

Abstract
Histone deacetylase (HDAC) inhibitors, which are approved for the treatment of cutaneous T cell lymphoma and multiple myeloma, are undergoing evaluation in other lymphoid neoplasms. How they kill susceptible cells is incompletely understood. Here we show that trichostatin A, romidepsin, and panobinostat induce apoptosis across a panel of malignant B cell lines, including lines that are intrinsically resistant to bortezomib, etoposide, cytarabine, and BH3 mimetics. Further analysis traces the pro-apoptotic effects of HDAC inhibitors to increased acetylation of the chaperone heat shock protein 90 (HSP90), causing release and degradation of the HSP90 client proteins RASGRP1 and CRAF, which in turn leads to downregulation of mitogen activated protein kinase pathway signaling and upregulation of the pro-apoptotic BCL2 family member BIM in vitro and in vivo. Importantly, these pro-apoptotic effects are mimicked by RASGRP1 siRNA or HSP90 inhibition and reversed by overexpression of constitutively active MEK1 or siRNA-mediated downregulation of BIM. Collectively, these observations not only identify a new HSP90 client protein, RASGRP1, but also delineate a complete signaling pathway from HSP90 acetylation through RASGRP1 and CRAF degradation to BIM upregulation that contributes to selective cytotoxicity of HDAC inhibitors in lymphoid malignancies.
INTRODUCTION
The histone deacetylase (HDAC) inhibitors romidepsin and vorinostat are approved for the treatment of cutaneous T cell lymphomas.1–3 These and additional HDAC inhibitors are currently undergoing clinical testing in various neoplasms, including B-cell lymphomas, where they have shown clinical activity.3–8 As a class, these agents inhibit deacetylation of lysines in multiple proteins, including histones, and enhance transcription of a wide range of genes.9–11 This altered transcription is widely thought to contribute to the antineoplastic effects of HDAC inhibitors, including in B cell neoplasms,6, 11 although other mechanisms have also been proposed.12
HDAC inhibitors have also been widely reported to induce apoptosis, which can be triggered by either the mitochondrial or the death receptor pathway in susceptible cells.9 The mitochondrial pathway is regulated by the BCL2 family of proteins, which is comprised of proapoptotic sensors (BH3-only proteins such as BIM, PUMA, NOXA, and truncated BID), apoptotic effectors (BAX and BAK), and apoptosis inhibitors (e.g., BCL2, BCLXL and MCL1). Interactions between these proteins regulate mitochondrial outer membrane integrity and cell survival. When cells encounter unfavorable conditions, one ore more of the BH3-only proteins is typically increased or activated and can then directly bind and activate the apoptotic effectors BAX or BAK.13–17 Upon activation, BAX and BAK permeabilize the mitochondrial outer membrane, triggering release of cytochrome c and activation of caspase 9, which initiates an intracellular protease cascade that results in the apoptotic phenotype.18, 19 Conversely, anti-apoptotic BCL2 family members such as BCL2, BCLXL, and MCL1 bind and neutralize activated BH3-only proteins as well as partially activated BAX and BAK.20–22 Importantly, increased BCL2 expression, which is seen in lymphomas with t(14;18 translocations),23, 24 or certain lymphoma-associated BCL2 mutations that result in increased affinity of BCL2 proteins for BAK,25 NOXA,26 or PUMA and BIM,27 are associated with increased resistance to apoptosis.
The death receptor pathway, the other major pathway for triggering apoptosis, involves sequential binding of cytokines such as CD95 ligand and tumor necrosis factor a TNF-related apoptosis inducing ligand (TRAIL) to cell surface receptors, recruitment of the adaptor protein Fas-associated death domain (FADD) to the intracellular domains of these receptors, activation of procaspase 8 by the oligomerized FADD, and subsequent caspase activation. Importantly, inactivating CD95 mutations have been identified in a subset of follicular lymphomas.28
HDAC inhibitors have been reported to induce apoptosis through multiple changes affecting both of these apoptotic pathways. In particular, HDAC inhibitors enhance expression of the TNFRSF10B gene encoding death receptor 5, which binds TRAIL and activates the death receptor pathway.29–32 In addition, HDAC inhibitors repress transcription of BCL2 and BCLXL33 as well as upregulate genes encoding the pro-apoptotic BCL2 family members BAX, BAK, and BIM in various cell types,34–36 potentially activating the mitochondrial pathway. Additional studies have demonstrated that BIM expression can also be increased by posttranslational mechanisms37, 38 that involve inhibition RSK2-mediated BIM phosphorylation and subsequent phosphorylation-dependent degradation39, 40 after treatment with MEK or farnesyltransferase inhibitors.41, 42 A separate line of investigation has identified the heat shock protein 90 (HSP90) chaperone as a critical target of HDAC inhibitors in malignant T cells.43 How these various observations are mechanistically linked, and which of these changes accounts for HDAC inhibitor-induced killing of malignant B cells, has been unclear.
Here we identify the cytotoxic pathway triggered by HDAC inhibitors in B-cell lymphoma and T-cell leukemia cells. In particular, we show for the first time that HDAC inhibitors act on HSP90 to downregulate RASGRP1, a lymphoid-specific guanine nucleotide exchange factor,44 as well as the RAS effector CRAF, leading to diminished mitogen activated kinase signaling, increased BIM expression, and apoptosis. These results not only identify cell type-specific signaling downstream of HSP90 inhibition that potentially contributes to selective cytotoxicity of HDAC inhibitors in malignant T cells, but also show that the same signaling is inhibited during HDAC inhibitor-induced killing of B-cell lymphoma cells.
MATERIALS AND METHODS
Materials
Commercial antibodies (Table S1) were obtained as follows: phospho-Thr202/Tyr204-ERK1/2, ERK1/2, phospho-Ser217/Ser221-MEK1/2, MEK1/2, BAX, BIM, BID, MCL1, BCLXL, cleaved caspase 3 and PARP1 from Cell Signaling Technology (Beverly, MA); HSP70 and NOXA from Enzo Life Sciences (Farmington, NY); BAK from Millipore (Billerica, MA); CRAF, PUMA and BCL2 (rabbit polyclonal) from Santa Cruz Biotechnology (Santa Cruz, CA); BCL2 (murine monoclonal) from Dako (Carpenteria, CA); acetylated tubulin and α-tubulin from Sigma-Aldrich (St. Louis, MO); acetylated lysine (AcK) antibody from Rockland (Limerick, PA); cytochrome c from BD Biosciences (Franklin Lakes, NJ); and cytochrome oxidase subunit IV (COX IV) from Thermo (Waltham, MA). Anti-HSP90 (clone H9010) was from from David Toft (Mayo Clinic, Rochester, MN). Antibody to RASGRP1 was generated and characterized as described.42 Reagents were purchased from the following suppliers: DNA oligonucleotides from Integrated DNA Technologies (Coralville, IA); 3-(4,5-dimethylthiazol-2-yl)-5(3-carboxymethonyphenol)-2-(4-sulfophenyl)-2H-tetrazolium (MTS) from Promega (Madison, WI); romidepsin and 17-allylamino-17-demethoxygeldanamycin (17AAG) were from the Drug Synthesis Branch of the National Cancer Institute and Toronto Research Chemicals (Toronto, ON); trichostatin A (TSA), etoposide and phenazine methosulfate from Sigma-Aldrich; the broad spectrum caspase inhibitor Q-VD-OPh from SM Biochemicals (Anaheim, CA); OSI-127, navitoclax, venetoclax and panobinostat from Chemietek (Indianapolis, IN); and bortezomib from Enzo Life Sciences. Short oligonucleotides targeting BAX (nucleotides 271–289, GenBank™ accession number NM_138761), BAK (nucleotides 913–931, NM_001188), BIM (nucleotides 325–343, NM_138621), PUMA (nucleotides 784–802, NM_014417), BIK (nucleotides 65–83, NM_001197), NOXA (nucleotides 272–290, NM_021127) and RASGRP1 (nucleotides 1555–1573, AF_081195) were from Ambion (Austin, TX). All other reagents were obtained as previously described.25
Cell culture
Cell lines obtained as described42, 45 were authenticated by short tandem repeat profiling and confirmed to be mycoplasma free. BCL2 mutation status as reported in the COSMIC database (http://www.sanger.ac.uk/genetics/CGPF/cosmic/) was confirmed by Sanger sequencing.27 Cell lines were propagated at densities of <1 × 106 cells/ml in RPMI 1640 medium containing 100 units/ml penicillin G, 100 μg/ml streptomycin, 2 mM glutamine and 15% (JB-6, I9.2, I2.1, JMR) or 10% (all other cell lines) heat-inactivated fetal bovine serum (medium A). Small interfering RNA (siRNA) was transfected by electroporation using a BTX 830 electroporator delivering a 10 msec pulse at 240 V (WSU) or 280 V (Jurkat cells) as described.42 Jurkat cells stably expressing MEK(DD), a constitutively active form of MEK1 (clones 10 and 12),46 were generated by electroporating 1.5 × 107 parental cells with 40 μg plasmid,25 selecting for stable integrants in 5 μg/ml puromycin after 48 h, and cloning the resulting puromycin-resistant cells by limiting dilution.
Detection of apoptosis by flow microfluorimetry
DNA fragmentation and annexin V binding were assayed as described previously.25, 26, 45
Immunoblotting
After treatment with HDAC inhibitors in the presence of 5 μM Q-VD-OPh (to inhibit changes in cellular proteins due to caspase-mediated cleavages) as indicated in the figure legends, cells were washed in serum-free RPMI-HEPES and solubilized in 6 M guanidine hydrochloride containing 250 mM Tris-HCl (pH 8.5 at 21 °C), 10 mM EDTA, 1% (v/v) ß-mercaptoethanol, and 1 mM α-phenylmethylsulfonyl fluoride (freshly added from a 100 mM stock in anhydrous isopropanol). Following reaction with iodoacetamide, cell lysates were dialyzed sequentially into 4 M urea followed by 0.1% (v/v) SDS.47 After lyophilization, aliquots were resuspended in SDS sample buffer at 5 mg protein/ml (assayed by the bicinchoninic acid method), separated by SDS-PAGE, transferred to nitrocellulose membranes, and probed with various antibodies.48
Cell fractionation
After treatment with diluent or 20 nM romidepsin for 24 hours in the presence of 5 μM Q-VD-OPh, cells were sedimented at 200 × g for 10 min, washed twice with ice-cold PBS, and lysed by swelling for 20 minutes at 4 °C in hypotonic buffer (25 mM HEPES, pH 7.5 at 4°C, 5 mM MgCl2, and 1 mM EGTA supplemented immediately before use with 1 mM α-phenylmethylsulfonyl fluoride, 10 μg/mL leupeptin, and 10 μg/mL pepstatin) followed by Dounce homogenization. After trypan blue staining confirmed that all cells were ruptured, samples were sedimented at 800 × g to remove nuclei. The postnuclear supernatant was sedimented at 4000 × g to isolate crude mitochondria, which were resuspended in hypotonic buffer with 300 mM sucrose and sedimented at 4000 × g a second time. Fractions were prepared for SDS-PAGE as described under “Immunoblotting.”
Quantitative RT-PCR (qRT-PCR)
After total RNA was isolated from cells treated with diluent or romidepsin in the presence of 5 μM Q-VD-OPh, qRT-PCR was performed in triplicate employing 50 ng RNA and TaqMan One-Step RT-PCR Master Mix (Applied Biosystems, Carlsbad, CA). Using BIM (Hs00197982_m1) and PUMA (Hs00248075_m1) probe sets, PCR was performed on a ABI Prism 7900HT Real Time System using a program consisting of 48 °C for 30 min, 95 °C for 10 min, then 40 cycles of 95 °C for 15 sec and 60 °C for 1 min. Data were analyzed using the following standard equations: ΔCt = Ct(sample) − Ct(endogenous control); ΔΔCt = ΔCt(sample) −ΔCt(untreated); and Fold Change = 2−ΔΔCt, where Ct is the PCR cycle where the fluorescence exceeds the threshold.
Xenograft study
After approval by the Institutional Animal Care and Use Committee, SCID/Beige mice (4–5 weeks, Harlan) were implanted subcutaneously with 100 μl of a 1:1 slurry containing Matrigel (BD Bioscience) and 5 × 106 washed, log phase SuDHL-6 cells in the right flank as well as radiofrequency identification chips along the neck. Lymphoma volumes were calculated according to the formula ab2/2, where a is the diameter along the longest dimension and b is the orthogonal diameter. When xenografts reached an average volume of 100 mm3, mice were randomized for euthanasia 0, 24 or 48 h after treatment with a single intravenous dose of romidepsin at 1 mg/kg (dissolved in ethanol and diluted 1:39 in 5% dextrose).49
Samples of lymphoid malignancies
Clinical lymphoma samples from the University of Iowa/Mayo Lymphoma SPORE Biospecimens Core were studied with IRB approval. Mononuclear cells were isolated from excisional lymph node biopsies or splenectomy specimens by mechanical disruption over a wire mesh screen followed by Ficoll-Hypaque centrifugation. All patients provided informed consent for research use of specimens.
MTS assay
Aliquots containing ~2 × 104 cells in 120 μl medium A were incubated at 37° C with varying concentrations of romidepsin or TSA for 5 days. As a measure of the relative amounts of surviving cells,50 plates were treated with MTS and phenazine methosulfate as instructed by the supplier and incubated for 2–6 h to obtain an absorbance of 0.5–1.0 at 490 nm in control samples.
Statistical analysis
Full dose-response curves were performed in cell lines at least three times independently, with examples presented and bar graphs showing summary data. Error bars represent mean ± SD of 3 independent experiments and p values represent results of unpaired t tests.
RESULTS
Cross-resistance patterns of lymphoid cell lines to chemotherapeutic agents and BH3 mimetics
Building on earlier results showing broad cross-resistance of certain lymphoma lines to multiple agents,51 we examined sensitivity of a panel of 12 lymphoid cell lines (Table S2) to a variety of agents. In view of recent results showing that certain BCL2 mutations enhance the ability of BCL2 to inhibit apoptosis,25, 26 we included five lines that harbor BCL2 mutations (identified with black symbols in Fig. 1) and seven that do not (grey symbols). Baseline expression of BCL2 family members varied widely across these cell lines (Fig. 1a) as discussed below.
Figure 1. Drug sensitivity of human lymphoid cell lines.
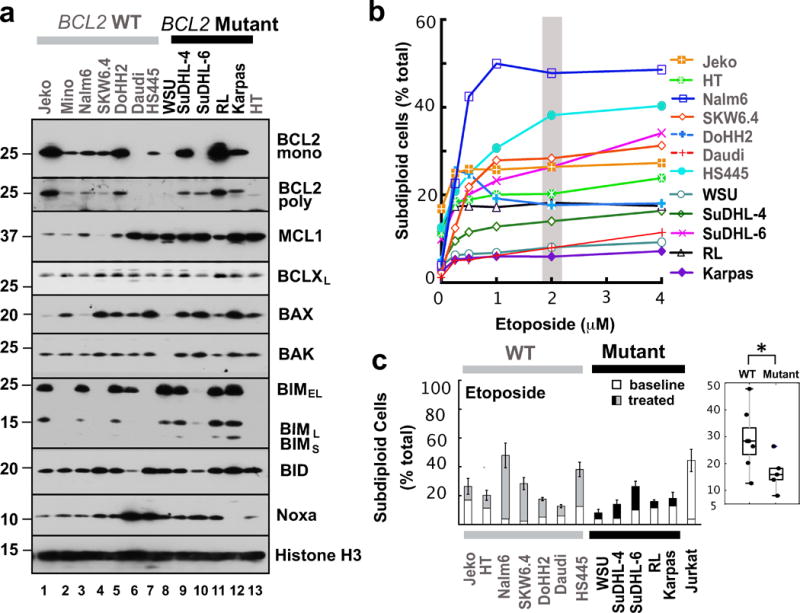
(a) Western blot showing protein levels for pro-apoptotic (BAX, BAK, BIM, BID, and NOXA) and anti-apoptotic (BCL2, BCLXL, and MCL1) BCL2 family members in human lymphoid cell lines with wild type (lanes 1–7, 13) or mutant BCL2 (lanes 8–12). Whole cell lysates were subjected to SDS-PAGE and probed with the indicated antibodies. The polyclonal anti-BCL2 antiserum was employed to assess the possibility that the paucity of signal with monoclonal anti-BCL2 antibody in certain BCL2-mutant lines results from mutation of the recognized epitope. Grey and black bars on top indicate cells lines with wild type (WT) or mutant BCL2, respectively. Histone H3 was used as loading control. (b) Cells were treated with etoposide for 24 h, stained with propidium iodide and subjected to flow microfluorimetry as illustrated in Fig. S1a. Curves shown are representative of 3 independent experiments and the grey bar indicates the drug concentration used for the bar graph and box plot in Fig. 1c. (c). Effect of etoposide (2 μM, 24 h) on the number of subdiploid cells. The Jurkat cell line, which is sensitive to all of these treatments, was used as positive control. Error bars indicate mean ± SD of three independent experiments. *, p<0.05.
As indicated in Figs. 1b, 1c and Supplemental Fig. S1a, these lines exhibited a wide range of sensitivities to the topoisomerase II poison etoposide, with apoptosis ranging from 5 to 50% after treatment with 2 μM for 48 h. There was more apoptosis in cells with wild type BCL2 compared to mutant BCL2 (Fig. 1c, right panel; p <0.05). Likewise, the cells exhibited varying sensitivities to ionizing radiation, the antimetabolite cytarabine, the proteasome inhibitor bortezomib, and the mTOR inhibitor OSI-027 (Figs. S1b–S1i). With ionizing radiation and cytarabine there was again diminished apoptosis in the BCL2 mutant cell lines (Figs. S1c and S1e, p <0.05), but this was not evident with bortezomib or OSI-027 (Figs. S1g and S1i).
A somewhat different pattern emerged when sensitivity to BH3 mimetics was examined. In particular, three lines showed striking hypersensitivity to the BCL2 selective BH3 mimetic venetoclax (Figs. 2a and 2b). This sensitivity did not track with BCL2 mutations. Instead, three of the most sensitive lines, Jeko, DoHH2 and RL, expressed the highest levels of BCL2 protein (Fig. 1a), Conversely, the majority of lines were resistant to venetoclax despite more modest BCL2 levels. Likewise, the majority of lines were relatively resistant to the BCL2/BCLXL targeted inhibitor navitoclax (Figs. 2c and S2a), with Jeko, DoHH2 and RL still being the most sensitive. These observations are consistent with previous observations that lymphoid cells with elevated BCL2 express more BIM and are particularly sensitive to BH3 mimetic drugs that inhibit BCL2.52 In contrast, more of the lines responded to obatoclax (Figs. S2b and S2c), which reportedly inhibits anti-apoptotic BCL2 family members more broadly53 but also might have off-target effects.54
Figure 2. Sensitivity of lymphoid cells to venetoclax and navitoclax.
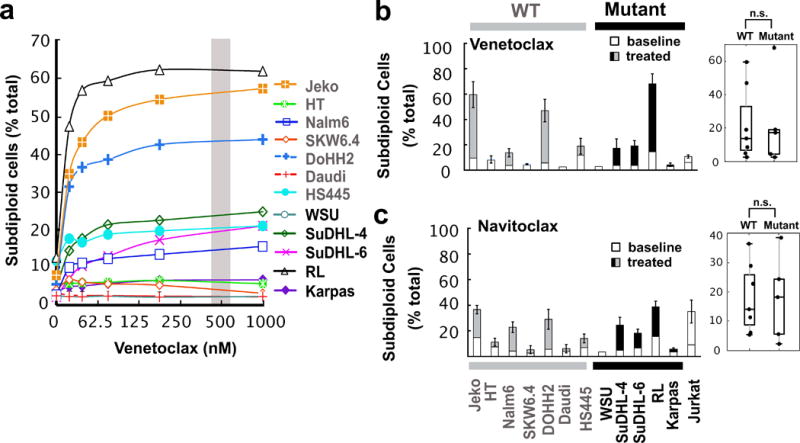
(a) Venetoclax dose-response profile for lymphoid cell lines. Jurkat cells were used as a control. Curves shown are representative of three independent experiments and the grey bar indicates the drug concentration used for the bar graph and box plot in Fig. 2b. Dose-response curves for navitoclax and obatoclax are found in Fig. S2a and S2b. (b–c) Induction of subdiploid cells at 48 h by treatment with venetoclax (500 nM) or navitoclax (500 nM), respectively. Error bars, mean ± SD of three independent experiments. n.s., not significant.
HDAC inhibitors are cytotoxic in a variety of neoplastic B cell lines
Based on previous reports that HDAC inhibitors upregulate components of the death receptor pathway, including both ligands and receptors,9, 30, 31 as well as induce other changes that activate the death receptor pathway,55 along with the demonstration that small increases in death receptor expression are sufficient to render death receptor signaling BCL2-independent,56 we examined the effect of three different HDAC inhibitors on the same lymphoid cell lines. Strikingly, all of the lines, including lines that were resistant to conventional agents and BH3 mimetics (e.g., WSU and Daudi, Figs. 1b, 1c, S1 and S2), were sensitive to the prototypic HDAC inhibitor TSA (Fig. S3) as well as clinically utilized HDAC inhibitors such as romidepsin (Figs. 3a and 3b) and panobinostat (Figs. 3c and S3a). Accordingly, further studies focused on the mechanism by which these agents were able to induce cell death even in the face of resistance to other agents.
Figure 3. Sensitivity of malignant B cell lines to HDAC inhibitors.
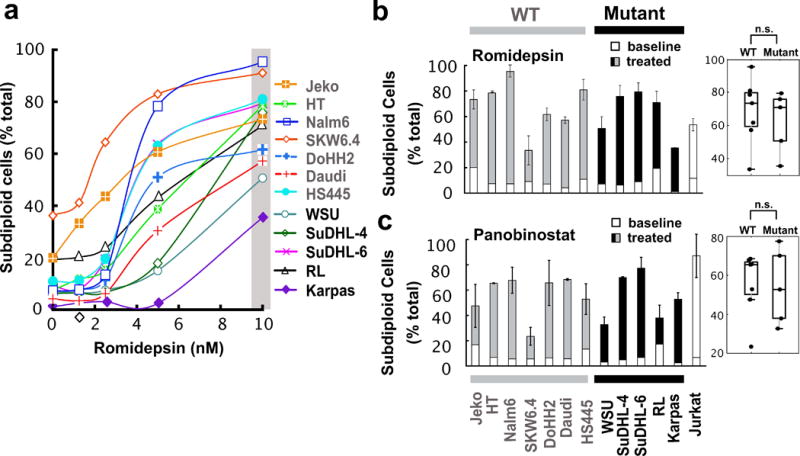
(a) Romidepsin dose-response profile after treatment for 48 h. Curves shown are representative of three independent experiments and the grey bar indicates the drug concentration used for the graph and box plot in panel b. Additional dose-response curves for other agents are found in Figs. S3a–b. (b–c) Effects of romidepsin (10 nM, 72 h) and panobinostat (25 nM, 48 h) on lymphoid cell lines. Jurkat cells were used as a positive control. Error bars, mean ± SD of three independent experiments. n.s., not significant.
Initial mechanistic experiments examined Jurkat cells, a T-cell acute lymphoblastic leukemia line, as a prototypic malignant lymphoid line because of the availability of isogenic lines with disabled death receptor or mitochondrial pathway signaling.26 Jurkat variants that lack procaspase 8 (I9.2) or FADD (I2.1), two critical components of the death receptor pathway (Fig. S4a), were just as sensitive as parental Jurkat cells to all three HDAC inhibitors (Fig. S4b). In contrast, Jurkat cells lacking procaspase 9 (JMR) or massively overexpressing BCL2 (JB-6) were resistant, indicating a predominant role for the mitochondrial pathway in romidepsin-, TSA- and panobinostat-induced cell death (Figs. S4a and S4b).
In further experiments, parental Jurkat cells were treated for 24 hours with increasing romidepsin concentrations in the presence of Q-VD-OPh to inhibit caspase-mediated cleavages downstream of mitochondrial pathway activation (Fig. S4c). Marked upregulation of PUMA and the three most abundant BIM isoforms was observed (Fig. S4d). Additional experiments demonstrated that the amount of BIM in mitochondria also increased (Fig. S4e). In contrast, the anti-apoptotic BCL2 family members MCL1, BCL2, and BCLXL were unchanged, as were the apoptotic effectors BAX and BAK (Fig. S4d). Similar results were observed in the B cell line Nalm6 (Fig. S4f).
To assess the contribution of the upregulated BH3-only proteins to killing, various proapoptotic BCL2 family members were knocked down with RNA oligonucleotides. Decreased BIM, like knockdown of the mitochondrial outer membrane permeabilizing protein BAK, markedly inhibited romidepsin-induced apoptosis (Figs. S4g and S5). In contrast, knockdown of other BH3-only proteins, including PUMA, BIK and NOXA, had little effect. Accordingly, further studies examined the cause of the BIM upregulation.
BIM in lymphoid cells is known to be sometimes upregulated by changes in transcription.57 qRT-PCR after treatment of Jurkat cells with romidepsin showed a small change in BIM mRNA (Fig. S6a) that appeared too limited to account entirely for the 5- to 10-fold BIM protein upregulation. Another mechanism of BIM upregulation involves decreased BIM degradation39, 40, 42 as a consequence of diminished signaling through the MAP kinase pathway.39, 41 When examined for potential involvement of this pathway, Jurkat cells exhibited a marked decrease in phosphorylation of ERK1/2 and their upstream kinases MEK1/2 during romidepsin treatment (Fig. 4a). Moving further up this signaling pathway, additional analysis (Fig. 4a) showed that this decreased signaling reflected HDAC inhibitor-induced diminution of the RAS effector kinase CRAF, which participates in MEK1/2 phosphorylation, and the RAS guanine nucleotide exchange factor RASGRP1, which participates in MAP kinase pathway activation in lymphoid cells.44 Additional investigation demonstrated that these effects reflected romidepsin-induced increases in HSP90 acetylation and accompanying decreases in binding of both CRAF and RASGRP1 to the HSP90 chaperone complex (Fig. 4b).
Figure 4. HDAC inhibitor diminishes MAP kinase signaling in T-cells.
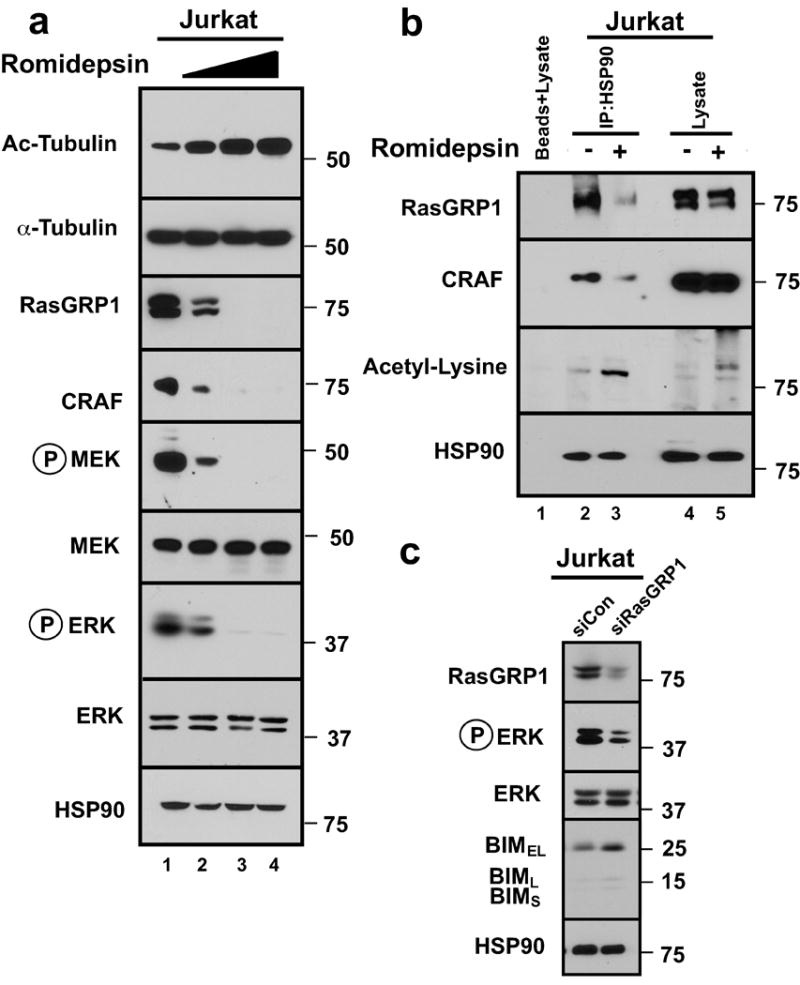
(a) Whole cell lysates from Jurkat cells treated for 24 h with romidepsin (0, 5, 10, 20 nM) were subjected to SDS-PAGE and probed with the indicated antibodies. HSP90 was used as loading control. Because antibodies against acetylated HSP90 gave a high background and recognized multiple bands of varying molecular weight, antibody to acetylated α-tubulin was employed as an indicator of HDAC inhibition in this and subsequent figures. (b) After Jurkat cells were treated with diluent or 20 nM romidepsin for 6 h (a time chosen to optimize HSP90 release of clients without their extensive degradation), immunoprecipitates prepared with anti-HSP90 antibody and subjected to SDS-PAGE and immunoblotting with the indicated antibodies. (c) Immunoblots of whole cell lysates prepared from Jurkat cells 48 h transfection with control siRNA control or RASGRP1 siRNA. HSP90 served as a loading control.
In further experiments, we examined the biochemical impact of RASGRP1 downregulation. After siRNA-mediated RASGRP1 knockdown, signaling through the MAP kinase pathway was markedly diminished and BIM was upregulated (Fig. 4c), suggesting that RASGRP1 downregulation contributes to the biochemical changes that are critical for HDAC inhibitor-induced apoptosis. Conversely, Jurkat cells stably transfected with constitutively active MEK1 (MEKDD), which partially protected from ERK dephosphorylation, exhibited diminished BIM induction (Fig. S6b) and less killing during romidepsin treatment (Fig. S6c) compared to the cells transfected with empty vector, again indicating the importance of MAP kinase pathway inhibition in the cytotoxicity of HDAC inhibitors.
Romidepsin action in neoplastic B cell lines
All of the critical HDAC inhibitor-induced changes in MAP kinase signaling and BCL2 family expression were also observed in malignant B cells. When SuDHL-4, SuDHL-6, and WSU cells were treated with romidepsin (Figs. 5a–c) or TSA (Fig. 5b) in the presence of Q-VD-OPh to again prevent secondary apoptotic effects, downregulation of RASGRP1 and CRAF, inhibition of CRAF→MEK→ERK signaling, and BIM upregulation were observed. This was accompanied by increased mitochondrial BIM accumulation and cytochrome c release (Fig. 6a). BIM knockdown, like BAK knockdown, was again protective against cell death, as illustrated by results in WSU cells (Fig. 6b). Importantly, similar effects, including RASGRP1 and CRAF downregulation, inhibition of MAP kinase pathway signaling, and BIM upregulation were observed in these lines after treatment with the HSP90 inhibitor 17AAG (Fig. 6c), supporting the view that disruption of HSP90/client protein interactions is sufficient to account for these changes.
Figure 5. BIM upregulation reflects inhibition of MAP Kinase signalling in multiple malignant B-cell lines.
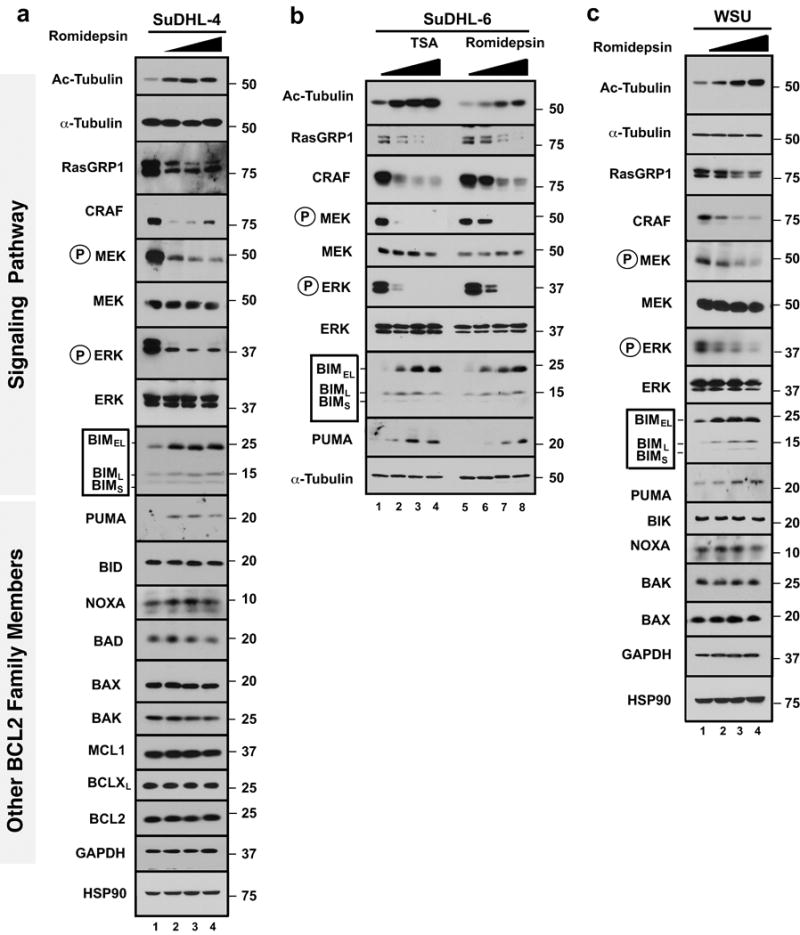
(a–c) SuDHL-4 (a), SuDHL-6 (b) or WSU cells (c) were treated for 24 h with romidepsin (0, 5, 10, 20 nM, panels a–c) or TSA (0, 50, 100, 200 nM, panel b) in the presence of 5 μM Q-VD-OPh. Whole cell lysates were subjected to SDS-PAGE and probed with the indicated antibodies. Acetylated tubulin (Ac-Tubulin) served as marker of HDAC inhibition. HSP90 and/or α-tubulin served as loading controls.
Figure 6. BIM upregulation contributes to HDAC inhibitor-induced killing in malignant B-cell lines.
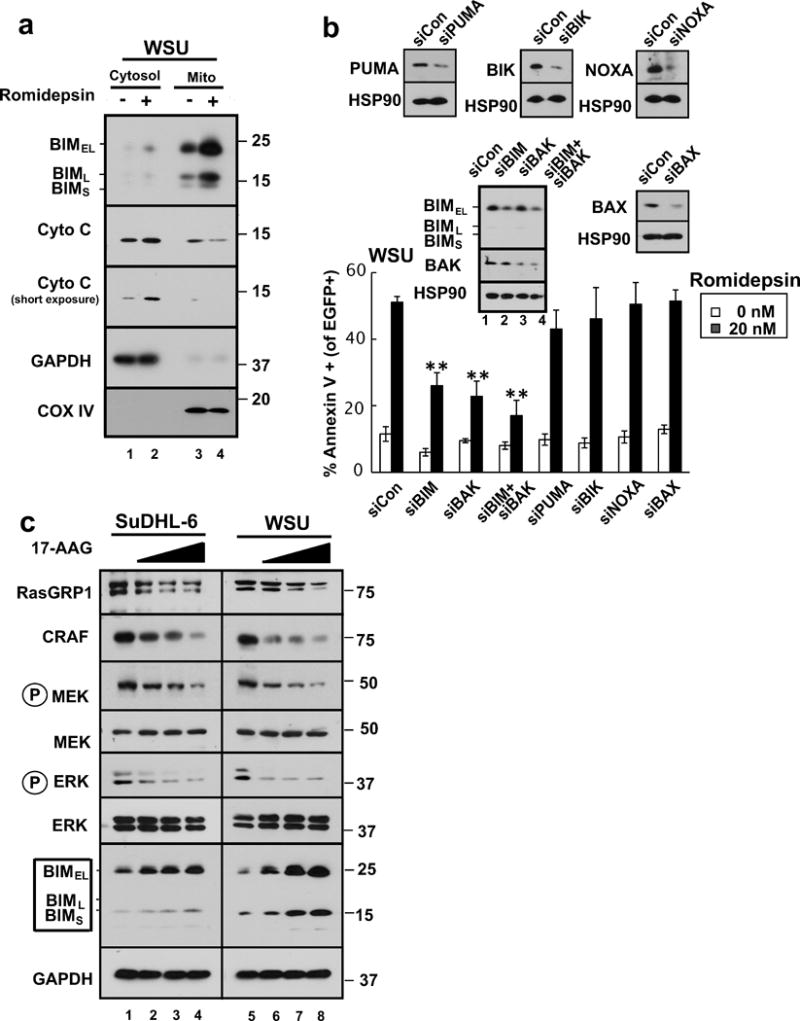
(a) After WSU cells were treated with diluent (0.1% DMSO, −) or 20 nM romidepsin (+) in the presence of 5 μM Q-VD-OPh for 24 h, the indicated subcellular fractions were isolated and subjected to immunoblotting. GAPDH served as a marker for cytosol and cytochrome c oxidase subunit IV (CoX IV) served as a marker for mitochondria. (b) 24 hours after transfection of the indicated siRNAs along with plasmid encoding EGFP-histone H2B (to mark successfully transfected cells), WSU cells were treated for 24 h with diluent or 20 nM romidepsin before staining with APC-conjugated annexin V and analysis by 2-color flow cytometry. Error bars, ± SD of three independent experiments. **, p <0.01. Inset: Immunoblots of whole cell lysates prepared from siRNA-treated cells incubated in drug-free medium in parallel with samples harvested for flow cytometry. HSP90 served as a loading control. (c) SuDHL-6 and WSU cells were treated for 48 h with diluent (lanes 1 and 5, respectively) or the HSP90 inhibitor 17AAG at 0.3, 1, or 3 μM (lanes 2–4 and 6–8, respectively) or diluent in the presence of 5 μM Q-VD-OPh. Whole cell lysates were subjected to SDS-PAGE and probed with the indicated antibodies. GAPDH served as a loading control.
In further experiments, we examined HDAC inhibitor sensitivity of clinical lymphoma specimens (Table S3) ex vivo. This analysis showed that a wide range of B-cell lymphoma specimens responded to to both romidepsin (Fig. 7a) and TSA (Fig. S7) at concentrations similar to those that affected T cell lymphoma. In particular, the IC50s for romidepsin and TSA were 1–10 nM and 30–700 nM, respectively.
Figure 7. Effects of HDAC inhibitors on malignant B cells ex vivo and in vivo.
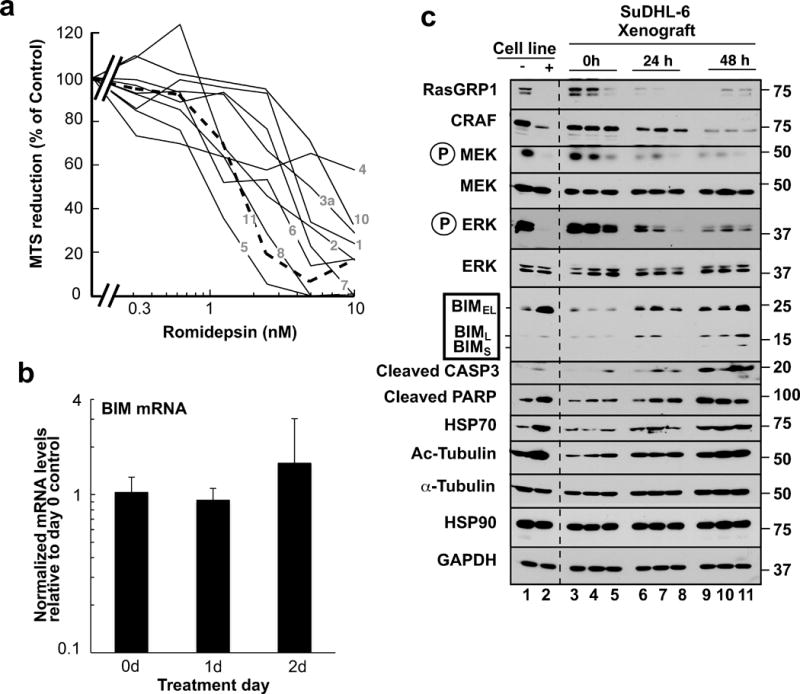
(a). B-cell lymphoma specimens (solid lines) or T-cell lymphoma specimens (dashed lines) were treated with the indicated concentrations of romidepsin for 5 days ex vivo and assayed for relative cell survival using MTS assays. (b, c) RT-PCR for BIM (b) and immunoblotting for the indicated antigens (c) in SuDHL-6 xenografts harvested from 9 mice immediately before, 24 h and 48 h after the first dose of romidepsin (lanes 3–11). SuDHL-6 cells treated for 48 h with diluent (−) or 10 nM romidepsin (+) ex vivo in the presence of 5 μM Q-VD-OPh served as a positive control for romidepsin-induced signaling changes (lanes 1–2).
To extend these results, the effects of romidepsin were assessed in SuDHL-6 xenografts in vivo. As was the case in vitro, romidepsin treatment of xenografts caused limited upregulation of BIM mRNA (Fig. 7b) but readily detectable increases in BIM protein that again accompanied decreases in the HSP90 client proteins RASGRP1 and CRAF as well as diminished signaling through the MAP kinase pathway (Fig. 7c). Thus, the results observed in tissue culture were readily observed 24–48 hours after romidepsin treatment in vivo.
DISCUSSION
Collectively, results of the present study suggest that HDAC inhibitors kill malignant B lymphocytes through a series of signaling events outlined in Fig. 8. In particular, working upstream from the observation that BIM plays a critical role in HDAC inhibitor-induced apoptosis, we have traced BIM upregulation to diminished MAP kinase pathway signaling, which in turn reflects HDAC inhibitor-induced downregulation of RASGRP1 and CRAF that follows release of these proteins upon acetylation of HSP90. Importantly, expression of constitutively active MEK (Fig. S6b), like downregulation of BIM or BAK (Figs. 6b and S4g), diminishes HDAC inhibitor-induced apoptosis in malignant T and B cell lines, suggesting the importance of this pathway for HDAC inhibitor-induced killing.
Figure 8. Summary of signaling elucidated in the present study.
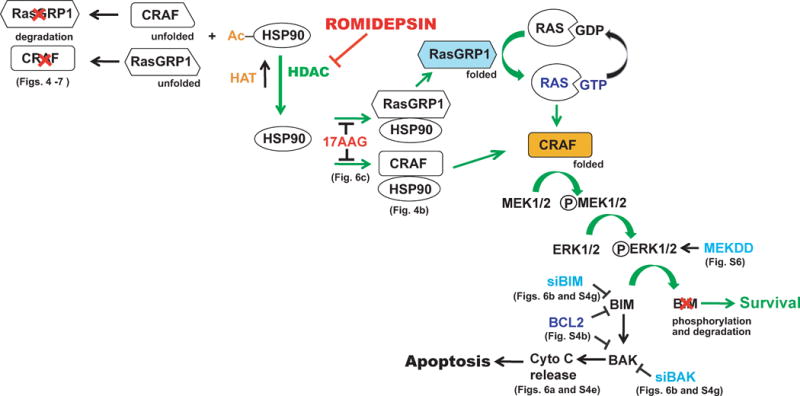
Under normal circumstances, RASGRP1 and CRAF are bound to the HSP90 chaperone complex, which stabilizes them and facilitates signaling through the MEK/ERK pathway to induce phosphorylation-dependent BIM degradation.37 Treatment with romidepsin shifts the equilibrium in favor of HSP90 acetylation, causing release and degradation of RASGRP1 and CRAF, inhibition of MEK/ERK signaling, elevation of BIM and apoptosis. Interventions performed in this paper to dissect the pathway (and the corresponding figures) are indicated.
Although inhibition of HSP9043 and upregulation of BIM36 have been previously observed during HDAC inhibitor treatment of susceptible T lymphocytes, steps tying these events together have been unclear. Results of the present study extend these prior studies in several important ways. First the present results identify RASGRP1, a lymphoid cell-specific guanine nucleotide exchange factor that facilitates activation of RAS and its effectors,44 as a novel client protein that is released from HSP90 upon HDAC inhibitor treatment. The present studies not only show that HDAC inhibitors promote release of RASGRP1 from HSP90 at early time points before total cellular levels have decreased (Fig. 4b), but also demonstrate that RASGRP1 levels decrease when cells are treated with the well established HSP90 inhibitor 17AAG (Fig. 6c). These are hallmarks of an HSP90 client. Second, the present results show that signaling changes downstream of RASGRP1 and CRAF loss (i.e., inhibition of MAP kinase signaling and consequent BIM upregulation) are observed in malignant B cells (Figs. 5–7) as well as T cells (Fig. S4). Third, the present observations extend the potential use of HDAC inhibitors to malignant B cells that are resistant to other agents through a variety of mechanisms. Results observed in several cell lines are particularly informative in this regard.
The SuDHL-6 line has low levels of BIM relative to other lymphoid cell lines (Fig. 1a) and, consistent with a previous study,51 is resistant to navitoclax (Fig. 2) as well as etoposide and cytarabine (Figs. 1 and S1). Interestingly, however, multiple HDAC inhibitors were able to upregulate BIM and induce apoptosis in this line (Figs. 3, 5b and S3), indicating that resistance due to low BIM levels can be pharmacologically reversed.
Likewise, the WSU line has low levels of BAX and BAK relative to other lymphoid lines (Fig. 1a), although these proteins can be detected upon prolonged exposure (Fig. 5c). Like SuDHL-6 cells, WSU cells are resistant to multiple conventional and experimental anti-lymphoma agents (Figs. 1, 2 and S1) but are able to upregulate BIM and undergo BIM-dependent apoptosis after HDAC inhibitor treatment (Figs. 3, 5c, 6 and S3).
Cell lines such as Jeko, DoHH2 and RL, which express extremely high concentrations of BCL2 (Fig. 1a), appeared dependent on this BCL2 overexpression for survival, as evidenced by their heightened sensitivity to the BCL2 antagonist venetoclax (Fig. 2b) and diminished sensitivity to agents that activate the mitochondrial apoptotic pathway, including etoposide, bortezomib and OSI-027 (Fig. S1). Despite high BCL2 levels, these cells were sensitive to romidepsin (Fig. 3b).
Consistent with recent studies demonstrating that some lymphoma-associated nonsynonymous nucleotide substitutions in the BCL2 gene enhance BCL2 anti-apoptotic function ex vivo,25–27 the lymphoid cell lines with BCL2 mutations were somewhat less sensitive to etoposide, ionizing radiation and cytarabine than lymphoma lines without BCL2 mutations (Fig. 1b, 1c, S1c and S1e). Importantly, these BCL2-mutant lines were just as sensitive as BCL2 wildtype lines to multiple HDAC inhibitors, including romidepsin and panobinostat (Figs. 3 and S3).
During HDAC inhibitor treatment of malignant lymphoid cells, BIM mRNA increased modestly, in agreement with previous studies.34–36 BIM protein, however, increased more than BIM message (Fig. 7). Moreover, constitutively active MEKK blunted the increase in BIM protein (Fig. S6b). Accordingly, HSP90 inhibition and the resulting downregulation of mitogen activated kinase signaling appear to contribute as least as much as transcriptional activation during HDAC inhibitor-induced killing of the lymphoid cell lines examined in the present study.
HDAC inhibitor-induced killing was observed in a wide range of malignant B cells (Fig. 7a). Importantly, however, the IC50s varied over a 10-fold range, suggesting that some lymphomas might be more sensitive than others. Further investigation is required to assess the cause of this heterogeneity. It is possible, for example, that varied sensitivities reflect differences in dependence of lymphoma cells on the HSP90•CRAF and HSP90•RASGRP1 complexes for suppression of BIM levels. If so, pretreatment assessment of signaling through the mitogen activated protein kinase pathway might help identify lymphomas that are more responsive to HDAC inhibitors.
In summary, we have observed that HDAC inhibitors such as romidepsin induce apoptosis in lymphoma cell lines relatively independent of their sensitivity to other agents. Although romidepsin is currently FDA approved for the treatment of cutaneous T cell lymphoma,1–3 it has also been shown to enhance CD20 expression in B cells.58 Moreover, previous preclinical studies have demonstrated its activity in B-cell CLL55, 59 and B-cell lymphomas60 alone and in combination with rituximab.58 The present results provide a mechanistic explanation for these findings by identifying the lymphoid-specific RAS guanine nucleotide exchange factor RASGRP1 as a new HSP90 client that is released and degraded together with CRAF during treatment with romidepsin, providing at least a partial explanation for the prominent clinical activity of this class of agent in lymphoid malignancies, including both T and B cell neoplasms.
Supplementary Material
Acknowledgments
These studies were supported by grants from the Leukemia & Lymphoma Society (6125-10), National Cancer Institute (P50 CA097274, R01 CA166741) and Predolin Foundation. We acknowledge gifts of HSP90 antibody from David Toft as well as romidepsin and 17AAG from the NCI Drug Synthesis Branch.
Footnotes
AUTHOR CONTRIBUTIONS
Conception and design: HD, SHK, GSN
Financial support: SHK
Collection and assembly of data: HD, KLP, CC, BK
Data analysis and interpretation: HD, CC, GSN, SHK
Manuscript writing: All authors
Final approval of manuscript: All authors
CONFLICT OF INTEREST
The authors have no conflict of interest.
References
- 1.Wilcox RA. Cutaneous T-cell lymphoma: 2014 update on diagnosis, risk-stratification, and management. Am J Hematol. 2014 Aug;89(8):837–851. doi: 10.1002/ajh.23756. [DOI] [PubMed] [Google Scholar]
- 2.Karlin L, Coiffier B. The changing landscape of peripheral T-cell lymphoma in the era of novel therapies. Semin Hematol. 2014 Jan;51(1):25–34. doi: 10.1053/j.seminhematol.2013.11.001. [DOI] [PubMed] [Google Scholar]
- 3.Bates SE, Robey RW, Piekarz RL. CCR 20th Anniversary Commentary: Expanding the Epigenetic Therapeutic Portfolio. Clin Cancer Res. 2015 May 15;21(10):2195–2197. doi: 10.1158/1078-0432.CCR-14-2555. [DOI] [PubMed] [Google Scholar]
- 4.Fowler N, Oki Y. Developing novel strategies to target B-cell malignancies. ASCO Educational book. 2013:366–372. doi: 10.14694/EdBook_AM.2013.33.366. [DOI] [PubMed] [Google Scholar]
- 5.Mondello P, Younes A. Emerging drugs for diffuse large B-cell lymphoma. Expert Rev Anticancer Ther. 2015 Apr;15(4):439–451. doi: 10.1586/14737140.2015.1009042. [DOI] [PubMed] [Google Scholar]
- 6.Lue JK, Amengual JE, O’Connor OA. Epigenetics and Lymphoma: Can We Use Epigenetics to Prime or Reset Chemoresistant Lymphoma Programs? Current Oncol Rep. 2015 Sep;17(9):40. doi: 10.1007/s11912-015-0464-y. [DOI] [PubMed] [Google Scholar]
- 7.Smolewski P, Witkowska M, Robak T. Treatment options for mantle cell lymphoma. Expert Opin Pharmacother. 2015;16(16):2497–2507. doi: 10.1517/14656566.2015.1087507. [DOI] [PubMed] [Google Scholar]
- 8.Morabito F, Recchia AG, Vigna E, De Stefano L, Bossio S, Morabito L, et al. Promising therapies for the treatment of chronic lymphocytic leukemia. Expert Opinn Invest Drugs. 2015 Jun;24(6):795–807. doi: 10.1517/13543784.2015.1021920. [DOI] [PubMed] [Google Scholar]
- 9.Marks PA. Histone deacetylase inhibitors: a chemical genetics approach to understanding cellular functions. Biochim Biophys Acta. 2010 Oct-Dec;1799(10–12):717–725. doi: 10.1016/j.bbagrm.2010.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang J, Zhong Q. Histone deacetylase inhibitors and cell death. Cell Mol Life Sci. 2014 Oct;71(20):3885–3901. doi: 10.1007/s00018-014-1656-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yazbeck VY, Grant S. Romidepsin for the treatment of non-Hodgkin’s lymphoma. Expert Opinion Invest Drugs. 2015;24(7):965–979. doi: 10.1517/13543784.2015.1041586. [DOI] [PubMed] [Google Scholar]
- 12.Bose P, Dai Y, Grant S. Histone deacetylase inhibitor (HDACI) mechanisms of action: emerging insights. Pharmacol Therap. 2014 Sep;143(3):323–336. doi: 10.1016/j.pharmthera.2014.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Walensky LD, Pitter K, Morash J, Oh KJ, Barbuto S, Fisher J, et al. A stapled BID BH3 helix directly binds and activates BAX. Mol Cell. 2006 Oct 20;24(2):199–210. doi: 10.1016/j.molcel.2006.08.020. [DOI] [PubMed] [Google Scholar]
- 14.Dai H, Smith A, Meng XW, Schneider PA, Pang Y-P, Kaufmann SH. Transient Binding of an Activator BH3 Domain to the Bak BH3-Binding Groove Initiates Bak Oligomerization. J Cell Biol. 2011;194:39–48. doi: 10.1083/jcb.201102027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dai H, Pang Y-P, Kaufmann SH. Evaluation of the BH3-Only Protein Puma as a Direct Bak Activator. J Biol Chem. 2014;289:89–99. doi: 10.1074/jbc.M113.505701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Czabotar PE, Westphal D, Dewson G, Ma S, Hockings C, Fairlie WD, et al. Bax Crystal Structures Reveal How BH3 Domains Activate Bax and Nucleate Its Oligomerization to Induce Apoptosis. Cell. 2013 Jan 31;152(3):519–531. doi: 10.1016/j.cell.2012.12.031. [DOI] [PubMed] [Google Scholar]
- 17.Brouwer JM, Westphal D, Dewson G, Robin AY, Uren RT, Bartolo R, et al. Bak Core and Latch Domains Separate during Activation, and Freed Core Domains Form Symmetric Homodimers. Mol Cell. 2014 Sep 18;55(6):938–946. doi: 10.1016/j.molcel.2014.07.016. [DOI] [PubMed] [Google Scholar]
- 18.Earnshaw WC, Martins LM, Kaufmann SH. Mammalian Caspases: Structure, Activation, Substrates and Functions During Apoptosis. Ann Rev Biochemy. 1999;68:383–424. doi: 10.1146/annurev.biochem.68.1.383. [DOI] [PubMed] [Google Scholar]
- 19.Taylor RC, Cullen SP, Martin SJ. Apoptosis: controlled demolition at the cellular level. Nat Rev Mol Cell Biol. 2008 Mar;9(3):231–241. doi: 10.1038/nrm2312. [DOI] [PubMed] [Google Scholar]
- 20.Llambi F, Moldoveanu T, Tait SW, Bouchier-Hayes L, Temirov J, McCormick LL, et al. A Unified Model of Mammalian BCL-2 Protein Family Interactions at the Mitochondria. Mol Cell. 2011 Nov 18;44(4):517–531. doi: 10.1016/j.molcel.2011.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Strasser A, Cory S, Adams JM. Deciphering the rules of programmed cell death to improve therapy of cancer and other diseases. EMBO J. 2011 Sep 14;30(18):3667–3683. doi: 10.1038/emboj.2011.307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dai H, Meng XW, Kaufmann SH. BCL2 family, mitochondrial apoptosis, and beyond. Cancer Translational Med. 2016;2(1):7–20. [Google Scholar]
- 23.Miyashita T, Reed JC. Bcl-2 Oncoprotein Blocks Chemotherapy-Induced Apoptosis in a Human Leukemia Cell Line. Blood. 1993;81(1):151–157. [PubMed] [Google Scholar]
- 24.Yang E, Korsmeyer SJ. Molecular Thanatopsis: A Discourse on the Bcl2 Family and Cell Death. Blood. 1996;88(2):386–401. [PubMed] [Google Scholar]
- 25.Dai H, Meng XW, Lee S-H, Schneider PA, Kaufmann SH. Context-dependent Bcl-2/Bak Interactions Regulate Lymphoid Cell Apoptosis. J Biol Chem. 2009;284:18311–18322. doi: 10.1074/jbc.M109.004770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Smith AJ, Dai H, Correia C, Lee S-H, Takahashi R, Kaufmann SH. Noxa/Bcl-2 Interactions Contribute to Bortezomib Resistance in Human Lymphoid Cells. J Biol Chem. 2011;286:17682–17692. doi: 10.1074/jbc.M110.189092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Correia C, Schneider PA, Dai H, Dogan A, Maurer MJ, Church AK, et al. BCL2 Mutations Are Associated with Increased Risk of Transformation and Shortened Survival in Follicular Lymphoma. Blood. 2015;125:658–667. doi: 10.1182/blood-2014-04-571786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pasqualucci L, Khiabanian H, Fangazio M, Vasishtha M, Messina M, Holmes AB, et al. Genetics of Follicular Lymphoma Transformation. Cell Rep. 2014 Jan 2;16:130–140. doi: 10.1016/j.celrep.2013.12.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Insinga A, Monestiroli S, Ronzoni S, Gelmetti V, Marchesi F, Viale A, et al. Inhibitors of histone deacetylases induce tumor-selective apoptosis through activation of the death receptor pathway. Nat Med. 2005;11:71–76. doi: 10.1038/nm1160. [DOI] [PubMed] [Google Scholar]
- 30.Frew AJ, Lindemann RK, Martin BP, Clarke CJ, Sharkey J, Anthony DA, et al. Combination therapy of established cancer using a histone deacetylase inhibitor and a TRAIL receptor agonist. Proc Natl Acad Sci U S A. 2008 Aug 12;105(32):11317–11322. doi: 10.1073/pnas.0801868105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jazirehi AR, Kurdistani SK, Economou JS. Histone deacetylase inhibitor sensitizes apoptosis-resistant melanomas to cytotoxic human T lymphocytes through regulation of TRAIL/DR5 pathway. J Immunol. 2014 Apr 15;192(8):3981–3989. doi: 10.4049/jimmunol.1302532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fulda S. Histone deacetylase (HDAC) inhibitors and regulation of TRAIL-induced apoptosis. Exp Cell Res. 2012 Jul 1;318(11):1208–1212. doi: 10.1016/j.yexcr.2012.02.005. [DOI] [PubMed] [Google Scholar]
- 33.Cao XX, Mohuiddin I, Ece F, McConkey DJ, Smythe WR. Histone deacetylase inhibitor downregulation of bcl-xl gene expression leads to apoptotic cell death in mesothelioma. Am J Resp Cell Mol Biol. 2001 Nov;25(5):562–568. doi: 10.1165/ajrcmb.25.5.4539. [DOI] [PubMed] [Google Scholar]
- 34.Wiegmans AP, Alsop AE, Bots M, Cluse LA, Williams SP, Banks KM, et al. Deciphering the molecular events necessary for synergistic tumor cell apoptosis mediated by the histone deacetylase inhibitor vorinostat and the BH3 mimetic ABT-737. Cancer Res. 2011 May 15;71(10):3603–3615. doi: 10.1158/0008-5472.CAN-10-3289. [DOI] [PubMed] [Google Scholar]
- 35.Xargay-Torrent S, Lopez-Guerra M, Saborit-Villarroya I, Rosich L, Campo E, Roue G, et al. Vorinostat-induced apoptosis in mantle cell lymphoma is mediated by acetylation of proapoptotic BH3-only gene promoters. Clin Cancer Res. 2011 Jun 15;17(12):3956–3968. doi: 10.1158/1078-0432.CCR-10-3412. [DOI] [PubMed] [Google Scholar]
- 36.Dai Y, Chen S, Venditti CA, Pei XY, Nguyen TK, Dent P, et al. Vorinostat synergistically potentiates MK-0457 lethality in chronic myelogenous leukemia cells sensitive and resistant to imatinib mesylate. Blood. 2008 Aug 1;112(3):793–804. doi: 10.1182/blood-2007-10-116376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wahner Hendrickson AE, Meng XW, Kaufmann SH. Anticancer Therapy: Boosting the Bang of Bim. J Clin Invest. 2008;118:3582–3584. doi: 10.1172/JCI37553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pinon JD, Labi V, Egle A, Villunger A. Bim and Bmf in tissue homeostasis and malignant disease. Oncogene. 2009;27:S41–S52. doi: 10.1038/onc.2009.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ley R, Balmanno K, Hadfield K, Weston C, Cook SJ. Activation of the ERK1/2 Signaling Pathway Promotes Phosphorylation and Proteasome-dependent Degradation of the BH3-only Protein, Bim. J Biol Chem. 2003;278:18811–18816. doi: 10.1074/jbc.M301010200. [DOI] [PubMed] [Google Scholar]
- 40.Dehan E, Bassermann F, Guardavaccaro D, Vasiliver-Shamis G, Cohen M, Lowes KN, et al. betaTrCP- and Rsk1/2-mediated degradation of BimEL inhibits apoptosis. Mol Cell. 2009 Jan 16;33(1):109–116. doi: 10.1016/j.molcel.2008.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cragg MS, Jansen ES, Cook M, Harris C, Strasser A, Scott CL. Treatment of B-RAF mutant human tumor cells with a MEK inhibitor requires Bim and is enhanced by a BH3 mimetic. J Clin Invest. 2008;118:3582–3584. doi: 10.1172/JCI35437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ding H, Hackbarth J, Schneider PA, Peterson KL, Meng XW, Dai H, et al. Cytotoxicity of Farnesyltransferase Inhibitors in Lymphoid Cells Mediated by MAPK Pathway Inhibition and Bim Upregulation. Blood. 2011;(118):4872–4881. doi: 10.1182/blood-2011-02-334870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bali P, Pranpat M, Bradner J, Balasis M, Fiskus W, Guo F, et al. Inhibition of histone deacetylase 6 acetylates and disrupts the chaperone function of heat shock protein 90: a novel basis for antileukemia activity of histone deacetylase inhibitors. J Biol Chem. 2005 Jul 22;280(29):26729–26734. doi: 10.1074/jbc.C500186200. [DOI] [PubMed] [Google Scholar]
- 44.Stone JC. Regulation of Ras in lymphocytes: get a GRP. Biochem Soc Transactions. 2006 Nov;34(Pt 5):858–861. doi: 10.1042/BST0340858. [DOI] [PubMed] [Google Scholar]
- 45.Meng X, Chandra J, Loegering D, Van Becelaere K, Kottke TJ, Gore SD, et al. Central role of FADD in Apoptosis Induction by the Mitogen Activated Activated Protein Kinase Kinase Inhibitor CI1040 (PD184352) in Acute Lymphocytic leukemia Cell Lines in Vitro. J Biol Chem. 2003;278:47326–47339. doi: 10.1074/jbc.M304793200. [DOI] [PubMed] [Google Scholar]
- 46.Boehm JS, Zhao JJ, Yao J, Kim SY, Firestein R, Dunn IF, et al. Integrative genomic approaches identify IKBKE as a breast cancer oncogene. Cell. 2007 Jun 15;129(6):1065–1079. doi: 10.1016/j.cell.2007.03.052. [DOI] [PubMed] [Google Scholar]
- 47.Kaufmann SH, Svingen PA, Gore SD, Armstrong DK, Cheng Y-C, Rowinsky EK. Altered Formation of Topotecan-Stabilized Topoisomerase I-DNA Adducts in Human Leukemia Cells. Blood. 1997;89:2098–2104. [PubMed] [Google Scholar]
- 48.Kaufmann SH. Reutilization of Immunoblots After Chemiluminescent Detection. Analytical Biochem. 2001;296:283–286. doi: 10.1006/abio.2001.5313. [DOI] [PubMed] [Google Scholar]
- 49.Karam JA, Fan J, Stanfield J, Richer E, Benaim EA, Frenkel E, et al. The use of histone deacetylase inhibitor FK228 and DNA hypomethylation agent 5-azacytidine in human bladder cancer therapy. Int J Cancer. 2007 Apr 15;120(8):1795–1802. doi: 10.1002/ijc.22405. [DOI] [PubMed] [Google Scholar]
- 50.Riss TL, Moravec RA, Niles AL, Benink HA, Worzella TJ, Minor L. Cell Viability Assays. In: Sittampalam GS, Coussens NP, Nelson H, Arkin M, Auld D, Austin C, et al., editors. Assay Guidance Manual. Bethesda (MD): 2004. [Google Scholar]
- 51.Deng J, Carlson N, Takeyama K, Dal Cin P, Shipp M, Letai A. BH3 profiling identifies three distinct classes of apoptotic blocks to predict response to ABT-737 and conventional chemotherapeutic agents. Cancer Cell. 2007 Aug;12(2):171–185. doi: 10.1016/j.ccr.2007.07.001. [DOI] [PubMed] [Google Scholar]
- 52.Merino D, Khaw SL, Glaser SP, Anderson DJ, Belmont LD, Wong C, et al. Bcl-2, Bcl-x(L), and Bcl-w are not equivalent targets of ABT-737 and navitoclax (ABT-263) in lymphoid and leukemic cells. Blood. 2012 Jun 14;119(24):5807–5816. doi: 10.1182/blood-2011-12-400929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Nguyen M, Marcellus RC, Roulston A, Watson M, Serfass L, Murthy Madiraju SR, et al. Small molecule obatoclax (GX15-070) antagonizes MCL-1 and overcomes MCL-1-mediated resistance to apoptosis. Proc Natl Acad Sci U S A. 2007 Dec 4;104(49):19512–19517. doi: 10.1073/pnas.0709443104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Vogler M, Weber K, Dinsdale D, Schmitz I, Schulze-Osthoff K, Dyer MJ, et al. Different forms of cell death induced by putative BCL2 inhibitors. Cell Death Differ. 2009 Jul;16(7):1030–1039. doi: 10.1038/cdd.2009.48. [DOI] [PubMed] [Google Scholar]
- 55.Aron JL, Parthun MR, Marcucci G, Kitada S, Mone AP, Davis ME, et al. Depsipeptide (FR901228) induces histone acetylation and inhibition of histone deacetylase in chronic lymphocytic leukemia cells concurrent with activation of caspase 8-mediated apoptosis and down-regulation of c-FLIP protein. Blood. 2003 Jul 15;102(2):652–658. doi: 10.1182/blood-2002-12-3794. [DOI] [PubMed] [Google Scholar]
- 56.Meng XW, Peterson KL, Dai H, Schneider P, Lee SH, Zhang JS, et al. High cell surface death receptor expression determines type I versus type II signaling. J Biol Chem. 2011 Aug 24;286:35823–35833. doi: 10.1074/jbc.M111.240432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gupta M, Wahner Hendrickson A, Seongseouk Y, Han JJ, Schneider PA, Stenson M, et al. Dual TORC1/TORC2 Inhibition Diminishes Akt Activation and Induces Puma-Dependent Apoptosis in Lymphoid Malignancies. Blood. 2012;119(2):476–487. doi: 10.1182/blood-2011-04-346601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Shimizu R, Kikuchi J, Wada T, Ozawa K, Kano Y, Furukawa Y. HDAC inhibitors augment cytotoxic activity of rituximab by upregulating CD20 expression on lymphoma cells. Leukemia. 2010 Oct;24(10):1760–1768. doi: 10.1038/leu.2010.157. [DOI] [PubMed] [Google Scholar]
- 59.Byrd JC, Shinn C, Ravi R, Willis CR, Waselenko JK, Flinn IW, et al. Depsipeptide (FR901228): a novel therapeutic agent with selective, in vitro activity against human B-cell chronic lymphocytic leukemia cells. Blood. 1999 Aug 15;94(4):1401–1408. [PubMed] [Google Scholar]
- 60.Newbold A, Lindemann RK, Cluse LA, Whitecross KF, Dear AE, Johnstone RW. Characterisation of the novel apoptotic and therapeutic activities of the histone deacetylase inhibitor romidepsin. Mol Cancer Therapeutics. 2008 May;7(5):1066–1079. doi: 10.1158/1535-7163.MCT-07-2256. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.