

Abstract
Eukaryotic genomes contain substantial amounts of repetitive DNA organized in the form of constitutive heterochromatin and associated with repressive epigenetic modifications, such as H3K9me3 and C5-cytosine methylation (5mC). In the fungus Neurospora crassa, H3K9me3 and 5mC are catalyzed, respectively, by a conserved SUV39 histone methyltransferase DIM-5 and a DNMT1-like cytosine methyltransferase DIM-2. Here we show that DIM-2 can also mediate Repeat-Induced Point mutation (RIP) of repetitive DNA in N. crassa. We further show that DIM-2-dependent RIP requires DIM-5, HP1, and other known heterochromatin factors, implying the role of a repeat-induced heterochromatin-related process. Our previous findings suggest that the mechanism of repeat recognition for RIP involves direct interactions between homologous double-stranded (ds) DNA segments. We thus now propose that, in somatic cells, homologous dsDNA/dsDNA interactions between a small number of repeat copies can nucleate a transient heterochromatic state, which, on longer repeat arrays, may lead to the formation of constitutive heterochromatin.
INTRODUCTION
A substantial proportion of nearly every eukaryotic genome is occupied by repetitive DNA that is organized in the form of constitutive heterochromatin and associated with repressive epigenetic modifications, such as histone H3 lysine-9 di-/trimethylation (H3K9me2/3) and C5-cytosine methylation (5mC)1–6. The process of de novo heterochromatin assembly has been characterized particularly well in the fungus Neurospora crassa, where H3K9me3 and 5mC are established, respectively, by a conserved SUV39 histone methyltransferase DIM-5 and a DNMT1-like cytosine methyltransferase DIM-27. In N. crassa, while the bulk of constitutive heterochromatin occurs at dedicated AT-rich sequences7, H3K9me3 and 5meC can also be detected at newly-introduced transgenic repeat arrays of sufficiently large size8–11. A similar threshold effect is observed in other organisms, including humans12–14. For example, in Facioscapulohumeral Muscular Dystrophy Type 1 (FSHD1), the contraction of the D4Z4 macrosatellite array from 11–100 to 1–10 copies leads to the loss of heterochromatin and the concomitant reactivation of the D4Z4-embedded DUX4 gene14. The existence of an apparent copy-number threshold raises an interesting possibility: heterochromatic modifications may be constantly installed and removed on short repeat arrays unless they are stabilized in the presence of a larger number of repeats.
The phenomenon of Repeat-Induced Point mutation (RIP) in N. crassa occurs in haploid germ-line nuclei that continue to divide by mitosis in preparation for karyogamy and ensuing meiosis15–16. During RIP, duplications of chromosomal DNA longer than a few hundred base-pairs are detected and subjected to strong cytosine-to-thymine (C-to-T) mutation. Duplicated DNA segments are recognized largely irrespective of their particular base-pair sequence, transcriptional capacity, or relative/absolute positions in the genome. Thus, RIP represents a process that is uniquely sensitive to DNA homology. Notably, in some filamentous fungi, an analogous process leads to cytosine methylation rather than mutation17. Previously, we showed that RIP does not involve the canonical homology-recognition pathway mediated by MEI-3 (the only RecA homolog in N. crassa) and that RIP can match weakly similar DNA sequences as long as those sequences shared a series of interspersed homologous base-pair triplets spaced at intervals of 11 or 12 base-pairs18–20. These and other results are consistent with a mechanism of recombination-independent homology recognition that involves interactions between co-aligned double-stranded DNA molecules18. Our current findings, below, raise the possibility that such homologous interactions may occur not only in pre-meiotic but also in somatic cells, where they may nucleate heterochromatin formation on repetitive DNA.
RESULTS
RIP can occur in the absence of RID
In our earlier work18, we developed a sensitive RIP tester construct comprising one endogenous and one closely-positioned (ectopic) copy of an arbitrarily-chosen 802-bp region of the Neurospora genome (Fig. 1a; Supplementary Fig. 1). This construct induces strong mutation in a wildtype genetic background. Thus, specifically, a sample of 24 progeny spores was found to contain 3163 mutations in the endogenous (“left”) repeat copy, 3153 mutations - in the ectopic (“right”) repeat copy, and 524 mutations - in the endogenous 600-bp segment of the linker region (Fig. 1b: “rid+/+, dim-2+/+”; data replotted from ref. 18). We now confirm that all of the above mutations, including those of the linker region, are induced by the presence of DNA homology: if the ectopic repeat copy is specifically omitted, no RIP activity can be detected (Fig. 1c).
Figure 1. Cytosine methyltransferase DIM-2 can mediate RIP.
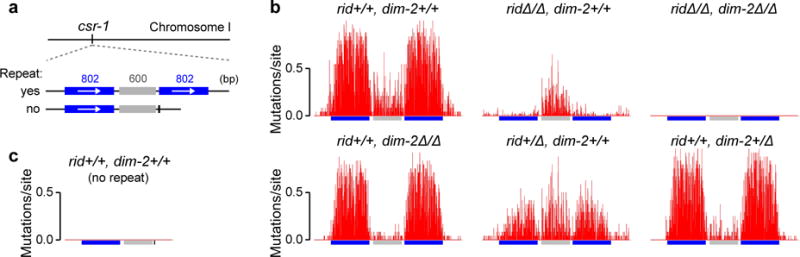
a, The 802-bp tester construct contains one endogenous (“left”) and one ectopic (“right”) copy of an 802-bp region of the Neurospora genome (ref. 18). The “no-repeat” construct specifically lacks the ectopic repeat copy.
b, RIP mutation profiles of the 802-bp construct. Top row (left to right): crosses X1{24}, X2{48} and X3{48}; bottom row (left to right): crosses X4{24}, X5{24} and X6{24}. The number of mutations is reported per site per spore. Cross X1 was published previously (ref. 18).
c, RIP does not occur in the absence of the ectopic repeat copy. Cross X7{48}.
The number of spores analyzed for each cross is provided in curly brackets. Strain and cross genotypes are provided in Supplementary Table 1 and 2, respectively.
In N. crassa, RIP has long been known to require a putative C5-cytosine methyltransferase RID (“RIP defective”)21. However, the genomes of several fungal species contain clear signatures of RIP-like mutation but do not encode any apparent homologs of RID, hinting at the possibility that RIP could be mediated by other factors22,23. In accord with this idea, we now find that our 802-bp tester construct can induce substantial mutation in the absence of RID (Fig. 1b: “ridΔ/Δ, dim-2+/+”). Intriguingly, while the expected (mean) number of mutations in the repeated regions is decreased by nearly two orders of magnitude in the ridΔ/Δ background, the linker region is still mutated at essentially the wildtype level (Supplementary Fig. 2a). The findings above (Fig. 1c) imply that all of these RID-independent mutations are induced by DNA homology.
RID-independent RIP is mediated by DIM-2
In addition to RID, N. crassa encodes another cytosine methyltransferase, DIM-2 (“Defective in methylation-2”)24, which catalyzes all known 5meC in the genome of this organism, including of newly-created repetitive transgenes8,11. We now find that all RID-independent RIP mutation of the 802-bp construct requires DIM-2: when both RID and DIM-2 are absent, RIP activity can no longer be detected (Fig. 1b: “ridΔ/Δ, dim-2Δ/Δ”). Furthermore, when DIM-2 is absent but RID is present, the pattern of effects is the reciprocal of that observed when DIM-2 is present but RID is absent (above): the number of mutations declines strongly (by the factor of 7.6) in the linker region and only moderately (by the factor of 1.3) in the repeated regions (Fig. 1b: “rid+/+, dim-2Δ/Δ”). Interestingly, both ridΔ and dim-2Δ appear haploinsufficient: when present in combination with a corresponding wildtype allele, each gene deletion decreases the number of mutations in the corresponding affected region(s) by the factor of 3 or more (Supplementary Fig. 2a).
The above findings show that: (i) RID and DIM-2 can each individually mediate RIP; (ii) RID and DIM-2 together account for all RIP; (iii) in the context of the 802-bp tester construct, RID-mediated mutation targets predominantly the repeated sequences, whereas DIM-2-mediated mutation targets predominantly the single-copy linker region; and (iv) the effects of RID and DIM-2 are additive. Taken together, these results suggest that the RIP processes mediated by RID and DIM-2, even though both triggered by the same DNA repeats, are nonetheless functionally distinct and, to a first approximation, independent of one another.
To confirm the distinct and complementary nature of the RID- and DIM-2-mediated pathways of RIP, we defined pair-wise correlations, on a per-spore basis, between the numbers of mutations occurring in different segments of the 802-bp tester construct (Supplementary Fig. 3). In situations where RID and DIM-2 activities are both strong, the total number of mutations in the left and in the right repeat copy of each individual spore clone are strongly correlated. That is, if one repeat copy exhibits a certain number of mutations, so does the other copy, on a per-spore basis. This pattern is expected if the two repeat copies are mutated by the same process. In contrast, the number of mutations in the linker region correlates less strongly with the number of mutations in either repeat copy, as expected if “linker” mutations and “repeat” mutations were mediated by two separate processes (i.e. mediated by DIM-2 and RID, respectively).
DIM-2-mediated RIP requires DIM-5
In vegetative cells of N. crassa, DIM-2 is recruited to DNA by Heterochromatin protein 1 (HP1), which recognizes H3K9me3 established by the SUV39 histone methyltransferase DIM-5 (Fig. 2a)15. The above observations raised the possibility that DIM-2-mediated mutation might also require these same factors. As one test of this possibility, we investigated the functional relevance of DIM-5 for RIP. Because female sexual development of N. crassa dim-5Δ strains is impaired in the presence of SET-7 (H3K37 methyltransferase encoded by the set-7 gene25,26) and because RIP occurs normally in the absence of SET-7 (Supplementary Fig. 4), the role of DIM-5 in RIP was tested using set-7Δ female strains (Supplementary Tables 1 and 2). We find that, when DIM-5 and RID are both absent, no mutation can be detected, even if DIM-2 is still available at the wildtype level (Fig. 2b). DIM-2-mediated mutation can be readily restored in the presence of a single functional dim-5+ allele (Fig. 2b).
Figure 2. DIM-2-mediated RIP requires the SUV39 histone methyltransferase DIM-5.
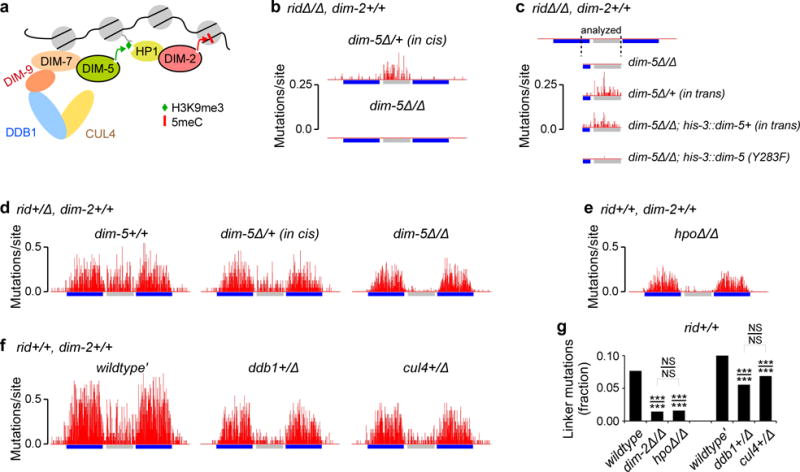
a, A cartoon representation of the canonical heterochromatin pathway in N. crassa (ref. 15).
b, RIP mutation profiles of the 802-bp construct. Crosses X8{60} (top) and X9{60} (bottom).
c, RIP mutation profiles of the 802-bp construct. Crosses X10{60}, X11{60}, X12{60} and X13{60} (top to bottom). Only the right-most portion of the endogenous repeat copy and the entire 600-bp segment of the linker region were sequenced.
d, RIP mutation profiles of the 802-bp construct. Crosses (left to right) X14{24}, X15{24} and X16{48}.
e, RIP mutation profile of the 802-bp construct. Cross X17{48}.
f, RIP mutation profiles of the 802-bp construct. Crosses (left to right) X18{24}, X19{24} and X20{48}.
g, A fraction of “linker” mutations reports the relative activity of DIM-2-mediated RIP. Each fraction value corresponds to the number of mutations in the 600-bp segment of the linker region normalized by the total number of mutations found in this 600-bp segment and in the repeated sequences. Analyzed crosses (left to right): X1, X4, X17, X18, X19, X20. The difference between any two fraction values is evaluated for significance using the chi-squared homogeneity test (P-value is indicated above the line) and the Fisher’s exact test (P-value is indicated below the line) on the actual mutation counts. *** P ≤ 0.001, NS P > 0.05. The number of spores analyzed for each cross is provided in curly brackets. Strain and cross genotypes are provided in Supplementary Table 1 and 2, respectively.
In the heterozygous dim-5Δ/+ cross analyzed above (Fig. 2b), the male parent provided the wildtype dim-5+ allele together with the 802-bp construct, whereas the female parent provided the null dim-5Δ allele. In such a configuration (where the dim-5+ allele and the tester construct were supplied “in cis”; Supplementary Fig. 1c), DIM-5 could, in principle, have acted on the repeat construct in vegetative cells, prior to fertilization and the onset of RIP. To investigate if the role of DIM-5 in RIP could be fulfilled after fertilization, we assayed the RIP-promoting activity of DIM-5 in a cross between a repeat-carrying maternal dim-5Δ strain and a repeat-less paternal strain that supplied the dim-5+ allele (i.e., the dim-5+ allele and the tester construct were provided “in trans”). We find that “cis” and “trans” configurations result in the comparable levels of RIP, regardless of whether the functional dim-5+ allele is present in the endogenous or in the ectopic location (Fig. 2c). These results imply that DIM-5 can act after fertilization. Furthermore, nearly all mutation observed in the “trans” configuration can be abolished by a single amino-acid change, Y283F, in DIM-5 that eliminates a catalytically important tyrosine residue27 (Fig. 2c). Moreover, the fact that the mutant DIM-5(Y283F) protein still exhibits very weak RIP-promoting activity in vivo corresponds to the fact that it also retains some weak catalytic activity in vitro27. Thus, DIM-5 appears to exert its role in RIP by its H3K9 methyltransferase activity.
We also examined the effect of decreasing DIM-5 levels in the presence of RID (Fig. 2d). The complete lack of DIM-5 confers a strong and specific phenotype in the linker region (a 24.3-fold decline) while having a much more modest impact on the repeated sequences (a 1.4-fold decline, similar to the 1.3-fold decline observed in repeats in the absence of DIM-2; Supplementary Fig. 2a,b). These findings provide further evidence that RID-mediated RIP and DIM-2-mediated RIP represent two largely independent processes that, nevertheless, respond to the same signal of DNA homology.
DIM-2-mediated RIP also requires HP1, CUL4 and DDB1
The finding that RIP could be mediated by the DIM-5/DIM-2 pathway raised the possibility that Neurospora HP1 (encoded by the hpo gene) might also be involved in RIP. In an attempt to test this idea, we found that the lack of HP1 precluded meiotic sporogenesis, even in the set-7Δ background that normally permitted sexual development in the absence of DIM-525,26. This phenotype likely reflects a broader requirement for HP1 during the sexual phase in N. crassa. However, we were able to find a combination of two hpoΔ strains that produced some limited amount of late-arising spores. One of these strains (C135.3, used as a female parent) lacked the type II topoisomerase-like protein SPO11, which catalyzes the formation of double-strand DNA breaks in meiosis and which is fully dispensable for RIP18,20. Our analysis shows that the apparent level of RIP mutation is strongly decreased in the hpoΔ/Δ background (Fig. 2e). However, (i) substantial mutation of the repeated sequences still occurs in the absence of HP1, and (ii) the relative levels of RIP in the linker region are diminished by the same amount in both hpoΔ/Δ and dim-2Δ/Δ crosses (Fig. 2g). Thus, we infer that HP1 is specifically involved in the DIM-5/DIM-2 pathway for RIP.
We next investigated the roles of two additional components of the Neurospora heterochromatin pathway: CUL4 (Cullin 4) and DDB1 (Damage specific DNA binding protein 1, also known as DNA damage-binding protein 1), which are are required for all DIM-5-mediated H3K9me3 in Neurospora vegetative cells15. We have found that the lack of either protein abrogates the normal development of perithecia (even when examined in various set-7Δ, spo11 backgrounds), thus precluding our standard analysis of RIP. However, instead, we were able to assay the role of CUL4 and DDB1 in the corresponding heterozygous conditions (Fig. 2f; Supplementary Fig. 2c). While the overall levels of RIP are decreased substantially in both situations, the pattern of changes is diagnostic of the specific defect in the DIM-5/DIM-2-mediated process, i.e. a stronger reduction of mutation in the linker region as compared to a more moderate reduction in the repeats (Fig. 2g). Taken together, the above results suggest that RIP can be mediated by a process that does not require RID, and in which DIM-5, CUL4/DDB1, HP1 and DIM-2 act in one pathway.
DIM-2-mediated RIP responds to weak interspersed homology
The discovery of a heterochromatin-related pathway of RIP now raises the issue of the precise nature of the homology requirements for this process. Our previous results provided no indication that the canonical recombination-mediated mechanism of homology recognition (mediated by MEI-3) played a role in either the RID- or the DIM-2-mediated pathways18. We have also previously shown that in the wildtype situation, when both pathways are active, RIP can respond to the presence of weak interspersed homology, as long as the latter comprises short islands of homology (≥3 base-pairs) spaced at regular intervals of 11 (or 12) base-pairs along a pair of participating DNA segments18. This and other results permitted a model in which homology recognition for RIP involved direct interactions between co-aligned DNA duplexes18. One particular series of experiments that led to this conclusion utilized repeat constructs in which 200-bp regions of interspersed homology were adjacent to a 220-bp region of perfect homology (Fig. 3a,b). These direct “200+220” repeats were linked by a single-copy region of 537 base-pairs. The 220-bp region of perfect homology was incorporated to provide a stable, permanent point of interaction, thereby facilitating the detection of weak effects induced by interspersed homologies. In this context, different homology patterns could be created by manipulating only the “right” 200-bp segment while leaving the rest of the construct unchanged.
Figure 3. DIM-2-mediated RIP responds to weak interspersed homology.
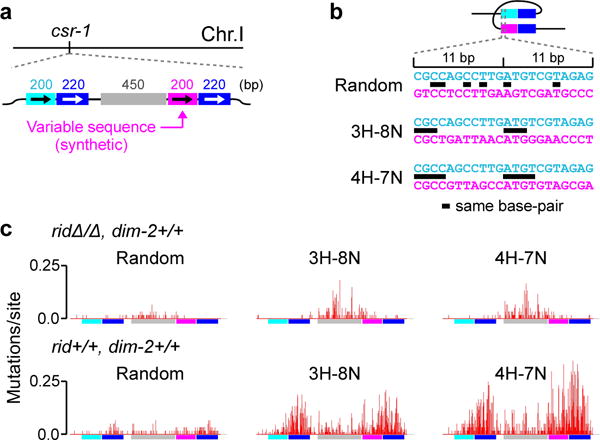
a, Different homology patterns are created by only varying the base-pair sequence of the magenta region (as described in ref. 18).
b, The assayed patterns of weak interspersed homology contain short units of three (“3H-8N”) and four (“4H-7N”) homologous base-pairs spaced at regular intervals of 11 base-pairs. An instance of “Random” homology corresponds to a 200-bp fragment of the GFP coding sequence.
c, RIP mutation profiles. Top row (left to right): crosses X21{60}, X22{60} and X23{60}; bottom row (left to right): crosses X24{60}, X25{90} and X26{60} (as reported previously in ref. 18).
We have now used this same repeat system to address specifically the homology requirements for DIM-2-mediated RIP. We first re-analyzed our published data18 (obtained in the rid+/+, dim-2+/+ background) with respect to mutation of the 450-bp segment of the linker region expected to differentially report the effects of the DIM-5/DIM-2-meditated process (Fig. 3a). The analyzed instances of interspersed homology involve homologous units of 3 or 4 base-pairs spaced at regular intervals of 11 base-pairs (Fig. 3b). We have found that the presence of these weak interspersed homologies corresponds to a significant increase in RIP mutation throughout the 450-bp linker segment (Fig. 3c; Supplementary Fig. 2d). We then asked whether these same homology patterns could promote RIP in the absence of RID, when only the DIM-5/DIM-2 pathway is active. Here we find again that RIP in the linker segment is increased significantly in the presence of the assayed interspersed homologies (Fig. 3c; Supplementary Fig. 2d). Moreover, the levels of mutation in the linker segment induced in the presence and in the absence of RID by any given homology pattern appear similar, suggesting that the majority of the previously18 reported “linker” mutations were mediated by DIM-2. Taken together these findings suggest that the heterochromatin-related pathway of RIP can respond to weak interspersed homology.
DIM-2-mediated RIP targets tandem repeat arrays
In all repeat constructs analyzed thus far, DIM-2-mediated RIP occurred mostly in the single-copy linker region between the repeated sequences, suggesting that the presence of relatively short direct repeats can direct this heterochromatin-related process to the adjoining single-copy region. To further explore this idea in a different context, we analyzed a construct containing four copies of the dsRed coding sequence (the “4x” array) inserted near the his-3 gene (Fig. 4a). As a baseline, we first examined mutation of the 4x array by RID-mediated RIP alone, in the absence of DIM-2. We find that all four repeat copies are strongly mutated, yet no spreading of mutations into the adjoining single-copy regions has occurred (Fig. 4b). We then examined mutation of the 4x array by the DIM-5/DIM-2 pathway, in the absence of RID. In a dramatic contrast, DIM-5/DIM-2-dependent mutations occur at comparable levels in the repeated and the adjoining non-repetitive regions (Fig. 4b). Taken together, these results suggest that (i) tandem repeat arrays represent a very potent homology trigger of RIP; and (ii) the DIM-5/DIM-2 pathway targets not only the repeated sequences (which are nevertheless essential for activating this pathway), but also the adjoining non-repetitive regions.
Figure 4. Recognition of short tandem repeat arrays by the DIM-5/DIM-2 pathway.
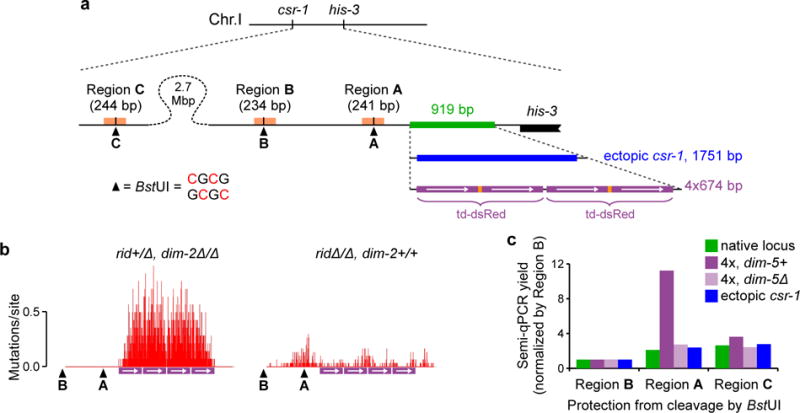
a, Four copies of the dsRed coding sequence (the “4x” array) are inserted by homologous recombination as the replacement of an endogenous genomic segment (green). The ectopic csr-1 copy is inserted analogously. All DNA segments are drawn to the same scale. The restriction site 5′-CGCG-3′ can only be cut by BstUI if none of its four cytosines is methylated (ref. 29). The distal BstUI site “C” is located near the csr-1 gene.
b, RIP mutation profiles of the 4x array. Crosses X27{14} (left) and X28{30} (right). The mean number of mutations in the entire sequenced region is 174.93± 30.84 and 15.20±1.69 for X26 and X27, respectively. Positions of the two proximal BstUI restriction sites (“A” and “B”, as shown in a) are indicated.
c, The assay detects the failure of cleavage by BstUI at the three specified sites (“A”, “B” and “C”, as shown in a). PCR yields for Regions A and C are normalized by the corresponding PCR yield for Region B. Unnormalized PCR yields are also provided in Supplementary Fig. 6b. The analyzed repeat constructs were never subjected to RIP. Strains: FGSC#9720 (native locus), T485.4h (4x, dim-5+), T486.3h (4x, dim-5Δ) and T402.1h (ectopic csr-1).
DIM-2-mediated RIP can target widely-separated repeats
All of the above analyses examined DIM-2-mediated RIP as triggered by closely-positioned repeats. It was of interest to determine whether the heterochromatin-related pathway of RIP could also mutate DNA repeats at widely-separated genomic positions. To address this question, we designed a sensitive genetic system that can detect RIP between a single pair of homologous sequences located 2.7 Mbp apart on the same chromosome (Fig. 5a; also ref. 20). The csr-1 gene encodes a cyclophilin protein that has a high affinity for cyclosporin A, and the presence of a single active copy of csr-1 is sufficient to render a Neurospora strain cyclosporin-sensitive28. Thus, if two csr-1 copies are present, one of which is active and the other is not, RIP mutation of the active copy (induced by the ectopic inactive copy) will generate cross progeny resistant to cyclosporin (Fig. 5b). We find that in the absence of the ectopic copy, no cyclosporin-resistant progeny could be detected among nearly 5×105 spores produced by a cross between two wildtype strains, implying that the frequency of spontaneous mutation of the csr-1 gene is extremely low. In contrast, in the presence of the ectopic copy, a large proportion of cross progeny becomes cyclosporin-resistant (Fig. 5c). This proportion decreases by nearly four orders of magnitude in the absence of both RID and DIM-2. All rare cyclosporin-resistant progeny that still emerge in the ridΔ/Δ, dim-2Δ/Δ condition carry the same G-to-A mutation that is already present in the parental ectopic copy. Thus, these rare events can likely be attributed to gene conversion between the endogenous and the ectopic copies. Finally, when DIM-2 is present and RID is absent, cyclosporin-resistant progeny are produced at a level that is one order of magnitude above the gene-conversion background. These progeny contain only C-to-T and G-to-A mutations at the csr-1 locus, confirming that these csr-1 alleles were produced by RIP (Fig. 5d). Similarly to the pattern of mutations at closely-positioned repeats (above), RID-mediated mutations at widely-separated repeats remain confined to the duplicated region, whereas DIM-2-mediated mutations tend to spread into the adjoining single-copy regions (Fig. 5d; Supplementary Fig. 5). An additional interesting difference between the two mutational pathways is the even stronger preference for CpA sites associated with DIM-2-mediated RIP (Fig. 5e). Taken together, these results demonstrate the capacity of the heterochromatin-related pathway to mediate mutation of DNA repeats that are separated by large genomic distances.
Figure 5. DIM-2-mediated RIP can target repeats at widely separated genomic positions.
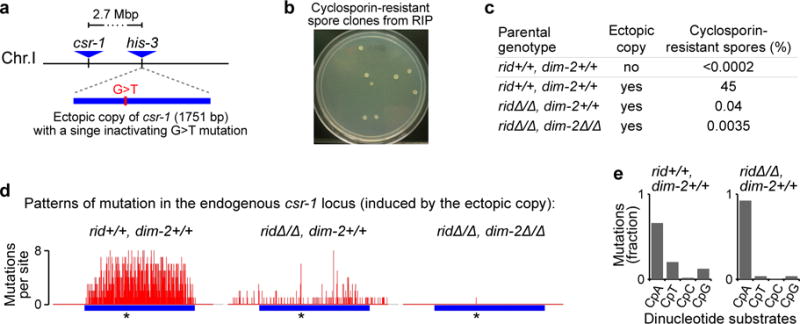
a, A pair of widely-separated repeats of the csr-1 gene: the inactive ectopic copy of csr-1 is inserted near the his-3 gene, 2.7 million base-pairs away from the endogenous csr-1 locus.
b, Inactivation of the endogenous csr-1 gene by RIP produces cyclosporin-resistant colonies originating from individual progeny spores.
c, The percentage of cyclosporin-resistant spores produced in different genetic backgrounds. Because the ectopic repeat is present in only one of the two parental strains, the maximum expected level of cyclosporin-resistant progeny is 50 per cent.
d, Patterns of RIP mutation in the endogenous csr-1 locus induced in the presence of the ectopic csr-1 copy (integrated near the his-3 gene). The per-site number of mutations is reported for sets of unique csr-1 alleles. “*” denotes the position of the existing G-to-A mutation in the ectopic csr-1 copy. The number of replica crosses, the total number of sequenced cyclosporin-resistant progeny, and the number of unique csr-1 alleles were as follows: X29 - 1 cross, 10 sequenced spores, 10 unique alleles; X30 - 9 crosses, 70 sequences spores, 34 unique alleles; X31 - 3 crosses, 28 sequenced spores, 1 unique allele. A cluster of 20 cyclosporin-resistant progeny spores was likely produced in the ridΔ/Δ, dim-2Δ/Δ background by a single gene-conversion event that have occurred early in the pre-meiotic lineage undergoing RIP.
e, Dinucleotide (CpN) context of RIP mutations.
Short repeat arrays induce transient somatic 5meC
The above results demonstrate that relatively short DNA repeats can trigger RIP by the heterochromatin-related pathway. Our previous studies18–20, also extended above, suggest that recognition of repeats for this pathway is mediated by direct homologous dsDNA/dsDNA interactions. Taken together, these findings raise the possibility that such dsDNA/dsDNA interactions may be involved in the formation of heterochromatin in vegetative cells of N. crassa, but result in cytosine methylation instead of mutation.
The most straightforward test of this idea involves asking whether, in vegetative cells, the DIM-5/DIM-2 pathway can mediate 5meC at short repeat arrays that also trigger DIM-2-mediated RIP in pre-meiotic cells (above). Stable heterochromatin formation on such short repeat arrays was not detected by earlier studies in vegetatively growing N. crassa9. However, previous approaches used bulk assays, e.g. Southern blots, which may not be sensitive enough to detect the presence of transient epigenetic effects associated with only a few repeat units. We, therefore, developed a sensitive PCR-based assay for measuring 5meC at a diagnostic restriction site, 5′-CGCG-3′, that can be cleaved by BstUI only if none of its four cytosines is methylated29. This specific site is located just to the “left” side of the integration position of the 4x array, in a single-copy genomic region that shows strong DIM-5/DIM-2 dependent RIP (Fig. 4a,b: site “A”).
We have analyzed 5meC at the BstUI site “A” under several conditions using two additional sites (“B” and “C”), which were not expected to undergo heterochromatinization, as normalization controls. We find that cleavage of the site “A” is partially inhibited when the 4x array is present, as indicated by the enrichment of the corresponding PCR product (Fig. 4c; raw data are provided in Supplementary Fig. 6). Importantly, this enrichment effect does not occur in the absence of DIM-5 or if a copy of the csr-1 gene is integrated instead of the 4x array. The level of the diagnostic PCR signal induced by the 4x array remains low and corresponds to the 5meC frequency of ~2–3%. We conclude that our assay is detecting rare (and thus likely transient) events of the repeat-induced heterochromatin formation in vegetative cells of N. crassa. These results have two important implications. First, they support the idea that transient local events involving small numbers of repeat copies may underlie the formation of stable heterochromatin on long repeat arrays. Second, they suggest that the rules for repeat recognition during RIP, e.g. pairwise interactions between co-aligned homologous dsDNA segments, could, analogously, underlie repeat recognition for heterochromatin assembly in vegetative/somatic cells.
A further question arises as to why our model 4x array elicits a robust RIP signal in pre-meiotic cells but only a weak 5meC signal, diagnostic of unstable heterochromatic marks, in vegetative cells. This difference likely reflects the combined effects of two factors. First, during RIP, a transient pairing interaction involving a small number of repeat copies produces a permanent record in the form of C-to-T mutation. Second, each pre-meiotic nucleus normally undergoes as many as 6–7 rounds of RIP over the course of several days. As a result, RIP mutations that we analyze represent an integrated form of many transient heterochromatinization events.
DISCUSSION
Current models of heterochromatin assembly on repetitive DNA invoke either cis-acting proteins (which recognize specific DNA sequence motifs) or RNAs that associate with newly-transcribed RNA at repetitive loci1,3,6. Yet the apparent connection between the repetitive nature of chromatin and its heterochromatic status was noted by Guido Pontecorvo as early as 1944 (ref. 30) and elaborated further by Douglas Dorer and Steven Henikoff in 1994 (ref. 31), with the suggestion that the presence of repetitive DNA per se was detected and served as a local signal for heterochromatin assembly. Based on our current findings, we propose that pairwise dsDNA/dsDNA interactions involving a small number of repeat units can provide such a signal (Fig. 6). Stable heterochromatin on long repeat arrays could then arise via multiple nucleation events of this type as well as by extending dsDNA/dsDNA interactions outward from nucleation site(s). Given the pairwise nature of repeat recognition, the number of potential nucleation events will scale more-or-less exponentially with the number of repeat copies, thus permitting an effective way of nucleating and extending heterochromatin over very large repetitive regions.
Figure 6. A general model for nucleation of heterochromatin by pairwise dsDNA/dsDNA interactions.
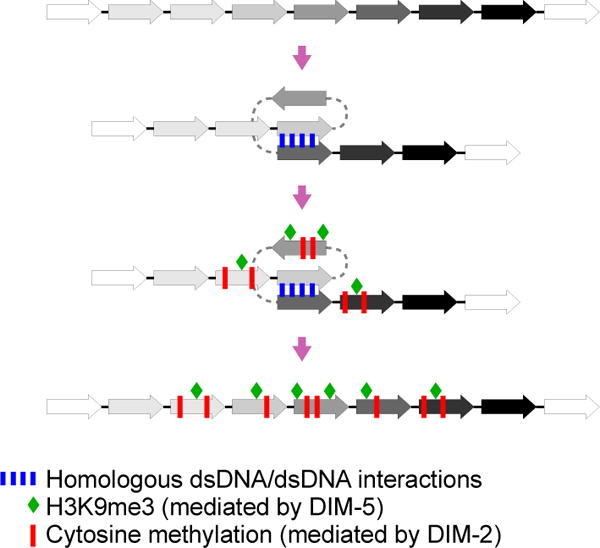
In vegetative/somatic cells, a direct homologous interaction between two co-aligned copies of a repeated DNA sequence induces a localized deposition of H3K9me3 (green diamonds) and 5meC (red bars). Such heterochromatin nucleation sites are intrinsically unstable. However, on longer repeat arrays, multiple concomitant nucleation events may be stabilized to yield constitutive heterochromatin. During the pre-meiotic phenomenon of RIP, the same transient localized interactions result in permanent C-to-T mutations, instead of 5meC. These mutations accumulate during several cell cycles over a period of several days to give a high level of RIP in analyzed spores.
The discovery that RIP required RID21 created a conundrum: RIP involves cytosine-to-thymine mutation, whereas RID is a putative C5-cytosine methyltransferase20,21. It was originally proposed that RIP might occur as a two-step process, in which cytosine methylation by RID was followed by deamination of 5meC by another enzymatic activity (discussed in ref. 21). This idea was supported by the fact that pre-meiotic recognition of repeats induces 5meC without mutation in another fungus Ascobolus immersus17,32. An alternative proposal was also put forward by which RID alone could mediate mutation, via modulation of its catalytic activity21. Our current findings reveal that RIP can be mediated by the canonical cytosine methyltransferase DIM-2. In vegetative cells of N. crassa, DIM-2 catalyzes all known cytosine methylation, without obvious deamination. Interestingly, DIM-2 methylates cytosines in all dinucleotide contexts33, whereas DIM-2-mediated mutation exhibits a very strong preference for CpA dinucleotides (Fig. 5e). These findings are fully compatible with a two-step mechanism. By this hypothesis, repeat recognition would trigger 5meC (by either DIM-2 or RID) that would be converted to mutation by a DNA deaminase that is active specifically during the pre-meiotic phase. It can be noted, however, that in vegetative cells, DIM-2 can also methylate non-repetitive parts of the genome34. If that same process takes place in pre-meiotic nuclei, then a two-step mechanism would further require the deamination step to be dependent on the presence of DNA homology as well. A one-step mechanism involving pre-meiotic modulation of the catalytic activity of DIM-2 would not have such a requirement.
Finally, we note that the characteristic spread of DIM-5/DIM-2-mediated genetic and epigenetic modifications from repeats into the neighboring non-repetitive genomic regions implicates this homology-directed pathway in transcriptional silencing and accelerated evolution of pathogenic genes that are often found in clusters with repetitive elements in the genomes of many filamentous fungi23,35.
ONLINE METHODS
Plasmids
Plasmids pEAG66, pEAG115A, pEAG186B, pEAG186K and pEAG186L are based on the plasmid pCSR136. Plasmids pEAG82B (inactive copy of the csr-1 gene), pEAG236A (the “4x” construct), pEAG244B (active copy of the dim-5 gene) and pEAG244G (inactive copy of the dim-5 gene) are based on the plasmid pMF28037 and pMF33438. Annotated maps of pEAG66, pEAG115A, pEAG186B, pEAG186K and pEAG186L were published previously18. Annotated maps of pEAG82B, pEAG236A, pEAG244B and pEAG244G are provided in Supplementary Data Set 1 (in GenBank format, individual plain-text files are compressed with tar/gz).
Manipulation of Neurospora strains
All strains used in this study are listed in Supplementary Table 1. All FGSC# strains were obtained from the Fungal Genetics Stock Center39. Transformation of the csr-1 locus using linearized plasmids pEAG66, pEAG115A, pEAG186B, pEAG186K and pEAG186L was carried out as described previously18. Transformation of the his-3 locus using linearized plasmids pEAG82B, pEAG236A, pEAG244B and pEAG244G was done analogously, by electroporation of macroconidia. Homokaryotic his-3+ strains were isolated from primary heterokaryotic transformants by macroconidiation. Crosses were setup on agar slants in capped culture tubes (15 mm × 125 mm) as described previously18. 0.25 mg/ml histidine was added to the crossing medium only if required by the female parent. All crosses analyzed in this study are listed in Supplementary Table 2.
Analysis of the cyclosporin-resistant phenotype
This assay was designed to detect very low levels of RIP activity involving two widely-separated repeats of the csr-1 gene20,28. A copy of the csr-1 gene (including the promoter region) was integrated into the his-3 locus, 2.7 million base-pairs away from the endogenous csr-1. The ectopic copy carries a single G-to-A mutation in the 5′ splice site, which confers resistance to cyclosporin. Thus, inactivation of the endogenous csr-1 allele was sufficient to confer a cyclosporin-resistant phenotype. All ejected spores (both early- and late-arising) were collected into 1 ml of distilled water. Spores were heat-shocked for 30 minutes at 60°C to induce germination. To estimate the total number of viable spores, 10 μl of the original 1-ml suspension were diluted 100-fold (back to 1 ml) and 50 μl of that 1:100 dilution were plated on non-selective sorbose agar solidified in standard Petri dishes (100 mm × 15 mm). 3 replica dishes were plated in total, consuming 3×50 μl of the 1:100 dilution. The number of viable spores was estimated as the average number of colonies per dish multiplied by the overall dilution factor. To estimate the number of cyclosporin-resistant progeny in crosses with strong RIP, additional 3×50 μl from the same 1:100 dilution were plated in triplicate on sorbose agar containing cyclosporin A (5 μg/ml; Sigma cat. no. 30024-25MG). The total number of cyclosporin-resistant progeny was estimated by the same formula. In cases of weak RIP (expected in the absence of RID or in the absence of the ectopic csr-1 copy), the remaining 0.99 ml of the original 1:1 suspension were plated directly on selective medium, and the number of cyclosporin-resistant progeny was determined by the total count of growing colonies.
Sampling RIP products for sequence analysis
RIP is likely activated in haploid parental nuclei that become assorted into dikaryotic ascogenous cells40. Approximately 6–7 rounds of RIP may occur as these pre-meiotic nuclei continue to divide by mitosis. After the nuclei undergo karyogamy and meiosis, the haploid meiotic products are packaged into progeny spores and ejected from the mature perithecium (the fruiting body). RIP mutation tends to be intrinsically weak in the “early-arising” spores (ejected within the first 1–3 days after the onset of sporogenesis) but then increases in “late-arising” spores (ejected over the next 7–10 days). 100–200 spore-producing perithecia can normally develop in a single test tube under our experimental conditions18. Each perithecium represents an autonomous anatomical structure; it hosts several independent lineages of ascogenous cells and ultimately produces several hundred progeny spores. All ejected spores correspond to one statistical population. Late-arising spores are sampled without any knowledge of their RIP status. Because the number of sampled spores (24–60 from each cross) is much smaller than the estimated number of ascogenous lineages, each spore effectively represents an independent measure of RIP activity. Despite the apparent spore-to-spore variability in RIP levels, our studies has indicated that the expected number of RIP mutations (the arithmetic mean) represents an accurate and useful measure of RIP activity18,19.
Genomic DNA extraction, PCR amplification, and sequencing
Genomic DNA was extracted from individual spore clones as described previously18. PCR products were purified using Omega Bio-tek kit and sequenced directly by primer walking. The following primers were used for PCR and sequencing (primers are provided in Supplementary Table 3): The 802-bp construct was amplified with primers P66_Seq3 and RIP2_R1; sequenced with primers P66_Seq1, P66_Seq3, LNK_SeqR1 and RIP2_R2. All (220+200)-bp constructs were amplified with primers P66_Seq3 and RIP2_R1; sequenced with primers P66_Seq17 and P66_Seq18. The entire 4x construct was amplified with primers NcHis3_R6 and NcHis3_F7; sequenced with primers NcHis3_F4, NcHis3_F7, NcHis3_R1, NcHis3_R4, NcHis3_R6, P236A_Seq1, P236A_Seq2, P236A_Seq4, P236A_Seq5 and P236A_Seq8. If the entire 4x construct could not be amplified as a single fragment, piece-wise applications were carried out using pair-wise combinations of the above sequencing primers. The endogenous csr-1 gene was amplified with primers P66_Seq10 and RIP2_R1; sequenced with primers P66_Seq10, P66_Seq12, CSR1_SeqF, CSR1_SeqR and RIP2_R1. Eight progeny spores with particularly strong levels of DIM-2-mediated mutation were selected for further sequence analysis of the adjoining genomic regions. For these eight clones, the extended 5.4-kbp region containing the endogenous csr-1 gene was amplified with primers P66_Seq3 and RIP2_R3; and sequenced with additional primers P66_Seq3, P66_Seq9, P66_Seq4, CSR1_SeqR2, CSR1_SeqR3 and RIP2_R3. If a primer site appeared affected by RIP, additional ad hoc primers were used instead. Sequencing reactions were read with an ABI3730xl DNA analyzer at the DNA Resource Core of Dana-Farber/Harvard Cancer Center (funded in part by NCI Cancer Center support grant 2P30CA006516-48). Individual chromatograms were assembled into contigs with Phred/Phrap. All assembled contigs were validated manually using Consed.
Sequence and statistical analysis
For each cross, assembled contigs were aligned with the parental (reference) sequence using ClustalW. All alignments analyzed in this study are provided in Supplementary Data Set 2 in ClustalW format, individual plain-text files are compressed with tar/gz. Mutations were detected and analyzed as described previously18. The absolute level of RIP in a given region of interest was expressed (i) as the expected number of mutations (the arithmetic mean) and, if necessary, (ii) as the percentage of mutated strands, where all C-to-T mutations were considered to be on the top strand and all G-to-A mutations - on the bottom strand18. Standard error of the mean (s.e.m.) was used as a measure of variation. The following regions were analyzed for the 802-bp construct: the endogenous “left” repeat copy (positions 348–1149), the 600-bp segment of the linker (positions 1227–1826), and the ectopic “right” repeat copy (positions 1879–2680). The following region was analyzed for the (220+200)-bp construct: the 450-bp segment of the linker region (positions 622–1071). Empirical distributions of mutation counts (i.e., the numbers of C-to-T and G-to-A mutations found together within a given region on per-spore basis) were compared by the Kolmogorov-Smirnov test18. The pair-wise differences in the percentage of mutated strands and the proportion of linker mutations were compared for significance as the original raw counts by (i) Pearson’s Chi-squared homogeneity test with Yates’ continuity correction and (ii) Fisher’s exact test (as implemented in R).
Analysis of cytosine methylation in vegetative cells of N. crassa
I. Purification of genomic DNA
Crude preparations of genomic DNA were obtained by the method used for extracting DNA for RIP analysis (above; ref. 18), except that phenol/chloroform was replaced with chloroform in all purification steps. DNA was precipitated with isopropanol, washed with ethanol, resuspended in 80 μl of water and mixed with 20 μl of the 5x loading buffer (20% v/v glycerol, 0.25% SDS, 5 mM EDTA, 1 mg/ml xylene cyanol). The entire sample (100 μl) was loaded into a single well of a 0.75% agarose mini-gel (NuSieve GTG Agarose, cat. no. 50080) containing ethidium bromide. The gel was run at 3 V/cm in 1x TAE (Tris-Acetate-EDTA) buffer for 1.5 hours. A region of the gel containing high-molecular weight DNA was excised with a sterile razor blade under long UV light and digested with β-Agarase I (NEB, cat. no. M0392L; 3 hours at 42°C). Following a single extraction with chloroform, genomic DNA was precipitated with ammonium acetate/ethanol, washed with ethanol, and resuspended in 50 μl of water. DNA concentrations were measured using NanoDrop 2000. A typical procedure yielded 0.5–1.0 μg of purified DNA (per one mycelial sample grown overnight in 2 ml of 1x Vogel’s culture medium).
II. Restriction digest with BstUI
BstUI recognition sequence (5′-CGCG-3′) contains four cytosines that can be potentially methylated by DIM-2 (two cytosines on the top strand, and two cytosines on the bottom strand); and methylation of any one (or more) of these cytosines protects the site from cleavage by BstUI29. Each restriction reaction was setup in the total volume of 50 μl, using 0.2 μg of purified genomic DNA from the previous step and 4 μl of BstUI (10 U/μl; NEB, cat. no. R0518S). Reactions were incubated for 3 hours at 50°C. BstUI was inactivated by Proteinase K (by adding 2 μl of Proteinase solution, 20 mg/ml, and incubating at 50°C for additional 1.5 hours). Proteinase K was heat-inactivated (95°C for 10 min) and the volume of each sample was adjusted to 200 μl (by adding 148 μl of water). Mock-digested samples were processed in exactly the same way, except that 4 μl of 50% glycerol were added to each reaction instead of BstUI.
III. Semi-quantitative PCR
Each PCR was run in the total volume of 10 μl. Three genomic regions were amplified separately for each DNA sample: Region A (primers REG_A_F1 and REG_A_R1), Region B (primers REG_B_F1 and REG_B_R1) and Region C (primers REG_C_F1 and REG_C_R1). DNA sample concentrations were adjusted (by no more than a factor of 1.5) to produce comparable PCR yields of Region B. No additional adjustments were implemented. Each DNA sample (corresponding to 20–36 μl of the 200-μl product from the previous step) was used to make one Principal Master Mix (PMM) for 18 individual PCRs. Each PMM also received 18 μl of the 10x reaction buffer (EconoTaq DNA Polymerase, Lucigen, cat. no. F93366-1), 7.2 μl of the dNTP solution (NEB, cat. no. N0447S), and 5.4 μl of EconoTaq polymerase (in the total volume of 162 μl). Each PMM was split equally into three 54-μl Region-Specific Master Mixes (RSMMs). Each RSMM received 6 μl of a primer solution (containing one pair of primers corresponding to a given PCR region, at 5 pmol/μl each), bringing its total volume to 60 μl. Each RSMM was then split into six individual 10-μl reactions.
All PCRs were run in a C1000 Touch thermal cycler (Bio-Rad) using the following program: 94°C-2′, {94°C-20″; 59°C-20″; 72°C-20″} × N, 72°C-2′. For all digested DNA samples, the value of N (the number of amplification cycles) was set to 27. For all mock-digested samples, the value of N was set to 22. The number of cycles was determined empirically to ensure that all PCRs remained in the exponential regime. Each PCR product was mixed with 2 μl of the 5x buffer (above) and loaded on a 2% regular agarose gel in the total volume of 12 μl. All PCRs corresponding to a given DNA sample were analyzed side-by-side on the same gel. Gels were run for 30 min in 0.5x TAE at 6 V/cm and scanned with the resolution of 50 microns using Molecular Imager FX (Bio-Rad). Luminosities of PCR products were measured in ImageJ (with the “Analyze → Measure” tool) using fixed region-of-interest parameters. A constant value corresponding to the image background (estimated separately based on five independent measurements) was subtracted from a raw luminosity value of each PCR band. The relative number of uncleavable BstUI sites in the digested DNA sample (in Region A) was estimated using a calibration curve based on a series of two-fold dilutions of the corresponding mock-digested DNA sample.
Supplementary Material
Acknowledgments
We thank Denise Zickler for critically reading the manuscript. This work was supported by the grant GM044794 from the National Institutes of Health to N.K. and research fellowships from The Helen Hay Whitney Foundation, The Howard Hughes Medical Institute and Charles A. King Trust to E.G.
Footnotes
DATA AVAILABILITY STATEMENT
All data generated or analysed during this study are included in this published article and its supplementary information files.
AUTHOR CONTRIBUTIONS
E.G. designed, performed and analyzed all experiments. E.G and N.K. wrote the manuscript.
COMPETING FINANCIAL INTERESTS STATEMENT
The authors declare no competing financial interests.
References
- 1.Almouzni G, Probst AV. Heterochromatin maintenance and establishment: lessons from the mouse pericentromere. Nucleus. 2011;2:332–338. doi: 10.4161/nucl.2.5.17707. [DOI] [PubMed] [Google Scholar]
- 2.Becker JS, Nicetto D, Zaret KS. H3K9me3-dependent heterochromatin: barrier to cell fate changes. Trends Genet. 2016;32:29–41. doi: 10.1016/j.tig.2015.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Martienssen R, Moazed D. RNAi and heterochromatin assembly. Cold Spring Harb Perspect Biol. 2015;7:a019323. doi: 10.1101/cshperspect.a019323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Padeken J, Zeller P, Gasser SM. Repeat DNA in genome organization and stability. Curr Opin Genet Dev. 2015;31:12–19. doi: 10.1016/j.gde.2015.03.009. [DOI] [PubMed] [Google Scholar]
- 5.Plohl M, Meštrović N, Mravinac B. Centromere identity from the DNA point of view. Chromosoma. 2014;123:313–325. doi: 10.1007/s00412-014-0462-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Saksouk N, Simboeck E, Déjardin J. Constitutive heterochromatin formation and transcription in mammals. Epigenetics Chromatin. 2015;8:3. doi: 10.1186/1756-8935-8-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Aramayo R, Selker EU. Neurospora crassa, a model system for epigenetics research. Cold Spring Harb Perspect Biol. 2013;5:a017921. doi: 10.1101/cshperspect.a017921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bull JH, Wootton JC. Heavily methylated amplified DNA in transformants of Neurospora crassa. Nature. 1984;310:701–704. doi: 10.1038/310701a0. [DOI] [PubMed] [Google Scholar]
- 9.Selker EU, Garrett PW. DNA sequence duplications trigger gene inactivation in Neurospora crassa. Proc Natl Acad Sci U S A. 1988;85:6870–6874. doi: 10.1073/pnas.85.18.6870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Freitag M, et al. DNA methylation is independent of RNA interference in Neurospora. Science. 2004;304:1939. doi: 10.1126/science.1099709. [DOI] [PubMed] [Google Scholar]
- 11.Chicas A, Forrest EC, Sepich S, Cogoni C, Macino G. Small interfering RNAs that trigger posttranscriptional gene silencing are not required for the histone H3 Lys9 methylation necessary for transgenic tandem repeat stabilization in Neurospora crassa. Mol Cell Biol. 2005;25:3793–3801. doi: 10.1128/MCB.25.9.3793-3801.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Garrick D, Fiering S, Martin DI, Whitelaw E. Repeat-induced gene silencing in mammals. Nat Genet. 1998;18:56–59. doi: 10.1038/ng0198-56. [DOI] [PubMed] [Google Scholar]
- 13.Preis JI, Downes M, Oates NA, Rasko JE, Whitelaw E. Sensitive flow cytometric analysis reveals a novel type of parent-of-origin effect in the mouse genome. Curr Biol. 2003;13:955–959. doi: 10.1016/s0960-9822(03)00335-x. [DOI] [PubMed] [Google Scholar]
- 14.van der Maarel SM, Tawil R, Tapscott SJ. Facioscapulohumeral muscular dystrophy and DUX4: breaking the silence. Trends Mol Med. 2011;17:252–258. doi: 10.1016/j.molmed.2011.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Galagan JE, Selker EU. RIP: the evolutionary cost of genome defense. Trends Genet. 2004;20:417–423. doi: 10.1016/j.tig.2004.07.007. [DOI] [PubMed] [Google Scholar]
- 16.Hane J, et al. Repeat-Induced Point mutation: a fungal-specific, endogenous mutagenesis process. Genetic Transformation Systems in Fungi. 2015;2:55–68. doi: 10.1007/978-3-319-10503-1_4. [DOI] [Google Scholar]
- 17.Rossignol JL, Faugeron G. Gene inactivation triggered by recognition between DNA repeats. Experientia. 1994;50:307–317. doi: 10.1007/BF01924014. [DOI] [PubMed] [Google Scholar]
- 18.Gladyshev E, Kleckner N. Direct recognition of homology between double helices of DNA in Neurospora crassa. Nat Commun. 2014;5:3509. doi: 10.1038/ncomms4509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gladyshev E, Kleckner N. Recombination-independent recognition of DNA homology for Repeat-Induced Point mutation (RIP) is modulated by the underlying nucleotide sequence. PLoS Genet. 2016;12:e1006015. doi: 10.1371/journal.pgen.1006015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gladyshev E, Kleckner N. Recombination-independent recognition of DNA homology for repeat-induced point mutation. Curr Genet. 2016 doi: 10.1007/s00294-016-0649-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Freitag M, Williams RL, Kothe GO, Selker EU. A cytosine methyltransferase homologue is essential for Repeat-Induced Point mutation in Neurospora crassa. Proc Natl Acad Sci U S A. 2002;99:8802–8807. doi: 10.1073/pnas.132212899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Clutterbuck AJ. Genomic evidence of repeat-induced point mutation (RIP) in filamentous ascomycetes. Fungal Genet Biol. 2011;48:306–326. doi: 10.1016/j.fgb.2010.09.002. [DOI] [PubMed] [Google Scholar]
- 23.Amselem J, Lebrun MH, Quesneville H. Whole genome comparative analysis of transposable elements provides new insight into mechanisms of their inactivation in fungal genomes. BMC Genomics. 2015;16:141. doi: 10.1186/s12864-015-1347-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kouzminova E, Selker EU. dim-2 encodes a DNA methyltransferase responsible for all known cytosine methylation in Neurospora. EMBO J. 2001;20:4309–4323. doi: 10.1093/emboj/20.15.4309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jamieson K, et al. Loss of HP1 causes depletion of H3K27me3 from facultative heterochromatin and gain of H3K27me2 at constitutive heterochromatin. Genome Res. 2016;26:97–107. doi: 10.1101/gr.194555.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Basenko EY, et al. Genome-wide redistribution of H3K27me3 is linked to genotoxic stress and defective growth. Proc Natl Acad Sci U S A. 2015;112:E6339–6348. doi: 10.1073/pnas.1511377112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang X, et al. Structure of the Neurospora SET domain protein DIM-5, a histone H3 lysine methyltransferase. Cell. 2002;111:117–127. doi: 10.1016/s0092-8674(02)00999-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tropschug M, Barthelmess IB, Neupert W. Sensitivity to cyclosporin A is mediated by cyclophilin Neurospora crassa and Saccharomyces cerevisiae. Nature. 1989;342:953–955. doi: 10.1038/342953a0. [DOI] [PubMed] [Google Scholar]
- 29.Roberts RJ, Vincze T, Posfai J, Macelis D. REBASE–a database for DNA restriction and modification: enzymes, genes and genomes. Nucleic Acids Res. 2015;43:D298–299. doi: 10.1093/nar/gku1046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pontecorvo G. Structure of heterochromatin. Nature. 1944;153:365–367. doi: 10.1038/153365a0. [DOI] [Google Scholar]
- 31.Dorer DR, Henikoff S. Expansions of transgene repeats cause heterochromatin formation and gene silencing in Drosophila. Cell. 1994;77:993–1002. doi: 10.1016/0092-8674(94)90439-1. [DOI] [PubMed] [Google Scholar]
- 32.Malagnac F, et al. A gene essential for de novo methylation and development in Ascobolus reveals a novel type of eukaryotic DNA methyltransferase structure. Cell. 1997;91:281–290. doi: 10.1016/s0092-8674(00)80410-9. [DOI] [PubMed] [Google Scholar]
- 33.Selker EU, Fritz DY, Singer MJ. Dense nonsymmetrical DNA methylation resulting from repeat-induced point mutation in Neurospora. Science. 1993;262:1724–1728. doi: 10.1126/science.8259516. [DOI] [PubMed] [Google Scholar]
- 34.Belden WJ, Lewis ZA, Selker EU, Loros JJ, Dunlap JC. CHD1 remodels chromatin and influences transient DNA methylation at the clock gene frequency. PLoS Genet. 2011;7:e1002166. doi: 10.1371/journal.pgen.1002166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ohm RA, et al. Diverse lifestyles and strategies of plant pathogenesis encoded in the genomes of eighteen Dothideomycetes fungi. PLoS Pathog. 2012;8:e1003037. doi: 10.1371/journal.ppat.1003037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bardiya N, Shiu PK. Cyclosporin A-resistance based gene placement system for Neurospora crassa. Fungal Genet Biol. 2007;44:307–314. doi: 10.1016/j.fgb.2006.12.011. [DOI] [PubMed] [Google Scholar]
- 37.Freitag M, Hickey PC, Raju NB, Selker EU, Read ND. GFP as a tool to analyze the organization, dynamics and function of nuclei and microtubules in Neurospora crassa. Fungal Genet Biol. 2004;41:897–910. doi: 10.1016/j.fgb.2004.06.008. [DOI] [PubMed] [Google Scholar]
- 38.Freitag M, Selker EU. Expression and visualization of red fluorescent protein (RFP) in Neurospora crassa. Fungal Genetics Newsletter. 2005;52:14–17. [Google Scholar]
- 39.McCluskey K, Wiest A, Plamann M. The Fungal Genetics Stock Center: a repository for 50 years of fungal genetics research. J Biosci. 2010;35:119–126. doi: 10.1007/s12038-010-0014-6. [DOI] [PubMed] [Google Scholar]
- 40.Arnaise S, Zickler D, Bourdais A, Dequard-Chablat M, Debuchy R. Mutations in mating-type genes greatly decrease repeat-induced point mutation process in the fungus Podospora anserina. Fungal Genet Biol. 2008;45:207–220. doi: 10.1016/j.fgb.2007.09.010. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.