

Abstract
Neuropathic pain is a debilitating pathological condition that is poorly understood. Recent evidence suggests that abnormal central processing occurs during the development of neuropathic pain induced by the cancer chemotherapeutic agent, paclitaxel. Yet, it is unclear what role neurons in supraspinal pain network sites, such as the periaqueductal gray, play in altered behavioral sensitivity seen during chronic pain conditions. To elucidate these mechanisms, we studied the spontaneous and thermally-evoked firing patterns of ventrolateral periaqueductal gray (vlPAG) neurons in awake behaving rats treated with paclitaxel to induce neuropathic pain. In the present study vlPAG neurons in naive rats exhibited either excitatory, inhibitory or neutral responses to noxious thermal stimuli, as previously observed. However, after development of behavioral hypersensitivity induced by the chemotherapeutic agent, paclitaxel, vlPAG neurons displayed increased neuronal activity and changes in thermal pain-evoked neuronal activity. This involved elevated levels of spontaneous firing and heightened responsiveness to non-noxious stimuli (allodynia) as well as noxious thermal stimuli (hyperalgesia) as compared to controls. Furthermore, after paclitaxel treatment only excitatory neuronal responses were observed to both non-noxious and noxious thermal stimuli. Systemic administration of gabapentin, a non-opioid analgesic, induced significant dose-dependent decreases in the elevated spontaneous and thermally-evoked vlPAG neuronal firing to both non-noxious and noxious thermal stimuli in rats exhibiting neuropathic pain but not in naïve rats. Thus, these results show a strong correlation between behavioral hypersensitivity to thermal stimuli and increased firing of vlPAG neurons in allodynia and hyperalgesia that occur in this neuropathic pain model.
Keywords: periaqueductal gray (PAG), chronic pain, paclitaxel, gabapentin, allodynia, hyperalgesia
Introduction
Chemotherapeutic agents, such as paclitaxel, are known to cause neuropathic pain, but the neuronal mechanisms that underlie neuropathic pain are not well understood [34; 51]. Several lines of evidence suggest that hyperexcitability in dorsal root ganglion (DRG) and spinal cord dorsal horn neurons may contribute to the development of thermal and mechanical hypersensitivity in animal models of chemotherapy-induced neuropathic pain [10; 66; 69]. However, it has been suggested that enhanced primary afferent discharge alone is not sufficient for maintaining neuropathic pain without the contribution of supraspinal mechanisms [32]. Despite the clinical importance of chemotherapy-induced neuropathic pain, we currently do not understand how alterations in supraspinal components of the pain pathway contribute to this pathological painful condition.
The ventrolateral periaqueductal gray (vlPAG) is an important brainstem structure, exerting both inhibitory and facilitatory effects on pain perception [22; 24; 30; 37; 38]. The vlPAG modulates spinal cord nociceptive transmission via projections to the rostral ventral medulla (RVM), forming the PAG-RVM-spinal descending pain inhibitory network [16; 22]. In the naïve state, midbrain processing of nociceptive information involves a homeostatic balance between descending inhibitory and facilitatory outputs in the vlPAG. vlPAG neurons are classified into three different subclasses based on the response to painful stimuli: “off cells” which show decreased firing rate, “on cells” which show increased firing rate, and “neutral cells” which show no change in firing rate [21; 54; 60]. Recent studies in chronic pain states have observed behavioral hypersensitivity and increased neuronal activity in the RVM that is strongly associated with a shift in descending inhibition to increased descending facilitation of spinal cord nociceptive transmission, leading to intensification of pain signals [5; 22; 50; 62]. Although vlPAG is a critical site in the descending pain modulatory matrix that is situated upstream of the RVM, it is unknown whether neuronal activity in the vlPAG is altered in neuropathic pain states. Altered activity in the descending pain inhibition circuitry might contribute to the development or maintenance of neuropathic pain. Currently, there are no studies investigating changes in vlPAG neuronal activity induced by neuropathic pain. Therefore, to understand how PAG neurons respond to non-noxious and noxious stimuli, we performed chronic microwire recordings from neurons in awake-behaving rats before and after the induction of neuropathic pain by paclitaxel treatment [49]. The present study investigated whether alterations in spontaneous and thermally-evoked neuronal firing in the vlPAG are induced by chemotherapy-induced neuropathic pain in rats. We also evaluated whether gabapentin, an agent with analgesic properties that is used to treat neuropathic pain [43], affects spontaneous and evoked activity in the vlPAG before and after chemotherapy-induced neuropathic pain.
Methods
Animals
Male Sprague-Dawley (SD) rats (250–350 g; Harlan Laboratories) were used in this study. All procedures performed in this study included appropriate measures to lessen pain, suffering, and also minimize animal usage, and were approved by the Southern Illinois University School of Medicine Laboratory Animal Care and Use Committee.
Surgery for microwire implantation recording
Chronically implanted microwire electrodes (NB Labs, Denison, TX) were used for all single unit recordings. Microwires consisted of eight 50-μm-diameter Teflon-insulated, stainless steel wires separated by 0.25 mm and were chronically implanted using a hydraulic microdrive (Kopf Instruments) under ketamine/xylazine anesthesia (85/5 mg/kg, i.p.). The rats were placed in a stereotaxic device, and the incisor bar was set at −3.3 mm in the vertical plane to hold the skull in the flat position. Microwire electrodes were implanted stereotaxically into the PAG (−7.6 to −8.7 mm posterior from bregma; ± 3.6 mm mediolateral from the midline at 10° angle) according to the atlas of Paxinos and Watson [48] and cemented into place with dental acrylic. The site was covered with dental acrylic, and the microwire assembly was secured to the skull with dental cement. Before the skull screws were encapsulated with dental acrylic, a wire from the electrode assembly was wrapped around the exposed screws for electrical grounding. The microdrive was used to prevent tissue damage only during electrode implantation, but it was not used subsequently, which is a common approach with this type of electrode [15; 46; 61]. Animals were allowed one week to recover from surgery before recording sessions were started. Seven to 14 days after the implantation surgery, we were able to record spontaneous single unit action potentials. Once we obtained reliable spontaneous single unit recordings on three consecutive days from the same electrodes, thermal stimuli were delivered and neuronal activity was recorded.
Noxious thermal stimulation
To record the effects of thermal stimulation on neuronal activity, rats were acclimatized in a Plexiglass® chamber (44×44×44 cm3) for 60 min every day for one week with an 8 channel JFET headstage connected to the implanted electrode. A modified hotplate test was developed using a computer feedback-controlled Peltier device [65], which was built into the floor of a Plexiglass® chamber to administer thermal stimuli. Plate temperature was maintained at 30°C as baseline for all the experiments conducted in this study. On the experimental days the rats were acclimatized for 30 min prior to all the experiments. We made sure that the rat’s hind paw was in contact with the plate when thermal stimulation was delivered. The animals were allowed to move freely within the chamber. After adaptation to the baseline recordings at 30°C, non-noxious (38° and 42°C) and noxious thermal stimuli (48° and 51°C) were applied to the paw, and the stimuli were terminated immediately after the rats exhibited a nocifensive response [hind paw licking and/or guarding (sustained lifting) behaviors]. The thermal stimulation was delivered to the paw at a rate of 12.5°C per s, until the temperature reached the target level. Paw withdrawal was consistently observed in response to noxious thermal stimuli but not to non-noxious thermal stimuli in vehicle treated rats. Our classification of non-noxious (38° and 42°C) and noxious thermal stimuli (48° and 51°C) was based on Banik and Kabadi, 2013 [1], who showed that the normal distribution to paw withdrawal in SD rats is between 44–52°C (comparable to the noxious withdrawals threshold temperature for human glabrous skin). However, after induction of neuropathic pain, thermal stimuli at 37.5°C could induce paw withdrawal in SD rats. In order to determine if the stimulus parameters used in our study induced sensitization of neuronal response, changes in paw withdrawal latency (PWL) were evaluated by applying the thermal stimulus three times at 15 min intervals. The thermal stimulus terminated automatically at 30 s to avoid tissue damage.
Experimental procedure for paclitaxel-induced neuropathic pain
After baseline and evoked neuronal activity were reliably recorded for three days from the naïve rats, the animals were treated with paclitaxel (2 mg/kg per ml) dissolved in dimethylsulfoxide (DMSO). The protocol involved four intraperitoneal (i.p.) injections of either vehicle or paclitaxel administered on alternate days (days 0, 2, 4, and 6) to induce a neuropathic pain state, as previously described by Flatters and Bennett, 2004 [17]. The experimenter was blind to the treatment. After termination of the treatment protocol, rats were tested for changes in PWL and vlPAG neuronal activity to non-noxious (38° and 42°C) and noxious (48° and 51°C) temperatures. To avoid sensitization of neuronal response, all the responses were examined by applying the thermal stimulus three times at 15-min intervals. All the neuronal recording experiments were performed at a baseline temperature of 30°C. We observed consistent hypersensitivity to thermal stimuli, based on changes in PWLs, starting 10 days after termination of paclitaxel treatment. Neuronal recording studies began on day 10 after paclitaxel treatment throughout this study.
Experimental procedure for gabapentin experiments
Gabapentin (10, 30 and 50 mg/kg) (Sigma-Aldrich, St. Louis, MO, U.S.A.) or the saline vehicle was injected intraperitoneally (i.p.). We evaluated the nociceptive responses to the non-noxious (38° and 42°C) and noxious (48° and 51°C) thermal stimuli in vehicle-treated rats and paclitaxel-treated rats at one h and 24 h. Injections of either saline or gabapentin (i.p.) were administered in vehicle-treated rats or paclitaxel-treated rats.
Electrophysiological data acquisition
To record neuronal activity, the animals’ implants were connected to an 8 channel JFET headstage (NB Labs) using a lightweight cable that allowed the animal unrestricted movement during recording. Extracellular multiunit activity was recorded using a multiple channel amplifier at 500× gain and 5.9–220 kHz band pass filters (Plexon Inc., Dallas, TX). The amplified signal from each electrode was digitized at 40 kHz sampling rate (PowerLab, AD Instruments), and the data were analyzed off-line. Spike sorting was performed with spike window discriminator software (Lab Chart, AD Instrument) to isolate extracellular single unit action potentials (APs). Principal component analysis (PCA) and waveform shape were used for spike sorting. Single units were identified as having 1) consistent waveform shape and 2) separable clusters defined by waveform parameters, and were analyzed with PCA (peak of AP, peak amplitude, and spike width). Electrical artifacts generated by movements were removed manually after comparison with single units, and the isolated single units were exported to NeuroExplorer® for data analysis. These single unit data were analyzed by generating rate meter histograms, as described below.
Burst analysis
Since burst firing is considered to be an important measure of increased excitation [63], the NeuroExplorer 3.16 (Nex Technologies) interval-specification algorithm was used to assess the presence and duration of bursting. This algorithm required six parameters to define a burst: 1) maximum allowable burst duration: 100 ms; 2) maximum interspike interval (ISI) at burst start: 500 ms; 3) maximum within-burst ISI: 500 ms; 4) minimum interval between bursts: 500 ms; 5) minimum burst duration: 100 ms; and 6) minimum number of spikes comprising a burst: 2. These classification parameters were optimized and validated by using the following procedures: 1) Neuronal recordings of 1800 s duration of spontaneous activity were used, and the mean of 9 independent bursting units were randomly selected; selection was blind with respect to animal and treatment group; 2) Continuous repetitions of the NeuroExplorer burst algorithm were run with parameter variations to obtain the best correlation between visually rated bursting and algorithm-determined bursting.
Data analysis
The recorded neuronal responses of each PAG neuron evoked by the thermal stimuli were characterized by performing rate meter histogram analysis (80 ms bin width) and expressed as a mean frequency (Hz) ± S.D. The criterion for whether a neuron was responsive to the stimulus was based on the change of firing rate in the 30 sec period immediately following the stimulus. We plotted the standard deviation of the percent change in neuronal firing rate from the pre-stimulus baseline following application of the thermal stimulus and set the criterion that any change of <10% over 3 trials was considered to be non-responsive in the present study. The change in neuronal firing evoked by the thermal stimulus for all excitatory and inhibitory vlPAG neuronal response was >75% change from the pre-stimulus baseline. All data sets were evaluated for normality using the D’Agostino and Pearson omnibus normality test. Pairwise comparisons were performed with either the paired Student’s t-test for before/after comparisons or the two-sample Student’s t-test to compare two groups. Comparisons between different treatments and stimuli were conducted using two-way ANOVA and Bonferroni post-hoc analysis (before/after comparisons) with time as a repeated measure. Statistical significance was set at p < 0.05.
Histology
At the end of the experiments, the rats were deeply anesthetized with pentobarbital (100 mg/kg, i.p.), and the microwire electrode tips were marked with lesions by passage of DC current (10 μA, 30 s). Brains were fixed by cardiac injection of normal saline followed by 10% formalin. The brains were subsequently removed and stored in formalin. Coronal sections (40 μm) were sectioned using a freezing microtome. The sections were stained with Neutral Red, and the sites of the electrode tips were verified microscopically. The anatomical distribution of the neurons studied in neuronal recording studies is shown in Fig. 1A. Coronal histological sections confirmed that the tips of the recording electrodes were located in the vlPAG (stereotaxic coordinates from bregma −7.6 to −8.7 mm) [48].
Fig. 1. Responses of vlPAG neurons to noxious stimuli.
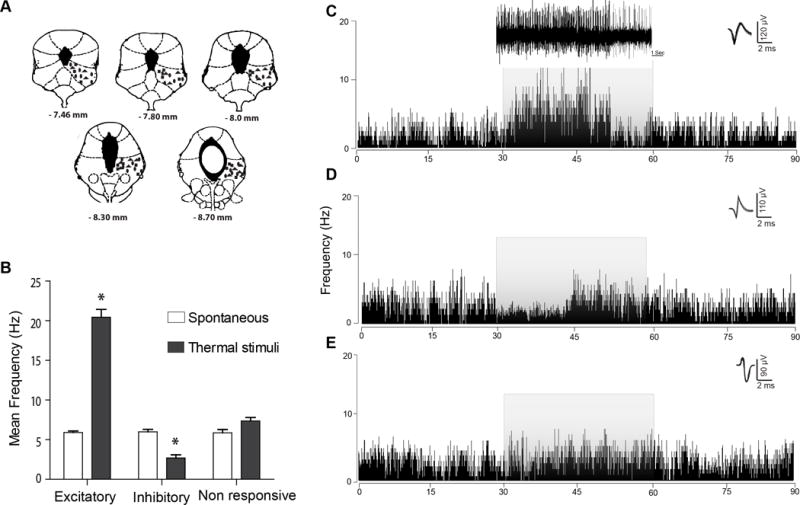
(A) Representative vlPAG microwire recording sites indicate that most electrode placements were localized to the vlPAG at the anterior-posterior coordinates listed, according to the atlas of Paxinos and Watson [48]. Rectangles indicate the locations of vlPAGon neurons; ovals indicate the locations of vlPAGoff neurons and triangles indicate the locations of vlPAGneutral neurons. (B) Mean frequency of vlPAG spontaneous and evoked neuronal firing in response to noxious thermal stimulation. We observed three subclasses of neuronal responses in the vlPAG: “on,” “off,” and “neutral.” (C) A typical example of the rate meter histogram (80 ms bin width) analysis of vlPAGon neurons that were excited by the thermal stimulus (51°C), which exhibited a significant increase in firing rate, as compared to the pre-stimulus spontaneous firing rate. (D) A typical example of the rate meter histogram (80 ms bin width) of vlPAGoff neurons that exhibited an inhibitory response to the noxious thermal stimulus characterized by significantly decreased firing rates, as compared to the spontaneous firing rate. (E) A typical example of the rate meter histogram (80 ms bin width) of vlPAGneutral neurons that did not exhibit any detectable change in the neuronal firing in response to the thermal stimulus. *P < 0.05 (Two way ANOVA); each value is the mean ± S.D; Mean frequency (Hz) represent the mean of three trials; vlPAGon responses (n=32), vlPAGoff responses (n=16) and vlPAGneutral responses (n=20). The grey overlay of each histogram represents the duration of the thermal stimulus administered to the paw. Waveforms of the action potentials that were used to generate the rate histograms are shown above each histogram to the right.
Results
Neuronal Responses to Noxious Thermal Stimulation
The characteristics of vlPAG neuronal firing in the awake-behaving rats were evaluated, using single unit extracellular action potentials in a total of 68 neurons in the initial study from 14 rats. All the vlPAG neurons detected (Fig. 1A) were spontaneously active with an overall mean firing rate of 6.6 ± 0.28 Hz. Three classes of vlPAG units were observed that exhibited different firing changes in response to 51°C noxious thermal stimulation. The majority (71%, 48/68) of vlPAG neurons exhibited firing changes to noxious thermal stimulus that exceeded 10%. Of the responsive vlPAG neurons 47% (n=32) exhibited excitatory responses (vlPAGon neurons), while 24% (n=16) were inhibited by the thermal stimulus (vlPAGoff neurons). The mean spontaneous discharge rates (prior to the thermal stimulus) of vlPAGon neurons (6.7 ± 0.2 Hz) and vlPAGoff neurons (6.1 ± 0.5 Hz) were not significantly different (Student’s t-test, t(19)=4.983, p > 0.05). A minority of vlPAG neurons (29%, n=20) did not exhibit a neuronal firing change that exceeded 10% (vlPAGneutral) in response to the thermal stimulus.
vlPAGon Cells
The 47% of vlPAG neurons that were excited (vlPAGon) by the noxious thermal stimulus (51°C) exhibited a significant mean increase in firing rate (18.1 ± 2.1 Hz), as compared with the pre-stimulus spontaneous firing rate (6.3 ± 0.8 Hz) (Fig. 1B, C; N=32, t(31)=6.891, p<0.05, Student t-test). The firing rate of vlPAGon neurons returned to spontaneous levels after termination of the stimulus. There was no significant difference in the mean spontaneous firing rate seen during the 30 s period after termination of the thermal stimulus (7.9 ± 1.2 Hz) as compared to the firing rate before the stimulus (6.3 ± 0.8 Hz, 30 s, t(31)=37.24, paired t-test). We did not observe any significant sensitization of neuronal responses to the thermal stimulus after presenting the stimulus to the paw in repeated trials [two way ANOVA, F (1, 31) = 2.84; p > 0.05]. Thus, the mean spontaneous firing for each of 3 consecutive days of testing ranged from 6.6 ± 0.9 to 7.7 ± 0.4 Hz, while the mean response to thermal stimulation ranged from 18.2 ± 1.9 to 19.9 ± 0.3 Hz.
vlPAGoff Cells
The 24% of vlPAG neurons that consistently exhibited inhibitory responses (vlPAGoff) to the thermal stimulus showed a significant decrease in firing rate from the mean spontaneous firing rate, (6.2 ± 1.2 Hz to a firing rate of 1.6 ± 0.6 Hz -,Fig. 1B, D; N=16, t(15)=10.67, p<0.05 by Student t-test). Although the stimulus was applied for 30 s, the decrease in firing rate to noxious thermal stimulus was observed only during the initial 15 s of the stimulation period, thus, during the initial 15 s period the firing rate was 1.6 ± 0.6 Hz, while the firing rate for the subsequent 15 s period was 4.7 ± 0.8 Hz, which was significantly different (t(15)=7.12, p<0.05, paired t-test). This might be indicative of acclimatization to the stimulus in these neurons. For the vlPAGoff neurons, there was no significant difference in the mean spontaneous firing rate during the 30 s period following termination of the thermal stimulus (6.9 ± 1.2 Hz) as compared with the firing rate before the stimulus (6.2 ± 1.2 Hz).
Effects of paclitaxel-induced neuropathic pain
Behavioral effects of paclitaxel treatment
We then investigated the effects of the paclitaxel protocol on the behavioral responses to thermal stimuli. The paclitaxel treatment regimen induced thermal allodynia and hyperalgesia as evidenced by changes in PWL that were observed 10 days after the start of the protocol, which is consistent with previous studies [17; 49]. In vehicle-treated rats, thermal stimulation at 38°C and 42°C did not evoke any consistent paw withdrawal responses during the 30 s stimulus period, and we classified these as non-noxious stimuli for the purposes of this study [1] (Fig. 2A). However, 10 days post paclitaxel treatment, thermal stimulation at these same intensities evoked paw withdrawal responses in paclitaxel-treated rats, as shown by the changes in mean PWL (Fig. 2A). Since PWL responses at these stimulus intensities were not seen in vehicle-treated rats, these findings indicate that paclitaxel treatment had induced thermal allodynia (Fig. 2A). Thermal stimulation at 48°C and 51°C did evoke PWLs in vehicle-treated rats and were classified as noxious stimuli (Fig. 2A). After day 10 of the protocol paclitaxel-treated rats exhibited significantly decreased mean PWLs at these stimulus intensities as compared with the pre-paclitaxel responses (Fig. 2A). The difference between these two groups indicate the development of paclitaxel-induced thermal hyperalgesia. Paclitaxel-induced thermal allodynia and hyperalgesia were observed initially at day 10 and continued to be present for 21 days after the initiation of treatment [F(2, 26) = 19.75, p<0.05; Repeated measures ANOVA], which was the last day of testing. Importantly, mean PWLs did not significantly differ from day 10 to 21 post-paclitaxel treatment, and all the neuronal recording experiments were conducted during this time period when the rats were in this hyperalgesic state.
Fig. 2. Response changes of vlPAGon neurons induced by paclitaxel administration.
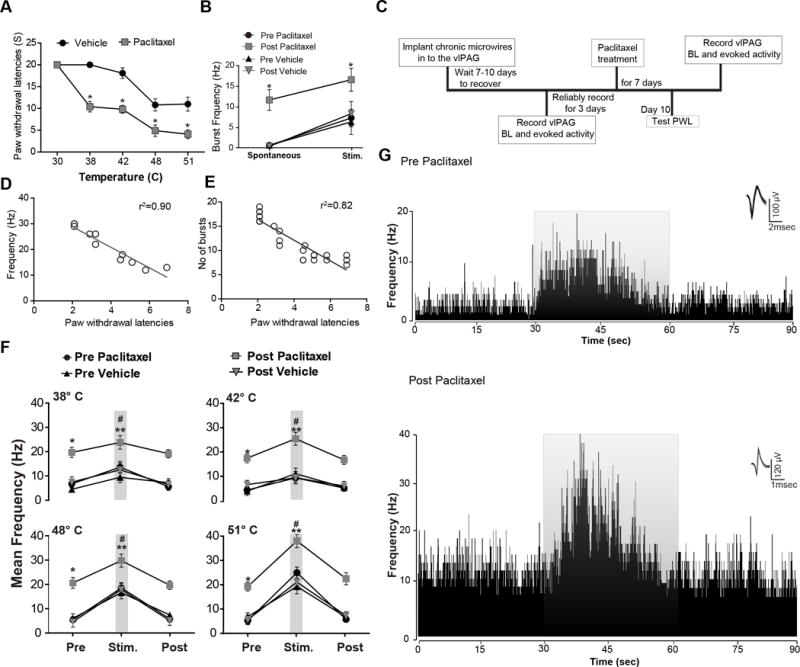
(A) Thermal stimulation at 38°C and 42°C did not evoke any consistent paw withdrawal responses during the 30 s stimulus period in rats prior to treatment with the paclitaxel. Whereas, noxious thermal stimulation at 48°C, and 51°C evoked paw withdrawal in all the rats. The paclitaxel protocol significantly reduced paw withdrawal latencies (PWLs) as compared to pre-treatment and vehicle groups. Significant hypersensitivity to non-noxious thermal stimulus and noxious thermal stimulus after paclitaxel treatment indicates the presence of thermal allodynia and hyperalgesia. (B) Induction of neuropathic pain resulted in the emergence of spontaneous burst firing pattern, whereas vehicle treated rats did not exhibit any spontaneous burst firing pattern. Noxious thermal stimulus at 51°C resulted in a significant increase of the burst firing frequency in paclitaxel-treated rats as compared to pre-treatment and vehicle treated rats. (C) Timeline of vlPAG recordings pre and post paclitaxel treatment. (D) Changes in spontaneous neuronal firing (F(1,8)= 77.25; p< 0.0001) is directly correlated with PWLs changes observed after neuropathic pain. (E) Burst firing (F(1,22) = 101.6; p< 0.0001) observed after the paclitaxel treatment directly correlates with changes in PWLs. (F) The presentation of non-noxious thermal stimulus to the paw did not evoke any changes in the vlPAGon neuronal responses of vehicle-treated rats. In the paclitaxel-induced hyperalgesic state vlPAGon neurons exhibited significantly increased neuronal excitation to previously non-noxious thermal stimulus. vlPAGon neurons showed significantly increased neuronal excitation to the noxious thermal stimulus as compared to the pre-treatment period, whereas in vehicle-treated rats we did not see any changes in neuronal response to noxious thermal stimulus compared to pretreatment. (G) Representation of the rate meter histograms (80 ms bin width) analysis of a typical vlPAGon neuronal response during noxious thermal stimulus (51°C) from vehicle (top trace) and paclitaxel-treated rats (lower trace). vlPAGon neurons in paclitaxel-treated rats showed significantly increased spontaneous and excitatory neuronal responses to the noxious thermal stimulus compared to vlPAGon neuronal responses recorded from the vehicle treated rats. The grey overlay represents the thermal stimulation. Waveforms of the action potentials that were used to generate the rate histograms are shown above each histogram to the right. * P < 0.05, ** P < 0.01; compared to pre-treatment levels, # P < 0.05 compared to vehicle treated. Two-way ANOVA followed by Bonferroni post hoc test. Each value is the mean ± SD; PWLs represent the mean of three trials; N=14 rats per group.
Effects of paclitaxel treatment on PAG neuronal firing patterns
We then investigated whether the neuropathic pain state that followed administration of paclitaxel caused changes in vlPAG neuronal excitability. The mean baseline pre-paclitaxel spontaneous neuronal firing rate was 6.6 ± 0.28 Hz. In paclitaxel-treated rats the mean spontaneous firing rate of vlPAG neurons was increased significantly 10 days after initiation of the paclitaxel treatment as compared to the pre-paclitaxel treatment levels (Fig. 2B, Table 1). vlPAG neurons exhibited an increased incidence of irregular bursting patterns after paclitaxel treatment, whereas we did not observe any evidence of burst firing in the vehicle-treated group or prior to paclitaxel treatment (Fig. 2B). Analysis of burst firing incidence revealed a significant increase in the mean number of bursts after paclitaxel treatment, as compared to pre-paclitaxel treatment (0 vs. 9.1 ± 1.9 bursts, t(9)=5.11, p<0.01, Fig. 2B, paired student- t test). Changes in spontaneous neuronal firing [r2=0.90, F(1,8)= 77.25; p< 0.0001, Fig. 2D] and burst firing [r2=0.82, F(1,22)= 101.6; p< 0.0001, Fig. 2E] were directly correlated with the changes in PWLs observed after neuropathic pain induced by paclitaxel.
Table 1.
Neuropathic pain induced changes in ventrolateral periaqueductal gray on (vlPAGon) neurons.
| Pre-paclitaxel | Post-paclitaxel | Pre-vehicle | Post-vehicle | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
| ||||||||||||
| Stim | Mean Freq (Hz) | No of neurons | Mean Freq (Hz) | No of neurons | P value | No of rats | Mean Freq (Hz) | No of neurons | Mean Freq (Hz) | No of neurons | P value | No of rats |
| Spont | 6.6 ± 0.28 | 52 | 18.8±0.1 | 38 | <0.01 | 14 | 6.8±0.9 | 64 | 6.1±1.6 | 64 | >0.05 | 14 |
|
| ||||||||||||
| Pre | 7.4±1.6 | 52 | 20.3±2.1 | 38 | <0.01 | 14 | 5.1±1.3 | 64 | 8.1±2.2 | 64 | >0.05 | 14 |
| 38° | 12.2±2.4 | 52 | 24.4±2.8 | 38 | <0.05 | 14 | 9.8±2.1 | 64 | 11.1±2.8 | 64 | >0.05 | 14 |
| Post | 6.2±1.5 | 52 | 19.8±1.7 | 38 | <0.01 | 14 | 7.8±1.5 | 64 | 7.1±1.7 | 64 | >0.05 | 14 |
|
| ||||||||||||
| Pre | 5.90±1.1 | 52 | 19.8±1.3 | 38 | <0.01 | 14 | 6.4±1.8 | 64 | 9.1±1.9 | 64 | >0.05 | 14 |
| 42° | 10.86±1.8 | 52 | 25.1±2.6 | 38 | <0.01 | 14 | 11.5±2.3 | 64 | 10.9±2.5 | 64 | >0.05 | 14 |
| Post | 6.70±1.3 | 52 | 17.2±1.8 | 38 | <0.05 | 14 | 7.9±1.6 | 64 | 8.5±1.8 | 64 | >0.05 | 14 |
|
| ||||||||||||
| Pre | 6.8±1.2 | 52 | 21.3±2.3 | 38 | <0.01 | 14 | 7.5±1.8 | 64 | 7.5±2.3 | 64 | >0.05 | 14 |
| 48° | 19.2±2.2 | 52 | 30.6±2.7 | 38 | <0.01 | 14 | 17.2±2.5 | 64 | 18.5±2.6 | 64 | >0.05 | 14 |
| Post | 7.3±1.5 | 52 | 20.3±1.8 | 38 | <0.01 | 14 | 8.8±1.3 | 64 | 6.5±1.9 | 64 | >0.05 | 14 |
|
| ||||||||||||
| Pre | 6.6±1.6 | 52 | 19.1±1.7 | 38 | <0.01 | 14 | 8.1±1.5 | 64 | 7.0±1.7 | 64 | >0.05 | 14 |
| 51° | 24.6±2.2 | 52 | 37.2±2.7 | 38 | <0.01 | 14 | 19.1±2.91 | 64 | 20.8±2.9 | 64 | >0.05 | 14 |
| Post | 7.1±1.3 | 52 | 22.3±2.4 | 38 | <0.01 | 14 | 7.9±1.8 | 64 | 8.5±1.5 | 64 | >0.05 | 14 |
Freq = frequency; Spont = spontaneous; Stim = stimulation
Neuropathic pain-induced changes in vlPAGon neuronal firing
To study how paclitaxel treatment affected thermally-evoked vlPAG neuronal responses, we administered thermal stimulation at 38°, 42°, 48° and 51°C to the rats (Fig. 2F). Presentation of the thermal stimulation at 38° and 42°C to the paw did not evoke any significant changes in the vlPAGon neuronal responses of vehicle-treated rats (Fig. 2F, N=52). On the other hand, vlPAGon neurons (N=38) of rats after the paclitaxel protocol exhibited significantly increased mean neuronal excitation to previously non-noxious (38° and 42°C) stimuli (Fig. 2F, Table 1). These results indicate that vlPAGon neurons exhibit lower thresholds in response to thermal stimuli, which suggests that these neurons may play an important role in the maintenance of thermal allodynia in neuropathic pain. vlPAGon neurons also showed significantly increased mean neuronal excitation to the previous noxious (48° and 51°C) temperatures, as compared to pre-treatment levels (Fig. 2F, 2G, Table 1). Vehicle-treated rats did not show any significant changes in the mean neuronal response to 48° and 51°C stimuli (Table 1). In paclitaxel-treated rats, the incidence of burst neuronal firing observed after noxious thermal stimulus increased significantly as compared to pre-paclitaxel treatment (5.8 ± 1.6 vs. 12.8 ± 2.1; t(9)=8.385, p<0.05, paired student t-test), suggesting that this increase in burst activity to thermal stimulation might underlie the thermal hypersensitivity. The magnitude of thermal stimulation-evoked mean neuronal responses of paclitaxel-treated rats at all stimulus intensities was significantly greater than that of pre-drug recordings (Fig. 2F, 2G, Table 1). Thus, in the paclitaxel-induced hyperalgesic state, vlPAGon neurons exhibited enhanced spontaneous and evoked excitability, which correlated with the increase in behavioral sensitivity.
Neuropathic pain-induced changes in vlPAGoff neuronal firing
Prior to paclitaxel treatment, vlPAGoff neurons did not exhibit any detectable neuronal inhibition to the thermal stimulation at 38° and 42°C in pre-paclitaxel recordings (Fig. 3A). However, stimulation at 48°C and 51°C evoked inhibitory neuronal responses in the vlPAGoff neurons (Fig. 3A). Vehicle treated rats did not exhibit any significant changes in mean vlPAGoff neuronal firing at all temperatures tested (Fig. 3A, Table 2). After paclitaxel treatment, surprisingly, thermal stimulation at 38°C and 42°C evoked excitatory neuronal responses from the same electrodes that had recorded non-responsive neurons at these stimulus intensities before the paclitaxel protocol. Prior to paclitaxel treatment, thermal stimulation at 48°C and 51°C evoked inhibitory neuronal responses in vlPAGoff neurons, whereas after paclitaxel treatment only excitatory neuronal responses were observed to thermal stimulation at all temperatures tested (Fig. 3B, 3C, Table 2). Waveforms show that the single units recorded post-paclitaxel treatment exhibited different waveform shapes, peak amplitudes, and spike widths from the neurons recorded from the same electrodes pre-paclitaxel treatment (Fig. 3B, 3C & 4E). Changes in evoked neuronal firing to the 51°C stimulus (r2=0.74, F(1,35)= 100.3; p< 0.0001, Fig. 3D) were directly correlated with PWL changes observed after neuropathic pain induction. These results suggest that a shift in homeostatic balance from descending inhibitory drive to descending facilitatory drive occurred in the vlPAG after paclitaxel treatment.
Fig. 3. Responses from electrodes that recorded vlPAGoff neuronal responses before and from the same electrode after the neuropathic pain protocol.
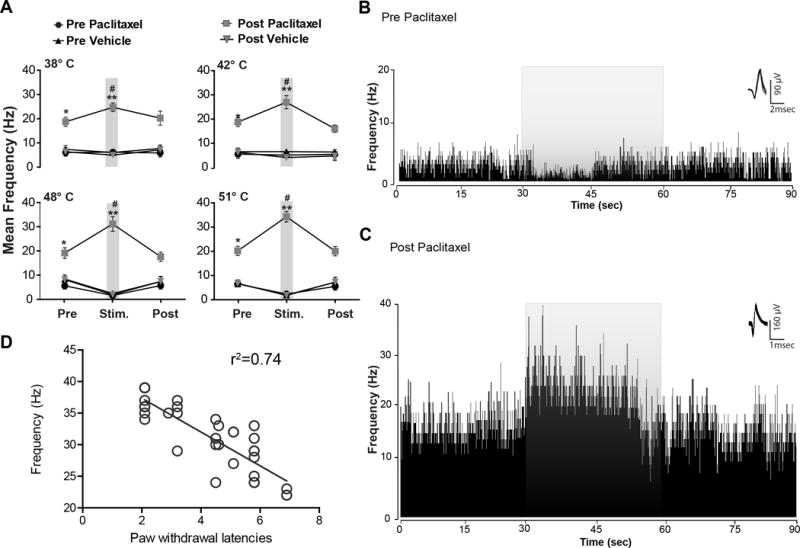
(A) Prior to the paclitaxel protocol (pre-paclitaxel) no detectable neuronal inhibition to the non-noxious thermal stimulus (38° and 42°C) vlPAGoff was seen (n=16). However, noxious thermal stimulus evoked vlPAGoff neuronal inhibitory responses (48° and 51°C, N=16 neurons). After paclitaxel treatment, noxious thermal stimulus did not evoke neuronal inhibition in the vlPAG neurons recorded from the same electrodes but excitatory responses were consistently seen. Prior to paclitaxel treatment, noxious thermal stimulus evoked inhibitory neuronal responses (N=16) in vlPAGoff neurons, whereas after paclitaxel treatment only excitatory neuronal responses was observed to both non-noxious thermal stimulus and noxious thermal stimulus from the same electrodes. (B) Example of the rate meter histogram (80 ms bin width) analysis of a typical vlPAGoff neuronal response during noxious thermal stimulus (51°C) from vehicle and (C) from the same electrode after paclitaxel treatment. Prior to paclitaxel treatment vlPAGoff neurons exhibited an inhibitory response to the noxious thermal stimulus. vlPAG neurons in paclitaxel-treated rats recorded from the same electrodes that previously recorded vlPAGoff neurons showed significantly increased spontaneous activity and excitatory neuronal responses to the noxious thermal stimulus. The grey overlay represents the thermal stimulation. Waveforms of the action potentials that were used to generate the rate histograms are shown above each histogram to the right. (D) Changes in thermal stimulus evoked neuronal firing (N=37 neurons, F(1,35)= 100.3; p< 0.0001) is directly correlated with PWLs changes observed after neuropathic pain. * P < 0.05, ** P < 0.01; compared to pre-treatment levels, # P < 0.05 compared to vehicle treated group. Two-way ANOVA followed by Bonferroni post hoc test. Each value is the mean ± SD; N=14 rats per group.
Table 2.
Neuropathic pain induced changes in ventrolateral periaqueductal gray off (vlPAGoff) neurons.
| Pre-paclitaxel | Post-paclitaxel | Pre-vehicle | Post-vehicle | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
|
| ||||||||||
| Stim | Mean Freq (Hz) | No of neurons | Mean Freq (Hz) | No of neurons | P value | Mean Freq (Hz) | No of neurons | Mean Freq (Hz) | No of neurons | P value |
| Pre | 6.9±1.5 | 16 | 19.6±1.9 | 12 | <0.05 | 8.1±1.5 | 16 | 7.1±1.8 | 16 | >0.05 |
| 38° | 7.1±1.1 | 16 | 23.6±1.8 | 12 | <0.01 | 6.9±0.9 | 16 | 5.8±0.9 | 16 | >0.05 |
| Post | 6.7±1.7 | 16 | 21.1±2.9 | 12 | <0.01 | 8.5±1.4 | 16 | 7.9±1.5 | 16 | >0.05 |
|
| ||||||||||
| Pre | 7.2±1.7 | 16 | 20.3±1.8 | 12 | <0.01 | 8.1±1.2 | 16 | 7.9±1.7 | 16 | >0.05 |
| 42° | 6.6±0.2 | 16 | 26.4±2.7 | 12 | <0.001 | 8.2±0.7 | 16 | 5.8±0.8 | 16 | >0.05 |
| Post | 7.1±1.9 | 16 | 17.6±1.5 | 12 | <0.05 | 8.0±1.1 | 16 | 6.4±1.8 | 16 | >0.05 |
|
| ||||||||||
| Pre | 7.1±1.4 | 16 | 20.6±2.2 | 12 | <0.05 | 8.9±1.5 | 16 | 8.6±1.8 | 16 | >0.05 |
| 48° | 1.2±0.3 | 16 | 31.6±3.0 | 12 | <0.0001 | 1.8±1.0 | 16 | 1.4±0.7 | 16 | >0.05 |
| Post | 6.2±1.5 | 16 | 19.1±1.9 | 12 | <0.01 | 7.9±2.1 | 16 | 8.9±1.9 | 16 | >0.05 |
|
| ||||||||||
| Pre | 6.9±1.2 | 16 | 20.3±1.5 | 12 | <0.05 | 8.0±1.2 | 16 | 7.1±1.4 | 16 | >0.05 |
| 51° | 2.8±0.9 | 16 | 34.2±2.1 | 12 | <0.0001 | 1.4±0.9 | 16 | 1.2±0.8 | 16 | >0.05 |
| Post | 5.9±1.5 | 16 | 21.6±1.2 | 12 | <0.01 | 8.26±2.0 | 16 | 8.5±1.5 | 16 | >0.05 |
Freq = frequency; Stim = stimulation
Fig. 4. Responses from electrodes that recorded vlPAGneutral neuronal responses before and from the same electrodes after the neuropathic pain protocol.
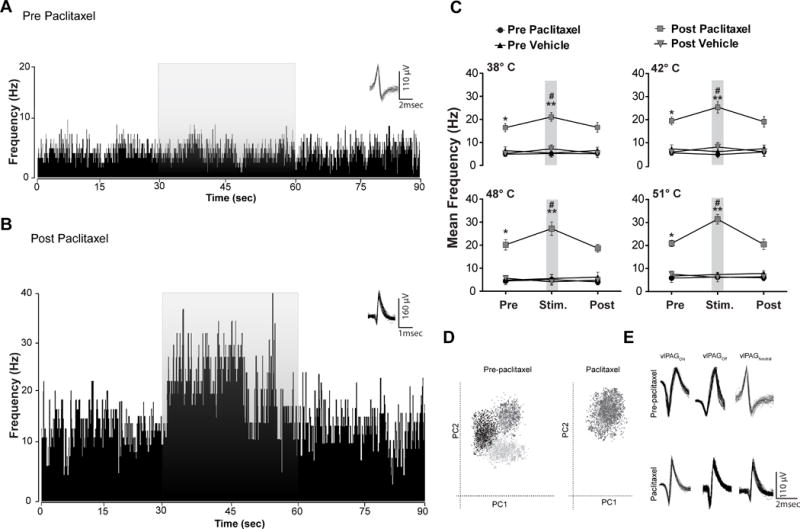
(A) An example of a typical rate meter histogram (80 ms bin width) of a vlPAGneutral neuronal response during noxious thermal stimulus (51°C) from vehicle and (B) paclitaxel treated rats recorded from the same electrode. Prior to paclitaxel treatment vlPAG neurons exhibited no change in neuronal activity to the noxious thermal stimulus. Paclitaxel-treated rats showed significantly increased spontaneous activity and excitatory neuronal responses to the noxious thermal stimulus unlike the vlPAG neuronal responses recorded from the vehicle treated rats. The grey overlay represents the thermal stimulation. Waveforms of the action potentials that were used to generate the rate histograms are shown above each histogram to the right. (C) Prior to paclitaxel treatment in vlPAGneutral neurons no detectable responses to the non-noxious thermal stimulus (38°C and 42°C) or noxious thermal stimulus (48°C and 51°C) were seen. However, after the paclitaxel treatment protocol excitatory neuronal responses to non-noxious thermal stimulus and noxious thermal stimulus in vlPAG neurons were observed from the same electrodes. (D) PCA shows prior to paclitaxel treatment, there are three different clusters of single units whereas post paclitaxel treatment only one cluster of single units was observed. (E) Representative example of waveforms recorded from the vlPAG before and after paclitaxel treatment. * P < 0.05, ** P < 0.01; compared to pre-treatment levels, # P < 0.05 compared to vehicle treated group. Two-way ANOVA followed by Bonferroni post hoc test. Each value is the mean ± SD; N=14 rats per group.
Neuropathic pain-induced changes in vlPAGneutral neuronal firing
vlPAGneutral neurons (N=20, 29%) did not exhibit responses to thermal stimulation at any temperatures tested (Fig. 4A). However, after paclitaxel treatment, we observed excitatory neuronal responses to thermal stimulation at all of these temperatures from the same electrodes (Fig. 4B, 4C, Table 3). Vehicle-treated rats did not exhibit any significant changes in mean vlPAGneutral neuronal firing at non-noxious (38° and 42°C) temperatures (Fig. 4C, Table 3) and noxious (48° and 51°C) stimuli (Fig. 4C, Table 3). Our PCA showed that prior to paclitaxel treatment, there were three different clusters of single units, whereas post paclitaxel treatment, only one cluster of single units was observed. Waveforms show that the single units recorded post-paclitaxel treatment exhibited different waveform shapes, peak amplitudes, and spike widths from the neurons recorded from the same electrodes pre-paclitaxel treatment, suggesting that the neurons recorded after paclitaxel treatment may be different from the ones recorded prior to paclitaxel treatment (Fig 4D and E). That is, we did not encounter vlPAGneutral neurons after paclitaxel treatment. Thus, neuropathic pain induces specific and persistent changes in vlPAG neuronal responses to thermal stimuli.
Table 3.
Neuropathic pain induced changes in ventrolateral periaqueductal gray neutral (vlPAGneutral) neurons.
| Pre-paclitaxel | Post-paclitaxel | Pre-vehicle | Post-vehicle | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
|
| ||||||||||
| Stim | Mean Freq (Hz) | No of neurons | Mean Freq (Hz) | No of neurons | P value | Mean Freq (Hz) | No of neurons | Mean Freq (Hz) | No of neurons | P value |
| Pre | 6.6±1.0 | 18 | 18.1±1.8 | 9 | <0.05 | 7.1±1.6 | 16 | 6.8±1.2 | 16 | >0.05 |
| 38° | 6.9±1.2 | 18 | 22.8±1.9 | 9 | <0.01 | 7.1±1.8 | 16 | 7.9±1.7 | 16 | >0.05 |
| Post | 6.8±1.1 | 18 | 18.3±2.1 | 9 | <0.01 | 7.2±1.4 | 16 | 7.0±1.8 | 16 | >0.05 |
|
| ||||||||||
| Pre | 6.3±1.2 | 18 | 18.9±1.9 | 9 | <0.01 | 6.8±1.5 | 16 | 6.5±1.6 | 16 | >0.05 |
| 42° | 6.8±0.8 | 18 | 27.9±1.6 | 9 | <0.001 | 7.1±1.2 | 16 | 7.0±1.2 | 16 | >0.05 |
| Post | 6.2±1.2 | 18 | 18.7±1.2 | 9 | <0.05 | 6.9±1.3 | 16 | 6.8±1.5 | 16 | >0.05 |
|
| ||||||||||
| Pre | 6.8±1.6 | 18 | 22.6±2.3 | 9 | <0.05 | 6.2±1.1 | 16 | 6.1±1.8 | 16 | >0.05 |
| 48° | 7.5±2.1 | 18 | 29.6±2.9 | 9 | <0.0001 | 6.9±1.7 | 16 | 6.5±1.5 | 16 | >0.05 |
| Post | 6.4±1.3 | 18 | 21.1±1.7 | 9 | <0.01 | 6.5±2.1 | 16 | 7.2±1.4 | 16 | >0.05 |
|
| ||||||||||
| Pre | 6.7±1.9 | 18 | 21.7±1.4 | 9 | <0.05 | 6.9±1.8 | 16 | 6.3±1.2 | 16 | >0.05 |
| 51° | 7.1±2.1 | 18 | 32.2±2.1 | 9 | <0.0001 | 6.2±0.9 | 16 | 6.6±1.1 | 16 | >0.05 |
| Post | 6.8±1.3 | 18 | 21.3±2.3 | 9 | <0.01 | 7.1±1.3 | 16 | 6.6±1.5 | 16 | >0.05 |
Freq = frequency; Stim = stimulation
Effect of gabapentin on behavioral hypersensitivity induced neuropathic pain
The timeline of vlPAG recordings used to evaluate the effects of gabapentin in pre- and post-paclitaxel treated rats is shown in Fig. 5A. Systemic administration of gabapentin (GBP) (10, 30, and 50 mg/kg) (Fig. 5B) in paclitaxel-treated rats resulted in dose-dependent effects. A gabapentin dose of 10 mg/kg did not show any significant effects on thermal hyperalgesia [F(1, 13) = 0.42; p > 0.05, Fig. 5B]. However, gabapentin at 30 mg/kg [Fig. 5B; F(1, 13) = 79.23; p < 0.05] and 50 mg/kg [Fig. 5B, 5C; F(1, 13) = 88.52; p < 0.05] significantly reduced thermal hyperalgesia in paclitaxel-treated rats in a dose-dependent manner. Anti-hyperalgesia effects were observed at 30 and 60 min [F(1, 13) = 29.57; p < 0.05; F (1, 13) = 44.08; p < 0.05]. Recovery from the analgesic effects of gabapentin occurred by 24 hr for all the doses. As noted above, after paclitaxel treatment, previously non-noxious thermal stimuli (38°C and 42°C) (Fig. 5C) evoked paw withdrawal responses indicative of thermal allodynia. In addition, after paclitaxel treatment, noxious thermal stimulation significantly decreased PWL in all the rats compared to the baseline levels prior to paclitaxel treatment at 48°C and 51°C, (Fig. 5C), which is indicative of thermal hyperalgesia. Vehicle-treated animals (n=14) showed no significant change in mean PWL as compared with baseline levels (Fig. 5C, N=14 per group).
Fig. 5. Effects of gabapentin on responses of vlPAGon neurons.
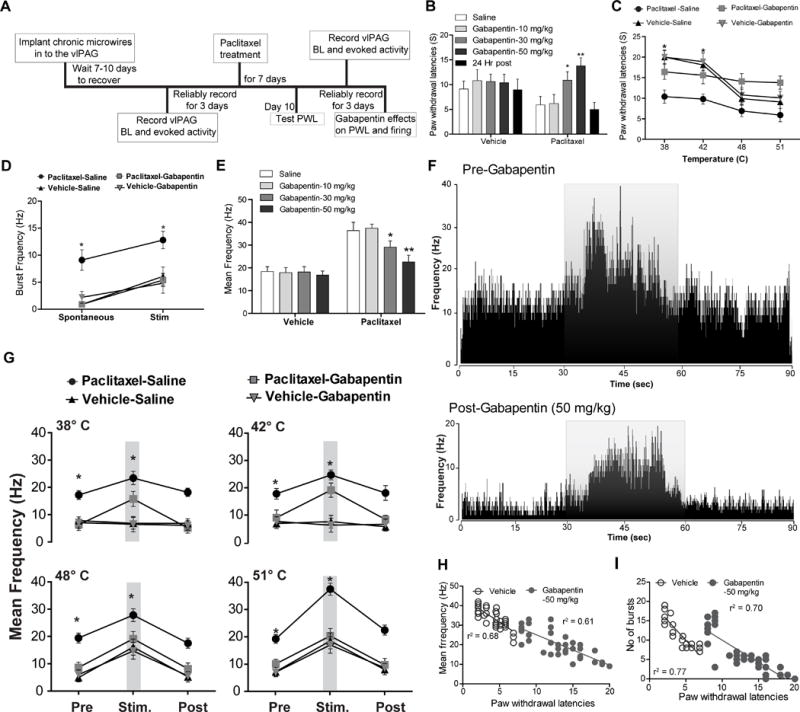
(A) Timeline of vlPAG recordings to evaluate effects of gabapentin in pre and post paclitaxel treated rats. (B) Systemic (intraperitoneal) administration of gabapentin resulted in dose-dependent antinociceptive effects on paw withdrawal latencies in paclitaxel treated rats but not in naïve (vehicle treated) rats. (C) Administration of gabapentin (50 mg/kg) reversed thermal hyperalgesia in paclitaxel treated rats but not in vehicle-treated rats. (D) Gabapentin administration (50 mg/kg) significantly reduced vlPAGon spontaneous and noxious thermal stimulus (51°C)-evoked burst neuronal firing in paclitaxel-treated rats but not in vehicle-treated rat. (E) Administration of gabapentin resulted in dose-dependent decrease in vlPAGon neuronal firing in paclitaxel treated rats but not in vehicle-treated rats. (F) Representative rate meter histograms show a significant reduction of spontaneous and noxious thermal stimulus (51°C) evoked activity in the vlPAGon neuron by gabapentin (50 mg/kg) i.p in the PAG neurons in paclitaxel-treated rats one hr after gabapentin treatment. The grey overlay represents the thermal stimulation. (G) Systemic administration of gabapentin (50 mg/kg) resulted in significant reduction in the vlPAGon neuronal firing evoked by the non-noxious thermal stimulus (38°C and 42°C) and noxious thermal stimulus (48°C and 51°C) in the paclitaxel-treated rats, but no effects were seen in vehicle-treated rats. (H) Gabapentin mediated attenuation of evoked neuronal firing (F(1, 37)= 56.44; p< 0.0001) and (I) burst firing (F(1,22)= 82.53; p< 0.0001) is directly correlated with elevated PWLs observed after gabapentin treatment in the paclitaxel-treated rats. * P < 0.05, ** P < 0.01; compared to pre-treatment levels. Two-way ANOVA followed by Bonferroni post hoc test. Each value is the mean ± SD; PWLs represent the mean of three trials; N=14 rats per group.
Gabapentin effects on vlPAG spontaneous neuronal firing in paclitaxel-treated rats
We examined whether the analgesic effect of gabapentin in paclitaxel-treated rats involved effects on vlPAG neuronal firing. We administered gabapentin systemically to determine effects of this agent on vlPAG spontaneous and evoked neuronal firing in vehicle vs. paclitaxel-treated rats. Gabapentin (10, 30, and 50 mg/kg i.p.) dose-dependently reduced the mean spontaneous vlPAG burst of neuronal firing in paclitaxel-treated rats (Fig. 5D, 5E and 5F, Table 4). However, gabapentin administration in the same doses in the vehicle-treated rats did not induce any significant changes in mean vlPAG neuronal firing. ANOVA analysis revealed that there was a significant interaction between gabapentin treatment (50 mg/kg, i.p) and paclitaxel treatment [F(2, 46) = 33.87; p < 0.05]. A dose of 10 mg/kg gabapentin did not have any significant effects on mean vlPAG spontaneous neuronal firing, [F(1, 37) = 0.54; p > 0.05, Table 4]. However, administration of the 30 and 50 mg/kg doses of gabapentin reduced the mean spontaneous vlPAG burst neuronal firing in the paclitaxel-treated rats significantly [F(1, 37) = 29.57; p < 0.05, F(1, 37) = 54.25; P < 0.05, Table 4]. These results suggest that gabapentin’s analgesic effects may be mediated, in part, by suppressing hyperactive vlPAG neuronal firing in the neuropathic pain conditions.
Table 4.
Effects of gabapentin on spontaneous and thermal stimulus evoked ventrolateral periaqueductal gray (vlPAG) neuronal firing in paclitaxel-treated rats.
| Saline | Gabapentin (10mg/kg) | Gabapentin (30mg/kg) | Gabapentin (50mg/kg) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
| ||||||||||||
| Stim | Mean Freq (Hz) | No of neurons | Mean Freq (Hz) | No of neurons | P value | Mean Freq (Hz) | No of neurons | P value | Mean Freq (Hz) | No of neurons | P value | |
| Vehicle | Spont | 5.9.±1.2 | 52 | 6.3±1.1 | 38 | >0.05 | 6.6±1.5 | 38 | p>0.05 | 6.1±1.2 | 38 | <0.01 |
| 38° | 7.3±1.5 | 52 | 7.9±1.6 | 38 | >0.05 | 7.8±1.3 | 38 | p>0.05 | 6.9±1.9 | 38 | <0.05 | |
| 42° | 7.9±1.7 | 52 | 7.7±1.8 | 38 | >0.05 | 8.1±1.1 | 38 | p>0.05 | 6.8±1.8 | 38 | <0.05 | |
| 48° | 17.9±1.4 | 52 | 17.2±1.8 | 38 | >0.05 | 19.1±1.5 | 38 | p>0.05 | 16.7±1.4 | 38 | <0.01 | |
| 51° | 18.4±2.1 | 52 | 17.9±2.2 | 38 | >0.05 | 18.2±2.3 | 38 | p>0.05 | 16.8±1.8 | 38 | <0.01 | |
|
| ||||||||||||
| Paclitaxel | Spont | 18.5.±2.0 | 52 | 19.1±1.6 | 38 | >0.05 | 14.4±1.1 | 38 | p<0.05 | 9.8±1.6 | 38 | <0.01 |
| 38° | 24.4±2.1 | 52 | 21.1±2.2 | 38 | >0.05 | 19.5±1.3 | 38 | p<0.05 | 16.8±1.8 | 38 | <0.05 | |
| 42° | 25.5±1.8 | 52 | 26.1±1.7 | 38 | >0.05 | 21.8±1.6 | 38 | p>0.05 | 19.5±2.6 | 38 | <0.05 | |
| 48° | 28.5±2.3 | 52 | 29.6±2.1 | 38 | >0.05 | 22.2±1.8 | 38 | p<0.05 | 19.9±2.6 | 38 | <0.01 | |
| 51° | 37.2±1.3 | 52 | 33.8±3.1 | 38 | >0.05 | 25.7±1.8 | 38 | p<0.01 | 20.1±2.7 | 38 | <0.01 | |
Freq = frequency; Spont = spontaneous; Stim = stimulation
Gabapentin effects on thermal stimulus-evoked vlPAG neuronal firing in paclitaxel-treated rats
Paclitaxel-treated rats showed significantly increased mean vlPAG neuronal responses to the previously non-noxious 38° and 42°C (Table 4) and noxious 48° and 51°C stimuli (Table 4) compared to vehicle-treated rats, which was consistent with our earlier observations. Gabapentin (10, 30 and 50 mg/kg; Fig. 5E) administration resulted in dose-dependent analgesic effects on the thermal evoked vlPAG neuronal responses in paclitaxel-treated rats, which was not seen in vehicle-treated rats. At a dose of 10 mg/kg, gabapentin did not have any effects on mean thermal evoked neuronal firing in paclitaxel-treated rats [F(1, 37) = 5.23; p > 0.05] or in vehicle-treated rats [F(1, 51) = 1.15; p > 0.05] (Table 4). Systemic administration of gabapentin (30 and 50 mg/kg) resulted in significant reductions in mean vlPAG neuronal firing evoked by the non-noxious thermal stimuli (38° and 42°C; Fig. 5G) and noxious thermal stimuli (48° and 51°C; Fig. 5G) in the paclitaxel-treated rats, but no effects of gabapentin were seen in vehicle-treated rats. The degree of suppression of vlPAG evoked responses was greatest with the 50 mg/kg dose of gabapentin [Fig. 5F, 5G, F(1, 37) = 58.93; p< 0.05]. Gabapentin-induced attenuation of evoked neuronal firing [r2=0.61, F(1, 37)= 56.44; p< 0.0001, Fig. 5H] and burst firing [r2=0.70, F(1,22)= 82.53; p< 0.0001, Fig. 5I] were directly correlated with elevated PWLs observed after gabapentin treatment in the paclitaxel-treated rats. We did not see any vlPAGoff or vlPAGneutral responses after gabapentin treatment in paclitaxel-treated rats. These results suggest that the analgesic effects of gabapentin in the paclitaxel-treated rats may involve decreased responsiveness of vlPAG neurons.
Discussion
The neuronal mechanisms that mediate neuropathic pain induced by chemotherapeutic agents, such as paclitaxel, are not well understood despite their clinical relevance. Increased excitability of dorsal root ganglion neurons has been shown to contribute to the persistent allodynia and hyperalgesia observed after treatment with paclitaxel [10; 49; 66]. The present study examined the contribution of vlPAG neurons in the transition from the acute to the neuropathic pain state by evaluating changes in the firing of vlPAG neurons induced by a paclitaxel protocol in awake-behaving rats. Our results show that systemic administration of paclitaxel induced a neuropathic pain state characterized by thermal allodynia and hyperalgesia of the paw to noxious and non-noxious stimuli, which is consistent with previous studies [11; 17; 39]. Ten days to three weeks after initiation of paclitaxel treatment, spontaneous firing rates in vlPAG neurons were significantly increased in treated rats as compared to the pre-paclitaxel and vehicle treated animals. This increase in activity correlated with the development of behavioral thermal hypersensitivity, which was also observed 10–21 days following initiation of the protocol. Paclitaxel treatment also resulted in a significant increase in spontaneous burst firing patterns in vlPAG neurons, which were not observed before the paclitaxel treatment. Thus, the induction of neuropathic pain paralleled the significant increase in spontaneous and burst firing in vlPAG neurons.
In the current study we recorded the responsiveness of vlPAG neurons to noxious thermal stimuli in awake-behaving rats. We confirmed that the vlPAG contains three distinct subpopulations of neurons that respond to noxious thermal stimuli applied to the paw. Thus, excitatory or “vlPAGon,” inhibitory or “vlPAGoff,” and neutral or “vlPAGneutral” neurons were observed in our unanesthetized rats, as seen previously in lightly anesthetized rats, although the percentage of neurons that were responsive to painful stimuli was higher in the unanesthetized animals [14; 19; 21; 53]. The majority of PAG neurons responded to noxious thermal stimuli with either increases or decreases of neuronal firing, and the firing patterns returned to spontaneous levels after termination of the stimuli. The responsiveness of PAG neurons to thermal pain in the present study is consistent with earlier studies that implicated the PAG in modulating nociception in humans and in rats [2; 6; 21; 50; 52; 60], which observed the presence of these three subclasses in response to painful stimuli [14; 19; 21; 53]. Notably, the present results are consistent with prior findings in the midbrain, where excitatory (48%) and inhibitory (25%) neuronal responses related to tail flick were observed in response to the noxious radiant heat, including animals anesthetized with an agent that also possesses stimulant properties (choralose) [14].
Single unit recording from vlPAG neurons prior to induction of neuropathic pain in awake-behaving rats offered a unique opportunity to monitor the electrophysiological properties of vlPAG neurons during transition from acute to chronic neuropathic pain, which to our knowledge was not investigated previously. In the present study, we demonstrate that after chemotherapy-induced neuropathy, vlPAGon neurons showed neuronal excitation to temperatures that were previously non-noxious thermal stimuli and significantly increased excitability to previous noxious thermal stimuli, as compared to the responses in pre-treatment and vehicle-treated rats. These results suggest that lowered response thresholds and increased responsiveness of vlPAGon neurons to non-noxious thermal stimuli and noxious thermal stimuli may contribute to maintenance of behavioral thermal allodynia and hyperalgesia, since the responsiveness of vlPAGon neurons to thermal stimuli is negatively correlated with the PWLs in these animals.
Surprisingly, in the present study, the inhibitory and neutral responses of vlPAG neurons seen during noxious thermal stimuli before paclitaxel treatment could not be observed following paclitaxel administration. Thus, after paclitaxel treatment, thermal stimulation only evoked excitatory neuronal responses from the same electrodes from which we previously recorded vlPAGoff and vlPAGneutral neurons. Waveform analysis and PCA showed differences in spike shape and separate clusters before and after development of neuropathic pain, suggesting that neurons recorded after development of neuropathic pain might be different from the ones that were recorded before paclitaxel treatment. Another possible explanation is that phenotypic changes in the vlPAGoff and vlPAGneutral to vlPAGon action potential waveforms occurred due to paclitaxel-induced alterations of neuronal membrane properties, since previous studies observed changes in neuronal phenotype in RVM neutral cells after acute inflammatory pain. In that study the neutral cells were reclassified as on- or off-like cells to noxious stimuli after acute inflammation [40]. It is also possible that the vlPAGoff and vlPAGneutral neurons became inactive, and we might be recording from other previously quiescent neurons in the area that became active after the development of neuropathic pain. The present results suggest that excitability changes in the vlPAG occur following the onset of neuropathic pain, which are correlated with development of behavioral hypersensitivity. It has been well-documented that altered discharges of spinal neurons contribute to chemotherapy-induced neuropathy [8; 10; 67]. It is possible that enhanced activity in spinal neurons after neuropathy could drive the excitability changes in the vlPAG, since these neurons receive direct projections from spinal cord neurons [7; 28; 44; 45].
In the naïve state, activation of the vlPAG recruits a descending inhibitory pathway via RVM, resulting in antinociception [2; 16; 22]. It has been previously reported that off-cells in the RVM that are associated with descending inhibitory analgesic pathways receive inputs from glutamatergic vlPAG projection neurons [33; 42; 59]. Numerous studies show a shift in homeostatic balance from descending inhibitory drive to descending facilitatory drive in the RVM that may contribute importantly to chronic pain states [9; 12; 25; 35; 40; 41; 55; 64; 68]. It is unclear whether this shift to descending facilitatory drive also occurs in the PAG, or involves upstream projections from the RVM. Our study shows that vlPAGon neurons exhibit significantly enhanced spontaneous and evoked excitability after the development of neuropathic pain. The absence of vlPAGoff and vlPAGneutral responses to thermal stimuli suggests a shift from descending inhibitory drive to increased descending facilitatory drive from the vlPAG. Recent studies have identified two mechanisms by which the vlPAG could increase descending facilitatory tone; either by increasing presynaptic GABA release [18], which inhibits vlPAG output neurons, or by decreased excitatory neurotransmission in the vlPAG due to decreased glutamate release in vlPAG output neurons [26]. It is possible that after development of neuropathic pain in the current study we are recording from presynaptic GABA neurons, which were shown to exhibit increased activity in animal models of neuropathic pain [26]. This sustained increase in GABAergic neuronal activity might lead to inhibition of vlPAG glutamatergic output neurons and contribute to the development or maintenance of neuropathic pain. Future experiments will need to address the functional roles of GABAergic and glutamatergic neurons in the vlPAG in the development and maintenance of neuropathic pain.
The present study found that systemic gabapentin exhibited anti-hyperalgesic effects in the paclitaxel-treated group, whereas gabapentin did not cause any changes in PWL in naïve rats [46; 57; 58]. We also observed that gabapentin exhibited dose-dependent decreases in the elevated spontaneous tonic firing and irregular bursting in the vlPAG neurons that were induced by the paclitaxel protocol. The elevated spontaneous neuronal firing in vlPAG also decreased to control levels after gabapentin treatment. No effects of gabapentin were observed prior to the paclitaxel protocol or in the vehicle control group. Gabapentin administration also exhibited dose-dependent decreases in the evoked vlPAG neuronal firing to both non-noxious and noxious thermal stimuli but only in the neuropathic pain state, which directly correlated with the anti-hyperalgesic effects of gabapentin. Gabapentin-mediated decreases in the excitability of vlPAG neurons might be due to effects on either spinal or supraspinal components of the pain pathway or both. The analgesic effect of gabapentin is thought to be due to its high affinity for the α2δ-1 subunit in N-type and P/Q-type calcium channels, which inhibits neurotransmitter release from the pre-synaptic terminal and contributes to the analgesic effects of gabapentin in neuropathic pain [13; 20; 27; 31; 56]. One possible mechanism for the effects seen in the current study might be that gabapentin binds to the up-regulated α2δ-1 subunit in the N- and P-type calcium channels in the vlPAG to inhibit presynaptic neurotransmitter release, decreasing vlPAG neuronal excitability in paclitaxel-treated rats [3; 4; 29].
In conclusion, our findings establish evidence for changes in vlPAG neuronal responses during the transition from acute to chronic pain. Such changes in supraspinal centers might be an important feature in the maintenance of neuropathic pain. These findings are consistent with previous studies, which observed increased excitability to non-noxious thermal stimuli and noxious thermal stimuli of RVMon neurons after inflammation in awake, freely moving rats [41]. Reduction in RVMon cell activity was associated with antinociceptive effects of opioids in awake freely moving rats [23; 36; 41]. We observed that the analgesic effects of the non-opioid analgesic, gabapentin, might be mediated, in part, by dampening the neuropathic elevation of excitability of the vlPAG, thus decreasing the tonic facilitatory drive in chemotherapy-induced neuropathic pain consistent with the model proposed for the RVM [23; 36; 41].
Acknowledgments
The authors wish to thank Marcus Randall for technical assistance and Gayle Stauffer for manuscript assistance. We thank Howard L. Fields, Robert W. Gereau IV, Daniel E. O’Brien, and Jose G. Grajales for manuscript comments. This work was supported by grants from National Institutes of Health and EAM award from SIUSOM.
Footnotes
The authors declare no competing financial interests.
References
- 1.Banik RK, Kabadi RA. A modified Hargreaves’ method for assessing threshold temperatures for heat nociception. J Neurosci Methods. 2013;219(1):41–51. doi: 10.1016/j.jneumeth.2013.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Basbaum AI, Fields HL. Endogenous pain control mechanisms: review and hypothesis. Ann Neurol. 1978;4(5):451–462. doi: 10.1002/ana.410040511. [DOI] [PubMed] [Google Scholar]
- 3.Bauer CS, Nieto-Rostro M, Rahman W, Tran-Van-Minh A, Ferron L, Douglas L, Kadurin I, Sri Ranjan Y, Fernandez-Alacid L, Millar NS, Dickenson AH, Lujan R, Dolphin AC. The increased trafficking of the calcium channel subunit alpha2delta-1 to presynaptic terminals in neuropathic pain is inhibited by the alpha2delta ligand pregabalin. J Neurosci. 2009;29(13):4076–4088. doi: 10.1523/JNEUROSCI.0356-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bauer CS, Rahman W, Tran-van-Minh A, Lujan R, Dickenson AH, Dolphin AC. The anti-allodynic alpha(2)delta ligand pregabalin inhibits the trafficking of the calcium channel alpha(2)delta-1 subunit to presynaptic terminals in vivo. Biochem Soc Trans. 2010;38(2):525–528. doi: 10.1042/BST0380525. [DOI] [PubMed] [Google Scholar]
- 5.Bee LA, Dickenson AH. Descending facilitation from the brainstem determines behavioural and neuronal hypersensitivity following nerve injury and efficacy of pregabalin. Pain. 2008;140(1):209–223. doi: 10.1016/j.pain.2008.08.008. [DOI] [PubMed] [Google Scholar]
- 6.Behbehani MM, Fields HL. Evidence that an excitatory connection between the periaqueductal gray and nucleus raphe magnus mediates stimulation produced analgesia. Brain Res. 1979;170(1):85–93. doi: 10.1016/0006-8993(79)90942-9. [DOI] [PubMed] [Google Scholar]
- 7.Bianchi R, Tredici G, Gioia M. The spinal terminals into the midbrain periaqueductal gray of the rat. A light and electron microscope study of the projections ascending via the ventro-lateral funiculus. J Hirnforsch. 1990;31(3):349–358. [PubMed] [Google Scholar]
- 8.Braz JM, Wang X, Guan Z, Rubenstein JL, Basbaum AI. Transplant-mediated enhancement of spinal cord GABAergic inhibition reverses paclitaxel-induced mechanical and heat hypersensitivity. Pain. 2015;156(6):1084–1091. doi: 10.1097/j.pain.0000000000000152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Carlson JD, Maire JJ, Martenson ME, Heinricher MM. Sensitization of pain-modulating neurons in the rostral ventromedial medulla after peripheral nerve injury. J Neurosci. 2007;27(48):13222–13231. doi: 10.1523/JNEUROSCI.3715-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cata JP, Weng HR, Chen JH, Dougherty PM. Altered discharges of spinal wide dynamic range neurons and down-regulation of glutamate transporter expression in rats with paclitaxel-induced hyperalgesia. Neuroscience. 2006;138(1):329–338. doi: 10.1016/j.neuroscience.2005.11.009. [DOI] [PubMed] [Google Scholar]
- 11.Cavaletti G, Cavalletti E, Oggioni N, Sottani C, Minoia C, D’Incalci M, Zucchetti M, Marmiroli P, Tredici G. Distribution of paclitaxel within the nervous system of the rat after repeated intravenous administration. Neurotoxicology. 2000;21(3):389–393. [PubMed] [Google Scholar]
- 12.Cleary DR, Heinricher MM. Adaptations in responsiveness of brainstem pain-modulating neurons in acute compared with chronic inflammation. Pain. 2013;154(6):845–855. doi: 10.1016/j.pain.2013.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Donovan-Rodriguez T, Dickenson AH, Urch CE. Gabapentin normalizes spinal neuronal responses that correlate with behavior in a rat model of cancer-induced bone pain. Anesthesiology. 2005;102(1):132–140. doi: 10.1097/00000542-200501000-00022. [DOI] [PubMed] [Google Scholar]
- 14.Eickhoff R, Handwerker HO, McQueen DS, Schick E. Noxious and tactile input to medial structures of midbrain and pons in the rat. Pain. 1978;5(2):99–113. doi: 10.1016/0304-3959(78)90032-5. [DOI] [PubMed] [Google Scholar]
- 15.Feng HJ, Faingold CL. Repeated generalized audiogenic seizures induce plastic changes on acoustically evoked neuronal firing in the amygdala. Brain Res. 2002;932(1–2):61–69. doi: 10.1016/s0006-8993(02)02282-5. [DOI] [PubMed] [Google Scholar]
- 16.Fields H. State-dependent opioid control of pain. Nat Rev Neurosci. 2004;5(7):565–575. doi: 10.1038/nrn1431. [DOI] [PubMed] [Google Scholar]
- 17.Flatters SJ, Bennett GJ. Ethosuximide reverses paclitaxel- and vincristine-induced painful peripheral neuropathy. Pain. 2004;109(1–2):150–161. doi: 10.1016/j.pain.2004.01.029. [DOI] [PubMed] [Google Scholar]
- 18.Hahm ET, Kim Y, Lee JJ, Cho YW. GABAergic synaptic response and its opioidergic modulation in periaqueductal gray neurons of rats with neuropathic pain. BMC Neurosci. 2011;12:41. doi: 10.1186/1471-2202-12-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Handwerker HO, Sack R. Single unit activity in the rat’s midbrain during nocifensive tail flick reaction. Neurosci Lett. 1982;30(1):79–84. doi: 10.1016/0304-3940(82)90016-7. [DOI] [PubMed] [Google Scholar]
- 20.Hayashida K, DeGoes S, Curry R, Eisenach JC. Gabapentin activates spinal noradrenergic activity in rats and humans and reduces hypersensitivity after surgery. Anesthesiology. 2007;106(3):557–562. doi: 10.1097/00000542-200703000-00021. [DOI] [PubMed] [Google Scholar]
- 21.Heinricher MM, Cheng ZF, Fields HL. Evidence for two classes of nociceptive modulating neurons in the periaqueductal gray. J Neurosci. 1987;7(1):271–278. doi: 10.1523/JNEUROSCI.07-01-00271.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Heinricher MM, Tavares I, Leith JL, Lumb BM. Descending control of nociception: Specificity, recruitment and plasticity. Brain Res Rev. 2009;60(1):214–225. doi: 10.1016/j.brainresrev.2008.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hellman KM, Mason P. Opioids disrupt pro-nociceptive modulation mediated by raphe magnus. J Neurosci. 2012;32(40):13668–13678. doi: 10.1523/JNEUROSCI.1551-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hemington KS, Coulombe MA. The periaqueductal gray and descending pain modulation: Why should we study them and what role do they play in chronic pain? J Neurophysiol. 2015 doi: 10.1152/jn.00998.2014. jn 00998 02014. [DOI] [PubMed] [Google Scholar]
- 25.Hirakawa N, Tershner SA, Fields HL, Manning BH. Bi-directional changes in affective state elicited by manipulation of medullary pain-modulatory circuitry. Neuroscience. 2000;100(4):861–871. doi: 10.1016/s0306-4522(00)00329-8. [DOI] [PubMed] [Google Scholar]
- 26.Ho YC, Cheng JK, Chiou LC. Hypofunction of glutamatergic neurotransmission in the periaqueductal gray contributes to nerve-injury-induced neuropathic pain. J Neurosci. 2013;33(18):7825–7836. doi: 10.1523/JNEUROSCI.5583-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hoppa MB, Lana B, Margas W, Dolphin AC, Ryan TA. alpha2delta expression sets presynaptic calcium channel abundance and release probability. Nature. 2012;486(7401):122–125. doi: 10.1038/nature11033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Keay KA, Feil K, Gordon BD, Herbert H, Bandler R. Spinal afferents to functionally distinct periaqueductal gray columns in the rat: an anterograde and retrograde tracing study. J Comp Neurol. 1997;385(2):207–229. doi: 10.1002/(sici)1096-9861(19970825)385:2<207::aid-cne3>3.0.co;2-5. [DOI] [PubMed] [Google Scholar]
- 29.Lana B, Schlick B, Martin S, Pratt WS, Page KM, Goncalves L, Rahman W, Dickenson AH, Bauer CS, Dolphin AC. Differential upregulation in DRG neurons of an alpha2delta-1 splice variant with a lower affinity for gabapentin after peripheral sensory nerve injury. Pain. 2014;155(3):522–533. doi: 10.1016/j.pain.2013.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lei J, Sun T, Lumb BM, You HJ. Roles of the periaqueductal gray in descending facilitatory and inhibitory controls of intramuscular hypertonic saline induced muscle nociception. Exp Neurol. 2014;257:88–94. doi: 10.1016/j.expneurol.2014.04.019. [DOI] [PubMed] [Google Scholar]
- 31.Li KW, Yu YP, Zhou C, Kim DS, Lin B, Sharp K, Steward O, Luo ZD. Calcium channel alpha2delta1 proteins mediate trigeminal neuropathic pain states associated with aberrant excitatory synaptogenesis. J Biol Chem. 2014;289(10):7025–7037. doi: 10.1074/jbc.M114.548990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liu X, Eschenfelder S, Blenk KH, Janig W, Habler H. Spontaneous activity of axotomized afferent neurons after L5 spinal nerve injury in rats. Pain. 2000;84(2–3):309–318. doi: 10.1016/s0304-3959(99)00211-0. [DOI] [PubMed] [Google Scholar]
- 33.Maione S, Bisogno T, de Novellis V, Palazzo E, Cristino L, Valenti M, Petrosino S, Guglielmotti V, Rossi F, Di Marzo V. Elevation of endocannabinoid levels in the ventrolateral periaqueductal grey through inhibition of fatty acid amide hydrolase affects descending nociceptive pathways via both cannabinoid receptor type 1 and transient receptor potential vanilloid type-1 receptors. J Pharmacol Exp Ther. 2006;316(3):969–982. doi: 10.1124/jpet.105.093286. [DOI] [PubMed] [Google Scholar]
- 34.Mantyh PW. Cancer pain and its impact on diagnosis, survival and quality of life. Nat Rev Neurosci. 2006;7(10):797–809. doi: 10.1038/nrn1914. [DOI] [PubMed] [Google Scholar]
- 35.Martenson ME, Halawa OI, Tonsfeldt KJ, Maxwell CA, Hammack N, Mist SD, Pennesi ME, Bennett RM, Mauer KM, Jones KD, Heinricher MM. A possible neural mechanism for photosensitivity in chronic pain. Pain. 2016;157(4):868–878. doi: 10.1097/j.pain.0000000000000450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Martin G, Montagne-Clavel J, Oliveras JL. Involvement of ventromedial medulla “multimodal, multireceptive” neurons in opiate spinal descending control system: a single-unit study of the effect of morphine in the awake, freely moving rat. J Neurosci. 1992;12(4):1511–1522. doi: 10.1523/JNEUROSCI.12-04-01511.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mason P. Medullary circuits for nociceptive modulation. Curr Opin Neurobiol. 2012;22(4):640–645. doi: 10.1016/j.conb.2012.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.McGaraughty S, Farr DA, Heinricher MM. Lesions of the periaqueductal gray disrupt input to the rostral ventromedial medulla following microinjections of morphine into the medial or basolateral nuclei of the amygdala. Brain Res. 2004;1009(1–2):223–227. doi: 10.1016/j.brainres.2004.02.048. [DOI] [PubMed] [Google Scholar]
- 39.Mielke S, Sparreboom A, Mross K. Peripheral neuropathy: a persisting challenge in paclitaxel-based regimes. Eur J Cancer. 2006;42(1):24–30. doi: 10.1016/j.ejca.2005.06.030. [DOI] [PubMed] [Google Scholar]
- 40.Miki K, Zhou QQ, Guo W, Guan Y, Terayama R, Dubner R, Ren K. Changes in gene expression and neuronal phenotype in brain stem pain modulatory circuitry after inflammation. J Neurophysiol. 2002;87(2):750–760. doi: 10.1152/jn.00534.2001. [DOI] [PubMed] [Google Scholar]
- 41.Montagne-Clavel J, Oliveras JL. Are ventromedial medulla neuronal properties modified by chronic peripheral inflammation? A single-unit study in the awake, freely moving polyarthritic rat. Brain Res. 1994;657(1–2):92–104. doi: 10.1016/0006-8993(94)90957-1. [DOI] [PubMed] [Google Scholar]
- 42.Morgan MM, Whittier KL, Hegarty DM, Aicher SA. Periaqueductal gray neurons project to spinally projecting GABAergic neurons in the rostral ventromedial medulla. Pain. 2008;140(2):376–386. doi: 10.1016/j.pain.2008.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Moulin D, Boulanger A, Clark AJ, Clarke H, Dao T, Finley GA, Furlan A, Gilron I, Gordon A, Morley-Forster PK, Sessle BJ, Squire P, Stinson J, Taenzer P, Velly A, Ware MA, Weinberg EL, Williamson OD, Canadian Pain S Pharmacological management of chronic neuropathic pain: revised consensus statement from the Canadian Pain Society. Pain Res Manag. 2014;19(6):328–335. doi: 10.1155/2014/754693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mouton LJ, Holstege G. Segmental and laminar organization of the spinal neurons projecting to the periaqueductal gray (PAG) in the cat suggests the existence of at least five separate clusters of spino-PAG neurons. J Comp Neurol. 2000;428(3):389–410. doi: 10.1002/1096-9861(20001218)428:3<389::aid-cne2>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]
- 45.Mouton LJ, Klop E, Holstege G. Lamina I-periaqueductal gray (PAG) projections represent only a limited part of the total spinal and caudal medullary input to the PAG in the cat. Brain Res Bull. 2001;54(2):167–174. doi: 10.1016/s0361-9230(00)00442-1. [DOI] [PubMed] [Google Scholar]
- 46.N’Gouemo P, Faingold CL. Audiogenic kindling increases neuronal responses to acoustic stimuli in neurons of the medial geniculate body of the genetically epilepsy-prone rat. Brain Res. 1997;761(2):217–224. doi: 10.1016/s0006-8993(97)00322-3. [DOI] [PubMed] [Google Scholar]
- 47.Narai Y, Imamachi N, Saito Y. Gabapentin augments the antihyperalgesic effects of diclofenac sodium through spinal action in a rat postoperative pain model. Anesth Analg. 2012;115(1):189–193. doi: 10.1213/ANE.0b013e31824e5da3. [DOI] [PubMed] [Google Scholar]
- 48.Paxinos G, Watson C. The rat brain in stereotaxic coordinates. Amsterdam Boston: Elsevier; 2007. [Google Scholar]
- 49.Polomano RC, Mannes AJ, Clark US, Bennett GJ. A painful peripheral neuropathy in the rat produced by the chemotherapeutic drug, paclitaxel. Pain. 2001;94(3):293–304. doi: 10.1016/S0304-3959(01)00363-3. [DOI] [PubMed] [Google Scholar]
- 50.Porreca F, Ossipov MH, Gebhart GF. Chronic pain and medullary descending facilitation. Trends Neurosci. 2002;25(6):319–325. doi: 10.1016/s0166-2236(02)02157-4. [DOI] [PubMed] [Google Scholar]
- 51.Quasthoff S, Hartung HP. Chemotherapy-induced peripheral neuropathy. J Neurol. 2002;249(1):9–17. doi: 10.1007/pl00007853. [DOI] [PubMed] [Google Scholar]
- 52.Rhodes DL. Periventricular system lesions and stimulation-produced analgesia. Pain. 1979;7(1):51–63. doi: 10.1016/0304-3959(79)90106-4. [DOI] [PubMed] [Google Scholar]
- 53.Sanders KH, Klein CE, Mayor TE, Heym C, Handwerker HO. Differential effects of noxious and non-noxious input on neurones according to location in ventral periaqueductal grey or dorsal raphe nucleus. Brain Res. 1980;186(1):83–97. doi: 10.1016/0006-8993(80)90257-7. [DOI] [PubMed] [Google Scholar]
- 54.Sharma R, Sinha R, Mathur R, Nayar U. Neuronal responses of periaqueductal gray to peripheral noxious stimulation. Indian J Physiol Pharmacol. 1999;43(4):449–457. [PubMed] [Google Scholar]
- 55.Staud R. Abnormal endogenous pain modulation is a shared characteristic of many chronic pain conditions. Expert Rev Neurother. 2012;12(5):577–585. doi: 10.1586/ern.12.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Suzuki R, Rahman W, Rygh LJ, Webber M, Hunt SP, Dickenson AH. Spinal-supraspinal serotonergic circuits regulating neuropathic pain and its treatment with gabapentin. Pain. 2005;117(3):292–303. doi: 10.1016/j.pain.2005.06.015. [DOI] [PubMed] [Google Scholar]
- 57.Takeuchi Y, Takasu K, Honda M, Ono H, Tanabe M. Neurochemical evidence that supraspinally administered gabapentin activates the descending noradrenergic system after peripheral nerve injury. Eur J Pharmacol. 2007;556(1–3):69–74. doi: 10.1016/j.ejphar.2006.10.059. [DOI] [PubMed] [Google Scholar]
- 58.Tanabe M, Murakami H, Honda M, Ono H. Gabapentin depresses C-fiber-evoked field potentials in rat spinal dorsal horn only after induction of long-term potentiation. Exp Neurol. 2006;202(2):280–286. doi: 10.1016/j.expneurol.2006.06.002. [DOI] [PubMed] [Google Scholar]
- 59.Tortorici V, Morgan MM. Comparison of morphine and kainic acid microinjections into identical PAG sites on the activity of RVM neurons. J Neurophysiol. 2002;88(4):1707–1715. doi: 10.1152/jn.2002.88.4.1707. [DOI] [PubMed] [Google Scholar]
- 60.Tryon VL, Mizumori SJ, Morgan MM. Analysis of morphine-induced changes in the activity of periaqueductal gray neurons in the intact rat. Neuroscience. 2016;335:1–8. doi: 10.1016/j.neuroscience.2016.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tupal S, Faingold CL. Audiogenic kindling induces plastic changes in the neuronal firing patterns in periaqueductal gray. Brain Res. 2011;1377:60–66. doi: 10.1016/j.brainres.2010.12.076. [DOI] [PubMed] [Google Scholar]
- 62.Urban MO, Zahn PK, Gebhart GF. Descending facilitatory influences from the rostral medial medulla mediate secondary, but not primary hyperalgesia in the rat. Neuroscience. 1999;90(2):349–352. doi: 10.1016/s0306-4522(99)00002-0. [DOI] [PubMed] [Google Scholar]
- 63.Valiante TA, Perez Velazquez JL, Jahromi SS, Carlen PL. Coupling potentials in CA1 neurons during calcium-free-induced field burst activity. J Neurosci. 1995;15(10):6946–6956. doi: 10.1523/JNEUROSCI.15-10-06946.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Vera-Portocarrero LP, Zhang ET, Ossipov MH, Xie JY, King T, Lai J, Porreca F. Descending facilitation from the rostral ventromedial medulla maintains nerve injury-induced central sensitization. Neuroscience. 2006;140(4):1311–1320. doi: 10.1016/j.neuroscience.2006.03.016. [DOI] [PubMed] [Google Scholar]
- 65.Wang N, Wang JY, Luo F. Corticofugal outputs facilitate acute, but inhibit chronic pain in rats. Pain. 2009;142(1–2):108–115. doi: 10.1016/j.pain.2008.12.016. [DOI] [PubMed] [Google Scholar]
- 66.Xiao WH, Bennett GJ. Chemotherapy-evoked neuropathic pain: Abnormal spontaneous discharge in A-fiber and C-fiber primary afferent neurons and its suppression by acetyl-L-carnitine. Pain. 2008;135(3):262–270. doi: 10.1016/j.pain.2007.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Yadav R, Yan X, Maixner DW, Gao M, Weng HR. Blocking the GABA transporter GAT-1 ameliorates spinal GABAergic disinhibition and neuropathic pain induced by paclitaxel. J Neurochem. 2015;133(6):857–869. doi: 10.1111/jnc.13103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Yarnitsky D. Conditioned pain modulation (the diffuse noxious inhibitory control-like effect): its relevance for acute and chronic pain states. Curr Opin Anaesthesiol. 2010;23(5):611–615. doi: 10.1097/ACO.0b013e32833c348b. [DOI] [PubMed] [Google Scholar]
- 69.Zhang H, Dougherty PM. Enhanced excitability of primary sensory neurons and altered gene expression of neuronal ion channels in dorsal root ganglion in paclitaxel-induced peripheral neuropathy. Anesthesiology. 2014;120(6):1463–1475. doi: 10.1097/ALN.0000000000000176. [DOI] [PMC free article] [PubMed] [Google Scholar]