

Summary
Background
Concurrent administration of the immune checkpoint inhibitors nivolumab and ipilimumab has shown greater efficacy than either agent alone in patients with advanced melanoma, albeit with more high-grade adverse events. We assessed whether sequential administration of nivolumab followed by ipilimumab, or the reverse sequence, could improve safety without compromising efficacy.
Methods
We did this randomised, open-label, phase 2 study at nine academic medical centres in the USA. Eligible patients (aged ≥18 years) with unresectable stage III or IV melanoma (treatment-naive or who had progressed after no more than one previous systemic therapy, with an Eastern Cooperative Oncology Group performance status of 0 or 1) were randomly assigned (1:1) to induction with intravenous nivolumab 3 mg/kg every 2 weeks for six doses followed by a planned switch to intravenous ipilimumab 3 mg/kg every 3 weeks for four doses, or the reverse sequence. Randomisation was done by an independent interactive voice response system with a permuted block schedule (block size four) without stratification factors. After induction, both groups received intravenous nivolumab 3 mg/kg every 2 weeks until progression or unacceptable toxicity. The primary endpoint was treatment-related grade 3–5 adverse events until the end of the induction period (week 25), analysed in the as-treated population. Secondary endpoints were the proportion of patients who achieved a response at week 25 and disease progression at weeks 13 and 25. Overall survival was a prespecified exploratory endpoint. This study is registered with ClinicalTrials.gov, number NCT01783938, and is ongoing but no longer enrolling patients.
Findings
Between April 30, 2013, and July 21, 2014, 140 patients were enrolled and randomly assigned to nivolumab followed by ipilimumab (n=70) or to the reverse sequence of ipilimumab followed by nivolumab (n=70), of whom 68 and 70 patients, respectively, received at least one dose of study drug and were included in the analyses. The frequencies of treatment-related grade 3–5 adverse events up to week 25 were similar in the nivolumab followed by ipilimumab group (34 [50%; 95% CI 37·6–62·4] of 68 patients) and in the ipilimumab followed by nivolumab group (30 [43%; 31·1–55·3] of 70 patients). The most common treatment-related grade 3–4 adverse events during the whole study period were colitis (ten [15%]) in the nivolumab followed by ipilimumab group vs 14 [20%] in the reverse sequence group), increased lipase (ten [15%] vs 12 [17%]), and diarrhoea (eight [12%] vs five [7%]). No treatment-related deaths occurred. The proportion of patients with a response at week 25 was higher with nivolumab followed by ipilimumab than with the reverse sequence (28 [41%; 95% CI 29·4–53·8] vs 14 [20%; 11·4–31·3]). Progression was reported in 26 (38%; 95% CI 26·7–50·8) patients in the nivolumab followed by ipilimumab group and 43 (61%; 49·0–72·8) patients in the reverse sequence group at week 13 and in 26 (38%; 26·7–50·8) and 42 (60%; 47·6–71·5) patients at week 25, respectively. After a median follow-up of 19·8 months (IQR 12·8–25·7), median overall survival was not reached in the nivolumab followed by ipilimumab group (95% CI 23·7–not reached), whereas over a median follow-up of 14·7 months (IQR 5·6–23·9) in the ipilimumab followed by nivolumab group, median overall survival was 16·9 months (95% CI 9·2–26·5; HR 0·48 [95% CI 0·29–0·80]). A higher proportion of patients in the nivolumab followed by ipilimumab group achieved 12-month overall survival than in the ipilimumab followed by nivolumab group (76%; 95% CI 64–85 vs 54%; 42–65).
Interpretation
Nivolumab followed by ipilimumab appears to be a more clinically beneficial option compared with the reverse sequence, albeit with a higher frequency of adverse events.
Introduction
Median overall survival for patients with advanced melanoma treated with chemotherapy is approximately 11 months.1 Immune checkpoint inhibitors, along with targeted agents against the BRAF–MEK pathway, have transformed the treatment approach for advanced melanoma in recent years. Ipilimumab, a fully human IgG1 monoclonal antibody that blocks the cytotoxic T lymphocyte antigen 4 (CTLA-4) receptor on T cells, was the first agent to show a long-term overall survival benefit in advanced melanoma with up to 10 years' follow-up in some patients and 3-year survival of 22%.2 Nivolumab is a fully human IgG4 monoclonal antibody that blocks the interaction of programmed death receptor-1 (PD-1) on T cells with its ligands programmed death ligand 1 (PD-L1) and programmed death ligand 2 on tumour cells or antigen-presenting cells.3 In a phase 3 study in previously untreated patients with BRAF wild-type advanced melanoma, nivolumab led to improved overall survival and 40% of patients achieved an objective response.1 Another phase 3 study in patients with advanced melanoma that had progressed on ipilimumab, with or without a BRAF inhibitor, reported 32% of patients treated with nivolumab achieving an objective response.4
Because CTLA-4 and PD-1 inhibit antitumour immunity via non-redundant signalling pathways,5 combination therapy with ipilimumab and nivolumab has been investigated. A phase 3 study6 in previously untreated patients with advanced melanoma showed that concurrent nivolumab and ipilimumab, or nivolumab monotherapy, is associated with a significantly higher proportion of patients achieving an objective response and longer progression-free survival than ipilimumab monotherapy (58% [95% CI 52·0–63·2] of patients treated with concurrent nivolumab and ipilimumab; median progression-free survival 11·5 months [95% CI 8·9–16·7] vs 44% treated with nivolumab monotherapy; median progression-free survival 6·9 months [4·3–9·5] vs 19% treated with ipilimumab monotherapy; median progression-free survival 2·9 months [2·8–3·4]); however, the incidence of treatment-related grade 3–4 adverse events was higher with concurrent therapy (55%) than with nivolumab monotherapy (16%) or ipilimumab monotherapy (27%).
Clinical findings with concurrent administration of nivolumab and ipilimumab6–8 have generated interest in the sequential administration of these agents to maintain a high level of antitumour activity but reduce toxicity. However, during the design of our study, it was unknown whether one sequence might be associated with greater toxicity or greater efficacy than the alternative sequence because the agent given first might change the host or tumour biology in such a way as to make the second agent more or less active. Ipilimumab has been postulated to lead to a more favourable tumour milieu for efficacy with concurrent or sequential anti-PD-1 therapy because it increases tumour-infiltrating lymphocytes and interferon-γ-inducible genes in the tumour microenvironment,9,10 which in turn could increase PD-L1 expression.11 This hypothesis on the mechanism is noteworthy because high tumour expression of PD-L1, the main ligand for PD-1, has been associated with an increased proportion of patients achieving an objective response and improved overall survival in patients treated with nivolumab as a single agent.4 In a phase 1 dose-escalation study of concurrent nivolumab and ipilimumab in a combined cohort of 93 patients, 38 (41%) patients had an objective response, whereas sequential ipilimumab followed by nivolumab 1 mg/kg or 3 mg/kg in two cohorts of 16 patients each led to an objective response in ten (63%) and three (19%) patients, respectively.12 However, nivolumab followed by ipilimumab has not been tested in a clinical trial. Therefore, we assessed the safety and efficacy of a planned switch from nivolumab to ipilimumab, or the reverse sequence, followed by nivolumab continuation therapy, in patients with advanced melanoma.
Methods
Study design and participants
In this randomised, open-label, phase 2 study, we recruited patients from nine academic medical centres in the USA (appendix, p5). Eligible patients were at least 18 years of age, had histologically confirmed unresectable stage III or stage IV melanoma, and were previously untreated or had progressed after no more than one previous systemic therapy. Criteria for determining progression on previous systemic therapy were based on investigator-assessed radiographic imaging. Patients were also required to have measurable disease by CT or MRI within 28 days prior to randomisation as per Response Evaluation Criteria in Solid Tumors version 1.1 (RECIST v1.1) criteria, an Eastern Cooperative Oncology Group (ECOG) performance status of 0 or 1, known BRAFV600E mutational status or consent to BRAFV600E mutation testing during the screening period, and suitable lesions available for baseline biopsies and biopsies at week 13 (eg, assessment of PD-L1). Patients with active brain metastases, autoimmune disease, medical conditions requiring systemic treatment with corticosteroids or other immunosuppressive drugs, and previous treatment with anti-PD-1, anti-PD-L1, or anti-CTLA-4 antibodies were excluded. BRAFV600E mutation status was ascertained according to local institutional standards, which included several different methods: ABI polymerase chain reaction (PCR; Applied Biosystems, Foster City, CA, USA), COBAS (Roche, Branchburg, NJ, USA), pyrosequencing, RGQ PCR (Qiagen, Manchester, UK), Sanger sequencing, SNaPshot (Applied Biosystems, Foster City, CA, USA), THxID (bioMérieux, Durham, NC, USA), and other real-time PCR. Patients with either BRAFV600E wild-type or BRAFV600E mutation-positive status were eligible.
All patients provided written informed consent. The protocol, amendments, and patient informed consent were approved by the institutional review board (IRB) or independent ethics committee (IEC) of the participating centre before initiation of the study at the site.
Randomisation and masking
Patients were randomly allocated (1:1) to receive nivolumab followed by ipilimumab or ipilimumab followed by nivolumab (appendix p 6). When each patient had signed their informed consent form and was registered, they were assigned a patient number through an interactive voice response system (IVRS). The investigator or designee registered the patient for enrolment. Once enrolled in the IVRS, patients were randomised. An independent vendor managed the IVRS for randomisation, which used a permuted block schedule in a 1:1 ratio with a block size of four (2:2). There were no stratification factors. This study was open label; the investigators, site staff, and patients were aware of the treatment assignments before and during the study.
Procedures
Patients assigned to the nivolumab followed by ipilimumab group received nivolumab (Bristol-Myers Squibb, Princeton, NJ, USA) at 3 mg/kg as a 60-min intravenous infusion every 2 weeks for up to six doses during weeks 1 to 13 in the first induction period, followed by a planned switch to ipilimumab (Bristol-Myers Squibb) 3 mg/kg as a 90-min intravenous infusion every 3 weeks for up to four doses during weeks 13–25 in the second induction period. Patients assigned to the ipilimumab followed by nivolumab group received the reciprocal planned switch sequence. The time interval between drug sequences was 2 weeks for nivolumab followed by ipilimumab whereas it was 3 weeks for ipilimumab followed by nivolumab (dosing intervals were different for the two strategies because the agents have different frequencies of administration). After induction, all patients in both groups who completed the second induction period with the second immunotherapy agent and had clinical benefit were eligible to enter the continuation period and receive nivolumab 3 mg/kg every 2 weeks for up to 2 years or longer until progression, unacceptable toxicity, or withdrawal of consent.
During all treatment periods, dose reductions and escalations were not permitted, dose interruptions of up to 6 weeks were allowed for infusion reactions, and treatment modifications (eg, dose omissions) were based on specific laboratory and adverse event criteria. Dose omissions (but not dose delays) were allowed during the two induction periods. During the continuation period, dose delays with nivolumab were permitted. Dose omissions during the induction periods or dose delays during the continuation period were allowed in the following situations: any grade 2 non-skin treatment-related adverse event (excluding grade 2 treatment-related fatigue or laboratory abnormalities); any grade 3 skin treatment-related adverse event; any grade 3 treatment-related laboratory abnormality (with exceptions for lymphopenia, leukopenia, and raised concentrations of aspartate aminotransferase, alanine aminotransferase, total bilirubin, or asymptomatic amylase or lipase); and any adverse event, laboratory abnormality, or intercurrent illness which, in the judgment of the investigator, warranted omitting the dose.
Adverse events and laboratory values were graded according to the National Cancer Institute Common Terminology Criteria for Adverse Events version 4.0. Laboratory tests included chemistry and haematology tests, and were done in the nivolumab followed by ipilimumab group during the first induction period at weeks 1, 3, 5, 7, 9, and 11, and during the second induction period at weeks 13, 16, 19, and 22; and in the ipilimumab followed by nivolumab group during the first induction period at weeks 1, 4, 7, and 10, and during the second induction period at weeks 13, 15, 17, 19, 21, and 23; and in both groups during the continuation period every 2 weeks (plus or minus 3 days) starting at week 25. Select adverse events (ie, those with a potential immunological cause) were assessed and recorded by the investigator at each site according to organ category, as in previous studies.1,4,6 Patients had tumour assessments (per investigator assessment) as defined by modified RECIST v1.1 criteria13 with CT, MRI, or both, before dosing at baseline, weeks 13, 25, 33, and 41, and then every 12 weeks beginning at week 49. The same assessment method and technique, as specified by RECIST v1.1, were used to characterise each lesion at baseline and during follow-up. If the information required for assessment was unavailable or incomplete, the response in the patient's lesion was categorised as “unable to determine”. Patients who had clinical or radiological progressive disease before completing the first or second induction periods were allowed to remain on their current study treatment for the remainder of the induction period, as long as they were tolerating study treatment and did not have rapid clinical deterioration.
Permanent discontinuation of one study drug did not preclude patients from receiving the other immune checkpoint inhibitor, assuming recovery to baseline or resolution of the adverse event. However, patients who had a response status of progressive disease according to investigator-assessed RECIST v1.113 at week 13, and who had an additional 20% or greater increase in the sum of longest diameters, including all target lesions and measurable new lesions at week 25 compared with week 13, were discontinued from the study.
For the exploratory analysis of pharmacodynamic changes in immune markers, baseline tumour PD-L1 expression was assessed in a central laboratory (Mosaic Laboratories, Lake Forest, CA, USA) with an automated Bristol-Myers Squibb–Dako (Dako, Carpinteria, CA, USA) validated immunohistochemistry assay14 and scored by a qualified pathologist at Mosaic Laboratories. High PD-L1 expression was defined as at least 5% of tumour cells exhibiting cell-surface PD-L1 staining of any intensity in a section containing at least 100 evaluable cells.
Outcomes
Our research hypothesis was that a 24-week sequential treatment regimen of nivolumab followed by ipilimumab or ipilimumab followed by nivolumab would be associated with a clinically acceptable frequency of grade 3–5 treatment-related adverse events in patients with advanced or metastatic melanoma. The primary endpoint of this study was the occurrence of treatment-related grade 3–5 adverse events during the induction period (ie, up to week 25) in all patients who received sequential induction treatment and was investigator-assessed. Secondary endpoints were the proportion of patients with a confirmed response (partial or complete response) at week 25 and the proportion of patients with progressive disease at weeks 13 and 25. Duration of response was also reported in a post-hoc analysis. Prespecified exploratory endpoints included assessment of safety during the two induction periods and during the continuation period, overall survival, and an assessment of pharmacodynamic changes in immune markers from baseline and correlates with efficacy at weeks 13 and 25. These data will be reported elsewhere.
Patients were assessed for safety if they had received either study drug. The primary endpoint of treatment-related grade 3–5 adverse events during the induction period was defined as the number of patients who had at least one treatment-related grade 3–5 adverse event with an onset date on or after the first day of the induction period and not later than 30 days after the last dose from the induction period, divided by the number of treated patients. Adverse events with an onset date after the start of subsequent anticancer therapy or the start date of continuation period treatment were excluded from the primary endpoint analysis.
To assess the proportion of patients who achieved a response, RECIST v1.1 response criteria were modified in this study to disregard the week 13 assessment, which only reflects the activity of the first agent in the treatment sequence, at all post-week 13 assessment timepoints. Therefore, confirmed response at week 25 was defined as the number of patients with a complete or partial response at week 25, irrespective of the tumour assessment at week 13, with confirmatory imaging at week 33. The proportion of patients who achieved a response and best overall response during the overall study were assessed with additional follow-up during the continuation period, also disregarding the week 13 assessment. Progressive disease was defined at week 13 with RECIST v1.1 criteria, and at week 25 with modified RECIST v1.1 criteria (data at week 25 were compared with baseline data, discounting data from the week 13 scan).
The trial is ongoing but no longer enrolling patients; in this Article, we report the primary analysis for the primary and secondary endpoints, but overall survival follow-up is ongoing.
Statistical analysis
A sample size of about 140 patients (70 per group) was not based on power considerations and was chosen to provide a reasonable level of precision for estimating adverse event incidence for the treatment sequences. No statistical hypothesis testing was planned to assess differences between groups because the primary endpoint was safety. The population assessed for all primary, secondary, and exploratory outcomes was prespecified as all treated patients—ie, the as-treated population. The primary analysis of the frequency of treatment-related grade 3–5 adverse events during the first and second induction periods in both groups was done in all treated patients (ie, those who had received at least one dose of study treatment). The subset of patients who received at least one dose of study treatment in the second induction period was used as a sensitivity analysis of this primary endpoint. The proportions of patients reporting grade 3–5 adverse events with 95% CIs are presented for each treatment group. In determining the reliability of the adverse event estimates for the primary analysis, the exact 95% CI width was estimated to be 24% for a grade 3–5 adverse event rate of 40% in either cohort, assuming n=68. This sample size also provided for adequate samples of tumour tissues and peripheral immune cells to achieve stable estimates for exploratory biomarker analyses.
The proportion of patients achieving a response at week 25 was defined as the number of patients who had a complete response or partial response at week 25 per modified RECIST v1.1 criteria, with confirmation on the scheduled scan at week 33 (or any subsequent scan done at least 4 weeks after the week 25 scan), divided by the total number of treated patients. The proportion of patients with a response and corresponding 95% CIs were computed by group in all treated patients with the Clopper–Pearson method.
The exploratory endpoints of overall survival and response duration were estimated by the Kaplan-Meier method. Hazard ratios (HRs) and associated 95% CIs with a Cox-proportional hazards model, and median overall survival and corresponding two-sided 95% CIs, were also calculated on a post-hoc basis. We report an analysis of overall survival after 46% of patients had died. An update of the overall survival analysis is planned after 65% of the patients have died or after 2 years of follow-up from the last patient randomised, whichever occurs first.
The primary database lock occurred on May 22, 2015, after all treated patients had completed or discontinued the induction period. All week 13 and week 25 endpoints were analysed at this database lock. Safety, efficacy, and overall survival during the overall study, including the continuation period, were updated using a subsequent database lock on Nov 13, 2015, capturing a minimum follow-up of 15 months. SAS version 9.2 was used for all statistical analyses.
This study is registered with ClinicalTrials.gov, number NCT01783938.
Role of the funding source
The study was designed by the lead and corresponding investigators (JSW and FSH). Data collected by the funder were analysed in collaboration with all authors. The funder of the study provided funding for writing and editorial support for this report. The lead and corresponding authors (JSW and FSH) had full access to all the data in the study and had final responsibility for the decision to submit for publication.
Results
Between April 30, 2013, and July 21, 2014, we enrolled 177 patients at nine sites in the USA; 37 were excluded and 140 were randomly assigned to the two treatment groups (figure 1). Between May 9, 2013, and Aug 12, 2014, 70 patients were randomly allocated to nivolumab followed by ipilimumab and 70 patients to the reverse sequence (ie, ipilimumab followed by nivolumab). 68 patients were treated in the nivolumab followed by ipilimumab group (because two patients were excluded for unrelated serious adverse events before the first dose) and 70 patients were treated with the reverse sequence (figure 1). Demographic and baseline characteristics were generally balanced between the groups (table 1), apart from higher proportions of patients with ECOG performance status of 0, PD-L1 expression of 5% or higher, and a history of brain metastases in the nivolumab followed by ipilimumab group. Most patients were men, with ECOG performance status of 0, but with a predominance of M1c disease and a substantial proportion with raised lactate dehydrogenase concentrations. Previous systemic cancer therapy was received by ten (15%) of 68 patients in the nivolumab followed by ipilimumab group and eight (11%) of 70 patients in the ipilimumab followed by nivolumab group.
Figure 1. Trial profile.
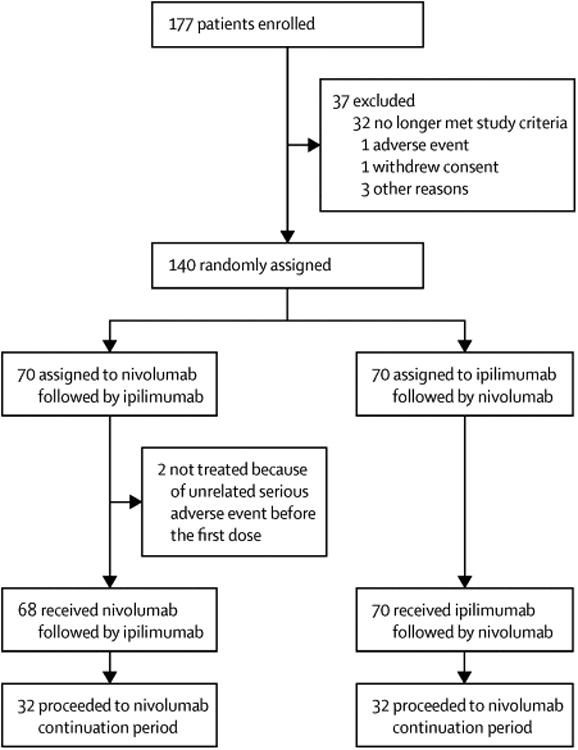
Table 1. Baseline characteristics.
| Nivolumab followed by ipilimumab (n=68) | Ipilimumab followed by nivolumab (n=70) | |
|---|---|---|
| Age (years) | 605 (46·5-70·0) | 63·0 (52·0-73·0) |
|
| ||
| Sex | ||
| Men | 46 (68%) | 46 (66%) |
| Women | 22 (32%) | 24 (34%) |
|
| ||
| Ethnic origin | ||
| White | 65 (96%) | 66 (94%) |
| Black or African–American | 1 (1%) | 2 (3%) |
| Other | 2 (3%) | 2 (3%) |
|
| ||
| ECOG performance status | ||
| 0 | 47 (69%) | 37 (53%) |
| 1 | 21 (31%) | 33 (47%) |
|
| ||
| AJCC stage at study entry | ||
| III | 6 (9%) | 12 (17%) |
| IV | 62 (91%) | 58 (83%) |
|
| ||
| M stage | ||
| M0 | 0 | 3 (4%) |
| M1a | 3 (4%) | 7 (10%) |
| M1b | 14 (21%) | 8 (11%) |
| M1c | 45 (66%) | 43 (61%) |
| Not reported | 6 (9%) | 9 (13%) |
|
| ||
| Baseline LDH | ||
| ≤ULN | 45 (66%) | 41 (59%) |
| >ULN | 23 (34%) | 29 (41%) |
| ≤2 × ULN | 59 (87%) | 58 (83%) |
| >2 × ULN | 9 (13%) | 12 (17%) |
|
| ||
| BRAF status | ||
| BRAFV600E mutant | 19 (28%) | 20 (29%) |
| Wild type | 44 (65%) | 43 (61%) |
| Not reported | 5 (7%) | 7 (10%) |
|
| ||
| Previous systemic therapy for metastatic disease | ||
| Any previous systemic therapy | 10 (15%) | 8 (11%) |
| Previous interleukin 2 | 4 (6%) | 2 (3%) |
|
| ||
| PD-L1 expression ≥5%* | 22/53 (42%) | 10/44 (23%) |
|
| ||
| History of brain metastases | ||
| Yes | 9 (13%) | 2 (3%) |
| No | 53 (78%) | 60 (86%) |
| Not reported | 6 (9%) | 8 (11%) |
Data are median (IQR), n (%), or n/N (%). ECOG=Eastern Cooperative Oncology Group. AJCC=American Joint Committee on Cancer. LDH=lactate dehydrogenase. PD-L1=programmed death ligand 1. ULN=upper limit of normal.
The proportion of patients with PD-L1 expression ≥5% among those with quantifi able PD-L1 (ie, 53 patients in the nivolumab followed by ipilimumab group and 44 patients in the ipilimumab followed by nivolumab group).
Patients in the nivolumab followed by ipilimumab group received a mean of 17·4 (SD 16·7) doses of nivolumab and 2·4 (1·37) doses of ipilimumab and a median of six (IQR 6·0–36·0) doses of nivolumab and three (1·5–4·0) doses of ipilimumab. Patients in the ipilimumab followed by nivolumab group received a mean of 3·2 (SD 1·05) doses of ipilimumab and 13·9 (16·8) doses of nivolumab and a median of four (IQR 2·0–4·0) doses of ipilimumab and four (1·0–30·0) doses of nivolumab. 32 (47%) of 68 patients assigned to nivolumab followed by ipilimumab received at least six doses of nivolumab compared with 29 (41%) of 70 assigned to the reverse sequence. More patients in the nivolumab followed by ipilimumab group than patients assigned to the reverse sequence continued on to at least one dose in the second induction period (58 [85%] of 68 in the nivolumab followed by ipilimumab group vs 53 [76%] of 70 in the ipilimumab followed by nivolumab group), although similar numbers of patients subsequently received at least one dose of nivolumab in the continuation period (32 [47%] vs 32 [46%], respectively; appendix p 1). Nine (13%) of 68 patients in the nivolumab followed by ipilimumab group remained on study treatment at database lock on Nov 13, 2015, compared with ten (14%) of 70 in the ipilimumab followed by nivolumab group; the most common reasons for discontinuation in the nivolumab followed by ipilimumab group were study drug toxicity (24 [35%] of 68 patients) and disease progression (18 [26%]), whereas in the ipilimumab followed by nivolumab group the most common reasons were disease progression (39 [56%] of 70 patients) and study drug toxicity (12 [17%]; appendix p 1). A higher proportion of patients assigned to nivolumab followed by ipilimumab received subsequent systemic anticancer therapy (25 [37%] of 68) than in the reverse sequence group (14 [20%] of 70; appendix p 2).
The proportions of patients with treatment-related grade 3–5 adverse events at the primary assessment timepoint at week 25 were similar between groups (34 [50%; 95% CI 37·6–62·4] of 68 patients in the nivolumab followed by ipilimumab group vs 30 [43%; 31·1–55·3] of 70 patients in the ipilimumab followed by nivolumab group; figure 2 [adverse events were counted only once for both induction periods]). No treatment-related deaths occurred during this study. In the prespecified sensitivity analysis in treated patients who received at least one study drug dose in the second induction period, the frequency of drug-related grade 3–5 adverse events during the induction periods (ie, up to week 25) was similar between groups (31 [53%] of 58 in the nivolumab followed by ipilimumab group vs 27 [51%] of 53 in the ipilimumab followed by nivolumab group). In the nivolumab followed by ipilimumab group, a higher rate of treatment-related grade 3–4 adverse events occurred during the second induction period and the continuation period compared with the first induction period (tables 2, 3, 4, figure 2). For the ipilimumab followed by nivolumab group, rates of treatment-related grade 3–4 adverse events were consistent throughout all three treatment periods (tables 2, 3, 4, figure 2). In patients with a grade 3–4 select adverse event (ie, those with a potential immunological cause) in the first induction period, only one patient subsequently developed a grade 3–4 select adverse event in the second induction period (a patient in the ipilimumab followed by nivolumab group who initially had a skin adverse event later developed a hepatic adverse event).
Figure 2. Treatment-related grade 3-5 adverse events by study period No treatment-related deaths were reported during any study period.
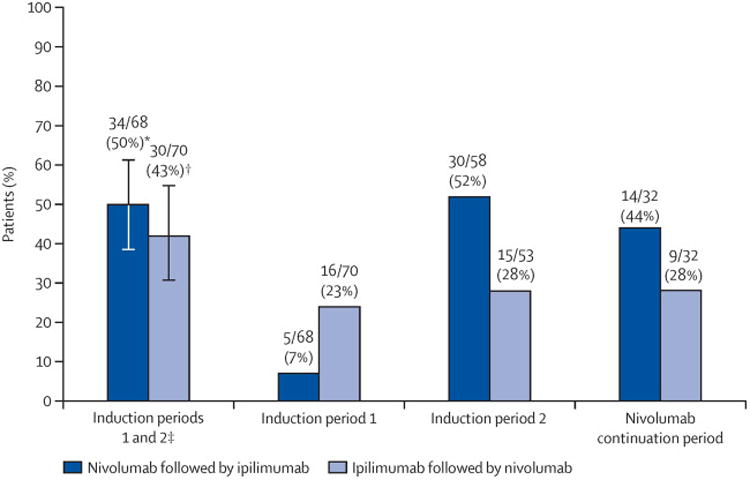
*95% CI 37·6-62·4%. †95% CI 31·1–55·3%. ‡Adverse events were counted only once for both induction periods. Error bars are 95% CIs.
Table 2. Treatment-related adverse events during the first induction period.
| Nivolumab followed by ipilimumab (n=68) | Ipilimumab followed by nivolumab (n=70) | |||||
|---|---|---|---|---|---|---|
| Grade 1–2 | Grade 3 | Grade 4 | Grade 1–2 | Grade 3 | Grade 4 | |
| Pruritus | 9 (13%) | 0 | 0 | 23 (33%) | 0 | 0 |
| Rash | 11 (16%) | 0 | 0 | 15 (21%) | 1 (1%) | 0 |
| Rash (maculopapular) | 5 (7%) | 0 | 0 | 9 (13%) | 0 | 0 |
| Fatigue | 26 (38%) | 0 | 0 | 24 (34%) | 1 (1%) | 0 |
| Chills | 3 (4%) | 0 | 0 | 9 (13%) | 0 | 0 |
| Diarrhoea | 8 (12%) | 0 | 0 | 23 (33%) | 1 (1%) | 0 |
| Nausea | 7 (10%) | 0 | 0 | 11 (16%) | 2 (3%) | 0 |
| Colitis | 0 | 0 | 0 | 3 (4%) | 9 (13%) | 0 |
| Autoimmune colitis | 0 | 0 | 0 | 0 | 1 (1%) | 0 |
| Diarrhoea (haemorrhagic) | 0 | 0 | 0 | 0 | 1 (1%) | 0 |
| Increased ALT | 4 (6%) | 0 | 0 | 7 (10%) | 1 (1%) | 0 |
| Increased AST | 4 (6%) | 0 | 0 | 7 (10%) | 0 | 0 |
| Increased lipase | 1 (1%) | 1 (1%) | 0 | 7 (10%) | 1 (1%) | 0 |
| Decreased neutrophil count | 1 (1%) | 0 | 0 | 0 | 0 | 1 (1%) |
| Decreased white blood cell count | 1 (1%) | 0 | 0 | 0 | 1 (1%) | 0 |
| Muscular weakness | 2 (3%) | 0 | 0 | 1 (1%) | 1 (1%) | 0 |
| Headache | 3 (4%) | 1 (2%) | 0 | 3 (4%) | 0 | 0 |
| Demyelinating polyneuropathy | 0 | 0 | 1 (2%) | 0 | 0 | 0 |
| Myasthenia gravis | 0 | 0 | 1 (2%) | 0 | 0 | 0 |
| Dyspnoea | 4 (6%) | 1 (1%) | 0 | 1 (1%) | 0 | 0 |
| Pneumonitis | 1 (1%) | 1 (1%) | 0 | 0 | 0 | 0 |
| Macular oedema | 2 (3%) | 0 | 0 | 0 | 1 (1%) | 0 |
| Mental status changes | 0 | 0 | 0 | 0 | 1 (1%) | 0 |
| Renal failure | 0 | 0 | 0 | 0 | 1 (1%) | 0 |
Data are n (%). ALT=alanine aminotransferase. AST=aspartate aminotransferase. No treatment-related deaths were recorded.
Table 3. Treatment-related adverse events during the second induction period.
| Nivolumab followed by ipilimumab (n=58) | Ipilimumab followed by nivolumab (n=53) | |||||
|---|---|---|---|---|---|---|
| Grade 1–2 | Grade 3 | Grade 4 | Grade 1–2 | Grade 3 | Grade 4 | |
| Increased ALT | 8 (14%) | 6 (10%) | 1 (2%) | 12 (23%) | 1 (2%) | 0 |
| Increased AST | 8 (14%) | 5 (9%) | 0 | 12 (23%) | 1 (2%) | 0 |
| Increased lipase | 6 (10%) | 1 (2%) | 0 | 9 (17%) | 4 (8%) | 1 (2%) |
| Increased amylase | 5 (9%) | 0 | 0 | 7 (13%) | 2 (4%) | 0 |
| Increased blood alkaline phosphatase | 4 (7%) | 2 (3%) | 0 | 2 (4%) | 0 | 0 |
| Decreased lymphocyte count | 1 (2%) | 1 (2%) | 0 | 1 (2%) | 0 | 0 |
| Increased blood creatinine | 0 | 1 (2%) | 0 | 0 | 0 | 0 |
| Increased hepatic enzyme | 0 | 0 | 1 (2%) | 0 | 0 | 0 |
| Pruritus | 16 (28%) | 1 (2%) | 0 | 5 (9%) | 0 | 0 |
| Rash | 14 (24%) | 2 (3%) | 0 | 5 (9%) | 1 (2%) | 0 |
| Rash (maculopapular) | 6 (10%) | 1 (2%) | 0 | 4 (8%) | 1 (2%) | 0 |
| Dermatitis (exfoliative) | 0 | 1 (2%) | 0 | 0 | 0 | 0 |
| Diarrhoea | 17 (29%) | 7 (12%) | 0 | 9 (17%) | 1 (2%) | 0 |
| Nausea | 13 (22%) | 2 (3%) | 0 | 2 (4%) | 0 | 0 |
| Colitis | 1 (2%) | 9 (16%) | 0 | 0 | 5 (10%) | 0 |
| Vomiting | 4 (7%) | 2 (3%) | 0 | 4 (8%) | 0 | 0 |
| Abdominal pain | 4 (7%) | 1 (2%) | 0 | 3 (6%) | 0 | 0 |
| Crohn's disease | 0 | 1 (2%) | 0 | 0 | 0 | 0 |
| Enterocolitis | 0 | 0 | 0 | 0 | 1 (2%) | 0 |
| Fatigue | 20 (34%) | 1 (2%) | 0 | 11 (21%) | 3 (6%) | 0 |
| Pyrexia | 10 (17%) | 0 | 0 | 1 (2%) | 0 | 0 |
| Headache | 8 (14%) | 2 (3%) | 0 | 4 (8%) | 1 (2%) | 0 |
| Hypothyroidism | 7 (12%) | 0 | 0 | 5 (9%) | 0 | 0 |
| Hypophysitis | 4 (7%) | 0 | 0 | 1 (2%) | 2 (4%) | 0 |
| Adrenal insufficiency | 1 (2%) | 1 (2%) | 0 | 1 (2%) | 0 | 0 |
| Decreased appetite | 7 (12%) | 0 | 0 | 4 (8%) | 1 (2%) | 0 |
| Dehydration | 2 (3%) | 1 (2%) | 0 | 1 (2%) | 1 (2%) | 0 |
| Polydipsia | 0 | 0 | 0 | 0 | 1 (2%) | 0 |
| Muscular weakness | 2 (3%) | 1 (2%) | 0 | 1 (2%) | 0 | 0 |
| Respiratory failure | 0 | 0 | 1 (2%) | 0 | 0 | 0 |
| Hepatitis | 0 | 3 (5%) | 0 | 0 | 0 | 0 |
| Hepatotoxicity | 0 | 0 | 1 (2%) | 0 | 0 | 0 |
| Autoimmune disorder | 0 | 1 (2%) | 0 | 0 | 0 | 0 |
| Acute kidney injury | 0 | 1 (2%) | 0 | 0 | 0 | 0 |
| Renal failure | 0 | 0 | 1 (2%) | 0 | 0 | 0 |
| Hypotension | 0 | 1 (2%) | 0 | 0 | 0 | 0 |
Data are n (%). ALT=alanine aminotransferase. AST=aspartate aminotransferase. No treatment-related deaths were recorded.
Table 4. Treatment-related adverse events during the continuation period.
| Nivolumab followed by ipilimumab (n=32) | Ipilimumab followed by nivolumab (n=32) | |||||
|---|---|---|---|---|---|---|
| Grade 1–2 | Grade 3 | Grade 4 | Grade 1–2 | Grade 3 | Grade 4 | |
| Increased lipase | 5 (16%) | 5 (16%) | 2 (6%) | 6 (19%) | 4 (13%) | 2 (6%) |
| Increased amylase | 8 (25%) | 1 (3%) | 1 (3%) | 4 (13%) | 1 (3%) | 0 |
| Increased ALT | 7 (22%) | 0 | 0 | 6 (19%) | 0 | 0 |
| Increased AST | 6 (19%) | 0 | 0 | 3 (9%) | 0 | 0 |
| Increased bloodthyroid-stimulatinghormone | 2 (6%) | 0 | 0 | 4 (13%) | 0 | 0 |
| Pruritus | 8 (25%) | 0 | 0 | 10 (31%) | 0 | 0 |
| Vitiligo | 8 (25%) | 0 | 0 | 10 (31%) | 0 | 0 |
| Rash | 5 (16%) | 0 | 0 | 7 (22%) | 0 | 0 |
| Rash (maculopapular) | 6 (19%) | 0 | 0 | 3 (9%) | 1 (3%) | 0 |
| Diarrhoea | 9 (28%) | 1 (3%) | 0 | 2 (6%) | 1 (3%) | 0 |
| Nausea | 7 (22%) | 0 | 0 | 3 (9%) | 0 | 0 |
| Vomiting | 4 (13%) | 0 | 0 | 4 (13%) | 0 | 0 |
| Abdominal pain | 2 (6%) | 0 | 0 | 3 (9%) | 2 (6%) | 0 |
| Colitis | 1 (3%) | 0 | 0 | 0 | 1 (3%) | 0 |
| Acute pancreatitis | 0 | 0 | 0 | 0 | 2 (6%) | 0 |
| Hypothyroidism | 7 (22%) | 0 | 0 | 8 (25%) | 0 | 0 |
| Adrenal insufficiency | 1 (3%) | 1 (3%) | 1 (3%) | 2 (6%) | 0 | 0 |
| Fatigue | 13 (41%) | 2 (6%) | 0 | 4 (13%) | 0 | 0 |
| Arthralgia | 5 (16%) | 1 (3%) | 0 | 4 (13%) | 0 | 0 |
| Back pain | 0 | 1 (3%) | 0 | 1 (3%) | 0 | 0 |
| Muscle spasms | 1 (3%) | 1 (3%) | 0 | 0 | 0 | 0 |
| Hyperglycaemia | 4 (13%) | 1 (3%) | 0 | 0 | 2 (6%) | 0 |
| Cough | 4 (13%) | 0 | 0 | 1 (3%) | 0 | 0 |
| Pneumonitis | 3 (9%) | 1 (3%) | 0 | 1 (3%) | 0 | 0 |
| Headache | 4 (13%) | 0 | 0 | 2 (6%) | 0 | 0 |
| Encephalitis | 0 | 0 | 1 (3%) | 0 | 0 | 0 |
| Hypertension | 0 | 1 (3%) | 0 | 0 | 0 | 0 |
| Cardiac failure | 0 | 1 (3%) | 0 | 0 | 0 | 0 |
| Deafness | 0 | 1 (3%) | 0 | 0 | 0 | 0 |
| Hearing impaired | 0 | 1 (3%) | 0 | 0 | 0 | 0 |
Data are n (%). ALT=alanine aminotransferase. AST=aspartate aminotransferase. No treatment-related deaths were recorded.
During the whole study period (a minimum follow-up of 15 months), grade 3–4 treatment-related adverse events were reported in 43 (63%) of 68 patients in the nivolumab followed by ipilimumab group (any grade: 65 [96%]) and in 35 (50%) of 70 patients in the ipilimumab followed by nivolumab group (any grade: 64 [91%]; appendix p 3). Treatment-related grade 3–4 increases in alanine aminotransferase and aspartate aminotransferase were more frequent in those treated with nivolumab followed by ipilimumab than in those treated with the reverse sequence (appendix p 3). The most common treatment-related grade 3–4 adverse events during the whole study period were colitis (ten [15%] in the nivolumab followed by ipilimumab group vs 14 [20%] in the reverse sequence group), increased lipase (ten [15%] vs 12 [17%]), and diarrhoea (eight [12%] vs five [7%]).
Types and frequencies of treatment-related adverse events leading to discontinuation during the whole study period were similar between groups (any grade: 25 [37%] of 68 in the nivolumab followed by ipilimumab group vs 23 [33%] of 70 in the ipilimumab followed by nivolumab group; grade 3–4: 17 (25%) vs 19 [27%], respectively). The most frequently reported treatment-related adverse events of any grade leading to discontinuation were colitis (six [9%] of 68 in the nivolumab followed by ipilimumab group vs 12 [17%] of 70 in the ipilimumab followed by nivolumab group), increases in alanine aminotransferase (three [4%] vs two [3%]), and AST (two [3%] vs two [3%]), and diarrhoea (two [3%] vs two [3%]).
During the whole study period, the most frequently reported treatment-related adverse events with a potential immunological cause of any grade in both groups were diarrhoea (32 [47%] of 68 in the nivolumab followed by ipilimumab group vs 31 [44%] of 70 in the ipilimumab followed by nivolumab group), pruritus (24 [35%] vs 30 [43%]), rash (27 [40%] vs 23 [33%]), raised alanine aminotransferase (24 [35%] vs 17 [24%]), raised AST (21 [31%] vs 17 [24%]), and hypothyroidism (15 [22%] vs 15 [21%]; appendix p 4). The most frequent treatment-related adverse events with a potential immunological cause of grade 3–4 in both groups were colitis (ten [15%] of 68 in the nivolumab followed by ipilimumab group vs 14 [20%] of 70 in the ipilimumab followed by nivolumab group), diarrhoea (eight [12%] vs five [7%]), and raised alanine aminotransferase (seven [10%] vs two [3%]).
Systemic corticosteroids were used in 57 (84%) of 68 patients assigned to nivolumab followed by ipilimumab and in 36 (51%) of 70 assigned to the reverse sequence; infliximab was used in four (6%) versus four (6%) patients, and mycophenolic acid in two (3%) versus no patients, respectively. In both treatment groups, most treatment-related adverse events with a potential immunological cause resolved after management in all categories, apart from the endocrine-related adverse events with a potential immunological cause. Most adverse events belonging to the endocrine select adverse event category were not regarded as resolved, even when well controlled, because these patients still needed hormone replacement therapy.
The proportion of patients who achieved a response at week 13 was higher in the nivolumab followed by ipilimumab group than in the ipilimumab followed by nivolumab group (24 [35%] of 68 vs seven [10%] of 70, respectively). The proportion of patients who had a confirmed response during induction (ie, up to week 25) was also higher in those treated with nivolumab followed by ipilimumab than in those treated with the reverse sequence (28 [41%; 95% CI 29·4–53·8] of 68 vs 14 [20%; 11·4–31·3] of 70, respectively; table 5). With additional follow-up, and with the efficacy assessment shifted from a simple timepoint analysis to best overall response analysis, a greater proportion of patients in the nivolumab followed by ipilimumab group achieved a complete or partial response (table 5).
Table 5. Response to treatment.
| Week 25 | Entire study period | |||
|---|---|---|---|---|
|
|
|
|||
| Nivolumab followed by ipilimumab (n=68) | Ipilimumab followed by nivolumab (n=70) | Nivolumab followed by ipilimumab (n=68) | Ipilimumab followed by nivolumab (n=70) | |
| Best overall response | ||||
|
| ||||
| Complete response | ·· | ·· | 8/68 (12%) | 4/70 (6%) |
| Partial response | ·· | ·· | 30/68 (44%) | 18/70 (26%) |
| Stable disease | ·· | ·· | 2/68 (3%) | 6/70 (9%) |
| Progressive disease | ·· | ·· | 15/68 (22%) | 20/70 (29%) |
| Unable to determine | ·· | ·· | 13/68 (19%) | 22/70 (31%) |
| Overall response, n/N (%; 95% CI) | 28/68 14/70 (41%; 29·4-53·8)* | (20%; 11·4-31·3)* | 38/68 (56%; 43·3-67·0) | 22/70 (31%; 20·9-43·6) |
| Median duration of overall response, months (IQR) | ·· | ·· | NR (8·4-19·3) | NR (7·5-17·2) |
|
| ||||
| Overall response by PD-L1 expression, n/N evaluable (%; 95% CI) | ||||
|
| ||||
| PD-L1 <5% | 8/31 (26%; 11·9–44·6) | 3/34 (9%; 1·9-237) | 13/31 (42%; 24·5-60·9) | 6/34 (18%; 6·8-34·5) |
| PD-L1 ≥5% | 12/22 (55%; 32·2-75·6) | 4/10 (40%; 12·2-73·8) | 16/22 (73%; 49·8-89·3) | 6/10 (60%; 26·2-87·8) |
Data are n/N (%) unless otherwise indicated. NR=not reached. PD-L1=programmed death ligand 1. The assessment of best overall response disregarded the week 13 tumour assessment. Reasons for patients judged as “unable to determine” include discontinuation, start of a subsequent anticancer therapy, or death before the week 25 assessment; and no evaluable tumour assessment done beyond the week 13 timepoint.
Confirmed with scan at week 33 (or any subsequent scan done ≥4 weeks after the week 25 scan).
Median duration of response was not reached in either group during the study; most responding patients remained in response (27 [71%] of 38 in the nivolumab followed by ipilimumab group vs 20 [90%] of 22 in the ipilimumab followed by nivolumab group) at an overall median duration of follow-up of 18·6 months (IQR 8·4–25·6). During the whole study period, a higher proportion of patients achieving a response was reported in patients with baseline PD-L1 expression of 5% or more versus less than 5% in both groups (table 5). However, a greater proportion of patients in the nivolumab followed by ipilimumab group than in the ipilimumab followed by nivolumab group were evaluable for PD-L1 expression at baseline (53 [78%] of 68 vs 44 [63%] of 70) and had baseline PD-L1 expression of 5% more (22 [42%] of 53 vs 10 [23%] of 44).
At week 13, disease progression was reported in 26 (38%; 95% CI 26·7–50·8) of 68 patients in the nivolumab followed by ipilimumab group and in 43 (61%; 49·0–72·8) of 70 patients in the ipilimumab followed by nivolumab group. At week 25, the proportion of patients with disease progression was unchanged for the nivolumab followed by ipilimumab group (26 [38%; 95% CI 26·7–50·8] of 68); in the ipilimumab followed by nivolumab group, one patient who showed progression at week 13 had a partial response at week 25, decreasing the progression rate to 60% (42/70; 47·6–71·5). The depth of tumour response, assessed as the percentage reduction in target lesion, improved from week 13 to week 25 and with additional follow-up (figure 3).
Figure 3. Tumour burden change from baseline (A) at week 13 and (B) at week 25, and (C) best tumour burden change during the entire study period.
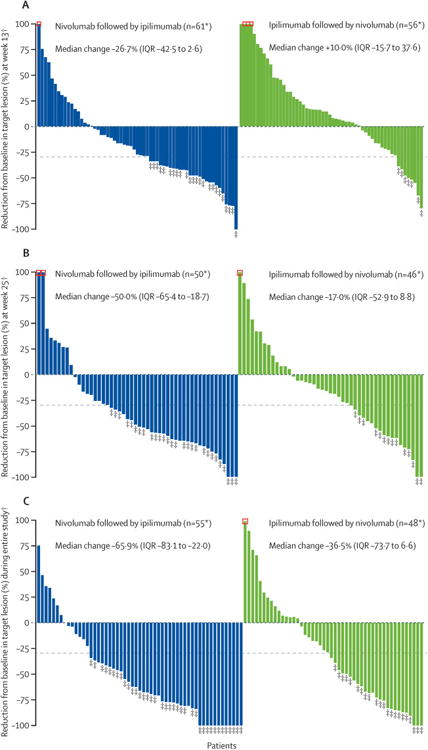
*Patients with target lesion at baseline and at least one tumour assessment (A) at week 13 or (B) at week 25 or (C) on treatment. †Negative/positive value means maximum tumour reduction/minimum tumour increase. Reduction is based on evaluable target lesion measurements up to the start of subsequent therapy. Horizontal dotted reference line indicates the 30% reduction consistent with a Response Evaluation Criteria in Solid Tumors version 1.1 response. ‡Responders (A) at week 13 or (B) at week 25 or (C) during the entire study period. Square symbol represents percentage change truncated to 100%.
After a median follow-up of 19·8 months (IQR 12·8–25·7) in the nivolumab followed by ipilimumab group, median overall survival was not reached (95% CI 23·7–not reached), whereas over a median follow-up of 14·7 months (IQR 5·6–23·9) in the ipilimumab followed by nivolumab group, median overall survival was 16·9 months (95% CI 9·2–26·5; HR 0·48 [95% CI 0·29–0·80], figure 4). A higher 12-month overall survival was recorded in patients assigned to nivolumab followed by ipilimumab than in those patients assigned to the reverse sequence (76% [95% CI 64–85] vs 54% [42–65%]; figure 4). In a post-hoc multivariate analysis of overall survival adjusting for the imbalances between groups in ECOG performance status, history of brain metastases, and baseline PD-L1 expression, the adjusted HR was consistent with the primary analysis in favouring nivolumab followed by ipilimumab relative to the reverse sequence (adjusted HR 0·57 [95% CI 0·33–0·99]). Figure 5 shows the results of the post-hoc subgroup analyses.
Figure 4. Overall survival.
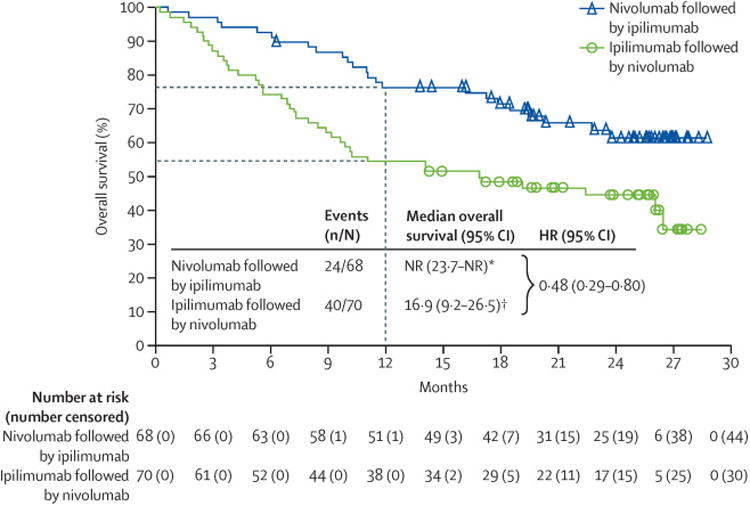
HR=hazard ratio. NR=not reached. *Median follow-up in the nivolumab followed by ipilimumab group was 19·8 months (IQR 12·8–257). †Median follow-up in the ipilimumab followed by nivolumab group was 147 months (56–23·9).
Figure 5. Subgroup survival analysis.
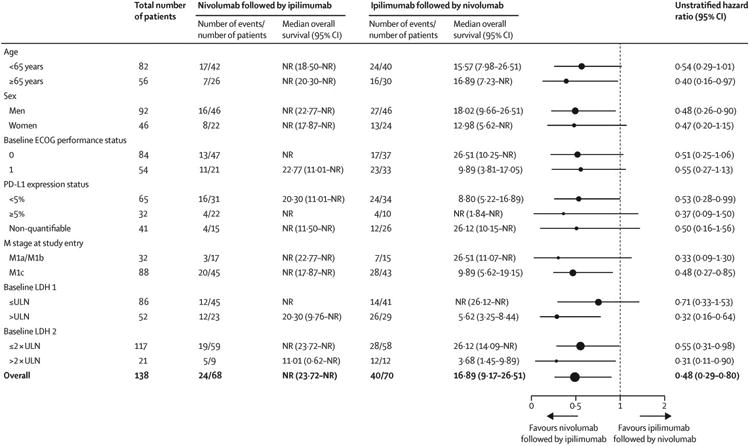
NR=not reached. ECOG=Eastern Cooperative Oncology Group. PD-L1=programmed death-ligand 1. LDH=lactate dehydrogenase. ULN=upper limit of normal.
Discussion
In this open-label, randomised, phase 2 study, with a median follow-up of 18 • 6 months, patients treated with nivolumab followed by ipilimumab had a similar frequency of treatment-related grade 3–5 adverse events up to week 25 as those treated with the reverse sequence. Grade 3–4 adverse events leading to treatment discontinuation were also similar between groups. Nivolumab followed by ipilimumab was associated with a lower proportion of patients with disease progression at weeks 13 and 25, a higher proportion of patients with a confirmed response at week 25 and throughout the entire study period, and longer median overall survival than the reverse sequence of ipilimumab followed by nivolumab. Sequential administration of immune checkpoint inhibitors with complementary mechanisms of action has drawn attention recently because of trial data showing the clinical benefits of concurrent combination therapy with nivolumab and ipilimumab compared with either agent given alone in patients with advanced melanoma.6–8 Different dosing sequences with immune checkpoint inhibitors could result in distinct efficacy and toxicity profiles, offering patients new therapeutic options. We report, for the first time to our knowledge, the safety and efficacy of a planned switch from nivolumab to ipilimumab, or the reverse sequence, in patients with advanced melanoma. Nivolumab followed by ipilimumab versus the reverse sequence was associated with a higher overall frequency of adverse events but with improved efficacy. Taken together, and consistent with the higher response rate of nivolumab compared with ipilimumab as monotherapy, these data suggest a benefit–risk profile that favours the sequence of nivolumab followed by ipilimumab in patients with advanced melanoma.
The frequency of treatment-related grade 3–4 adverse events was similar between groups during the induction periods up to week 25. A difference between groups in the frequency of treatment-related grade 3–4 adverse events was seen over time. When looking at the safety profile during each study period, we saw a higher than expected toxicity of ipilimumab when given in close proximity after nivolumab. In the nivolumab followed by ipilimumab group, the frequency of treatment-related grade 3–4 adverse events was low in the first induction period with nivolumab monotherapy but increased with ipilimumab monotherapy during the second induction period to a rate near that seen with concomitant nivolumab and ipilimumab,8 and decreased only slightly during the nivolumab continuation period. By contrast, there was little change in the frequency of treatment-related grade 3–4 adverse events in the ipilimumab followed by nivolumab group from ipilimumab monotherapy during the first induction period to nivolumab monotherapy during the second induction period and the continuation period. Additionally, systemic corticosteroids were used more frequently in patients treated with nivolumab followed by ipilimumab than in those treated with the reverse sequence, which probably represents the investigator's recognition of immune-mediated toxicity that might not have been fully captured by standard Common Terminology Criteria for Adverse Events grading criteria and its management using established guidelines. Furthermore, a higher proportion of patients discontinued treatment because of progressive disease in the ipilimumab followed by nivolumab group, which indicates that fewer patients in this group continued on therapy and had the potential to develop toxicity requiring discontinuation, which might have magnified the difference in corticosteroid use between treatment groups.
A novel feature of the design of this study was that patients who discontinued the first immune checkpoint inhibitor because of toxicity were allowed, upon resolution of the adverse event, to continue and receive the second immune checkpoint inhibitor. Our findings did not show any exacerbation of adverse events, irrespective of the severity of the initial event, within or between the select adverse event categories, in patients treated sequentially with nivolumab and ipilimumab in both treatment groups. Of patients with a grade 3–4 select adverse event with the first agent, only one subsequently developed a grade 3–4 select adverse event with the second agent, suggesting that toxicity with one immune checkpoint inhibitor does not predict toxicity with a subsequent immune checkpoint inhibitor. Whether toxicity with one immune checkpoint inhibitor is likely to predispose a patient to toxicity with another immune checkpoint inhibitor is an important question for future trials.
The nature of the treatment-related adverse events and those leading to permanent discontinuation was consistent with what has been reported in previous studies of ipilimumab and nivolumab given as monotherapy or concurrent combination therapy.1,4,6,8,15,16 However, the frequency of adverse events was higher in our study than with either agent as monotherapy, and slightly lower than or similar to that with concurrent ipilimumab and nivolumab. As in previous studies, select adverse events (ie, those with a potential immunological cause) were manageable with standard treatment algorithms (ie, that included systemic corticosteroids). Of the treatment-related select adverse events, hepatic and endocrine adverse events occurred more frequently in patients treated with nivolumab followed by ipilimumab than in those treated with the reverse sequence, whereas the frequencies of gastrointestinal adverse events were similar between groups.
With a planned treatment switch irrespective of week 13 response, a consistently improved clinical benefit with nivolumab followed by ipilimumab versus ipilimumab followed by nivolumab suggested that nivolumab followed by ipilimumab might be a more beneficial option than the reverse sequence. Switching of treatments seemed to increase the depth of response as shown by the higher proportion of patients with confirmed responses between weeks 13 and 25, and in some patients, even converted the patient from being a non-responder to a responder.
A higher proportion of patients with baseline PD-L1 expression of 5% or more versus those with less than 5% achieved a response in both treatment groups, and the proportion of patients who achieved a response was higher in the nivolumab followed by ipilimumab group than in the ipilimumab followed by nivolumab group irrespective of PD-L1 expression. However, a greater proportion of patients in the nivolumab followed by ipilimumab group than in the ipilimumab followed by nivolumab group were evaluable for PD-L1 expression at baseline and had baseline PD-L1 expression of 5% or more, and these imbalances might partly explain the low response rate in patients with PD-L1 expression of less than 5% in the ipilimumab followed by nivolumab group.
Although this study provides important information about the sequential use of nivolumab and ipilimumab in the setting of advanced melanoma, the results should be interpreted with caution because of the limitations of the trial. First, the study used an open-label, randomised, phase 2 design with little power to detect differences in the outcome. Additionally, the absence of stratification led to imbalances in baseline characteristics between the treatment groups (eg, a greater percentage of patients with ECOG performance status 0, PD-L1 expression ≥5%, and a history of brain metastases in the nivolumab followed by ipilimumab group than in the other group). Moreover, the subgroup analyses were post-hoc and were limited by a scarcity of events. The planned treatment switch, which occurred irrespective of disease response, does not typically represent real-world practice, although patients who progress on one agent are usually switched rapidly to the next line of therapy. Furthermore, the safety profiles should be interpreted in the context of the different dosing schedules for nivolumab and ipilimumab and a study design that allowed patients to permanently discontinue during one study period or agent but still continue onto the next study period or agent. The concept of initiating a different immune checkpoint inhibitor after permanent discontinuation of another is an area of future research. The primary efficacy endpoint of response at week 25 might have occurred too early to represent the full benefit from immune checkpoint inhibition, as shown by improved response rates recorded with longer follow-up. Finally, there was an imbalance between groups in patients who received subsequent anticancer therapy, which is possibly indicative of the difference between groups in survival.
These results could influence further development of sequenced therapy with nivolumab and ipilimumab, in view of the difference in response and overall survival favouring nivolumab followed by ipilimumab, even when imbalances in prognostic factors are accounted for. Additionally, the original premise of the study was that sequential therapy would reduce the toxicity compared with concurrent therapy because drug concentrations of the initial agent in the first induction period, which might be associated with adverse events, would decrease during the second induction period. The rapid switch to the next treatment at week 13 apparently mitigated this decline and resulted in sufficiently high drug concentrations in the second induction period to account for the notable occurrence of adverse events that were similar to that seen with concurrent therapy. In fact, the proportion of patients who achieved a response and the level of toxicity in the nivolumab followed by ipilimumab group were similar to those seen with the FDA-approved, concurrent nivolumab and ipilimumab regimen, suggesting that there is little advantage to sequential as opposed to concurrent therapy.6,8,17 The differences in safety and efficacy between the two sequential regimens might also be attributable to differences in the pharmacodynamic properties of the individual agents. For example, because an anti-CTLA-4 antibody bound to CTLA-4 might be internalised rapidly by the cell, whereas an anti-PD-1 antibody might occupy the PD-1 receptor for a prolonged period,18–21 treatment with nivolumab before ipilimumab might be similar to receiving both agents concurrently. Alternatively, our results might be simply related to ipilimumab being more toxic and less efficacious than nivolumab. In support of this hypothesis, nivolumab and the anti-PD-1 antibody pembrolizumab given as single agents have been shown to be associated with better efficacy than ipilimumab monotherapy in patients with advanced melanoma, with fewer high-grade adverse events.6,22
In conclusion, a planned treatment switch from nivolumab to ipilimumab showed improved efficacy outcomes compared with a planned switch from ipilimumab to nivolumab, but with a higher overall frequency of adverse events. Because overall survival data for patients treated with nivolumab followed by ipilimumab are immature, any indirect comparison with concurrent therapy awaits long-term follow-up; as per protocol, a 2-year follow-up of overall survival is planned. Furthermore, the clinical relevance of the higher toxicity profile noted over time for nivolumab followed by ipilimumab remains unclear. The mechanism that accounts for the improved clinical outcomes with nivolumab followed by ipilimumab compared with the reverse sequence is unknown, although detailed correlative analyses of tumour and blood samples are being done to assess what factors might account for the difference. Future prospective studies will hopefully provide insight into important mechanisms by which immune checkpoint inhibition mediates tumour regression, how one immune checkpoint inhibitor affects another, and the development of new sequential treatment regimens.
Research in context.
Evidence before this study
To identify studies of combination therapy of a programmed death receptor-1 (PD-1) inhibitor and a cytotoxic T-lymphocyte antigen 4 (CTLA-4) inhibitor in advanced melanoma, we searched PubMed and congress abstracts from the annual meetings of the American Society of Clinical Oncology, European Society of Medical Oncology/European Cancer Congress, and Society for Melanoma Research, between April 1, 2008, and April 30, 2013. Our search terms were “ipilimumab”, “melanoma”, “nivolumab”, and “pembrolizumab”. Our search identified three (phase 1, 2, and 3) clinical studies of nivolumab given concurrently with ipilimumab, a phase 1 safety trial of pembrolizumab given concurrently with ipilimumab, and a small retrospective case series of nivolumab or pembrolizumab given sequentially with ipilimumab in patients with advanced melanoma. These studies showed that concurrent combination therapy with nivolumab and ipilimumab improves the proportion of patients achieving a response versus either agent alone, albeit with more high-grade adverse events. Ipilimumab, nivolumab, and pembrolizumab are approved by the US Food and Drug Administration and the European Commission for the treatment of patients with metastatic melanoma as single agents; nivolumab and ipilimumab are also approved as concurrent combination therapy. Clinical findings with concurrent administration of nivolumab and ipilimumab have triggered interest in sequential administration of these agents to potentially maintain a high amount of antitumour activity and minimise toxicity.
Added value of this study
For the patient population investigated in this study, treatment options are needed that improve existing approved therapies, from both efficacy and safety perspectives. Our data suggest that administration of nivolumab followed by a planned switch to ipilimumab is associated with a higher overall frequency of treatment-related grade 3–4 adverse events, but provides improved efficacy, compared with administration of ipilimumab followed by a planned switch to nivolumab. No previous study has shown benefit from a clear temporal approach to the administration of checkpoint inhibitors.
Implications of all the available evidence
Results from this study could help to inform the choice of initial treatment if sequential approaches in advanced melanoma are used, and a planned analysis could provide insights into the mechanism of action and correlative science of sequential therapy with nivolumab and ipilimumab.
Acknowledgments
Financial support for the study was provided by Bristol-Myers Squibb. We thank the patients and their families, and the participating study teams, for making this study possible. We also thank Allen Chen of Bristol-Myers Squibb for early protocol development. Medical writing and editing support were provided by Mark Palangio and Cara Hunsberger of StemScientific, an Ashfield Company (Lyndhurst, New Jersey, USA), and funded by Bristol-Myers Squibb.
Funding Bristol-Myers Squibb.
Footnotes
Declaration of interests: JSW reports consulting or advisory fees from Bristol-Myers Squibb, Genentech, GlaxoSmithKline, Merck, and Roche; honoraria from Bristol-Myers Squibb, Genentech, GlaxoSmithKline, Merck, and Roche; stock ownership in Celldex Therapeutics, Altor BioScience, and cCAM Biotherapeutics; and research funding from Bristol-Myers Squibb, Genentech, GlaxoSmithKline, Merck, Macrogenics, and Roche. GG reports consulting or advisory fees from Novartis; and consulting or advisory fees from Bristol-Myers Squibb, Genentech, and Novartis. RJS reports consulting or advisory fees from Astex Pharmaceuticals, Biodesix, Boehringer Ingelheim, Novartis, and Prometheus. CLS reports consulting or advisory fees from Immatics Biotechnologies and Polynoma; honoraria from Castle Biosciences; research funding from 3M, Bristol-Myers Squibb, GlaxoSmithKline, Merck, and Polynoma; and patents or royalties from U.Va. Licensing and Ventures Group. TFL reports research funding from Abbott Laboratories, Acceleron Pharma, Abraxis BioScience, Amgen, Argos Therapeutics, AstraZeneca, AVEO, BioVex, Bristol-Myers Squibb, Cerulean, Eisai, EMD Serono, GlaxoSmithKline, Immatics, Lilly, Merck, Novartis, Pfi zer, Prometheus, Roche, Sunta, Threshold Pharmaceuticals, Millennium, and TRACON Pharma; and consulting/advisory fees from Prometheus. SN reports research funding from Bristol-Myers Squibb and Celldex Therapeutics. LF reports research funding or grant support from Bristol-Myers Squibb, Genentech/Roche, Merck, Sanofi -Aventis, and SU2C-MRA; consulting or advisory fees from Bristol-Myers Squibb; and honoraria from ASCO, DAVA Oncology, First Consult, GlaxoSmithKline, Imedex, and Via Oncology. EIB reports consulting or advisory fees from Castle Bioscience and Immune Design. EB, MR, and CH report employment and stock ownership with Bristol-Myers Squibb. GK and JJ report employment with Bristol-Myers Squibb. FSH reports consulting/advisory fees and research funding from Bristol-Myers Squibb; and patents/royalties from ImmuneTarget. JAS, DPL, and LMS declare no competing interests.
Contributors: JSW contributed to the conception and design of the study, patients' treatment, data acquisition, and writing of the report. GG contributed to the conception and design of the study, patients' treatment, and data acquisition. RJS, JAS, CLS, DPL, TFL, LMS, SN, LF, and EIB contributed to patients' treatment and data acquisition. EB was the study director. MR was the medical monitor. EB and MR contributed to the data cleaning and analysis. GK was the lead statistician for the study and the primary data analysis. JJ was the statistician for the updated analysis. CH was the biomarker lead for the study. FSH contributed to the conception and design of the study, patients' treatment, data acquisition, and writing of the report.
References
- 1.Schadendorf D, Hodi FS, Robert C, et al. Pooled analysis of long-term survival data from phase II and phase III trials of ipilimumab in unresectable or metastatic melanoma. J Clin Oncol. 2015;33:1889–94. doi: 10.1200/JCO.2014.56.2736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wang C, Thudium KB, Han M, et al. In vitro characterization of the anti-PD-1 antibody nivolumab, BMS-936558, and in vivo toxicology in non-human primates. Cancer Immunol Res. 2014;2:846–56. doi: 10.1158/2326-6066.CIR-14-0040. [DOI] [PubMed] [Google Scholar]
- 3.Robert C, Long GV, Brady B, et al. Nivolumab in previously untreated melanoma without BRAF mutation. N Engl J Med. 2015;372:320–30. doi: 10.1056/NEJMoa1412082. [DOI] [PubMed] [Google Scholar]
- 4.Weber JS, D'Angelo SP, Minor D, et al. Nivolumab versus chemotherapy in patients with advanced melanoma who progressed after anti-CTLA-4 treatment (CheckMate 037): a randomised, controlled, open-label, phase 3 trial. Lancet Oncol. 2015;16:375–84. doi: 10.1016/S1470-2045(15)70076-8. [DOI] [PubMed] [Google Scholar]
- 5.Okazaki T, Chikuma S, Iwai Y, Fagarasan S, Honjo T. A rheostat for immune responses: the unique properties of PD-1 and their advantages for clinical application. Nat Immunol. 2013;14:1212–18. doi: 10.1038/ni.2762. [DOI] [PubMed] [Google Scholar]
- 6.Larkin J, Chiarion-Sileni V, Gonzalez R, et al. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N Engl J Med. 2015;373:23–34. doi: 10.1056/NEJMoa1504030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wolchok JD, Kluger H, Callahan MK, et al. Nivolumab plus ipilimumab in advanced melanoma. N Engl J Med. 2013;369:122–33. doi: 10.1056/NEJMoa1302369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Postow MA, Chesney J, Pavlick AC, et al. Nivolumab and ipilimumab versus ipilimumab in untreated melanoma. N Engl J Med. 2015;372:2006–17. doi: 10.1056/NEJMoa1414428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ji RR, Chasalow SD, Wang L, et al. An immune-active tumor microenvironment favors clinical response to ipilimumab. Cancer Immunol Immunother. 2012;61:1019–31. doi: 10.1007/s00262-011-1172-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tarhini AA, Edington H, Butterfi eld LH, et al. Immune monitoring of the circulation and the tumor microenvironment in patients with regionally advanced melanoma receiving neoadjuvant ipilimumab. PLoS One. 2014;9:e87705. doi: 10.1371/journal.pone.0087705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Taube JM, Young GD, McMiller TL, et al. Differential expression of immune-regulatory genes associated with PD-L1 display in melanoma: implications for PD-1 pathway blockade. Clin Cancer Res. 2015;21:3969–76. doi: 10.1158/1078-0432.CCR-15-0244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kluger H, Sznol M, Callahan MK, et al. Survival, response duration, and activity by BRAF mutation status in a phase 1 trial of nivolumab (anti-PD-1, BMS-936558, ONO-4538) and ipilimumab concurrent or sequenced therapy in advanced melanoma. 39th European Society for Medical Oncology Congress; Madrid, Spain. Sept 26–30, 2014. [Google Scholar]
- 13.Eisenhauer EA, Therasse P, Bogaerts J, et al. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1) Eur J Cancer. 2009;45:228–47. doi: 10.1016/j.ejca.2008.10.026. [DOI] [PubMed] [Google Scholar]
- 14.Phillips T, Simmons P, Inzunza HD, et al. Development of an automated PD-L1 immunohistochemistry (IHC) assay for non-small cell lung cancer. Appl Immunohistochem Mol Morphol. 2015;23:541–49. doi: 10.1097/PAI.0000000000000256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hodi FS, O'Day SJ, McDermott DF, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363:711–23. doi: 10.1056/NEJMoa1003466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Robert C, Thomas L, Bondarenko I, et al. Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. N Engl J Med. 2011;364:251726. doi: 10.1056/NEJMoa1104621. [DOI] [PubMed] [Google Scholar]
- 17.OPDIVO (nivolumab) package insert. Princeton, NJ: Bristol-Myers Squibb Company; Mar, 2015. [Google Scholar]
- 18.Brahmer JR, Drake CG, Wollner I, et al. Phase I study of single-agent anti-programmed death-1 (MDX-1106) in refractory solid tumors: safety, clinical activity, pharmacodynamics, and immunologic correlates. J Clin Oncol. 2010;28:3167–75. doi: 10.1200/JCO.2009.26.7609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Qureshi OS, Kaur S, Hou TZ, et al. Constitutive clathrin-mediated endocytosis of CTLA-4 persists during T cell activation. J Biol Chem. 2012;287:9429–40. doi: 10.1074/jbc.M111.304329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pentcheva-Hoang T, Chen L, Pardoll DM, Allison JP. Programmed death-1 concentration at the immunological synapse is determined by ligand affinity and availability. Proc Natl Acad Sci USA. 2007;104:17765–70. doi: 10.1073/pnas.0708767104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shiratori T, Miyatake S, Ohno H, et al. Tyrosine phosphorylation controls internalization of CTLA-4 by regulating its interaction with clathrin-associated adaptor complex AP-2. Immunity. 1997;6:583–89. doi: 10.1016/s1074-7613(00)80346-5. [DOI] [PubMed] [Google Scholar]
- 22.Robert C, Schachter J, Long GV, et al. Pembrolizumab versus ipilimumab in advanced melanoma. N Engl J Med. 2015;372:2521–32. doi: 10.1056/NEJMoa1503093. [DOI] [PubMed] [Google Scholar]