evaluate the potential roles of selected phage and bacterial-encoded genes on systemic and colonic disease.
Methods
Mice
Male and female germ-free Swiss Webster mice were raised in the University of Michigan Laboratory of Animal Medicine germ-free colony, housed in soft-sided bubble isolators, and fed autoclaved water and laboratory chow ad libitum. The University of Michigan germ-free Swiss Webster colony was originally obtained from Taconic Biosciences (Hudson, NY) and has been maintained at the University of Michigan for 12 years. In 1 experiment, MyD88/TRIF double knockout mice on a C57BL/6 background (kindly supplied by Gabriel Nunez)18 were used. Unless otherwise indicated, mice were orally inoculated with EHEC, EHEC mutants, or control bacteria at 4 to 5 weeks of age and euthanized 1 to 11 days later. Mice were infected orally with ~106 cfu of LB-cultured bacteria.
Following inoculation, mice were placed in sterile microisolator cages within the germ-free isolators. Microisolator cages were then aseptically removed from the isolators and kept in a laminar flow hood for the duration of the experiment. These mice received sterile food, water, and bedding as did the mice in the germ-free isolators, and they remained bacteriologically sterile (except for the infecting E. coli strains) through-out the course of the experiment. In some figures, archival data from previously published experiments are included for comparison. Archival data were derived from mice treated with the same protocols.1,8,39 At necropsy, body and cecal weights were determined, urine was collected for specific gravity determination, cecal contents were snap-frozen in liquid nitrogen, and tissues for histology were collected as described below. Urine specific gravity was determined with a refractometer. All animal experiments were conducted with the approval of the University of Michigan Animal Care and Use Committee.
Bacterial Strains
Enterohemorrhagic E. coli strains used were EDL933, 86-24, or 185 (kindly supplied by Alison Weiss)13,14 or their mutants. In some archival experiments, EHEC strain 104:H4 (supplied by Shannon Manning)1 and PA2 (supplied by Edward Dudley)16 were also used. Nonpathogenic E. coli control strains included K12, HS, and F129 (kindly contributed by Alison Weiss).14 Deletion mutants included EDL933Δstx2, 86-24Δstx2, EDL933ΔN, EDL933Δint, and EDL933ΔescN. They were constructed using the λ Red recombination system as previously described,8 using pKD46 as the supplier of the λ Red functions and EDL933 or 86-24 as the host bacterium. Deletion mutant strains and their properties are summarized in Supplemental Table 1.
The induction-deficient mutant, EDL933cIind1, has been previously described.39 This mutant carries a mutation in the C1 region, rendering it uninducible. It is nonpathogenic in germ-free mice.39 K12933W was constructed by transforming the 933W prophage from EDL933 into a laboratory K12 E. coli strain. This strain produced Stx2 when induced in vitro.21
For mouse inoculation, bacteria were cultured overnight on LB agar plates, inoculated into LB broth, and incubated at 37°C with shaking until OD600 reached approximately 0.8. Bacteria were diluted to approximately 1×107 cfu/ml and mice were given 0.1 ml by oral gavage. Inocula were then quantified by plate dilution to verify inoculum concentration and purity.
Ultrastructure
Approximately 1-mm3 sections of colon were immersion-fixed in 3.0% gluteraldehyde, postfixed in 1% osmium tetroxide, and embedded in epoxy resin. Ultrathin sections were stained with uranyl acetate and lead citrate and were examined with a JEOL100 CXII electron microscope.
Light Microscopy
Tissues were emersion-fixed in 10% neutral buffered formalin and embedded in paraffin, and 5-micron sections were stained with hematoxylin and eosin. For scoring, the entire colon was removed, flushed with formalin, and embedded as a “Swiss roll.” Right and left kidneys were bisected transversely or longitudinally, respectively, and both were examined. Spleens were bisected longitudinally and examined in their entirety.
Renal tubular necrosis was scored semiquantitatively from 0 to 3 according to the presence and severity of tubular epithelial necrosis, as previously described.8 Briefly, 0 = no detectable lesions, 1 = occasional necrotic tubules, 2 = acute tubular necrosis clearly evident in most microscopic fields, and 3 = severe tubular necrosis involving most of the renal cortex. Splenic hematopoietic necrosis was scored as absent (score = 0), present (score = 1, detectable loss and/or apoptosis of hematopoietic cells, often associated with neutrophilic splenitis), or severe (score = 2, no evidence of hematopoietic tissue remaining in the red pulp, often associated with lymphoid necrosis or apoptosis).
Colonic epithelial cell death and neutrophilic colitis were scored separately. For this, each 200× microscopic field was scored for the presence and severity of either epithelial cell death (necrosis or apoptosis) or neutrophilic infiltration into the lamina propria. Severity was scored from 0 (none) to 3 (severe) according to the criteria in Supplemental Table 2. For each lesion, a weighted average percentage was calculated as follows:
Immunohistochemistry
Formalin-fixed, paraffin-embedded sections were stained for activated caspase-3 using a Biocare Medical Intellipath™ immunochemical stainer. All reagents were from Biocare Medical (Concord, CA). Primary antibody was antimouse activated caspase-3 (9661-T1000) at 1:1000 dilution, secondary antibody was peroxidase-linked polyclonal rabbit antimouse, and DAB was the chromogen. Slides were counterstained with hematoxylin. All reagents and the Intellipath protocol are engineered for use in mouse tissue.
Stx2 ELISA
At necropsy, samples of cecal contents were weighed and frozen at −20°C until use. Thawed samples were pelleted and supernatants were diluted 1:20 in phosphate-buffered saline. Stx2 was quantified using the Premier STX ELISA kit according to the manufacturer’s instructions. Quantification was based on a standard curve generated using purified Stx2, as previously described.8
Statistical Analysis
Groups were compared by the Mann–Whitney U-test or the Kruskal–Wallis test for multiple groups. Categorical data were compared by chi-square analysis. All statistics and charts were calculated using GraphPad Prism software. In scatter plots, each dot represents 1 mouse and horizontal bars indicate the median. Unless otherwise indicated, mice were euthanized 1 week after inoculation. In line plots, error bars indicate the standard error of the mean.
Results
Bacterial Colonization
In all mouse groups, bacterial colonization density ranged from about 109 to 1011 cfu/g of cecal contents. Colonization by EDL933 (2.1 ± 0.4 × 1010 cfu/g) was significantly different from colonization by 86-24 (8.5 ± 1.3 × 109 cfu/g, P = .0150) and HS (2.4 ± 0.6 × 109 cfu/g, P = .0172) but was not significantly different from any other strains or mutants.
In the current study, examination of the entire length of the colon of infected mice demonstrated that, although rare, foci of close association between bacteria and colonic epithelial cells occur in mice and can be physically associated with colonic epithelial necrosis and apoptosis as well as neutrophilic colitis (Figs. 1, 2). In addition to light microscopic evidence of adherence, ultrastructural examination revealed occasional pedestal-like structures in the colon (Fig. 3). In all instances of adhesion, epithelial cells were degenerate, and both ultrastructural and histologic examination suggested that adherence may be limited to necrotic or sloughing epithelial cells. It was not possible to determine if necrosis was a cause or a consequence of bacterial adherence.
Figure 1.
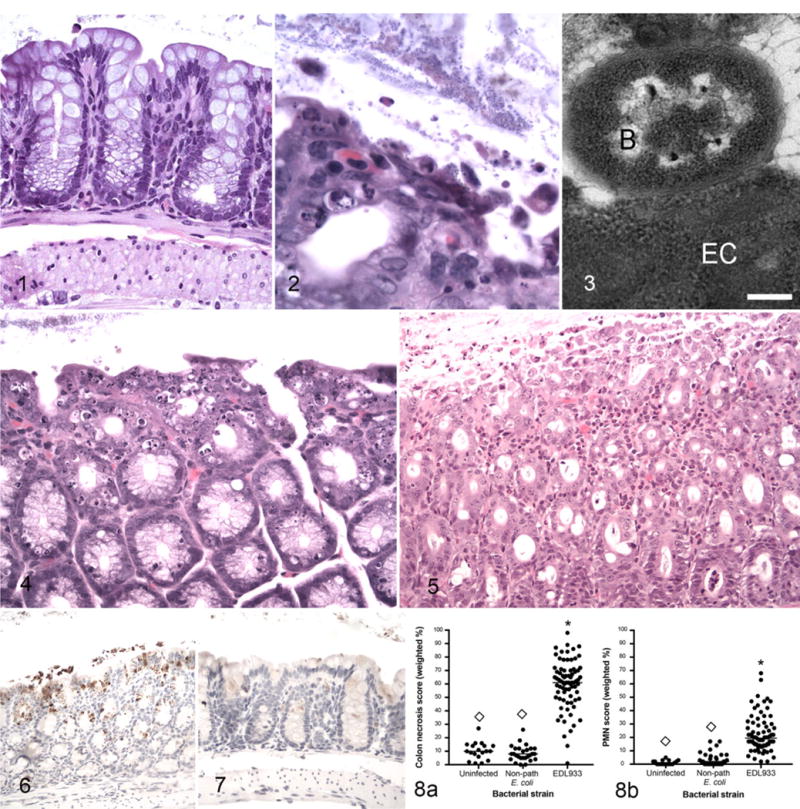
Uninfected germ-free control mouse, normal colon. Hematoxylin and eosin (HE). Figures 2–6. Enterohemorrhagic Escherichia coli (EHEC)-monocolonized mice, colon. Figure 2. A focus of colon epithelial necrosis associated with large numbers of adherent bacteria. Short bacilli adhere to both superficial and sloughed epithelial cells. HE. Figure 3. There is an adhesion-pedestal-like structure and close association between a degenerate epithelial cell (EC) and a bacterium (B). Bar = 250 nM. Transmission electron microscopy. Figure 4. Severe colonic epithelial necrosis and apoptosis compared to normal colon from a germ-free mouse (see Fig. 1). HE. Figure 5. Necrosis and sloughing of necrotic cells into the lumen, and neutrophilic infiltration in the lamina propria. HE. Figure 6. Activated caspase-3 immunoreactivity in the colonic epithelium is characterized by brown granular staining in surface cells, extending into the glandular epithelial cells, and in sloughed cells within the lumen. Immunoperoxidase-DAB. Figure 7. Uninfected germ-free mouse, colon. Caspase-3 immunoreactivity is not detectable in epithelial cells. Immunoperoxidase-DAB. Figure 8. Colonic epithelial necrosis (a) and neutrophil infiltration scores (PMN score) (b) in uninfected germ-free mice and in gnotobiotic mice monocolonized with nonpathogenic E. coli or with EHEC strain EDL933 and euthanized 1 week after bacterial colonization. *Significantly different from uninfected. ◊Significantly different from EDL933.
Colonic Epithelial Necrosis and Colitis
In contrast to morphologic evidence of bacterial adhesion, colonic epithelial necrosis and/or apoptosis was present in all EHEC-infected mice and was sometimes extensive (Fig. 4). In addition to epithelial lesions, colonic epithelial cell death in infected mice was accompanied by mild-to-moderate infiltration of neutrophils in the lamina propria (Fig. 5). Apoptosis was confirmed by immunolabeling of activated caspase-3 (Fig. 6) and not present in colon from germ-free mice (Fig. 7).
Epithelial necrosis and neutrophilic colitis were scored semiquantitatively as described in the methods. A weighted average percentage was calculated based on both severity and extent of the lesions. Both lesions were present in EHEC-monocolonized mice but not in uninfected mice or in mice infected with nonpathogenic E. coli (Fig. 8).
Systemic Disease
Mice colonized by wild-type EHEC (EDL933, 86-24, or 185) developed weight loss (infected: 14.0 ± 0.5 g vs uninfected: 29.4 ± 0.8 g; P < .0001) and dilute urine (urine specific gravity, infected: 1.011 ± 0.001 (infected) vs uninfected: 1.049 ± 0.0001; P < .001). Acute tubular necrosis developed in infected mice 5 to 7 days after inoculation (Figs. 9–11). Lesions were present in all EHEC-monocolonized mice, and scores ranged from 1 to 3 (median = 2). None of the uninfected mice or mice monocolonized by nonpathogenic E. coli had renal tubular necrosis. In addition to renal lesions, the most severely affected mice developed moderate to severe splenic necrosis principally affecting the red pulp. In some cases, hematopoietic tissue was absent (Figs. 12, 13). Splenic necrosis was detectable in 83% of infected mice and was severe in 7% (Supplemental Table 3). It was not present in uninfected mice or in mice colonized by nonpathogenic E. coli.
Figures 9–11.
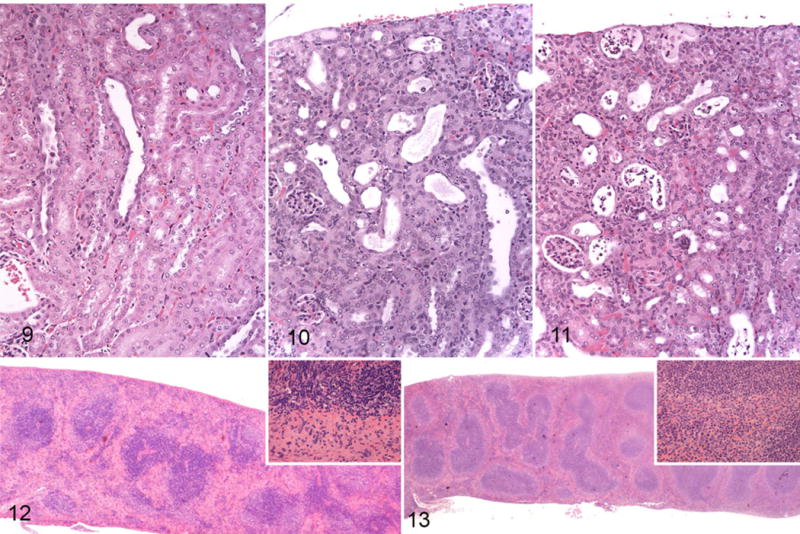
Enterohemorrhagic Escherichia coli (EHEC)-monocolonized mice, tubular necrosis, kidney. Hematoxylin and eosin (HE). Figure 9. Tubular necrosis score 1. Few dilated tubules with epithelial atrophy and rare sloughed cells. Figure 10. Score 2. Several dilated tubules with epithelial atrophy and regeneration, luminal protein, and sloughed epithelial cells. Figure 11. Score 3. Most tubules are dilated with flattened epithelium and contain sloughed epithelial cells. Figure 12. EHEC-monocolonized mouse, spleen. There is marked loss of cellularity in the red pulp. Lymphoid tissue is mildly depleted. Inset: junction between red pulp (bottom) and white pulp (top) with marked loss of hematopoietic cells and scattered karyorrhexis in the red pulp and scattered apoptosis in the white pulp. HE. Figure 13. Uninfected germ-free mouse, normal spleen. Inset: junction of red pulp (bottom) and white pulp (top). HE.
Chronology of Disease
Chronology of clinical and renal disease was documented over the course of the first 11 days of infection (Fig. 14). Loss of urine concentrating ability and morphologic evidence of renal disease gradually increased over the first 7 to 11 days of infection (Fig. 14a, b). Colonic epithelial necrosis and colitis (necrotizing colitis) peaked at 1 to 4 days PI and plateaued, gradually decreasing thereafter (Fig. 14c). Stx2 levels in the intestinal contents rose over the course of 7 days (Fig. 14d). Most mice became moribund or died by 7 days PI, at about the peak of cecal Stx2 concentration. Archival data8,39 corroborated the current study showing that clinical disease in EHEC-infected mice peaked at 5 to 7 days PI and that some mice survive acute disease (Fig. 15).
Figure 14.
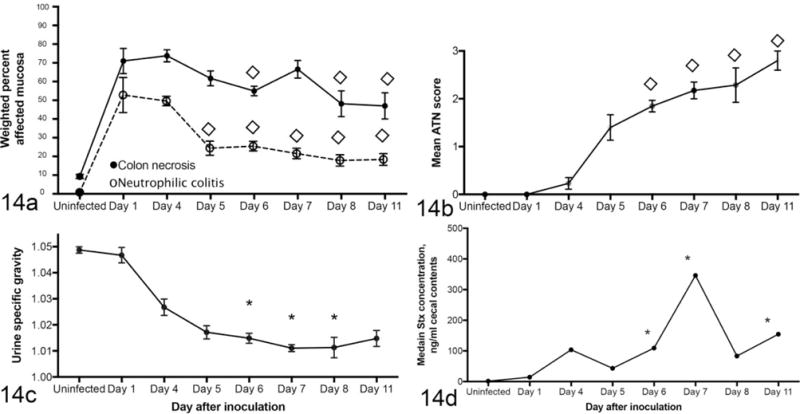
Chronology of disease in enterohemorrhagic Escherichia coli-monocolonized gnotobiotic mice. (a) Neutrophilic colitis is present 1 day after infection and plateaus thereafter. (b) Acute tubular necrosis (ATN) is detectable in some mice by day 4 PI and continues to increase in severity as urine specific gravity decreases (c). (d) Concentration of Stx in feces peaks at day 11 PI. The figure includes previously published data (days 8 and 11)8,39 as well as data from this study. *Significantly different from uninfected. ◊Significantly different from day 4.
Figure 15.
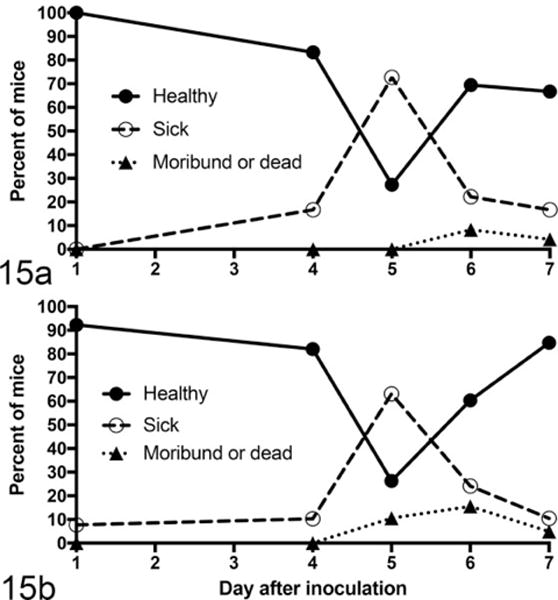
Percentage of enterohemorrhagic Escherichia coli (EHEC)-monocolonized gnotobiotic mice at each sacrifice interval that were clinically healthy, were sick (showed signs of illness but were not moribund), or had become moribund or were found dead. Clinical data are drawn from 25 experiments involving 193 mice monocolonized with EDL933 (a) or with other pathogenic EHEC strains (b). Clinically observable disease peaked at 5 days after inoculation. Some numerical data are taken from previously published studies.8,39
Stx2 Dependence of Systemic vs Intestinal Disease
We previously showed that clinical and renal disease are dependent on Stx2 production.8,39 In the current study, we used several different Stx-negative mutants to confirm that systemic disease, including renal disease, splenic necrosis, and loss of body weight, are Stx2 dependent in this model. Regardless of specific mutation, all EHEC variants that did not express Stx2, including EDL933Δstx2, EDL933ΔN, and EDL933cIind1, failed to cause systemic disease, whereas EDL933ΔescN and EDL933Δint, mutants that retained Stx2 expression, were not significantly different from wild-type EHEC (Fig. 16).
Figure 16.
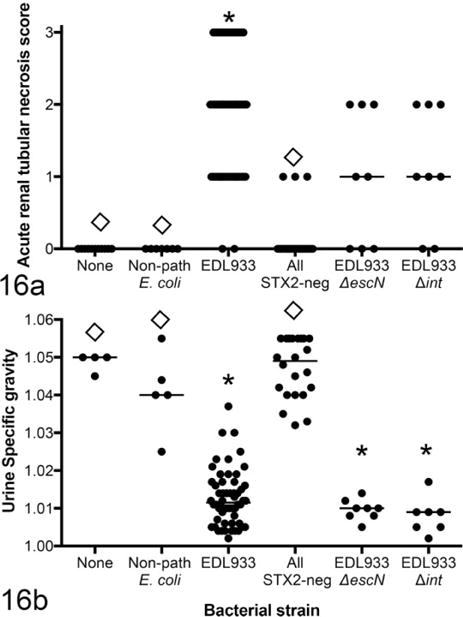
Renal disease as defined by acute tubular necrosis (a) and loss of urine concentrating ability (b) in mice monocolonized by EDL933 or by Stx-negative and Stx-positive mutants and euthanized 1 week after colonization. Stx-negative mutants include EDL933Δstx2, EDL933ΔN, and EDL933cIind1. EDL933ΔescN and EDL933Δint express Stx2. *Significantly different from uninfected mice. ◊Significantly different from EDL933 monocolonized mice. Non-path: nonpathogenic.
In contrast to systemic disease, necrotizing colitis was not entirely dependent on Stx2 production. Stx2-negative mutants induced at least some colitis, albeit somewhat less severe than the lesions induced by EDL933 (Fig. 17). Mutants that retained Stx2 expression (EDL933ΔescN and EDL933Δint) did not differ from wild-type EDL933 in their effect on necrotizing colitis (Fig. 18b, d). Thus, systemic disease (renal disease and splenic necrosis) was Stx2 dependent, but colonic disease (necrotizing colitis) was only partially dependent on Stx2.
Figure 17.
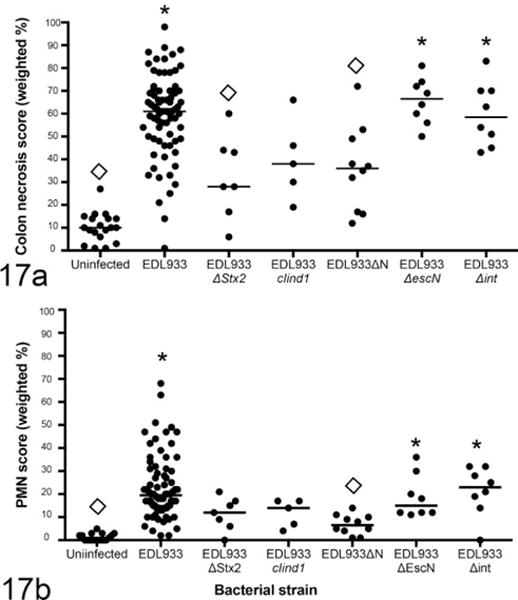
Colonic epithelial necrosis (a) and neutrophilic colitis (b) in mice monocolonized with EDL933 or mutant strains and euthanized 1 week after colonization. Loss of stx2 resulted in a decrease but not elimination of colonic lesions. Loss of escN or int had no effect. *Significantly different from uninfected mice. ◊Significantly different from EDL933 monocolonized mice.
Figure 18.
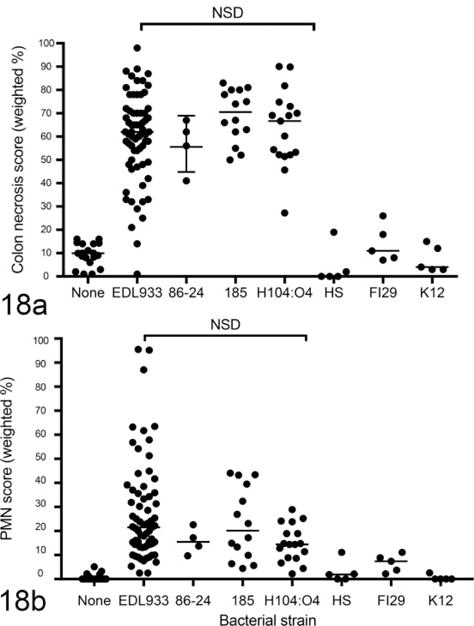
Colonic epithelial necrosis (a) and neutrophilic colitis (b) in mice monocolonized with enterohemorrhagic Escherichia coli (EHEC) strains or with nonpathogenic E. coli and euthanized 1 week after colonization. EHEC strains (EDL933, 86-24, 185, H104:O4) caused neutrophilic colitis, independent of Stx1 expression. Nonpathogenic E. coli (HS, F129, K12) did not induce colonic lesions.
One possible explanation for the incomplete dependence of necrotizing colitis on Stx2 is that Stx1 could contribute to colonic epithelial damage. EDL933 expresses both Stx1 and Stx2, and it is possible that in this model, Stx1 causes colonic epithelial necrosis but does not cause systemic disease. To evaluate the potential role of Stx1 in colitis, we first compared EHEC strains that either express Stx2 only (86-84 and O104:H4)4,17 or express both Stx1 and Stx2 (EDL933 and 185).13,14,36 There was no difference in the severity of necrotizing colitis between the strains that expressed both Stx2 and Stx1 and those that expressed only Stx2 (Fig. 18). Thus, the absence of Stx1, at least on a variable EHEC strain background, did not alter the severity of colonic disease.
Next, we evaluated the role of Stx2 in the absence of Stx1 in 2 ways. First, E. coli K12 was engineered to express Stx2 only. Necrotizing colitis induced by this strain was indistinguishable from that induced by EDL933 (Fig. 19), suggesting that Stx2 alone induces the lesions. Then, we deleted the stx2 genes from 86-84, an EHEC strain that expresses only Stx2. In this experiment, MyD88/TRIF-deficient mice were used, because absence of TLR signaling has been shown to enhance susceptibility of mice to EHEC-induced colitis.6 Colonic necrosis scores for EDL933-infected MyD88/TRIF-deficient mice were 0.94 ± 0.11, compared to 0.58 ± 0.21 in C57BL/6 mice (P = .0004). Thus, the higher scores in MyD88/TRIF-deficient mice were more likely to reveal small differences between EHEC strains than the scores in nonmutant mice. As expected, 86-24Δstx2 caused colitis that was less severe than did 86-24. However, unlike K12 in the absence of Stx2, 86-24Δstx2 induced mild colonic epithelial necrosis that was significantly greater than in uninfected mice (Fig. 19). This suggests that in contrast to nonpathogenic E. coli, at least some EHEC strains cause damage to colonic epithelial cells even in the absence of both Stx1 and Stx2 expression. In summary, all tested E. coli strains that expressed Stx2 (including EDL933, 86-24, and E. coli K12933W) caused necrotizing colitis. In the absence of Stx2, both EHEC strains EDL933 (expressing Stx1) and 86-24 (Stx1 negative) expressed mild colitis, but K12 did not cause colitis in the absence of Stx2.
Figure 19.
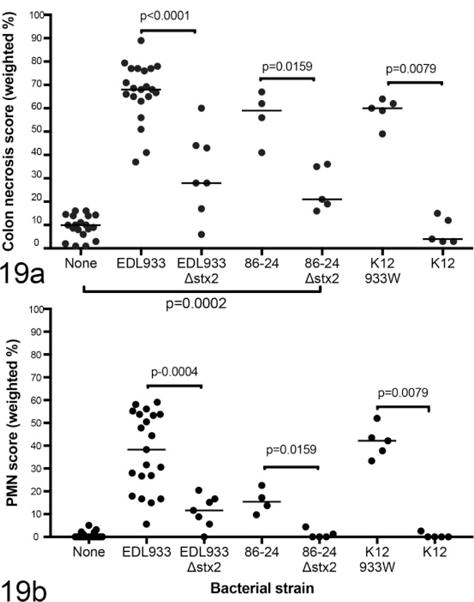
Colonic epithelial necrosis (a) and neutrophilic colitis (b) in mice monocolonized with Escherichia coli expressing Stx1, Stx2, or both Stx1 and Stx2 and euthanized 1 week after colonization. EDL933 expresses both Stx1 and Stx2, 84-24 expresses Stx2 only, and K12933W is a nonpathogenic E. coli strain engineered to express Stx2 only.
Role of the T3SS and Prophage Excision in Disease
In contrast to Stx2, T3SS function was not necessary for either systemic or intestinal disease due to EHEC (Figs. 16, 17). EDL933ΔescN, a mutant that does not express the ATPase function of the T3SS but does express Stx2, induced disease that was indistinguishable from that induced by wild-type EDL933. Like T3SS function, prophage excision was not necessary for disease induction. EDL933Δint, an excision-defective mutant, caused systemic disease of similar severity to wild-type EDL933. This mutant produces phage and Stx2, but the prophage fails to excise from the bacterial chromosome.
Discussion
The current study demonstrated that in addition to renal disease, EHEC infection in mice was associated with colonic epithelial necrosis and neutrophilic colitis (collectively referred to here as necrotizing colitis). Colonic lesions have been reported in other mouse models of EHEC2,5,37 but were incompletely described and were not quantified. In this study, we quantified both colonic epithelial necrosis/apoptosis and neutrophilic inflammation and showed that Stx2 contributed to the lesions but that mild colonic disease occurred in EHEC-monocolonized mice even in the absence of Stx2. In addition, we showed that, although rare, direct adherence of EHEC to colonic epithelial cells occurred in mice. Furthermore, as in human patients who develop HUS following EHEC-associated hemorrhagic colitis,38 EHEC-associated disease in mice progresses from acute colitis to renal failure. Thus, although mice do not show clinical signs of colitis, the progression of disease in mice, as in humans, is consistent with a pathogenesis in which damage to colonic epithelium permits translocation of Stx2 into the systemic circulation, followed by secondary damage to internal organs such as kidney and (in the mouse) spleen.
The precise mechanism of damage to colonic epithelial cells is not known and was not determined in the current study. The decreased severity of damage in mice colonized by Stx2-negative EHEC compared to wild-type EHEC indicates that Stx2 plays at least some role. Published studies support this conclusion. Zumbrun et al43 showed that intestinal epithelial cells express receptors for the Stx2B subunit and that, at least in vitro, Stx2 damages intestinal epithelial cells directly, possibly by a GB3-independent mechanism.34 Thus, there is some support for direct damage due to Stx2. Nevertheless, our finding that mild necrotizing colitis occurs even in the absence of Stx2 suggests that there are additional contributing factors.
One possible contributing factor is Stx1. The mild colitis in mice that were monocolonized with EDL933Δstx2 could be due to Stx1, which is expressed by this mutant. However, 86-24Δstx2 does not express either Stx1 or Stx2 and still causes mild colon damage that is not present in mice colonized by nonpathogenic EHEC. This suggests that Stx-independent EHEC virulence determinants may contribute to colon damage. Support for this possibility comes from published studies showing that, in vitro at least, such EHEC factors as H7 flagellin,3 hemorrhagic coli pili,20 and other Stx-independent mechanisms33 can induce pro-inflammatory mediators or cause direct epithelial damage. Identification and in vivo verification of additional colitis-inducing EHEC virulence factors will await further study.
In addition to documenting necrotizing colitis, the current study confirmed our previous work8,39 showing that systemic, HUS-like disease in EHEC-infected germ-free mice is dependent on the expression of Stx2 and that prophage induction is enhanced in vivo.39 We extended those findings to show not only that renal disease was Stx2 dependent but also that the most severely affected mice develop severe splenic necrosis, which is also Stx2 dependent. The splenic necrosis is of interest, given the association of HUS with anemia in humans. We did not document anemia in our previous study8 or in this study (not shown), but in the most severely affected mice, early stage erythropoietic cells were almost completely absent from the spleen. In human patients, anemia is generally attributed to destruction of red blood cells associated with microthrombosis. In mice infected with EHEC, microthrombosis is minimal if described at all8,30,35,41 and, in any case, would not be expected to cause necrosis of erythropoietic cells in the spleen. Thus, although the relationship between anemia in human patients and hematopoietic necrosis in mice remains unclear, our findings suggest that in mice colonized by EHEC, Stx2 may have a direct toxic effect on erythropoietic cells and possibly other splenocytes. It should be noted that, in contrast to at least 1 published study,19 we did not detect bacteremia in our mice (not shown). Thus, bacteremia was unlikely to account for the splenic lesions, although endotoxemia could not be ruled out.
In addition to Stx, we examined the potential roles of escN and int in contributing to disease. EscN is the ATPase moiety of the type III secretion system, which is dependent on EscN for its function.23 The finding that deletion of escN does not affect the outcome of EHEC infection suggests that, in germ-free mice at least, type III secretion is not essential for disease due to EHEC. As has been described by others,15 we did detect morphologic evidence of pedestal-like structures and adherence to epithelial cells both by electron and by light microscopy, but adherence appeared to be exclusively to dead or dying epithelial cells, and we were not able to determine if adherence was the cause of cell death or if bacteria simply adhere better to dead cells. Regardless, the presence of adherence-like pedestals suggests that the T3SS is functional in mouse colon but is not necessary for disease.
Loss of the int gene also did not affect disease outcome. int is a phage gene encoding a protein that catalyzes excision of the prophage from the bacterial chromosome during prophage induction.9 Loss of int does not prevent expression of phage functions including Stx2 production. The observed pathogenicity in the absence of int indicates that any loss of toxin production in the absence of prophage excision is not sufficient to prevent disease.
In summary, the current study described the progression of necrotizing colitis, renal tubular necrosis, and splenic necrosis in EHEC-infected mice, confirmed Stx2 as the cause of systemic disease in mice, and showed that a factor or factors in addition to Stx2 may contribute to colitis. We also showed that a functional T3SS was not necessary for disease due to EHEC in mice.
Supplementary Material
Acknowledgments
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Support was provided in part by PHS grant R21 AI094097 from the National Institutes of Health and by the University of Michigan Germ-Free Mouse Core.
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Supplemental material for this article is available on the Veterinary Pathology website at http://journals.sagepub.com/doi/suppl/10.1177/0300985817691582.
References
- 1.Al Safadi R, Abu-Ali GS, Sloup RE, et al. Correlation between in vivo biofilm formation and virulence gene expression in Escherichia coli O104:H4. PLoS One. 2012;7(7):e41628. doi: 10.1371/journal.pone.0041628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bekassy ZD, Calderon Toledo C, Leoj G, et al. Intestinal damage in enterohemorrhagic Escherichia coli infection. Pediatr Nephrol. 2011;26(11):2059–2071. doi: 10.1007/s00467-010-1616-9. [DOI] [PubMed] [Google Scholar]
- 3.Berin MC, Darfeuille-Michaud A, Egan LJ, et al. Role of EHEC O157:H7 virulence factors in the activation of intestinal epithelial cell NF-kappaB and MAP kinase pathways and the upregulated expression of interleukin 8. Cell Microbiol. 2002;4(10):635–648. doi: 10.1046/j.1462-5822.2002.00218.x. [DOI] [PubMed] [Google Scholar]
- 4.Bielaszewska M, Mellmann A, Zhang W, et al. Characterisation of the Escherichia coli strain associated with an outbreak of haemolytic uraemic syndrome in Germany, 2011: a microbiological study. Lancet Inf Dis. 2011;11(9):671–676. doi: 10.1016/S1473-3099(11)70165-7. [DOI] [PubMed] [Google Scholar]
- 5.Brando RJF, Miliwebsky E, Bentancor L, et al. Renal damage and death in weaned mice after oral infection with Shiga toxin 2-producing Escherichia coli strains. Clin Exper Immunol. 2008;153(2):297–306. doi: 10.1111/j.1365-2249.2008.03698.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Calderon Toledo C, Rogers TJ, Svensson M, et al. Shiga toxin-mediated disease in MyD88-deficient mice infected with Escherichia coli O157:H7. Am J Pathol. 2008;173(5):1428–1439. doi: 10.2353/ajpath.2008.071218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Conlan JW, Bardy SL, KuoLee R, et al. Ability of Escherichia coli O157:H7 isolates to colonize the intestinal tract of conventional adult CD1 mice is transient. Can J Microbiol. 2001;47(1):91–95. [PubMed] [Google Scholar]
- 8.Eaton KA, Friedman DI, Francis GJ, et al. Pathogenesis of renal disease due to enterohemorrhagic Escherichia coli in germ-free mice. Infect Immun. 2008;76(7):3054–3063. doi: 10.1128/IAI.01626-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Echols E, Guarneros G. Control of integration and excision. In: Hendrix RW, Roberts JW, Stahl FW, et al., editors. Lambda I. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1983. pp. 75–92. [Google Scholar]
- 10.Elliott SJ, Sperandio V, Giron JA, et al. The locus of enterocyte effacement (LEE)-encoded regulator controls expression of both LEE- and non-LEE-encoded virulence factors in enteropathogenic and enterohemorrhagic Escherichia coli. Infect Immun. 2000;68(11):6115–6126. doi: 10.1128/iai.68.11.6115-6126.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Friedman DI, Court DL. Bacteriophage lambda: alive and well and still doing its thing. Curr Opin Microbiol. 2001;4(2):201–207. doi: 10.1016/s1369-5274(00)00189-2. [DOI] [PubMed] [Google Scholar]
- 12.Fujii J, Kita T, Yoshida S, et al. Direct evidence of neuron impairment by oral infection with verotoxin-producing Escherichia coli O157:H- in mitomycin-treated mice. Infect Immun. 1994;62(8):3447–3453. doi: 10.1128/iai.62.8.3447-3453.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gamage SD, Patton AK, Strasser JE, et al. Commensal bacteria influence Escherichia coli O157:H7 persistence and Shiga toxin production in the mouse intestine. Infect Immun. 2006;74(3):1977–1983. doi: 10.1128/IAI.74.3.1977-1983.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gamage SD, Strasser JE, Chalk CL, et al. Nonpathogenic Escherichia coli can contribute to the production of Shiga toxin. Infect Immun. 2003;71(6):3107–3115. doi: 10.1128/IAI.71.6.3107-3115.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Girard F, Frankel G, Phillips AD, et al. Interaction of enterohemorrhagic Escherichia coli O157:H7 with mouse intestinal mucosa. FEMS Microbiol Lett. 2008;283(2):196–202. doi: 10.1111/j.1574-6968.2008.01166.x. [DOI] [PubMed] [Google Scholar]
- 16.Goswami K, Chen C, Xiaoli L, et al. Coculture of Escherichia coli O157:H7 with a nonpathogenic E. coli strain increases toxin production and virulence in a germfree mouse model. Infect Immun. 2015;83(11):4185–4193. doi: 10.1128/IAI.00663-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gunzer F, Bohn U, Fuchs S, et al. Construction and characterization of an isogenic slt-ii deletion mutant of enterohemorrhagic Escherichia coli. Infect Immun. 1998;66(5):2337–2341. doi: 10.1128/iai.66.5.2337-2341.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Harder J, Franchi L, Muñoz-Planillo R, et al. Activation of the NLRP3 inflammasome by Streptococcus pyogenes requires streptolysin O and NF-kappa B activation but proceeds independently of TLR signaling and P2X7 receptor. J Immunol. 2009;183(9):5823–5829. doi: 10.4049/jimmunol.0900444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Karpman D, Connell H, Svensson M, et al. The role of lipopolysaccharide and Shiga-like toxin in a mouse model of Escherichia coli O157:H7 infection. J Infect Dis. 1997;175(3):611–620. doi: 10.1093/infdis/175.3.611. [DOI] [PubMed] [Google Scholar]
- 20.Ledesma MA, Ochoa SA, Cruz A, et al. The hemorrhagic coli pilus (HCP) of Escherichia coli O157:H7 is an inducer of proinflammatory cytokine secretion in intestinal epithelial cells. PLoS One. 2010;5(8):e12127. doi: 10.1371/journal.pone.0012127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Livny J, Friedman DI. Characterizing spontaneous induction of Stx encoding phages using a selectable reporter system. Mol Microbiol. 2004;51(6):1691–1704. doi: 10.1111/j.1365-2958.2003.03934.x. [DOI] [PubMed] [Google Scholar]
- 22.Manning SD, Motiwala AS, Springman AC, et al. Variation in virulence among clades of Escherichia coli O157:H7 associated with disease outbreaks. Proc Natl Acad Sci U S A. 2008;105(12):4868–4873. doi: 10.1073/pnas.0710834105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mellies JL, Barron AMS, Carmona AM. Enteropathogenic and enterohemorrhagic Escherichia coli virulence gene regulation. Infect Immun. 2007;75(9):4199–4210. doi: 10.1128/IAI.01927-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Melton-Celsa AR. Shiga toxin (Stx) classification, structure, and function. Microbiol Spectr. 2014;2(4) doi: 10.1128/microbiolspec.EHEC-0024-2013. EHEC–0024–2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Melton-Celsa A, Mohawk K, Teel L, et al. Pathogenesis of Shiga-toxin producing Escherichia coli. Curr Top Microbiol Immunol. 2012;357:67–103. doi: 10.1007/82_2011_176. [DOI] [PubMed] [Google Scholar]
- 26.Neupane M, Abu-Ali GS, Mitra A, et al. Shiga toxin 2 overexpression in Escherichia coli O157:H7 strains associated with severe human disease. Microb Pathog. 2011;51(6):466–470. doi: 10.1016/j.micpath.2011.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nguyen Y, Sperandio V. Enterohemorrhagic E. coli (EHEC) pathogenesis. Front Cell Infect Microbiol. 2012;2:290. doi: 10.3389/fcimb.2012.00090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Panda A, Tatarov I, Melton-Celsa AR, et al. Escherichia coli O157:H7 infection in Dutch belted and New Zealand white rabbits. Comp Med. 2010;60(1):31–37. [PMC free article] [PubMed] [Google Scholar]
- 29.Paton JC, Paton AW. Survival rate of mice after transient colonization with Escherichia coli clones carrying variant Shiga-like toxin type II operons. Microb Pathog. 1996;20(6):377–383. doi: 10.1006/mpat.1996.0035. [DOI] [PubMed] [Google Scholar]
- 30.Porubsky S, Federico G, Müthing J, et al. Direct acute tubular damage contributes to Shigatoxin-mediated kidney failure. J Pathol. 2014;234(1):120–133. doi: 10.1002/path.4388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ritchie JM. Animal models of enterohemorrhagic Escherichia coli infection. Microbiol Spectr. 2014;2(4) doi: 10.1128/microbiolspec.EHEC-0022-2013. EHEC-0022-2013. [DOI] [PubMed] [Google Scholar]
- 32.Robinson CM, Sinclair JF, Smith MJ, et al. Shiga toxin of enterohemorrhagic Escherichia coli type O157:H7 promotes intestinal colonization. Proc Natl Acad Sci U S A. 2006;103(25):9667–9672. doi: 10.1073/pnas.0602359103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Roxas JL, Koutsouris A, Bellmeyer A, et al. Enterohemorrhagic E. coli alters murine intestinal epithelial tight junction protein expression and barrier function in a Shiga toxin independent manner. Lab Invest. 2010;90(8):1152–1168. doi: 10.1038/labinvest.2010.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schüller S, Frankel G, Phillips AD. Interaction of Shiga toxin from Escherichia coli with human intestinal epithelial cell lines and explants: Stx2 induces epithelial damage in organ culture. Cell Microbiol. 2004;6(3):289–301. doi: 10.1046/j.1462-5822.2004.00370.x. [DOI] [PubMed] [Google Scholar]
- 35.Shimizu K, Tanaka K, Akatsuka A, et al. Induction of glomerular lesions in the kidneys of mice infected with vero toxin-producing Escherichia coli by lipopolysaccharide injection. J Infect Dis. 1999;180(4):1374–1377. doi: 10.1086/315008. [DOI] [PubMed] [Google Scholar]
- 36.Strockbine NA, Marques LR, Newland JW, et al. Two toxin-converting phages from Escherichia coli O157:H7 strain 933 encode antigenically distinct toxins with similar biologic activities. Infect Immun. 1986;53(1):135–140. doi: 10.1128/iai.53.1.135-140.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Taguchi H, Takahashi M, Yamaguchi H, et al. Experimental infection of germ-free mice with hyper-toxigenic enterohaemorrhagic Escherichia coli O157:H7, strain 6. J Med Microbiol. 2002;51(4):336–343. doi: 10.1099/0022-1317-51-4-336. [DOI] [PubMed] [Google Scholar]
- 38.Tserenpuntsag B, Chang HG, Smith PF, et al. Hemolytic uremic syndrome risk and Escherichia coli O157:H7. Emerg Infect Dis. 2005;11(12):1955–1957. doi: 10.3201/eid1112.050607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tyler JS, Beeri K, Reynolds JL, et al. Prophage induction is enhanced and required for renal disease and lethality in an EHEC mouse model. PLoS Pathog. 2013;9(3):e1003236. doi: 10.1371/journal.ppat.1003236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wadolkowski EA, Burris JA, O‘Brien AD. Mouse model for colonization and disease caused by enterohemorrhagic Escherichia coli O157:H7. Infect Immun. 1990;58(8):2438–2445. doi: 10.1128/iai.58.8.2438-2445.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wadolkowski EA, Sung LM, Burris JA, et al. Acute renal tubular necrosis and death of mice orally infected with Escherichia coli strains that produce Shiga-like toxin type II. Infect Immun. 1990;58(12):3959–3965. doi: 10.1128/iai.58.12.3959-3965.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zangari T, Melton-Celsa AR, Panda A, et al. Enhanced virulence of the Escherichia coli O157:H7 spinach-associated outbreak strain in two animal models is associated with higher levels of Stx2 production after induction with ciprofloxacin. Infect Immun. 2014;82(12):4968–4977. doi: 10.1128/IAI.02361-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zumbrun SD, Hanson L, Sinclair JF, et al. Human intestinal tissue and cultured colonic cells contain globotriaosylceramide synthase mRNA and the alternate Shiga toxin receptor, globotetraosylceramide. Infect Immun. 2010;78(11):4488–4499. doi: 10.1128/IAI.00620-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.