

Abstract
We previously developed a model of opioid-induced neuroplasticity in the peripheral terminal of the nociceptor that could contribute to opioid-induced hyperalgesia, type II hyperalgesic priming. Repeated administration of mu-opioid receptor (MOR) agonists, such as DAMGO, at the peripheral terminal of the nociceptor, induces long-lasting plasticity expressed, prototypically as opioid-induced hyperalgesia and prolongation of prostaglandin-E2-induced hyperalgesia. In this study, we evaluated the mechanisms involved in the maintenance of type II priming. Opioid receptor antagonist, naloxone, induced hyperalgesia in DAMGO-primed paws. When repeatedly injected, naloxone induced hyperalgesia, and hyperalgesic priming, supporting the suggestion that maintenance of priming involves changes in mu-opioid receptor (MOR) signaling. However, the knockdown of MOR with oligodeoxynucleotide antisense did not reverse priming. Mitogen-activated protein kinase (MAPK) and focal adhesion kinase, which are involved in the Src signaling pathway, previously implicated in type II priming, also inhibited the expression, but not maintenance of priming. However, when Src and MAPK inhibitors were co-administered, type II priming was reversed, in male rats. A second model of priming, latent sensitization, induced by complete Freund’s adjuvant (CFA) was also reversed, in males. In females, the inhibitor combination was only able to inhibit the expression and maintenance of DAMGO-induced priming when knockdown of G-protein-coupled estrogen receptor 30 (GPR30) in the nociceptor was performed. These findings demonstrate that the maintenance of DAMGO-induced type II priming and latent sensitization is mediated by an interaction between, Src and MAP kinases, which in females is GPR30 dependent.
Keywords: hyperalgesic priming, hyperalgesia, Src, MAPK, chronic pain, GPR30, latent sensitization
Introduction
Chronic use of opioids has been shown to produce neuroplastic changes in peripheral sensory neurons such that opioid analgesics come to induce hyperalgesia [8; 21; 37; 46; 56], opioid-induced hyperalgesia (OIH). We recently developed a model of OIH, referred to as type II hyperalgesic priming [9], in which repeated administration of a mu-opioid receptor (MOR) agonist, prototypically DAMGO, at the peripheral terminal of the nociceptor, induces mechanical hyperalgesia, and a long lasting increased response to prohyperalgesic mediators such as prostaglandin E2 (PGE2)-induced hyperalgesia, in male and female rats [9]. Latent sensitization is a model similar to type II priming and, was initially found to develop in the setting of opioid-induced hyperalgesia, triggered by the repeated administration of opiates like heroin, morphine [19; 47], fentanyl [12; 64; 65] and remifentanil [18]. Recently, it has been suggested that administration of complete Freund’s adjuvant (CFA) in the rat hindpaw also induces latent sensitization, since the CFA-induced peripheral inflammation causes an increase in the constitutive MOR activity in the spinal cord, as a consequence of the augmented release of endogenous opioids [22; 57]. Thus, in both models, DAMGO-induced type II priming [9] and CFA-induced latent sensitization [22; 57] share similar features, including production of hyperalgesia in response an opioid receptor antagonist and their long lasting permanence [3; 22; 57]. However, the mechanism responsible for the maintenance of these primed states is unknown.
Regarding the mechanisms of maintenance of neuroplasticity, in contrast to type I priming [27], type II is not reversed by a protein translation inhibitor [9; 10]. In fact, so far no treatment has been able to reverse this type II condition, i.e., interrupt its maintenance. Thus, inhibitors of second messengers such as H-89 (a protein kinase A inhibitor [9]), SU 6656 (a Src inhibitor [9]), U0126 (MAPK inhibitor) or the focal adhesion kinase (FAK) inhibitor produced transient inhibition of priming, in male rats.
Based on studies in non-neuronal cells demonstrating that the simultaneous inhibition of Src and MAPK/ERK can reverse plastic changes in cell function [25; 26; 59], we investigated if the administration of inhibitors of Src and MAPK would affect the maintenance of type II hyperalgesic priming, since these two molecules have been shown to play a role, in series [44; 52; 53] or parallel [36; 75], in signaling pathways. Moreover, considering previous reports showing marked differences between the sexes in the mechanisms involved in models of chronic pain [15; 30; 40; 41], we performed our experiments in male and female rats.
Methods
Animals
Experiments were performed on 230–280 g male and female Sprague–Dawley rats (Charles River Laboratories, Hollister, CA, USA). Experimental animals were housed in a controlled environment in the animal care facility at the University of California, San Francisco, under a 12-h light/dark cycle. Food and water were available ad libitum. The experimental protocols were approved by the Institutional Animal Care and Use Committee at UCSF and adhered to the guidelines of the American Association of Laboratory Animal Care, the National Institutes of Health (NIH), and the Committee for Research and Ethical Issues of the International Association for the Study of Pain (IASP), for the use of animals in research. During the design of experiments, all efforts were made to minimize the number of animals used and their suffering.
Nociceptive threshold testing
Mechanical nociceptive threshold was quantified using an Ugo Basile Analgesymeter® (Randall-Selitto paw-withdrawal test, Stoelting, Chicago, IL), which applies a linearly increasing mechanical force to the dorsum of the rat’s hindpaw, as previously described [32; 72; 73]. Rats were placed in cylindrical acrylic restrainers designed to provide adequate comfort and ventilation, allow extension of the hind leg from lateral parts in the cylinder, and minimize restraint stress. To acclimatize rats to the testing procedure, they were placed in the restrainer for 1 h prior to starting each experiment and for 30 min prior to experimental manipulations. The nociceptive threshold was defined as the force, in grams, at which the rat withdrew its paw. Baseline paw-pressure nociceptive threshold was defined as the mean of the three readings taken before the test agents were injected. Each paw was treated as an independent measure, and each experiment performed on a separate group of rats.
Drugs and their administration
The chemicals used in this study were: PGE2 (a hyperalgesic agent that sensitizes nociceptors directly), complete Freund’s adjuvant (CFA; [57]), DAMGO [[D-Ala2, N-Me-Phe4, Gly5-ol]-enkephalin acetate salt, a MOR agonist; [9]], naltrexone (NTX, a MOR antagonist; [57]), naloxone (a MOR antagonist), FAK inhibitor 14 (focal adhesion kinase inhibitor), U0126 (MAPK/ERK inhibitor), and SU 6656 [a Src family kinase inhibitor; [9; 10]], all from Sigma-Aldrich (St. Louis, MO, USA).
The stock solution of PGE2 (1 μg/μL) was prepared in 10% ethanol and additional dilutions made with physiological solution (0.9% NaCl), yielding a final ethanol concentration <1%. DAMGO, naloxone, and FAK inhibitor were dissolved in saline. All other drugs were dissolved in 100% DMSO (Sigma-Aldrich) and further diluted in saline containing 2% Tween 80 (Sigma-Aldrich). The final concentration of DMSO and Tween 80 was ~2%. All the intradermal administration of the drugs on the dorsum of the hindpaw was performed using a 30-gauge hypodermic needle adapted to a 50 μL Hamilton syringe. SU 6656 and U0126 were diluted to a concentration of 1 μg/2 μL, and the combination of these inhibitors was injected by adding 2 μL of each of them, into the syringe, separated by a bubble of air, to avoid mixing in the same syringe. The administration of all drugs, including the combination of SU 6656 and U0126, except PGE2, DAMGO or naloxone, was preceded by a hypotonic shock to facilitate cell permeability to these agents (1 μL of distilled water separated by a bubble of air to avoid mixing in the same syringe), to facilitate the entry of compounds into the nerve terminal [14; 17].
Oligodeoxynucleotide antisense to mu-opioid receptor and estrogen receptors alpha, beta and, GPR30 mRNA
To investigate whether the mu-opioid receptor (MOR) plays a role in the maintenance of type II priming, in male rats, and whether the estrogen receptor (ER) subtype alpha (ER-α), beta (ER-β) or G-protein-coupled receptor 30 (GPR30) regulates the effect of a combination of Src and MAPK inhibitors on the expression and maintenance of type II priming in female rats, oligodeoxynucleotides (ODN) antisense (AS) for MOR and ER-α mRNA were used [4; 7; 24; 31; 43; 49]. The AS-ODN sequence for MOR was, 5′-CGC-CCC-AGC-CTC-TTC-CTC-T-3′, for ER-α, 5′-CAT GGT CAT GGT CAG-3, for ER-β, 5′-GAA TGT CAT AGC TGA-3′, and for GPR30, 5′- ATG TTC AGA GAG GTC CCC AG-3′ (Invitrogen Life Technologies, Carlsbad, CA, USA), were directed against a unique region of mu-opioid receptor or estrogen receptor alpha sequence, in the rat [UniProtKB database entry P33535 (OPRM_RAT) antisense sequence should block translation and downregulate the gene expression of all 8 known isoforms (MOR); GeneBank accession numbers NM_012689.1 (ER-α), NM_012754.1 (ER-β) and NM_133573 (GPR30)]. The ODN mismatch (MM) sequences, 5′-CGC-CCC-GAC-CTC-TTC-CCT-T-3′ for MOR, 5′-ATC GTG GAT CGT GAC-3′ for ERα, for ER-β, and 5′-AGG TCC AGA AAG ATG CCA AG-3′ for GPR30 were a scrambled version of the antisense sequence that has the same base pairs and GC ratio, but the order was scrambled, with little or no homology to any mRNA sequences posted at GeneBank.
Before use, ODNs were reconstituted in nuclease-free 0.9% NaCl and then administered intrathecally at a dose of 2 μg/μL in a volume of 20 μL. Type II priming was induced by repeated (hourly × 4) intradermal injections of DAMGO (1 μg) and, 24 hrs later MM or AS-ODN was injected, for 5 consecutive days, when at the 6th day (around 17 hrs after the last injection of ODN), PGE2, [in MM or AS-ODN for MOR-treated groups] or a combination of Src and MAPK inhibitors and/or PGE2, [in MM or AS-ODN for ER-α, ER-β or GPR30-treated groups] were intradermally injected, and the presence of mechanical hyperalgesia evaluated. As described previously [1], rats were anesthetized with isoflurane (2.5% in O2), and ODN injected using a microsyringe with a 30-gauge needle, inserted into the subarachnoid space, between the L4 and L5 vertebrae. A total of 40 μg of ODN, in a volume of 20 μL, was then injected. The intrathecal site of injection was confirmed by a sudden flick of the rat’s tail, a reflex that is evoked by subarachnoid space access and bolus injection [58]. Animals regained consciousness approximately 1 minute after completion of the injection. The use of AS-ODN to manipulate the expression of proteins, essential for their role in nociceptor sensitization, is well supported by previous studies by others [63; 69–71], as well as our group [7; 9; 10; 13; 31; 60].
MOR agonist DAMGO induced changes in nociceptor function
We had shown previously that, when the selective MOR agonist DAMGO was repeatedly injected at the site of mechanical nociceptive threshold testing, it produces changes in nociceptor function such that it no longer produces anti-hyperalgesia, but now produces mechanical hyperalgesia [2; 3; 9]. The repeated injection of DAMGO also induces a latent state of hyper-responsiveness to subsequent injection of pro-algesic mediators [9; 42], referred to as type II hyperalgesic priming [9]. This model of neuroplasticity is expressed as prolongation of the mechanical hyperalgesia induced by PGE2, lasting at least 4 hrs, as opposed to the injection of PGE2 in naive paws, in which hyperalgesia fully dissipated by 2 hrs [5]. To investigate the changes in nociceptor function produced by previous repeated injection of DAMGO or naloxone—measured as prolonged response to a hyperalgesic mediator at a point in time when the mechanical nociceptive threshold was not different from pre-DAMGO or naloxone baseline—PGE2 (100 ng) was injected at the same site and the hyperalgesia, evaluated after 30 min and again at 4 hrs. The presence of hyperalgesia at the 4th h is characteristic of priming [6; 9–11; 28]. To investigate the second messengers involved in the expression of the neuroplasticity induced by repeated exposure to MOR agonist DAMGO, inhibitors were administered 10 min before the injection of PGE2 in the primed paw (inhibition protocol). If the inhibitors were able to inhibit the expression of type II priming, we evaluated their role in the maintenance of the neuroplasticity, when PGE2 was injected again 10–15 days later, or at a time point when the inhibitors were no longer present (reversal protocol, 30 days). Importantly, although the activation of spinal descending systems modulates, in parallel, the response to noxious stimuli in both hind paws, our measurements were performed at the peripheral site of the nociceptor, where the phenomenon (neuroplasticity produced by repeated injections of DAMGO) is expressed.
Complete Freund’s adjuvant (CFA) model of chronic pain (latent sensitization) in male rats
Inflammation (e.g., induced by complete Freund’s adjuvant [CFA]) produces constitutive MOR activity that chronically suppressed nociceptive signaling in the spinal cord for months [22; 57]. Pharmacological blockade during the post-hyperalgesia state with MOR inverse agonists, such as naltrexone, reinstated central pain sensitization [22; 57], a phenomenon referred to as “latent pain sensitization” [12; 18] or “latent sensitization” [48; 57]. We used CFA-induced latent sensitization to evaluate if its maintenance shares the second messenger signaling pathway as MOR agonist-induced type II priming. Immediately following baseline assessment of mechanical nociceptive thresholds, male rats were anesthetized with isoflurane (2.5% in O2) and injected with CFA (50 μL, non-diluted) subcutaneously into the plantar (i.pl.) surface in one hindpaw [57]. A 25 gauge needle was inserted at an oblique angle from the heel in the middle of the hindpaw [57]. Mechanical nociceptive threshold was measured by Randall-Selitto paw-withdrawal test, on the dorsum of the hindpaws, on days 1, 3, 5, 7, 14, 21, 28 after the CFA injection. The mechanical nociceptive threshold decreased dramatically on day 1 and progressively returned to baseline values, at day 28. On day 28, rats were anesthetized with isoflurane (2.5% in O2), and a MOR antagonist naltrexone (NTX) or a combination of Src and MAPK inhibitors followed 5 min later by NTX were injected using a microsyringe with a 30-gauge needle, inserted into the subarachnoid space, between the L4 and L5 vertebrae. The combination of Src (SU 6656; 1 μg/5 μL) and MAPK (U0126; 1 μg/5 μL) inhibitors (final volume of 10 μL) or a total of 1 μg of NTX (in a volume of 20 μL or 10 μL when injected 5 min after the combination of Src and MAPK inhibitors), was then injected. NTX was diluted in sterile saline, and the combination of Src and MAPK inhibitors was diluted in 100% DMSO (Sigma-Aldrich) and further diluted in saline containing 2% Tween 80 (Sigma-Aldrich). The intrathecal site of injection was confirmed by a sudden flick of the rat’s tail, a reflex that is evoked by subarachnoid space access and bolus injection [58]. The mechanical nociceptive threshold was evaluated at 15, 30, 45, 60, 90 and 120 min after NTX injection [57].
Statistics
All behavioral data are presented as mean ± standard error of the mean (SEM) of N independent observations. Statistical comparisons were made using GraphPad Prism 5.0 statistical software (GraphPad Software, Inc., La Jolla, CA). A p-value < 0.05 was considered statistically significant. In experiments, the dependent variable was changed in mechanical threshold paw-withdrawal, expressed as percentage change from baseline. No significant difference in the mechanical nociceptive threshold was observed before the repeated (hourly × 4) injections of naloxone or DAMGO and immediately before injection of naloxone or inhibitors of second messengers followed 10 min later by PGE2 (average mechanical nociceptive threshold before priming stimuli: 125.2 ± 1.3 g for male and 122.6 ± 1.0 g for female rats; average mechanical nociceptive threshold before naloxone, inhibitors or PGE2 injection: 124.8 ± 1.2 g for male and 122.8 ± 1.1 g for female rats; N = 90 paws (males) and 120 paws (females); paired Student’s t-test, t89 = 0.0874, p = 0.9307 [males] and t119 = 0.1838, p = 0.8549 [females]). Of note, where ODN treatments were performed, we used 6 rats (12 paws) for each group; the left and right hindpaws were used and considered as independent subjects [29]. Also, 12 male rats received an intraplantar injection of CFA in one hindpaw, but both paws were evaluated [57]. The total number of rats used in this study was 96 males and 66 females. As specified in the figure legends, one- or two-way repeated-measures analysis of variance (ANOVA), followed by Bonferroni post-hoc test, was performed to compare the mechanical hyperalgesia induced by naloxone, naltrexone, PGE2 or the prolongation of the PGE2-induced hyperalgesia, with the control (vehicle)- and inhibitor-treated groups, at the indicated time points.
Results
The MOR inverse agonist naloxone induces hyperalgesia in DAMGO-primed paws in male rats
We have shown that the repeated (hourly × 4) intradermal injections of an MOR agonist DAMGO induced hyperalgesia, after the 4th injection and, prolongation of PGE2-induced hyperalgesia [9]. To test if this neuroplasticity is dependent on the MOR, we tested if naloxone would produce hyperalgesia in male rats primed 1 week before, with repeated exposure to DAMGO. A single intradermal injection of naloxone (1 μg) induced a robust decrease in the mechanical nociceptive threshold, observed at 30 min and 1 h after injection, in DAMGO-primed (Fig. 1A, black bar) but not in vehicle-treated paws (Fig. 1A, dotted bars), indicating that naloxone, can reinstate the hyperalgesia in DAMGO-primed paws.
Figure 1. Naloxone induces hyperalgesia in DAMGO-primed paws and after repeated exposure to naloxone, in male rats.
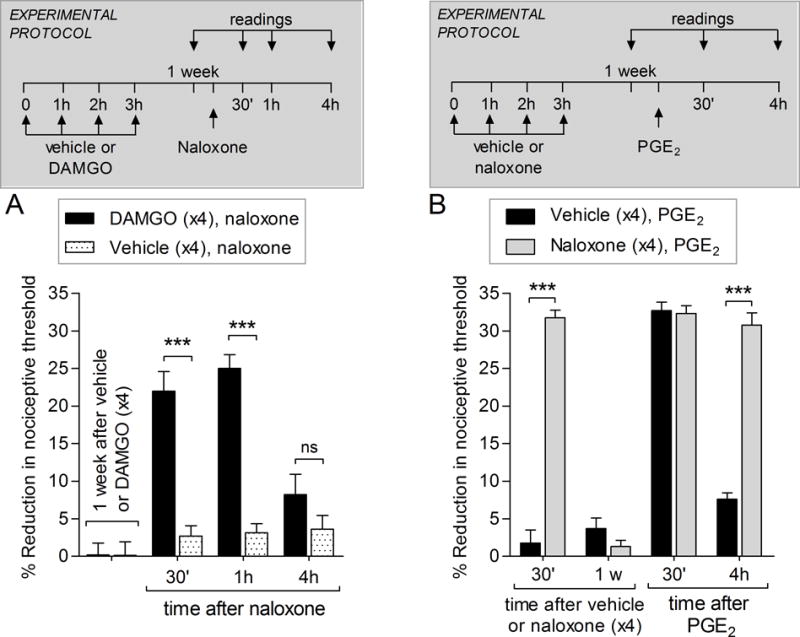
A. Male rats received repeated (hourly x4) intradermal injection of vehicle (5 μL) or DAMGO (1 μg) on the dorsum of the hindpaw. One week later (1 w), naloxone (1 μg) was injected at the same site and, the mechanical nociceptive threshold, evaluated. In the group primed by repeated injections of DAMGO, naloxone induced robust mechanical hyperalgesia (black bars), observed 30 min and 1 h after its injection (F1,30; *** p < 0.0001, when DAMGO-primed and the vehicle groups that received naloxone are compared; two-way repeated-measures ANOVA followed by Bonferroni post hoc test), indicating that naloxone, an MOR inverse agonist, can reinstate the hyperalgesia in DAMGO-primed paws. (N = 6 paws per group)
B. A different group of male rats received repeated (hourly × 4) intradermal injections of vehicle (5 μL) or naloxone (1 μg) on the dorsum of the hindpaw. Thirty minutes after the 4th injection of vehicle or naloxone, the mechanical nociceptive thresholds were evaluated, and significant mechanical hyperalgesia was observed in the naloxone-treated group (F1,10 = 111.76; *** p < 0.0001, when the naloxone- and the vehicle-treated groups are compared; two-way repeated-measures ANOVA followed by Bonferroni post hoc test). One week later, when PGE2 (100 ng) was injected, at the same site as naloxone or vehicle, mechanical hyperalgesia was observed 30 min after its injection in both groups. In the group that received repeated injections of naloxone, PGE2-induced hyperalgesia was still present at the 4th h after injection (F1,20 = 75.18; *** p < 0.0001, when naloxone- and vehicle-treated groups are compared; two-way repeated-measures ANOVA followed by Bonferroni post hoc test), suggesting that changes in MOR signaling maintain the nociceptor plasticity. (N = 6 paws per group)
Repeated exposure to naloxone induces hyperalgesia and hyperalgesic priming
The induction of hyperalgesia by naloxone, which does not induce hyperalgesia in naïve paws [3], in DAMGO-primed paws suggested that DAMGO (hourly × 4), might have induced changes in MOR signaling.
To determine if naloxone, which also acts as an inverse agonist on MOR, can also induce type II priming, we tested if its repeated injection also induces hyperalgesia and/or prolongation of PGE2-induced hyperalgesia. Male rats received repeated (hourly × 4) intradermal injections of naloxone. Thirty min after the 4th injection, a robust decrease in the mechanical nociceptive threshold (Fig. 1B, gray bar) was observed, similar to the effect of MOR agonist DAMGO [9]. One week after the repeated exposure to vehicle (black bars) or naloxone (gray bars), PGE2 was injected at the same site. Mechanical hyperalgesia was observed in both groups 30 min after the injection of PGE2 (Fig. 1B). The prolongation of PGE2-induced hyperalgesia was, however, observed only in the group previously treated with repeated injection of naloxone (Fig. 1B, gray bar). This data supports the idea that changes in MOR contribute to type II hyperalgesic priming.
Knockdown of MOR does not reverse DAMGO-induced type II priming in male rats
To determine if the presence of type II priming is due to changes in the MOR, affecting its downstream signaling pathway, we evaluated whether the knockdown of the mu-opioid receptor (MOR) in primed nociceptors would affect the expression of type II priming induced by repeated (hourly × 4) injections of DAMGO. Type II priming was induced in male rats and, 24 hrs later, intrathecal treatment with ODN mismatch (MM) or antisense (AS) for MOR mRNA was administered daily for 5 consecutive days. On the 6th day, ~17 hrs after the last ODN injection, PGE2 was injected in the dorsum of the hindpaw. Treatment with MOR AS-ODN did not affect the prolongation of PGE2-induced hyperalgesia, indicating that MOR does not play a role in the expression of type II priming (Fig. 2). In addition, when we injected a single dose of DAMGO, 7 days after the last injection of MM-ODN or AS-ODN, it produced hyperalgesia in both groups, indicating that MOR did not participate in the maintenance of type II priming. The treatment with AS-ODN for MOR also did not affect the magnitude of PGE2 hyperalgesia [43].
Figure 2. Mu-opioid receptor does not play a role in the expression of type II priming.
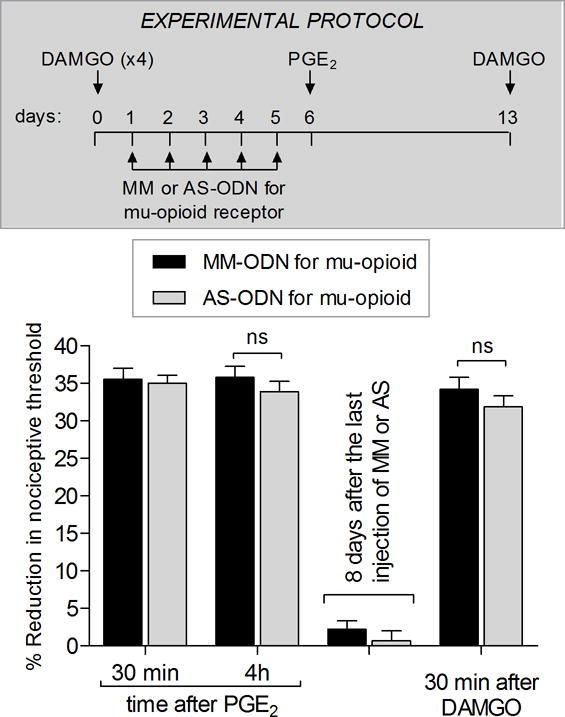
Male rats received repeated (hourly × 4) intradermal injections of DAMGO (1 μg) on the dorsum of the hindpaw and, 24 hrs after the last injection of DAMGO, treatment with intrathecal injections of MM-ODN (40 μg/20 μL/day; black bars) or AS-ODN (40 μg/20 μL/day; white bars) for mu-opioid receptor mRNA was performed once a day, for 5 consecutive days. On the sixth day, ~17 hrs after the last injection of MM-ODN or AS-ODN, PGE2 (100 ng) was injected on the dorsum of the hindpaw and, the mechanical nociceptive threshold, evaluated 30 min and 4 hrs later. PGE2-induced mechanical hyperalgesia was present at 30 min and 4 hrs, in both MM-ODN- and AS-ODN-treated groups, indicating that mu-opioid receptor does not play a role in the expression of type II priming (F1,22 = 0.22, p = 0.6410, ns, when the MM-ODN and AS-ODN-treated groups are compared; two-way repeated-measures ANOVA followed by Bonferroni post hoc test). To determine whether the treatment with AS-ODN for mu-opioid receptor affected the maintenance of type II priming, we injected a single dose of DAMGO (1 μg, intradermally) 7 days after the last injection of the ODNs. In both groups DAMGO induced hyperalgesia, indicating the presence of type II priming (F1,22 = 2.91, p = 0.1018, ns, when the MM-ODN and AS-ODN-treated groups are compared; two-way repeated-measures ANOVA followed by Bonferroni post hoc test). (N = 12 paws per group)
Second messengers involved in the expression and maintenance of type II priming in male rats
In our previous study of DAMGO-induced priming, inhibitors of PKA (H-89) and Src (SU 6656) [9] inhibited the expression of priming in male rats. However, these inhibitors did not affect the maintenance of type II priming [9]. Here, we evaluated the participation of mitogen-activated protein kinase (MAPK) and focal adhesion kinase (FAK), two kinases involved in the signaling pathways in which PKA and Src participate [20; 35; 50; 51]. We found that treatment with MAPK inhibitor U0126, 10 min before PGE2, inhibited the expression of type II priming (Fig. 3A). However, when PGE2 was injected again, 10 days after the MAPK inhibitor, the prolongation of PGE2-induced hyperalgesia was present, indicating that priming has not been reversed (Fig. 3B). When we evaluated the participation of FAK in the expression of type II priming, we found that treatment with a FAK inhibitor only partially attenuated the hyperalgesia induced by PGE2 at 30 min and 4 hrs after its injection (Fig. 3C), showing a small role of FAK in the expression of type II priming.
Figure 3. Second messengers involved in the expression of type II priming: MAPK and FAK.
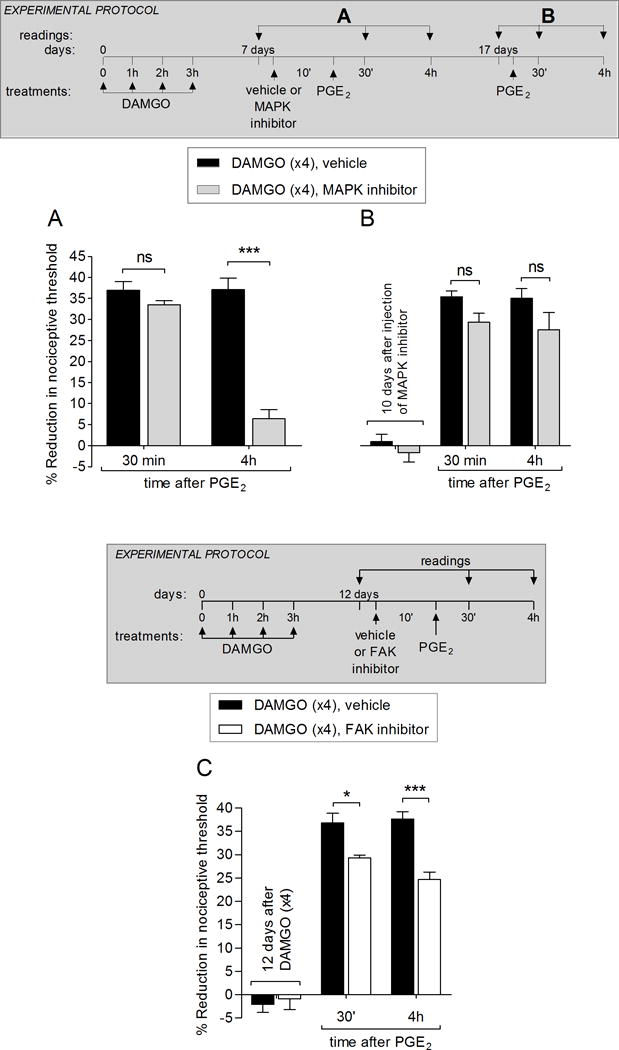
A. Male rats received repeated (hourly × 4) intradermal injections of DAMGO (1 μg) on the dorsum of the hindpaw. Seven days later, vehicle (5 μL, black bars) or the MAPK inhibitor U0126 (1 μg; gray bars) was injected at the same site, followed 10 min later by PGE2 (100 ng). In the group treated with U0126, PGE2-induced hyperalgesia was almost completely inhibited at the 4th h (F1,8 = 58.30; *** p < 0.0001, when the vehicle and MAPK inhibitor groups are compared; two-way repeated-measures ANOVA followed by Bonferroni post hoc test), indicating that the MAPK plays a role in the expression of type II priming.
B. To determine whether the inhibition of type II priming by the MAPK inhibitor was permanent, PGE2 (100 ng) was injected again at the same site, 10 days after the injection of U0126. At this time, PGE2-induced hyperalgesia was present at the 4th h, indicating that MAPK does not play a role in the maintenance of type II priming (F1,16 = 6.34; ns, when the vehicle and MAPK inhibitor groups are compared; two-way repeated-measures ANOVA followed by Bonferroni post hoc test).
C. Male rats received repeated (hourly × 4) intradermal injections of DAMGO (1 μg) on the dorsum of the hindpaw. Twelve days later, vehicle (5 μL, black bars) or the FAK inhibitor (1 μg; white bars) was injected at the same site, followed 10 min later by PGE2 (100 ng). In the group treated with the FAK inhibitor, PGE2-induced hyperalgesia was attenuated at 30 min and 4 hrs (F1,12 = 19.42; *p < 0.05 and *** p < 0.0001, when the vehicle and FAK inhibitor groups are compared; two-way repeated-measures ANOVA followed by Bonferroni post hoc test), indicating a small participation of FAK in the expression of type II priming. (N = 6 paws per group)
Combination of a Src and MAPK inhibitors reverses type II priming and latent sensitization in male rats
We have previously shown that the Src inhibitor, SU 6656, blocked the expression, but not the maintenance, of type II priming in male rats [9]. Here, we found that the MAPK inhibitor also inhibited the expression, but did not reverse, type II priming (Fig. 3A). We next combined these second messenger inhibitors, which had effects on the expression of type II priming, to test its effect on the neuroplasticity produced by repeated exposure to MOR agonist DAMGO [9].
In male rats, pretreatment with the combination of Src and MAPK inhibitors attenuated the PGE2-induced hyperalgesia, measured 30 min after injection, and completely blocked the prolongation of PGE2-induced hyperalgesia in DAMGO-primed paws (Fig. 4A) and also, in naloxone-primed paws (data not shown). This effect was still present when the tests with PGE2 were performed again after 15 (Fig. 4B) and 30 days (Fig. 4C) indicating that, when both Src and MAPK are inhibited, DAMGO or naloxone-induced priming can be reversed in male rats.
Figure 4. The combination of a Src and MAPK inhibitors reversed DAMGO-induced priming in male rats.
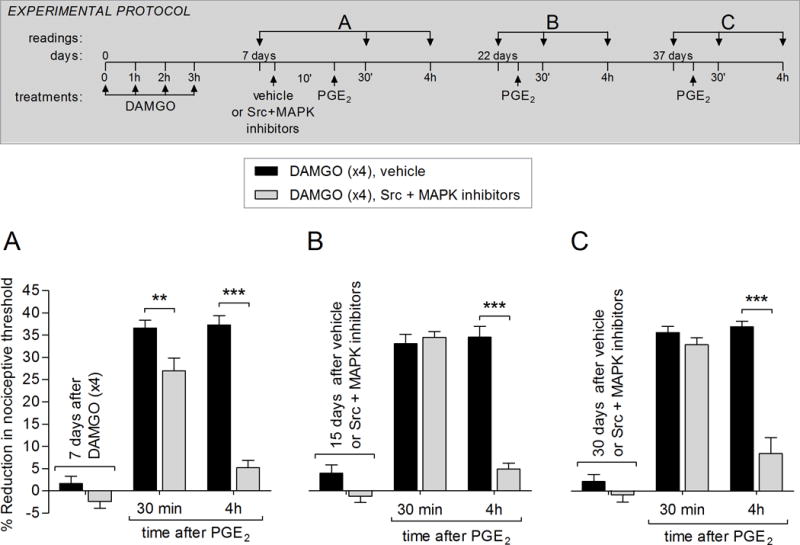
A. Rats received repeated (hourly × 4) intradermal injections of DAMGO (1 μg) on the dorsum of the hindpaw. Seven days later, vehicle (5 μL; black bars) or a combination of Src (SU 6656, 1 μg) and MAPK (U0126, 1 μg) inhibitor (gray bars) was injected at the same site. Ten min later, PGE2 (100 ng; intradermally) was injected and the mechanical nociceptive threshold, evaluated 30 min and 4 hrs after injection. In the group treated with the combination of Src and MAPK inhibitors PGE2-induced hyperalgesia, at 30 min, was significantly attenuated and its prolongation, evaluated at the 4th h, completely blocked (F1,20 = 133.49, ** p < 0.01 and ***p < 0.0001, when the vehicle and Src + MAPK inhibitors groups are compared; two-way repeated-measures ANOVA followed by Bonferroni post hoc test), indicating that Src and MAPK play a role in the expression of DAMGO-induced type II priming.
B. Fifteen days later, PGE2 (100 ng) was injected again at the same site. Although in all groups PGE2-induced mechanical hyperalgesia was present 30 min after injection, in the Src + MAPK inhibitors-treated group, it was markedly blocked at the 4th h (F1,20 = 73.73, *** p < 0.0001, when the vehicle and Src + MAPK inhibitors groups are compared; two-way repeated-measures ANOVA followed by Bonferroni post hoc test), supporting the suggestion that both Src and MAPK are required for the maintenance of type II priming.
C. To determine whether treatment with the combination of Src and MAPK inhibitors permanently reversed priming, PGE2 was injected again at the same site 30 days later. In the group previously treated with the inhibitors (gray bars), PGE2-induced mechanical hyperalgesia was not observed at the 4th h (F1,20 = 27.98, *** p = 0.0004, when the vehicle and Src + MAPK inhibitors groups are compared; two-way repeated-measures ANOVA followed by Bonferroni post hoc test), demonstrating that inhibition of Src and MAPK, at the same time, reversed type II priming in male rats. (N = 6 paws per group)
It has been demonstrated that CFA, injected in the paw, induces a latent sensitization [22; 57] and, a pharmacological blockade during the post-hyperalgesia state with MOR inverse agonists, such as naloxone or naltrexone, reinstated pain [22; 57]. We injected CFA (50 μL) into the plantar surface of one hindpaw (ipsilateral paw), and the mechanical nociceptive threshold was evaluated, in both paws, until the nociceptive threshold had returned to baseline values, 28 days after injection (Fig. 5A, black circles). On day 28, male rats received an intrathecal injection of the MOR antagonist naltrexone (NTX), or a combination of Src and MAPK inhibitors, followed 5 min later by NTX (Fig. 5B and Fig. 5C). Intrathecal NTX induced a robust decrease in mechanical nociceptive threshold, in both the ipsi and contralateral paws (Fig. 5B and Fig. 5C, white and black circles). However, when the combination of Src and MAPK inhibitors was injected 5 min before NTX, the decrease in mechanical nociceptive threshold induced by NTX was blocked, in both paws (Fig. 5B and Fig. 5C, white and black square). These data support the suggestion that the constitutive MOR activity induced by intraplantar CFA is dependent on Src and MAP kinase second messengers as was found for type II priming.
Figure 5. CFA-induced latent sensitization, a model of type II hyperalgesic priming, depends on Src and MAPK in male rats.
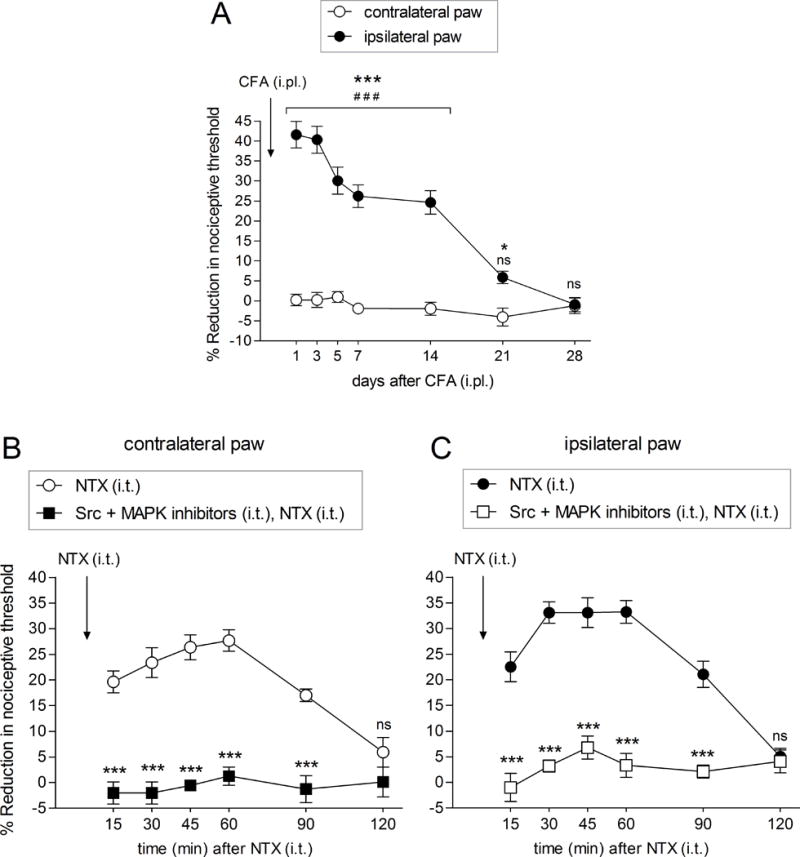
Mechanical nociceptive threshold was evaluated by Randall-Selitto paw-withdrawal test, in both hindpaws of male rats, before and on days 1, 3, 5, 7, 14, 21 and 28 after an intraplantar injection of CFA (50 μL, non-diluted), in one hindpaw (ipsilateral paw). A. In that paw (black circles), the mechanical nociceptive threshold decreased dramatically already on day 1 (F = 72.47;### p < 0.0001, when days after CFA were compared to baseline [before CFA injection]; one-way repeated measure ANOVA followed by Bonferroni post hoc test. F1,60 = 114.97; *** p < 0.0001 or * p < 0.05, when ipsilateral [black circles] were compared to contralateral [white circles] paws; two-way repeated-measures ANOVA followed by Bonferroni post hoc test), progressively returning to baseline values, days 21 (ns) and 28 (ns). Change in the mechanical nociceptive threshold was not observed in the contralateral paws (A; white circles; F = 1.253; p = 0.3016; one-way repeated measure ANOVA followed by Bonferroni post hoc test).
B – C. Twenty-eight days CFA injection, when the mechanical nociceptive threshold returned to baseline values in the ipsilateral paw, rats received an intrathecal injection of naltrexone (NTX; 1 μg/20 μL), and the mechanical nociceptive threshold was evaluated 15, 30, 45, 60, 90 and 120 min after injection. A robust reduction in the mechanical nociceptive threshold was observed in both the contralateral [B; white circles; F = 2.798; *** p < 0.0001, when time (min) after NTX was compared to baseline (before NTX); one-way repeated measure ANOVA followed by Bonferroni post hoc test] and in the ipsilateral [C; black circles; F = 45.21; *** p < 0.0001, when time (min) after NTX was compared to baseline (before NTX); one-way repeated measure ANOVA followed by Bonferroni post hoc test] paws. 120 min after the injection of NTX, the mechanical nociceptive threshold returned to baseline, in both paws (ns; B, white circles; C, black circles). (N = 6 rats). A different group of male rats that received CFA in one hindpaw, 28 days before, was treated with an intrathecal injection of the combination of Src (SU 6656, 10 μg/5 μL) and MAPK (U0126; 10 μg/5 μL) inhibitors, 5 min before of the intrathecal injection of NTX (1 μg/10 μL). At 15, 30, 45, 60, 90 and 120 min after the injection of NTX, we observed a complete inhibition of NTX-induced mechanical hyperalgesia in both paws, contralateral [B; F1,50 = 84.72; *** p < 0.0001, when NTX group (white circles) was compared to Src and MAPK inhibitors followed by NTX group (black square); two-way repeated-measures ANOVA followed by Bonferroni post hoc test; F = 0.9309; p = 0.4873, when time (min) after NTX was compared to baseline (before NTX); one-way repeated measure ANOVA followed by Bonferroni post hoc test] and ipsilateral [C; F1,50 = 149.49; *** p < 0.0001, when NTX group (black circles) was compared to Src and MAPK inhibitors followed by NTX group (white square); two-way repeated-measures ANOVA followed by Bonferroni post hoc test; F = 1.675; p = 0.1616, when time (min) after NTX was compared to baseline (before NTX); one-way repeated measure ANOVA followed by Bonferroni post hoc test]. These findings support the suggestion that the CFA-induced latent sensitization is also dependent on Src and MAPK second messengers. (N = 6 rats)
Combination of Src and MAPK inhibitors does not reverse type II priming in females
Our previous study demonstrated that repeated exposure to DAMGO also induced type II priming in female rats [9]. However, when we tested the effect of the combination of Src and MAPK inhibitors in females, primed with DAMGO, 7 days prior, we observed the PGE2-induced hyperalgesia at 30 min and 4 hrs, (Fig. 6A), also present after 10 days (Fig. 6B), demonstrating no effect of the combination of Src and MAPK inhibitors in the expression (Fig. 6A) or maintenance (Fig. 6B) of type II priming in females. These findings indicate that the intracellular signaling pathway involved in the expression and maintenance of type II priming is sexually dimorphic.
Figure 6. Expression of type II priming does not depend on Src and MAPK in female rats.
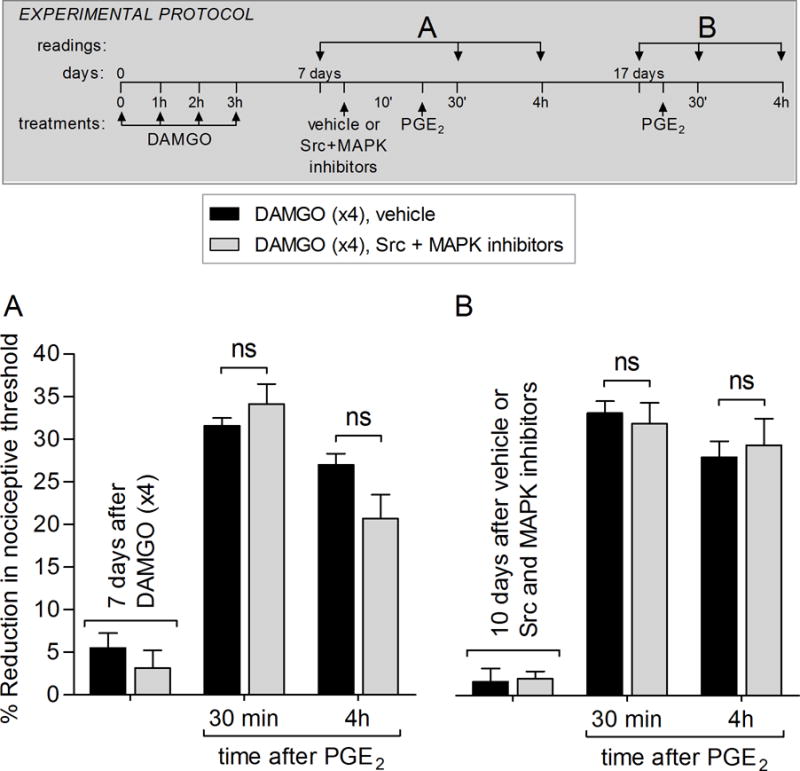
Female rats received repeated (hourly × 4) intradermal injections of DAMGO (1 μg) on the dorsum of the hindpaw. A. Seven days later, vehicle (5 μL; black bar) or a combination of Src (SU 6656, 1 μg) and MAPK (U0126; 1 μg) inhibitors (gray bar) was intradermally injected on the dorsum of the hindpaw, followed by injection of PGE2 (100 ng), at the same site. Mechanical nociceptive threshold was evaluated 30 min and 4 hrs after PGE2 injection. Two-way repeated-measures ANOVA followed by Bonferroni post hoc test showed PGE2-induced hyperalgesia at 30 min, which was still present at the 4th h after injection, in both groups, with no significant (ns) difference between the groups (F1,20 = 2.10; p = 0.1783, when vehicle and Src + MAPK inhibitors groups are compared; two-way repeated-measures ANOVA followed by Bonferroni post hoc test).
B. Ten days after the injection with Src and MAPK inhibitors, PGE2 (100 ng; intradermally) was again injected. PGE2-induced hyperalgesia was present 30 min and 4 hrs later, in both groups (ns, when vehicle and Src + MAPK inhibitors groups are compared; two-way repeated-measures ANOVA followed by Bonferroni post hoc test), indicating that Src and MAPK do not play a role in the expression or maintenance of priming in female rats, possibly due to sexual dimorphic role of Src and MAPK in this type of neuroplasticity. (N = 6 paws per group).
Involvement of the G-protein coupled estrogen receptor (GPR30) in the mechanism of type II priming in females
To evaluate if estrogen-dependent mechanisms mediate the resistance to the effect of the combination of Src and MAPK inhibitors in female rats, type II priming was induced by repeated DAMGO injections and, 24 hrs later, intrathecal treatment with ODN mismatch (MM) or ODN antisense (AS) for ER-α (Fig. 7A), ER-β (Fig. 7B) or GPR30 (Fig. 7C) mRNA was initiated, for 5 consecutive days (once a day). On the 6th day, ~17 hrs after the last injection of MM- or AS-ODN, the combination of the Src and MAPK inhibitors was injected followed, 10 min later, by injection of PGE2 at the same site. While treatment with AS-ODN for ER-α did not impact the effect of the combination of Src and MAPK inhibitors on the expression of type II priming, as PGE2-induced hyperalgesia was still present at the 4th h after injection (Fig. 7A), in the rats treated with AS-ODN for ER-β the combination of Src and MAPK inhibitors attenuated the mechanical hyperalgesia induced by PGE2 30 min after its injection (Fig. 7B, left panel) and inhibited the prolongation of PGE2-induced hyperalgesia at the 4th h (Fig. 7B, left panel). To determine whether the attenuation of type II priming by the combination of Src and MAPK inhibitors in these rats was permanent, PGE2 was injected again, 7 days after the injection of inhibitors (Fig. 7B, right panel). At this point, PGE2-induced mechanical hyperalgesia was still present at the 4th h, in all groups (Fig. 7B, right panel), indicating that ER-β regulates the effect of Src and MAPK inhibitors only on the expression, but not in the maintenance, of DAMGO-induced type II priming in females. When PGE2 was injected in primed females previously treated with AS-ODN for GPR30 that received the combination of Src and MAPK inhibitors, we observed attenuation of mechanical hyperalgesia induced by PGE2 30 min after its injection (Fig. 7C, left panel) and the prolongation of PGE2-induced hyperalgesia at the 4th h was completely blocked (Fig. 7C, left panel), demonstrating that GPR30 also regulates the inhibitory effect of Src and MAPK in the expression of DAMGO-induced type II priming in female rats. However, when PGE2 was injected again 7 (Fig. 7C, middle panel) or 14 (Fig. 7C, right panel) days after the injection of a combination of Src and MAPK inhibitors, we observed that the PGE2-induced hyperalgesia was not present at the 4th h (Fig. 7C, middle and right panel), indicating that GPR30 regulates the inhibitory effect of the combination of Src and MAPK inhibitors in DAMGO-induced type II priming in female rats.
Figure 7. Knockdown of GPR30 uncovers sensitivity to the combination of Src and MAPK inhibitors in females.
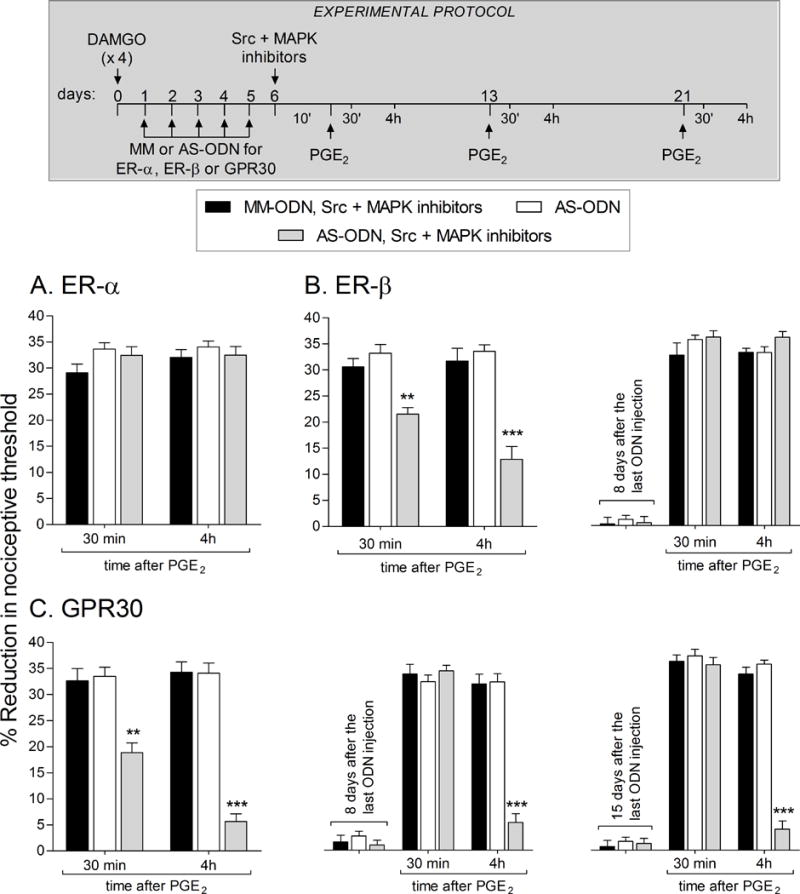
Female rats were treated hourly (x 4) with intradermal injections of DAMGO (1 μg) on the dorsum of the hindpaw. Twenty-four hours later, daily spinal intrathecal injection of MM-ODN (40 μg/20 μL/day; black bars) or AS-ODN (40 μg/20 μL/day; white and dotted bars) for ER-α (A), ER-β (B) or GPR30 (C) were performed for 5 consecutive days. On the sixth day, ~ 17 hrs after the last injection of MM-ODN or AS-ODN, rats received intradermal injection of the combination of Src (SU 6656; 1 μg) and MAPK (U0126, 1 μg) inhibitors (black and gray bars) on the dorsum of the hindpaw, followed, 10 min later, by PGE2 (100 ng) at the same site. The mechanical nociceptive threshold was evaluated 30 min and 4 hrs after PGE2 injection.
A. PGE2-induced hyperalgesia was present 30 min and 4 hrs later, in all groups (F1,33 = 1.36; p = 0.2523, when the groups are compared; two-way repeated-measures ANOVA followed by Bonferroni post hoc test), indicating that the knockdown of ER-α did not affect the inability of the combination of Src and MAPK inhibitors to reverse priming in female rats.
B (left panel). PGE2-induced hyperalgesia at 30 min was partially attenuated in the ER-β-treated AS-ODN-treated group, that received the combination of Src and MAPK inhibitors (gray bar; ** p < 0.01, when the MM-ODN- (black bar) and AS-ODN (white bar) groups are compared). At the 4th h, the prolongation of PGE2-induced hyperalgesia was still attenuated in the ER-β AS-ODN-treated group that received Src and MAPK inhibitors (gray bar; F2,33 = 38.54; *** p < 0.001; two-way repeated-measures ANOVA followed by Bonferroni post hoc test); Right panel. To determine whether the reversal of DAMGO-induced priming by treatment with AS-ODN for ER-β in rats that received the combination of Src and MAPK inhibitors, was permanent, PGE2 was injected again at the same site 8 days after the last ODN injection. We observed that PGE2-induced mechanical hyperalgesia was still present at the 4th h, indicating the presence of type II priming (F2,66 = 2.64; p = 0.0862, when the groups are compared; two-way repeated-measures ANOVA followed by Bonferroni post hoc test).
C (left panel). PGE2-induced hyperalgesia was inhibited at 30 min and 4 hrs in the group that had previously been treated with AS-ODN for GPR30 and received the combination of Src and MAPK inhibitors (gray bars; F2,33 = 62.37; ** p < 0.001 and *** p < 0.0001, when AS-ODN, Src and MAPK inhibitors is compared to AS-ODN or MM-ODN, Src and MAPK inhibitors; two-way repeated-measures ANOVA followed by Bonferroni post hoc test); Middle panel. To determine whether the reversal of DAMGO-induced priming by the treatment with AS-ODN for GPR30 that received the combination of Src and MAPK inhibitors was permanent, PGE2 was injected again at the same site 8 days after the last ODN injection. PGE2-induced mechanical hyperalgesia was attenuated at the 4th h in the group previously treated with AS-ODN for GPR30 that received the combination of Src and MAPK inhibitors (gray bar; F2,66 = 31.76; *** p < 0.0001, when AS-ODN, Src and MAPK inhibitors is compared to AS-ODN or MM-ODN, Src and MAPK inhibitors; two-way repeated-measures ANOVA followed by Bonferroni post hoc test); Right panel. Additionally, when PGE2 (100 ng; intradermally) was injected again, 15 days after the last injection of ODNs and 14 days after the injection of the combination of Src and MAPK inhibitors, PGE2-induced mechanical hyperalgesia was attenuated at the 4th h in the group previously treated with AS-ODN for GPR30 that received the combination of Src and MAPK inhibitors (gray bar; F2,66 = 85.64; *** p < 0.0001, when AS-ODN, Src and MAPK inhibitors is compared to the AS-ODN or MM-ODN, Src and MAPK inhibitors; two-way repeated-measures ANOVA followed by Bonferroni post hoc test) indicating that GPR30 regulates the inhibitory effect of the combination of Src and MAPK inhibitors on DAMGO-induced type II priming, in female rats.
Of note, the mechanical nociceptive thresholds before the treatment with MM- or AS-ODN antisense were not different when compared to the mechanical nociceptive thresholds ~17 hrs after the last injection of ODNs. In addition, the treatment with AS-ODN alone (white bars) was not able to inhibit the expression and/or maintenance of DAMGO-induced type II priming. (N = 12 paws per group)
Discussion
In the present experiments, we studied the maintenance mechanism of type II hyperalgesic priming [9] and compared it to that for the model of latent sensitization [22; 48; 57]. Given that naloxone induced mechanical hyperalgesia in DAMGO-primed paws, and itself induced hyperalgesic priming, the involvement of MOR was considered with respect to the expression and maintenance of type II priming. However, treatment with AS-ODN for MOR failed to inhibit the expression of DAMGO-induced type II priming (i.e., prolongation of PGE2-induced hyperalgesia) and DAMGO induced hyperalgesia, tested 8 days after the last injection of AS-ODN, confirming the presence of priming. Thus, we concluded that MOR is not necessary for either the expression or the maintenance of DAMGO-induced type II priming.
In our previous study of type II priming induced by repeated exposure to DAMGO, inhibitors of PKA (H-89), Src (SU 6656), and AS-ODN for PLC-β3 mRNA [9; 10] inhibited its expression, but did not reverse it in male rats [9; 10]. In this report, we evaluated the role of two other second messengers, MAPK and FAK, shown to be involved in PKA and Src signaling [20; 35; 50; 51]. FAK is considered as an important scaffolding protein that interacts with several adaptors and signaling proteins at focal adhesions [23; 61; 80]. For example, the autophosphorylation of FAK creates a binding site for Src via the SH2 domain and activates it, not only enhancing FAK activity but also phosphorylating FAK-binding proteins to trigger a wide range of signaling pathways [20; 35; 50]. Another important molecule involved in FAK signaling is MAPK/ERK [67]; phosphorylated FAK can stimulate ERK1/2 [39; 68], thus activating MAPK signaling pathways [38]. Similarly, FAK regulates cell adhesion disassembly and migration, by recruiting MAPK/ERK [16; 78]. In our experiments, both FAK inhibitor and the MAPK inhibitor U0126 inhibited the expression of type II priming, but not its maintenance. While this data confirms the participation of FAK and MAPK in the manifestation of the neuroplasticity, alone their activity cannot account for its maintenance. However, recent studies focused on cell signaling pathways in non-neuronal cells have shown a requirement for simultaneous inhibition of Src and MAPK/ERK in the transition from dormant to latent state [25; 26; 59]. Of note, similar to our observations in the present study for type II priming, this latent state phenomenon in non-neuronal cells is also FAK-dependent [16; 78].
Therefore, we evaluated the effect of a combination of Src and MAPK inhibitors in our model of DAMGO-induced type II priming [9]. We also tested this combination in the model of CFA-induced latent sensitization [57]. Of note, it has been suggested that the latent nociceptor sensitization observed in this model is produced by increased constitutive MOR activity, by augmented release of endogenous opioids in the spinal cord [22; 57]. Since this additional production of opioids could increase the activation of MOR at the central terminal of the nociceptor, it is possible that the latent sensitization observed is, actually, similar in mechanism to the one observed in our model of type II priming, also produced by increased activation of MOR [9; 22; 42; 57]. In fact, we found that the combination of the Src and MAPK inhibitor was able to permanently reverse both the prolongation of PGE2-induced hyperalgesia in rats submitted to the DAMGO-primed paws and the NTX-induced decrease in mechanical nociceptive threshold in the model of latent sensitization. In addition, also in the models of CFA-induced latent sensitization and DAMGO-induced priming, a MOR inverse agonist, produced hyperalgesia, suggesting that changes in the MOR could contribute to this plasticity. However, our finding that naloxone induces priming by itself, and also produces hyperalgesia in DAMGO-primed paws, indicated that the latent sensitization mechanism may not involve tonic opioid-induced repression of nociceptive signaling, but rather a neuroplastic mechanism at the nociceptor, as demonstrated by the effect of the combination of Src and MAPK inhibitors, which also reversed naloxone-induced priming (data not shown). The central role of the Src/MAPK pathway, in our model of opioid-induce neuroplasticity in the peripheral terminal of the primary afferent nociceptor and in the model of CFA-induced latent sensitization can be considered, thus a target for therapeutic interventions against painful conditions caused by chronic use of opioids.
Several studies have reported sexual dimorphism in mechanisms that play a role in the etiology of chronic pain [15; 30; 40; 41], emphasizing the action of estrogen, in females [15; 30; 41]. Considering this possibility in our model of type II priming, we also evaluated the effect of the combination of Src and MAPK inhibitors on the maintenance of DAMGO-induced type II priming in female rats. Unexpectedly, the simultaneous inhibition of Src and MAPK did not reverse type II priming in females. Hence, we next evaluated if this lack of effect of the combination of Src and MAPK inhibitors on type II priming was due to a regulatory role of estrogen through the receptors ER-α, ER-β, and GPR30. We found that the treatment of female rats with AS-ODN for ER-β or GPR30, but not ER-α, restored the inhibitory effect of the combination of Src and MAPK inhibitors on the expression of DAMGO-induced type II priming. However, only in the GPR30 AS-ODN-treated group the combination of inhibitors was able to permanently reverse priming. This data suggests the participation of estrogen, acting through GPR30, in the regulation of the second messengers that play a role in the maintenance of type II priming, induced by repeated exposure to MOR agonist DAMGO, in female rats. In good agreement with our findings, it has been demonstrated that 17β-estradiol induces cell proliferation, a process that is GPR30- and Src-mediated [79]. At the cellular level, GPR30 influences growth factor signaling pathways including trans-activation of signals such as epidermal growth factor (EGF) receptor, intracellular Ca2+ mobilization, phosphoinositide-3-kinase translocation, Src activation, and MAP/ERK kinase activation and to modulate downstream transcription factor networks [33; 34; 54; 55; 62; 66; 76; 77]. Interestingly, the knockdown of GPR30 alone did not affect the maintenance of type II priming. One possible explanation is that the repeated activation of MOR might generate superactivation of Src and MAPK, which might be regulated by estrogen in females. Consequently, the knockdown of GPR30 allowed the simultaneous inhibition of Src and MAPK to reverse DAMGO-induced type II hyperalgesic priming, which is not necessary in males. Another possibility considers that the stimulation of GPR30 by estrogen is coupled to the activation of the same signaling molecules that participate in most membrane-initiated signaling cascades as opioid receptors, e.g., protein kinase A, protein kinase B, protein kinase C, phospholipase C, inositol triphosphate, MAPK, ERK, tyrosine kinases, etc. [45]. Due to the overlapping of the secondary messenger pathways, activation of GPR30 by estrogen is postulated to influence the signaling cascades of the opioid receptors, leading to the sex differences in the effects of opioids because of different GPR30 expression patterns between males and females [45]. However, the nature of the interaction of Src and MAPK/ERK mediating the state switch, in any cell type, as well as sex differences, remains to be elucidated.
In conclusion, the maintenance of type II hyperalgesic priming induced by repeated exposure to an inhibitory G-protein coupled receptor agonist DAMGO and the latent sensitization, are dependent on mechanisms mediated by an interaction between two second messengers, Src and MAPK [53; 74], which in female rats is regulated by GPR30 signaling (Fig. 8). Understanding the mechanisms responsible for the maintenance of opioid-induced hyperalgesia (OIH), a form of neuroplasticity in the peripheral terminal of the nociceptor, may provide useful information for the design of drugs with improved therapeutic profiles to treat chronic pain.
Figure 8. Schematic representation of the mechanisms involved in the maintenance of DAMGO-induced type II hyperalgesic priming in the male and female rat.
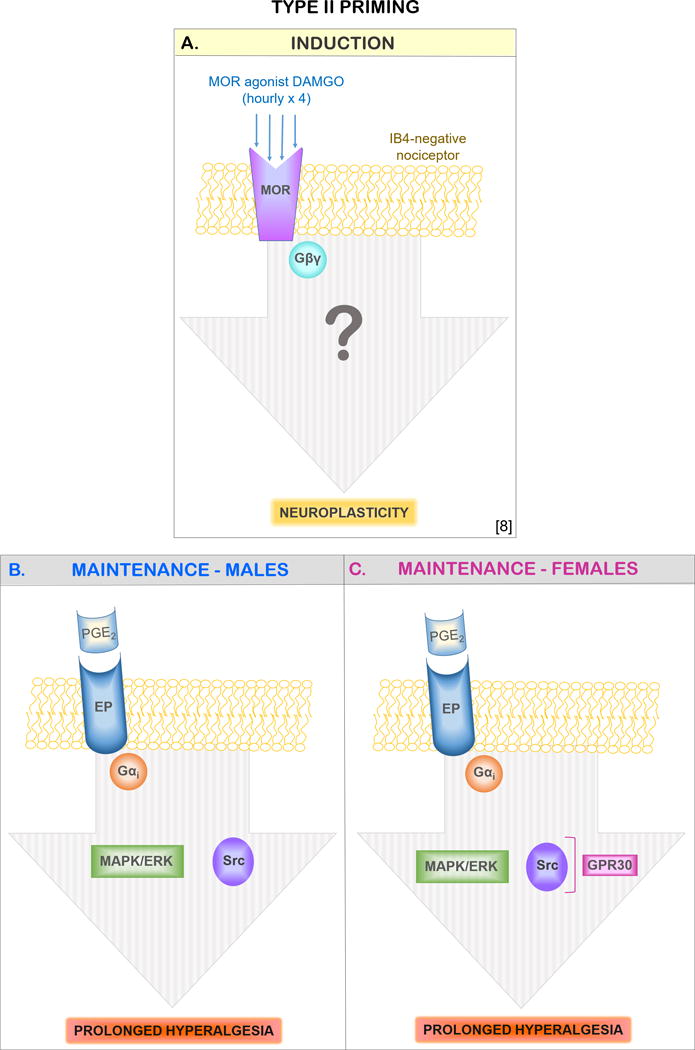
In (A) the induction of type II priming by repeated (hourly × 4) stimulation of mu-opioid receptor in IB4-negative nociceptors is illustrated. The repeated administration of DAMGO (a mu-opioid receptor agonist) stimulates a yet-to-be-determined pathway that produces neuroplastic changes in the nociceptor expressed as prolongation of PGE2-induced hyperalgesia [9]. The maintenance of DAMGO-induced type II hyperalgesic priming is markedly dependent on both Src and MAPK/ERK in male (B), which is regulated through GPR30 in female (C) rats. Abbreviations: EP, prostaglandin E2 receptor; GPCR, G-protein-coupled estrogen receptor 30; MOR, mu-opioid receptor; Gαi, inhibitory G-protein α subunit; IB4, isolectin B4; MAPK/ERK/, mitogen-activated protein kinase (MAPK)/extracellular signal-related kinase (ERK); PGE2, prostaglandin E2; Src, Src tyrosine kinase.
Acknowledgments
The authors would like to thank Marie Kern for technical assistance. This study was funded by a grant from the National Institutes of Health (NIH), NS084545.
Footnotes
Conflict of Interest: The authors declare no competing financial interests.
References
- 1.Alessandri-Haber N, Yeh JJ, Boyd AE, Parada CA, Chen X, Reichling DB, Levine JD. Hypotonicity induces TRPV4-mediated nociception in rat. Neuron. 2003;39(3):497–511. doi: 10.1016/s0896-6273(03)00462-8. [DOI] [PubMed] [Google Scholar]
- 2.Aley KO, Green PG, Levine JD. Opioid and adenosine peripheral antinociception are subject to tolerance and withdrawal. J Neurosci. 1995;15(12):8031–8038. doi: 10.1523/JNEUROSCI.15-12-08031.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Aley KO, Levine JD. Dissociation of tolerance and dependence for opioid peripheral antinociception in rats. J Neurosci. 1997;17(10):3907–3912. doi: 10.1523/JNEUROSCI.17-10-03907.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Aley KO, Levine JD. Multiple receptors involved in peripheral alpha 2, mu, and A1 antinociception, tolerance, and withdrawal. J Neurosci. 1997;17(2):735–744. doi: 10.1523/JNEUROSCI.17-02-00735.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Aley KO, Levine JD. Role of protein kinase A in the maintenance of inflammatory pain. J Neurosci. 1999;19(6):2181–2186. doi: 10.1523/JNEUROSCI.19-06-02181.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Aley KO, Messing RO, Mochly-Rosen D, Levine JD. Chronic hypersensitivity for inflammatory nociceptor sensitization mediated by the epsilon isozyme of protein kinase C. J Neurosci. 2000;20(12):4680–4685. doi: 10.1523/JNEUROSCI.20-12-04680.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Alvarez P, Bogen O, Levine JD. Role of nociceptor estrogen receptor GPR30 in a rat model of endometriosis pain. Pain. 2014;155(12):2680–2686. doi: 10.1016/j.pain.2014.09.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Angst MS, Clark JD. Opioid-induced hyperalgesia: a qualitative systematic review. Anesthesiology. 2006;104(3):570–587. doi: 10.1097/00000542-200603000-00025. [DOI] [PubMed] [Google Scholar]
- 9.Araldi D, Ferrari LF, Levine JD. Repeated Mu-Opioid Exposure Induces a Novel Form of the Hyperalgesic Priming Model for Transition to Chronic Pain. J Neurosci. 2015;35(36):12502–12517. doi: 10.1523/JNEUROSCI.1673-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Araldi D, Ferrari LF, Levine JD. Adenosine-A1 receptor agonist induced hyperalgesic priming type II. Pain. 2016;157(3):698–709. doi: 10.1097/j.pain.0000000000000421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Araldi D, Ferrari LF, Levine JD. Gi-protein-coupled 5-HT1B/D receptor agonist sumatriptan induces type I hyperalgesic priming. Pain. 2016;157(8):1773–1782. doi: 10.1097/j.pain.0000000000000581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bessiere B, Richebe P, Laboureyras E, Laulin JP, Contarino A, Simonnet G. Nitrous oxide (N2O) prevents latent pain sensitization and long-term anxiety-like behavior in pain and opioid-experienced rats. Neuropharmacology. 2007;53(6):733–740. doi: 10.1016/j.neuropharm.2007.08.003. [DOI] [PubMed] [Google Scholar]
- 13.Bogen O, Alessandri-Haber N, Chu C, Gear RW, Levine JD. Generation of a pain memory in the primary afferent nociceptor triggered by PKCepsilon activation of CPEB. J Neurosci. 2012;32(6):2018–2026. doi: 10.1523/JNEUROSCI.5138-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Borle AB, Snowdowne KW. Measurement of intracellular free calcium in monkey kidney cells with aequorin. Science. 1982;217(4556):252–254. doi: 10.1126/science.6806904. [DOI] [PubMed] [Google Scholar]
- 15.Brown AS, Levine JD, Green PG. Sexual dimorphism in the effect of sound stress on neutrophil function. J Neuroimmunol. 2008;205(1–2):25–31. doi: 10.1016/j.jneuroim.2008.08.005. [DOI] [PubMed] [Google Scholar]
- 16.Brunton VG, Frame MC. Src and focal adhesion kinase as therapeutic targets in cancer. Curr Opin Pharmacol. 2008;8(4):427–432. doi: 10.1016/j.coph.2008.06.012. [DOI] [PubMed] [Google Scholar]
- 17.Burch RM, Axelrod J. Dissociation of bradykinin-induced prostaglandin formation from phosphatidylinositol turnover in Swiss 3T3 fibroblasts: evidence for G protein regulation of phospholipase A2. Proc Natl Acad Sci U S A. 1987;84(18):6374–6378. doi: 10.1073/pnas.84.18.6374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Campillo A, Cabanero D, Romero A, Garcia-Nogales P, Puig MM. Delayed postoperative latent pain sensitization revealed by the systemic administration of opioid antagonists in mice. Eur J Pharmacol. 2011;657(1–3):89–96. doi: 10.1016/j.ejphar.2011.01.059. [DOI] [PubMed] [Google Scholar]
- 19.Celerier E, Rivat C, Jun Y, Laulin JP, Larcher A, Reynier P, Simonnet G. Long-lasting hyperalgesia induced by fentanyl in rats: preventive effect of ketamine. Anesthesiology. 2000;92(2):465–472. doi: 10.1097/00000542-200002000-00029. [DOI] [PubMed] [Google Scholar]
- 20.Chacon MR, Fazzari P. FAK: dynamic integration of guidance signals at the growth cone. Cell Adh Migr. 2011;5(1):52–55. doi: 10.4161/cam.5.1.13681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chu LF, Angst MS, Clark D. Opioid-induced hyperalgesia in humans: molecular mechanisms and clinical considerations. Clin J Pain. 2008;24(6):479–496. doi: 10.1097/AJP.0b013e31816b2f43. [DOI] [PubMed] [Google Scholar]
- 22.Corder G, Doolen S, Donahue RR, Winter MK, Jutras BL, He Y, Hu X, Wieskopf JS, Mogil JS, Storm DR, Wang ZJ, McCarson KE, Taylor BK. Constitutive mu-opioid receptor activity leads to long-term endogenous analgesia and dependence. Science. 2013;341(6152):1394–1399. doi: 10.1126/science.1239403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cox BD, Natarajan M, Stettner MR, Gladson CL. New concepts regarding focal adhesion kinase promotion of cell migration and proliferation. J Cell Biochem. 2006;99(1):35–52. doi: 10.1002/jcb.20956. [DOI] [PubMed] [Google Scholar]
- 24.Edinger KL, Frye CA. Androgens’ effects to enhance learning may be mediated in part through actions at estrogen receptor-beta in the hippocampus. Neurobiol Learn Mem. 2007;87(1):78–85. doi: 10.1016/j.nlm.2006.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.El Touny LH, Vieira A, Mendoza A, Khanna C, Hoenerhoff MJ, Green JE. Combined SFK/MEK inhibition prevents metastatic outgrowth of dormant tumor cells. J Clin Invest. 2014;124(1):156–168. doi: 10.1172/JCI70259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ferguson J, Arozarena I, Ehrhardt M, Wellbrock C. Combination of MEK and SRC inhibition suppresses melanoma cell growth and invasion. Oncogene. 2013;32(1):86–96. doi: 10.1038/onc.2012.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ferrari LF, Bogen O, Chu C, Levine JD. Peripheral administration of translation inhibitors reverses increased hyperalgesia in a model of chronic pain in the rat. J Pain. 2013;14(7):731–738. doi: 10.1016/j.jpain.2013.01.779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ferrari LF, Bogen O, Levine JD. Second messengers mediating the expression of neuroplasticity in a model of chronic pain in the rat. J Pain. 2014;15(3):312–320. doi: 10.1016/j.jpain.2013.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ferrari LF, Bogen O, Reichling DB, Levine JD. Accounting for the delay in the transition from acute to chronic pain: axonal and nuclear mechanisms. J Neurosci. 2015;35(2):495–507. doi: 10.1523/JNEUROSCI.5147-13.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ferrari LF, Khomula EV, Araldi D, Levine JD. Marked Sexual Dimorphism in the Role of the Ryanodine Receptor in a Model of Pain Chronification in the Rat. Sci Rep. 2016;6:31221. doi: 10.1038/srep31221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ferrari LF, Khomula EV, Araldi D, Levine JD. Marked Sexual Dimorphism in the Role of the Ryanodine Receptor in a Model of Pain Chronification in the Rat. Sci Rep. 2016;8(31221) doi: 10.1038/srep31221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ferrari LF, Levine JD. Plasma membrane mechanisms in a preclinical rat model of chronic pain. J Pain. 2015;16(1):60–66. doi: 10.1016/j.jpain.2014.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Filardo EJ, Quinn JA, Bland KI, Frackelton AR., Jr Estrogen-induced activation of Erk-1 and Erk-2 requires the G protein-coupled receptor homolog, GPR30, and occurs via trans-activation of the epidermal growth factor receptor through release of HB-EGF. Mol Endocrinol. 2000;14(10):1649–1660. doi: 10.1210/mend.14.10.0532. [DOI] [PubMed] [Google Scholar]
- 34.Filardo EJ, Thomas P. GPR30: a seven-transmembrane-spanning estrogen receptor that triggers EGF release. Trends Endocrinol Metab. 2005;16(8):362–367. doi: 10.1016/j.tem.2005.08.005. [DOI] [PubMed] [Google Scholar]
- 35.Girault JA, Costa A, Derkinderen P, Studler JM, Toutant M. FAK and PYK2/CAKbeta in the nervous system: a link between neuronal activity, plasticity and survival? Trends Neurosci. 1999;22(6):257–263. doi: 10.1016/s0166-2236(98)01358-7. [DOI] [PubMed] [Google Scholar]
- 36.Hales TG. Arresting the development of morphine tolerance and dependence. Br J Anaesth. 2011;107(5):653–655. doi: 10.1093/bja/aer294. [DOI] [PubMed] [Google Scholar]
- 37.Hay JL, White JM, Bochner F, Somogyi AA, Semple TJ, Rounsefell B. Hyperalgesia in opioid-managed chronic pain and opioid-dependent patients. J Pain. 2009;10(3):316–322. doi: 10.1016/j.jpain.2008.10.003. [DOI] [PubMed] [Google Scholar]
- 38.Hecker TP, Ding Q, Rege TA, Hanks SK, Gladson CL. Overexpression of FAK promotes Ras activity through the formation of a FAK/p120RasGAP complex in malignant astrocytoma cells. Oncogene. 2004;23(22):3962–3971. doi: 10.1038/sj.onc.1207541. [DOI] [PubMed] [Google Scholar]
- 39.Hecker TP, Grammer JR, Gillespie GY, Stewart J, Jr, Gladson CL. Focal adhesion kinase enhances signaling through the Shc/extracellular signal-regulated kinase pathway in anaplastic astrocytoma tumor biopsy samples. Cancer Res. 2002;62(9):2699–2707. [PubMed] [Google Scholar]
- 40.Joseph EK, Levine JD. Sexual dimorphism for protein kinase c epsilon signaling in a rat model of vincristine-induced painful peripheral neuropathy. Neuroscience. 2003;119(3):831–838. doi: 10.1016/s0306-4522(03)00203-3. [DOI] [PubMed] [Google Scholar]
- 41.Joseph EK, Parada CA, Levine JD. Hyperalgesic priming in the rat demonstrates marked sexual dimorphism. Pain. 2003;105(1–2):143–150. doi: 10.1016/s0304-3959(03)00175-1. [DOI] [PubMed] [Google Scholar]
- 42.Joseph EK, Reichling DB, Levine JD. Shared mechanisms for opioid tolerance and a transition to chronic pain. J Neurosci. 2010;30(13):4660–4666. doi: 10.1523/JNEUROSCI.5530-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Khasar SG, Gold MS, Dastmalchi S, Levine JD. Selective attenuation of mu-opioid receptor-mediated effects in rat sensory neurons by intrathecal administration of antisense oligodeoxynucleotides. Neurosci Lett. 1996;218(1):17–20. doi: 10.1016/0304-3940(96)13111-6. [DOI] [PubMed] [Google Scholar]
- 44.Kramer HK, Simon EJ. mu and delta-opioid receptor agonists induce mitogen-activated protein kinase (MAPK) activation in the absence of receptor internalization. Neuropharmacology. 2000;39(10):1707–1719. doi: 10.1016/s0028-3908(99)00243-9. [DOI] [PubMed] [Google Scholar]
- 45.Lee CW, Ho IK. Sex differences in opioid analgesia and addiction: interactions among opioid receptors and estrogen receptors. Mol Pain. 2013;9:45. doi: 10.1186/1744-8069-9-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lee M, Silverman SM, Hansen H, Patel VB, Manchikanti L. A comprehensive review of opioid-induced hyperalgesia. Pain Physician. 2011;14(2):145–161. [PubMed] [Google Scholar]
- 47.Li X, Angst MS, Clark JD. Opioid-induced hyperalgesia and incisional pain. Anesth Analg. 2001;93(1):204–209. doi: 10.1097/00000539-200107000-00040. [DOI] [PubMed] [Google Scholar]
- 48.Lian B, Vera-Portocarrero L, King T, Ossipov MH, Porreca F. Opioid-induced latent sensitization in a model of non-inflammatory viscerosomatic hypersensitivity. Brain Res. 2010;1358:64–70. doi: 10.1016/j.brainres.2010.08.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Liang YQ, Akishita M, Kim S, Ako J, Hashimoto M, Iijima K, Ohike Y, Watanabe T, Sudoh N, Toba K, Yoshizumi M, Ouchi Y. Estrogen receptor beta is involved in the anorectic action of estrogen. Int J Obes Relat Metab Disord. 2002;26(8):1103–1109. doi: 10.1038/sj.ijo.0802054. [DOI] [PubMed] [Google Scholar]
- 50.Lietha D, Cai X, Ceccarelli DF, Li Y, Schaller MD, Eck MJ. Structural basis for the autoinhibition of focal adhesion kinase. Cell. 2007;129(6):1177–1187. doi: 10.1016/j.cell.2007.05.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Luttrell DK, Luttrell LM. Not so strange bedfellows: G-protein-coupled receptors and Src family kinases. Oncogene. 2004;23(48):7969–7978. doi: 10.1038/sj.onc.1208162. [DOI] [PubMed] [Google Scholar]
- 52.Luttrell LM, Ferguson SS, Daaka Y, Miller WE, Maudsley S, Della Rocca GJ, Lin F, Kawakatsu H, Owada K, Luttrell DK, Caron MG, Lefkowitz RJ. Beta-arrestin-dependent formation of beta2 adrenergic receptor-Src protein kinase complexes. Science. 1999;283(5402):655–661. doi: 10.1126/science.283.5402.655. [DOI] [PubMed] [Google Scholar]
- 53.Luttrell LM, Hawes BE, van Biesen T, Luttrell DK, Lansing TJ, Lefkowitz RJ. Role of c-Src tyrosine kinase in G protein-coupled receptor- and Gbetagamma subunit-mediated activation of mitogen-activated protein kinases. J Biol Chem. 1996;271(32):19443–19450. doi: 10.1074/jbc.271.32.19443. [DOI] [PubMed] [Google Scholar]
- 54.Maggiolini M, Picard D. The unfolding stories of GPR30, a new membrane-bound estrogen receptor. J Endocrinol. 2010;204(2):105–114. doi: 10.1677/JOE-09-0242. [DOI] [PubMed] [Google Scholar]
- 55.Maggiolini M, Vivacqua A, Fasanella G, Recchia AG, Sisci D, Pezzi V, Montanaro D, Musti AM, Picard D, Ando S. The G protein-coupled receptor GPR30 mediates c-fos up-regulation by 17beta-estradiol and phytoestrogens in breast cancer cells. J Biol Chem. 2004;279(26):27008–27016. doi: 10.1074/jbc.M403588200. [DOI] [PubMed] [Google Scholar]
- 56.Mao J. Opioid-induced abnormal pain sensitivity: implications in clinical opioid therapy. Pain. 2002;100(3):213–217. doi: 10.1016/S0304-3959(02)00422-0. [DOI] [PubMed] [Google Scholar]
- 57.Marvizon JC, Walwyn W, Minasyan A, Chen W, Taylor BK. Latent sensitization: a model for stress-sensitive chronic pain. Curr Protoc Neurosci. 2015;71:95051–14. doi: 10.1002/0471142301.ns0950s71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mestre C, Pelissier T, Fialip J, Wilcox G, Eschalier A. A method to perform direct transcutaneous intrathecal injection in rats. J Pharmacol Toxicol Methods. 1994;32(4):197–200. doi: 10.1016/1056-8719(94)90087-6. [DOI] [PubMed] [Google Scholar]
- 59.Ochi N, Takigawa N, Harada D, Yasugi M, Ichihara E, Hotta K, Tabata M, Tanimoto M, Kiura K. Src mediates ERK reactivation in gefitinib resistance in non-small cell lung cancer. Exp Cell Res. 2014;322(1):168–177. doi: 10.1016/j.yexcr.2014.01.007. [DOI] [PubMed] [Google Scholar]
- 60.Parada CA, Yeh JJ, Reichling DB, Levine JD. Transient attenuation of protein kinase Cepsilon can terminate a chronic hyperalgesic state in the rat. Neuroscience. 2003;120(1):219–226. doi: 10.1016/s0306-4522(03)00267-7. [DOI] [PubMed] [Google Scholar]
- 61.Peng X, Guan JL. Focal adhesion kinase: from in vitro studies to functional analyses in vivo. Curr Protein Pept Sci. 2011;12(1):52–67. doi: 10.2174/138920311795659452. [DOI] [PubMed] [Google Scholar]
- 62.Prossnitz ER, Arterburn JB, Smith HO, Oprea TI, Sklar LA, Hathaway HJ. Estrogen signaling through the transmembrane G protein-coupled receptor GPR30. Annu Rev Physiol. 2008;70:165–190. doi: 10.1146/annurev.physiol.70.113006.100518. [DOI] [PubMed] [Google Scholar]
- 63.Quanhong Z, Ying X, Moxi C, Tao X, Jing W, Xin Z, Li W, Derong C, Xiaoli Z, Wei J. Intrathecal PLC(beta3) oligodeoxynucleotides antisense potentiates acute morphine efficacy and attenuates chronic morphine tolerance. Brain Res. 2012;1472:38–44. doi: 10.1016/j.brainres.2012.06.030. [DOI] [PubMed] [Google Scholar]
- 64.Rivat C, Laboureyras E, Laulin JP, Le Roy C, Richebe P, Simonnet G. Non-nociceptive environmental stress induces hyperalgesia, not analgesia, in pain and opioid-experienced rats. Neuropsychopharmacology. 2007;32(10):2217–2228. doi: 10.1038/sj.npp.1301340. [DOI] [PubMed] [Google Scholar]
- 65.Rivat C, Vera-Portocarrero LP, Ibrahim MM, Mata HP, Stagg NJ, De Felice M, Porreca F, Malan TP. Spinal NK-1 receptor-expressing neurons and descending pathways support fentanyl-induced pain hypersensitivity in a rat model of postoperative pain. Eur J Neurosci. 2009;29(4):727–737. doi: 10.1111/j.1460-9568.2009.06616.x. [DOI] [PubMed] [Google Scholar]
- 66.Sanden C, Broselid S, Cornmark L, Andersson K, Daszkiewicz-Nilsson J, Martensson UE, Olde B, Leeb-Lundberg LM. G protein-coupled estrogen receptor 1/G protein-coupled receptor 30 localizes in the plasma membrane and traffics intracellularly on cytokeratin intermediate filaments. Mol Pharmacol. 2011;79(3):400–410. doi: 10.1124/mol.110.069500. [DOI] [PubMed] [Google Scholar]
- 67.Schlaepfer DD, Hauck CR, Sieg DJ. Signaling through focal adhesion kinase. Prog Biophys Mol Biol. 1999;71(3–4):435–478. doi: 10.1016/s0079-6107(98)00052-2. [DOI] [PubMed] [Google Scholar]
- 68.Schlaepfer DD, Jones KC, Hunter T. Multiple Grb2-mediated integrin-stimulated signaling pathways to ERK2/mitogen-activated protein kinase: summation of both c-Src- and focal adhesion kinase-initiated tyrosine phosphorylation events. Mol Cell Biol. 1998;18(5):2571–2585. doi: 10.1128/mcb.18.5.2571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Song MJ, Wang YQ, Wu GC. Additive anti-hyperalgesia of electroacupuncture and intrathecal antisense oligodeoxynucleotide to interleukin-1 receptor type I on carrageenan-induced inflammatory pain in rats. Brain Res Bull. 2009;78(6):335–341. doi: 10.1016/j.brainresbull.2008.10.009. [DOI] [PubMed] [Google Scholar]
- 70.Su L, Wang C, Yu YH, Ren YY, Xie KL, Wang GL. Role of TRPM8 in dorsal root ganglion in nerve injury-induced chronic pain. BMC Neurosci. 2011;12:120. doi: 10.1186/1471-2202-12-120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Sun JL, Xiao C, Lu B, Zhang J, Yuan XZ, Chen W, Yu LN, Zhang FJ, Chen G, Yan M. CX3CL1/CX3CR1 regulates nerve injury-induced pain hypersensitivity through the ERK5 signaling pathway. J Neurosci Res. 2013;91(4):545–553. doi: 10.1002/jnr.23168. [DOI] [PubMed] [Google Scholar]
- 72.Taiwo YO, Bjerknes LK, Goetzl EJ, Levine JD. Mediation of primary afferent peripheral hyperalgesia by the cAMP second messenger system. Neuroscience. 1989;32(3):577–580. doi: 10.1016/0306-4522(89)90280-7. [DOI] [PubMed] [Google Scholar]
- 73.Taiwo YO, Levine JD. Prostaglandin effects after elimination of indirect hyperalgesic mechanisms in the skin of the rat. Brain Res. 1989;492(1–2):397–399. doi: 10.1016/0006-8993(89)90928-1. [DOI] [PubMed] [Google Scholar]
- 74.Tohgo A, Choy EW, Gesty-Palmer D, Pierce KL, Laporte S, Oakley RH, Caron MG, Lefkowitz RJ, Luttrell LM. The stability of the G protein-coupled receptor-beta-arrestin interaction determines the mechanism and functional consequence of ERK activation. J Biol Chem. 2003;278(8):6258–6267. doi: 10.1074/jbc.M212231200. [DOI] [PubMed] [Google Scholar]
- 75.Violin JD, Lefkowitz RJ. Beta-arrestin-biased ligands at seven-transmembrane receptors. Trends Pharmacol Sci. 2007;28(8):416–422. doi: 10.1016/j.tips.2007.06.006. [DOI] [PubMed] [Google Scholar]
- 76.Vivacqua A, Bonofiglio D, Albanito L, Madeo A, Rago V, Carpino A, Musti AM, Picard D, Ando S, Maggiolini M. 17beta-estradiol, genistein, and 4-hydroxytamoxifen induce the proliferation of thyroid cancer cells through the g protein-coupled receptor GPR30. Mol Pharmacol. 2006;70(4):1414–1423. doi: 10.1124/mol.106.026344. [DOI] [PubMed] [Google Scholar]
- 77.Vivacqua A, Bonofiglio D, Recchia AG, Musti AM, Picard D, Ando S, Maggiolini M. The G protein-coupled receptor GPR30 mediates the proliferative effects induced by 17beta-estradiol and hydroxytamoxifen in endometrial cancer cells. Mol Endocrinol. 2006;20(3):631–646. doi: 10.1210/me.2005-0280. [DOI] [PubMed] [Google Scholar]
- 78.Webb DJ, Donais K, Whitmore LA, Thomas SM, Turner CE, Parsons JT, Horwitz AF. FAK-Src signalling through paxillin, ERK and MLCK regulates adhesion disassembly. Nat Cell Biol. 2004;6(2):154–161. doi: 10.1038/ncb1094. [DOI] [PubMed] [Google Scholar]
- 79.Yang WR, Zhu FW, Zhang JJ, Wang Y, Zhang JH, Lu C, Wang XZ. I3K/Akt Activated by GPR30 and Src Regulates 17beta-Estradiol-Induced Cultured Immature Boar Sertoli Cells Proliferation. Reprod Sci. 2016 doi: 10.1177/1933719116649696. [DOI] [PubMed] [Google Scholar]
- 80.Zhao X, Guan JL. Focal adhesion kinase and its signaling pathways in cell migration and angiogenesis. Adv Drug Deliv Rev. 2011;63(8):610–615. doi: 10.1016/j.addr.2010.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]