

Abstract
This was a phase II study of linsitinib plus erlotinib versus placebo plus erlotinib in chemotherapy-naive patients with epidermal growth factor receptor-mutation positive, advanced, non—small-cell lung cancer. Linsitinib plus erlotinib resulted in inferior outcomes compared with erlotinib alone. Linsitinib combination was associated with increased adverse events that led to decreased erlotinib exposure. Results highlight the complexity of targeting insulin-like growth factor-1 receptor signaling.
Introduction
First-line epidermal growth factor receptor (EGFR) tyrosine kinase inhibitor treatment of advanced non—small-cell lung cancer with EGFR-activating mutations improves outcomes compared with chemotherapy, but resistance develops in most patients. Compensatory signaling through type 1 insulin-like growth factor 1 receptor (IGF-1R) may contribute to resistance; dual blockade of IGF-1R and EGFR may improve outcomes.
Patients and Methods
We performed a randomized, double-blind, placebo-controlled phase II study of linsitinib, a dual IGF-1R and insulin receptor tyrosine kinase inhibitor, plus erlotinib versus placebo plus erlotinib in chemotherapy-naive patients with EGFR-mutation positive, advanced non—small-cell lung cancer. Patients received linsitinib 150 mg twice daily or placebo plus erlotinib 150 mg once daily on continuous 21-day cycles. The primary end point was progression-free survival.
Results
After randomization of 88 patients (44 each arm), the trial was unblinded early owing to inferiority in the linsitinib arm. The median progression-free survival for the linsitinib versus the placebo group was 8.4 months versus 12.4 months (hazard ratio, 1.37; P = .29). Overall response rate (47.7% vs. 75.0%; P = .02) and disease control rate (77.3% vs. 95.5%; P = .03) were also inferior. Whereas most adverse events were ≤ grade 2, linsitinib plus erlotinib was associated with increased adverse events that led to decreased erlotinib exposure (median days, 228 vs. 305). No drug-drug interaction was suggested by pharmacokinetic and pharmacodynamic results.
Conclusion
Adding linsitinib to erlotinib resulted in inferior outcomes compared with erlotinib alone. Further understanding of the signaling pathways and a biomarker that can predict efficacy is needed prior to further clinical development of IGF-1R inhibitors in lung cancer.
Keywords: Combination, Epidermal growth factor receptor inhibitor, First-line therapy, Insulin-like growth factor-1 inhibitor, Tyrosine kinase inhibitor
Introduction
Lung cancer remains the most common cancer worldwide and the leading cause of cancer-related death.1 Modest improvement in outcomes has been achieved in the last decade, particularly in non—small-cell lung cancer (NSCLC), with advances in targeted therapy and immune checkpoint inhibitor therapy. Patients have benefited from the identification of epidermal growth factor receptor (EGFR) mutations and anaplastic lymphoma kinase translocations that can be effectively treated with small molecule tyrosine kinase inhibitors (TKIs), enabling personalized treatment selection based on tumor genotype. EGFR mutations are among the most commonly identified in NSCLC, seen in 10% to 15% of Caucasian patients and 30% to 35% of East Asian patients.2,3 First-line EGFR TKI therapy in this subgroup of patients is now standard of care, with multiple trials demonstrating improved response rates, progression-free survival (PFS), and quality of life compared with chemotherapy.4–6
Most patients who benefit from EGFR TKIs develop resistance, usually within 12 months. Resistance in more than one-half of patients is associated with secondary EGFR mutations, primarily T790M.7,8 Other mechanisms of resistance that have been identified include MET proto-oncogene (MET) amplification, high-grade neuroendocrine transformation, and signaling through alternate pathways.9–14 One such pathway is through the type 1 insulin-like growth factor receptor (IGF-1R).15–17 IGF-1R overexpression in NSCLC and other tumors is associated with decreased survival,18 and its increased activity leads to tumorigenesis in preclinical models.19–21 Collectively, this evidence has made IGF-1R a target of high interest for cancer therapy and the subject of active preclinical and clinical research. However, clinical trials of IGF-1R inhibition, primarily using anti-IGF-1R monoclonal antibodies, have generated negative results.22–25 It has been hypothesized that the lack of clinical efficacy may be due to alternative signaling through an aberrant form of the insulin receptor (IR) that is expressed in many cancers and has been shown to compensate for IGF-1R inhibition and enhance tumor activity.26–28 Preclinical data support this hypothesis, with co-inhibition of IGF1-R and IR providing enhanced antitumor activity, thought to be because of blockade of bidirectional crosstalk between the 2 receptors and simultaneous inhibition of their respective signaling pathways.19,20,27
Linsitinib is an orally bioavailable, dual inhibitor of IGF-1R and IR, developed based on the hypothesis that simultaneous inhibition of both IGF-1R and IR might improve the clinical activity of IGF-1R-targeted therapy.29 Linsitinib has shown antiproliferative effects in a variety of tumor cell lines and antitumor activity in xenograft models, including for lung cancer.30–33 In addition, synergistic effects have been noted with the combined inhibition of EGFR and IGF-1R inhibition.31 Limited efficacy was reported for single-agent linsitinib in patients with advanced solid tumors, with partial responses (PRs) observed in melanoma and adrenocortical carcinoma.34–36 However, its dual targeting of IGF-1R and IR make linsitinib a rational combination partner with EGFR inhibitors in patients with EGFR-mutant NSCLC where crosstalk between the IGF-1R, IR, and EGFR pathways may contribute to resistance.19,31,37 Phase I results showed that linsitinib could be combined with a therapeutic dose of erlotinib, resulting in unaltered plasma concentrations of either drug, a tolerable safety profile, and clinical activity consisting of 5 PRs (including 3 patients with NSCLC) and disease control (PR plus stable disease) in 51% of patients (n = 75).38
Here, we report the results of a randomized, double-blind, placebo-controlled phase II study comparing linsitinib plus erlotinib with placebo plus erlotinib as first-line therapy in chemotherapy-naive patients with EGFR mutation-positive, advanced NSCLC.
Materials and Methods
Patients
Eligible patients had treatment-naive, histologically confirmed, advanced stage IIIB or IV NSCLC (American Joint Committee on Cancer criteria, sixth edition) with tumor EGFR exon 19 deletion or exon 21 activating mutations. Also required were measurable disease by Response Evaluation Criteria in Solid Tumors (RECIST) 1.139; Eastern Cooperative Oncology Group performance status of 0 to 1; fasting glucose of ≤ 150 mg/dL; and adequate hematologic, hepatic, and renal function. Exclusion criteria included diabetes mellitus requiring insulinotropic or insulin therapy, history of major cardiovascular disease, a Friderica corrected QT interval > 450 ms, cerebrovascular accident within 6 months of randomization, and symptomatic brain metastases. Prior therapy with an IGF-1R inhibitor, EGFR inhibitor (eg, erlotinib, gefitinib, or cetuximab), or concurrent use of strong/moderate cytochrome P-450 (CYP) 1A2 and CYP3A4 inhibitors/inducers was not permitted.
The study was conducted in accordance with the International Conference on Harmonisation Good Clinical Practice with the ethical principles of Helsinki and approved by the independent ethics committee or institutional review board for each site. All patients provided written informed consent.
Study Design
This was a multicenter, randomized, double-blind, phase II study designed to compare the combination of linsitinib plus erlotinib with placebo plus erlotinib in chemotherapy-naive patients with EGFR mutation-positive, advanced NSCLC. Patients were randomized 1:1 and stratified by EGFR mutation (exon 19 vs. 21) and Eastern Cooperative Oncology Group performance status (0 vs. 1). Patients received oral linsitinib 150 mg twice daily (recommended phase II dose36) or matching placebo and erlotinib at the approved dose of 150 mg daily on continuous 21-day cycles. Dose modifications at the investigator’s discretion were permitted for toxicity of either drug, or both where the contribution of either drug was uncertain. Re-escalation was permitted for erlotinib only. A Data Monitoring Committee periodically evaluated the accumulating study results.
Efficacy and Safety Analyses
The primary end point was PFS, defined as time from randomization to disease progression based on RECIST v1.1 or death from any cause. Secondary efficacy end points included overall survival, calculated from randomization to death from any cause; overall response rate (ORR), defined as proportion of patients with complete response (CR) or PR according to RECIST v1.1; disease control rate (DCR) defined as CR, PR, or stable disease (SD) for ≥ 6 weeks; and duration of response, defined as time from the date of first documented CR or PR to documented progression or death. Safety, pharmacokinetics (PK), and pharmacodynamics (PD) of linsitinib were also assessed as secondary endpoints.
The PK population included all treated patients who had at least 1 blood sample collected for analysis of plasma concentrations of linsitinib, erlotinib, and OSI-420 (a metabolite of erlotinib). The PD population included all treated patients with sufficient blood or tissue (archival or fresh) samples for exploratory biomarker or PD analyses. E-cadherin, an epithelial marker previously shown to be required for activation of IGF-1R signaling,40 was compared with IGF-1 concentrations as a potential marker for improved response.41,42
All patients were included in the efficacy evaluation (intent-to-treat population). All patients who received at least 1 dose of treatment were included in safety evaluations, which were performed using National Cancer Institute Common Terminology Criteria for Adverse Events, v4.02.
Statistical Analysis
The primary efficacy analysis was to be performed on the full analysis set of 86 planned patients, with a 2-sided P-value test calculated using an unstratified log-rank. The HR of the treatment effect with 95% confidence interval (CI) was calculated using a Cox proportional hazard model. A prespecified sensitivity analysis was also performed using a stratified log-rank test for the stratification factors at randomization. PFS for a subgroup of patients who received 100% of planned dose intensity of both linsitinib and erlotinib until disease progression or study end was additionally analyzed as an exploratory analysis. The secondary endpoints response rates (ORR, DCR) were tested using the Fisher exact test. Demographics, safety, and PK analyses were summarized using descriptive statistics.
Results
Patients
This multicenter study was conducted in 29 sites worldwide, including the United States (13), South Korea (5), Canada (4), Thailand (4), Singapore (2), and Hong Kong (1). A total of 88 patients were randomized, 44 to linsitinib/erlotinib (linsitinib group) and 44 to placebo/erlotinib (placebo group). Each analysis set included 44 patients except for the group safety and PK sets, which each had 43 in the linsitinib group as 1 patient never received the study drug. The study was unblinded early, on February 15, 2013. This action was recommended by the Data Monitoring Committee because of inferiority in the experimental arm, observed after a routine evaluation when study enrollment had been completed and all patients had received at least 1 cycle of treatment. At this time, linsitinib treatment was discontinued for all patients. For patients considered to be benefiting from treatment, erlotinib treatment was permitted to continue, and, if warranted, to be increased to 150 mg daily. Efficacy analyses are therefore based on a February 2013 data cutoff date, whereas all patients were followed for safety until an October 2013 data cutoff date.
Baseline patient characteristics were balanced between the 2 treatment groups (Table 1). In the linsitinib and placebo groups (n = 44, respectively), 75.0% (n = 33) and 72.7% (n = 32) were never smokers and 59.1% (n = 26) and 56.8% (n = 25) had exon 19 deletion EGFR mutations, respectively. Patients (n = 88) had a median age of 60 years (range, 36–85 years) and the majority were female (70.5%; n = 62) and either white (52.3%; n = 46) or Asian (40.9%; n = 36). The median time from initial diagnosis was 1.7 months, and the majority of patients had stage IV disease (98.9%; n = 87) and adenocarcinoma histology (94.3%; n = 83). A larger proportion of patients in the placebo group received previous radiation (29.5% vs. 18.2%; n = 13 vs 8). Additional mutations (EGFR T790M, KRAS, and PIK3CA) were observed in an extremely small number of patients, precluding potential analysis (no KRAS and 1 PIK3CA and T790M mutation in the linsitinib group and 1 of each in the placebo group).
Table 1.
Baseline Patient Characteristics
| Linsitinib + Erlotinib (N = 44) N (%) |
Placebo + Erlotinib (N = 44) N (%) |
|
|---|---|---|
| Age, years, median (range) | 61.5 (44–82) | 57.5 (36–85) |
| Gender | ||
| Female | 30 (68.2) | 32 (72.7) |
| Male | 14 (31.8) | 12 (27.3) |
| Racea | ||
| White | 20 (45.5) | 26 (59.1) |
| Black | 4 (9.1) | 0 |
| Asian | 20 (45.5) | 16 (36.4) |
| ECOG performance score | ||
| 0 | 21 (47.7) | 21 (47.7) |
| 1 | 23 (52.3) | 23 (52.3) |
| Cigarette smoking history | ||
| Current or former smoker | 11 (25.0) | 12 (27.3) |
| Never smoker | 33 (75.0) | 32 (72.7) |
| NSCLC pathologic stage | ||
| Stage IIIB | 1 (2.3) | 0(0) |
| Stage IV | 43 (97.7) | 44 (100) |
| Histologic subtype | ||
| Adenocarcinoma | 41 (93.2) | 42 (95.5) |
| Squamous cell carcinoma | 1 (2.3) | 0(0) |
| Mixed histology | 2 (4.5) | 1 (2.3) |
| Other | 0(0) | 1 (2.3) |
| EGFR mutation status | ||
| Exon 19 deletion | 26 (59.1) | 25 (56.8) |
| Exon 21 single point mutation | 18 (40.9) | 19 (43.2) |
| Time from initial diagnosis, mos | ||
| Mean (SD) | 8.7 (16.4) | 7.2 (15.1) |
| Median (range) | 1.6 (0.7–61.1) | 1.8 (0.6–86.4) |
| Prior radiation therapy | 8 (18.2) | 13 (29.5) |
| Prior disease-related surgery | 13 (29.5) | 10 (22.7) |
| Prior regimen treatment | ||
| Neo-adjuvant | 1 (2.3) | 2 (4.5) |
| Adjuvant | 4 (9.1) | 3 (6.8) |
| Neo-adjuvant and adjuvant | 1 (2.3) | 0 (0) |
Abbreviations: ECOG = Eastern Cooperative Oncology Group; EGFR = epidermal growth factor receptor; NSCLC = non-small-cell lung cancer; SD = standard deviation.
In addition, 2 patients in the placebo/erlotinib arm were classified as having a race other than White, Black or Asian (4.5%).
Exposure to active treatment was substantially decreased in the linsitinib group. Patients had a median duration of 197 days on linsitinib and 228 days on erlotinib in the linsitinib group compared with 279 days on placebo and 305 days on erlotinib in the placebo group (see Supplemental Table 1 in the online version). Dose interruptions and dose reductions occurred more frequently in the linsitinib group. Additionally, more patients in the linsitinib group discontinued treatment (74.4% vs. 54.5%; n = 32 vs. 24). Of the patients who discontinued treatment in the linsitinib group (n = 32), 75.0% (n = 24) did so for disease progression and 15.6% (n = 5) for drug-related adverse events (AEs); for the placebo group (n = 24), 91.7% (n = 22) were for disease progression and none for drug-related AEs.
Efficacy
The prespecified primary analysis of PFS required at least 58 events, but because of the early unblinding, the actual number of events was 47. Median PFS for the linsitinib versus the placebo group was 8.4 months versus 12.4 months (hazard ratio [HR], 1.37; P = .29) (Figure 1, Table 2). Prespecified subgroup analyses for median PFS were generally consistent with the overall result: 8.4 months versus 12.9 months for the exon 19 deletion subgroup, 9.4 months versus 11.9 months for the exon 21 mutation subgroup, and 8.4 months versus 12.9 months for patients who never smoked (see Supplemental Table 2 in the online version). In the exploratory subgroup of patients (n = 25; 11 linsitinib and 14 placebo) who received full doses of the study drugs until disease progression or end of study, the difference in median PFS between the treatment arms was even greater in favor of the placebo group (2.8 months vs. 11.0 months; HR, 2.40; P = .10) (Figure 1B). Secondary efficacy end points were also worse for the linsitinib group (Table 2). Both ORR and DCR were significantly worse in the linsitinib group compared with the placebo group (47.7% vs. 75.0%; P = .02, and 77.3% vs. 95.5%; P = .03). Median duration of response was similar between the groups, with 9.9 months (95% CI, 6.9 months to not evaluable) and 9.7 (95% CI, 8.3 months-13.4 months) for the linsitinib and placebo groups, respectively. Because the study was stopped prematurely, overall survival estimates for the linsitinib group were immature and do not allow for a true estimate.
Figure 1. Kaplan-Meier Curves of Progression-Free Survival for the Full Analysis Set (A), Full Dose Exploratory Set (B), Exon 19 Subgroup (C), and Exon 21 Subgroup (D).
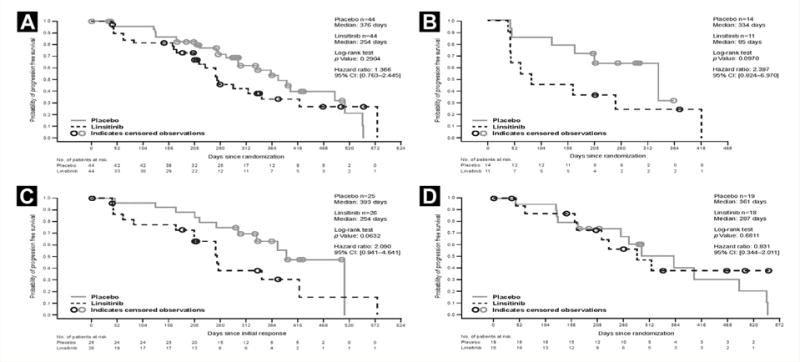
Abbreviation: CI = Confidence interval.
Table 2.
Summary of Efficacy
| Efficacy Endpoint | Linsitinib + Erlotinib (N = 44) | Placebo + Erlotinib (N = 44) | HR (95% CI) | P Value |
|---|---|---|---|---|
| Progression-free survival | ||||
| Number of events, n (%) | 23 (52.3) | 24 (54.5) | 1.37 | .29 |
| Median, mos (95% CI) | 8.4 (7.1–13.8) | 12.4 (9.7–16.8) | 0.76–2.45 | |
| Overall survival | ||||
| Number of events, n (%) | 5 (11.4) | 7 (15.9) | 0.77 | .65 |
| Median, mos (95% CI) | NR (NR, NR) | 19.5 (17.3-NR) | 0.24–2.42 | |
| Disease control rate,a n (%) | 34 (77.3) | 42 (95.5) | NA | .03 |
| 95% CI | 62.16–88.53 | 84.53–99.44 | ||
| Overall response rate,b n (%) | 21 (47.7) | 33 (75.0) | NA | .02 |
| 95% CI | 32.5–63.3 | 59.7–86.8 | ||
| Duration of responsec | ||||
| Median, mos (95% CI) | 9.9 (6.9-NR) | 9.7 (8.3–13.4) | NA | NA |
Abbreviations: CI = Confidence interval; HR = hazard ratio; NA = not applicable; NR = not reached.
Disease control rate = complete response + partial response + stable disease.
Overall response rate = complete response + partial response.
Duration of response = The time in months from the date of first documented complete or partial response to documented progression or death due to underlying cancer.
Safety
The most common drug-related AEs were skin rash (83.7%; n = 36 linsitinib; 97.7%; n = 43 placebo) and diarrhea (67.4%; n = 29 linsitinib; 75.0%; n = 33 placebo). Most drug-related AEs were grade 1/2 in severity, and there were no grade > 3 AEs except for 2 patients in the linsitinib group who had grade 4 elevations in alanine aminotransferase (ALT) (Table 3). However, there was an increased incidence of > grade 2 toxicity overall in the linsitinib group, which had a higher proportion of patients with any AEs > grade 2 (69.8% vs. 40.9%; n = 30 vs. 18), drug-related AEs > grade 2 (51.2% vs. 22.7%; n = 22 vs. 10), and drug-related serious AEs (18.6% vs. 6.8%; n = 8vs. 3). Furthermore, a higher proportion of patients in the linsitinib group had drug-related AEs that led to dose reductions and interruptions, with dose reductions in 32.6% (n = 14) versus 20.5% (n = 9) and temporary interruptions in 44.2% (n = 19) versus 20.5% (n = 9) of patients. Whereas there were no deaths during treatment, within 30 days of the last dose, 2 patients in the linsitinib group and 3 patients in the placebo group died. All 5 deaths were because of progressive disease and not considered related to the study drug.
Table 3.
Drug-Related Adverse Events Reported in ≥10% of Patients in Either Treatment Group
| Adverse Event, n (%) | Linsitinib + Erlotinib (N = 43) | Placebo + Erlotinib (N = 44) | ||||
|---|---|---|---|---|---|---|
| Grade 1/2 | Grade 3 | Grade 4 | Grade 1/2 | Grade 3 | Grade 4 | |
| Overall | 21 (48.8) | 20 (46.5) | 2 (4.7) | 34 (79.1) | 10 (22.7) | 0 |
| Skin rash | 33 (76.7) | 3(7.0) | 0 | 40 (90.9) | 3 (6.8) | 0 |
| Diarrhea | 28 (65.1) | 1 (2.3) | 0 | 29 (65.9) | 4 (9.1) | 0 |
| Dry skin | 20 (46.5) | 0(0) | 0 | 18 (40.9) | 0 | 0 |
| Fatigue | 12 (27.9) | 4 (9.3) | 0 | 15 (34.1) | 1 (2.3) | 0 |
| Nausea | 15 (34.9) | 4 (9.3) | 0 | 12 (27.3) | 0 | 0 |
| Decreased appetite | 16 (37.2) | 2 (4.7) | 0 | 8 (18.2) | 1 (2.3) | 0 |
| Paronychia | 8 (18.6) | 0 | 0 | 14 (31.8) | 0 | 0 |
| Alopecia | 5 (11.6) | 0 | 0 | 14 (31.8) | 0 | 0 |
| Stomatitis | 11 (25.6) | 0 | 0 | 7 (15.9) | 0 | 0 |
| Vomiting | 11 (25.6) | 2 (4.7) | 0 | 4 (9.1) | 0 | 0 |
| Pruritus | 4 (9.3) | 0 | 0 | 12 (27.3) | 0 | 0 |
| Increased ALT | 3(7.0) | 9 (20.9) | 2 (4.7) | 0 | 1 (2.3) | 0 |
| Increased AST | 7 (16.3) | 5 (11.6) | 0 | 1 (2.3) | 0 | 0 |
| Dysgeusia | 7 (16.3) | 0 | 0 | 6 (13.6) | 0 | 0 |
| Dry eye | 7 (16.3) | 0 | 0 | 5 (11.4) | 0 | 0 |
| Hyperglycemia | 7 (16.3) | 1 (2.3) | 0 | 1 (2.3) | 0 | 0 |
| Dry mouth | 5 (11.6) | 0 | 0 | 3 (6.8) | 0 | 0 |
| Increased blood bilirubin | 6 (14.0) | 1 (2.3) | 0 | 0 | 0 | 0 |
| Decreased weight | 4 (9.3) | 1 (2.3) | 0 | 2 (4.5) | 0 | 0 |
| Increased blood creatinine | 5 (11.6) | 0 | 0 | 0 | 0 | 0 |
| Skin fissures | 0 | 0 | 0 | 5 (11.4) | 0 | 0 |
Abbreviations: ALT = Alanine aminotransferase; AST = aspartate aminotransferase.
For the laboratory parameters measured, an increase in serum creatinine from baseline (grade 1 or 2) was observed in 31% (n = 13) versus 18.2% (n = 8) of patients in the linsitinib versus placebo groups. In addition, a higher proportion of patients in the linsitinib group had grade 3 or 4 increases in ALT (20.9% vs. 4.5%; n = 9 vs. 2) and aspartate aminotransferase (AST; 16.3% vs. 4.5%; n = 7 vs. 2). As expected and consistent with prior linsitinib studies, there was a higher proportion of patients with hyperglycemia in the linsitinib group (18.6% vs. 2.3%; n = 8 vs. 1). Only 1 hyperglycemic event was grade 3, and none was considered serious.
PK and PD
The plasma concentration of linsitinib from predose to 4 hours postdose in cycles 2 and 3 (steady-state levels) was similar to that observed in previous single-agent studies (Table 4). Whereas the mean erlotinib concentration values were slightly higher in the placebo arm, predose concentrations of steady-state erlotinib and OSI-420 (a metabolite of erlotinib) were statistically similar in both the linsitinib and placebo arms. In addition, erlotinib levels were similar to those required for efficacy.43,44 This suggests no significant effect of linsitinib on erlotinib PK, consistent with findings in the phase I combination trial.38
Table 4.
Plasma Concentrations of Erlotinib, OSI-420,a and Linsitinib
| Linsitinib + Erlotinib | Placebo + Erlotinib | |||||||
|---|---|---|---|---|---|---|---|---|
| N | Predose | N | 4-h Postdose | N | Predose | N | 4-h Postdose | |
| Plasma concentration of erlotinib, median, ng/mL (range) | ||||||||
| C 1, day 1 | 0 | NE | 31 | 993 (2–2260) | 0 | NE | 37 | 1160 (3–2700) |
| C 2, day 1 | 30 | 1000 (343–1670) | 32 | 994 (267–2790) | 35 | 1240 (163–3820) | 36 | 1460 (10–3180) |
| C 3, day 1 | 24 | 1018 (58–4160) | 24 | 996 (17–4210) | 36 | 1185 (189–2580) | 36 | 1305 (437–4200) |
| Plasma concentration of OSI-420, median, ng/mL (range) | ||||||||
| C 1, day 1 | 0 | NE | 30 | 102.5 (2.4–319.0) | 0 | NE | 35 | 89.7 (1.9–336.0) |
| C 2, day 1 | 30 | 104.0 (30.4–322.0) | 32 | 135.5 (13.6–430.0) | 35 | 141.0 (9.6–542.0) | 35 | 159.0 (34.5–359.0) |
| C 3, day 1 | 24 | 128.5 (3.6–604.0) | 23 | 108.0 (5.5–653.0) | 36 | 110.0 (10.2–648.0) | 36 | 143.5 (34.7–636.0) |
| Plasma concentration of linsitinib; median, ng/mL (range) | ||||||||
| C 1, day 1 | 0 | NE | 39 | 976 (135–4330) | NA | NA | ||
| C 2, day 1 | 31 | 746 (1.4–2890) | 32 | 1630 (256–4750) | NA | NA | ||
| C 3, day 1 | 24 | 590 (2.0–1940) | 24 | 1100 (191–3440) | NA | NA | ||
Abbreviations: C = Cycle; h = hour; NA = not applicable; NE = not estimated.
Metabolite of erlotinib.
Median plasma concentrations of IGF-1, an indirect indicator of IGF-1R inhibition, remained similar from cycles 1 to 5 (range, 149.0–166.8 ng/mL) in the placebo group (Table 5). In contrast, median IGF-1 concentrations were higher and increased from 171.8 ng/mL in cycle 1 to 272.3 ng/mL in cycle 5 in the linsitinib group, suggesting that sufficient concentrations of linsitinib were present to inhibit IGF-1R signaling in tissues involved in regulating IGF-1 expression in patients. E-cadherin levels showed no association with efficacy outcomes. The median PFS according to baseline tumor or blood E-cadherin levels above or below the median (n = 12 each group) was 7.1 versus 12.4 months (HR, 3.89; 95% CI, 1.2–12.5 months) for the linsitinib versus the placebo group, similar to the overall study results.
Table 5.
Predose Plasma Concentrations of Insulin-like Growth Factor-1
| Time point | Linsitinib + Erlotinib | Placebo + Erlotinib | ||
|---|---|---|---|---|
| N | Median (Range) ng/mL | N | Median (Range) ng/mL | |
| Cycle 1, day 1 | 33 | 171.8 (36.9–347.4) | 37 | 166.8 (64.6–364.3) |
| Cycle 2, day 1 | 28 | 244.6 (65.4–655.7) | 34 | 161.4 (71.7–371.5) |
| Cycle 3, day 1 | 25 | 217.3 (112.3–511.0) | 34 | 153.1 (65.3–318.6) |
| Cycle 4, day 1 | 21 | 257.3 (158.6–435.1) | 35 | 158.7 (77.6–292.4) |
| Cycle 5, day 1 | 19 | 272.3 (12.3–503.3) | 34 | 149.0 (75.2–258.7) |
Discussion
The scientific rationale for combination therapy with linsitinib and erlotinib in advanced NSCLC is supported by the literature. Preclinical evidence has shown reciprocal and compensatory signaling between the EGFR and IGF-1R pathways, with erlotinib enhancement of IGF-driven AKT activity and IGF-1R inhibition associated with upregulated EGFR signaling.19–21,31 The combination of IGF-1R targeted agents with erlotinib ablates this signaling in preclinical models, resulting in synergistic inhibition of cancer cell growth in vitro and tumor inhibition in vivo.19–21,31 Further preclinical data also suggest a role for IGF-1R in mediating resistance to EGFR-targeted therapies,15–17 as well as an interaction between IGF-1R and IR that might contribute to resistance to agents, such as monoclonal antibodies, that selectively target IGF-1R alone.26–28 Previously, phase I results in a 91-patient advanced solid tumor study showed acceptable tolerability for combined linsitinib plus erlotinib, with no evidence of significant drug-drug interaction, as well as initial evidence of efficacy in patients with advanced solid tumors.38
Despite this seemingly well-supported scientific rationale and a positive signal from the phase I study, our randomized study demonstrated an unfavorable effect that led to reduced efficacy in chemotherapy-naive patients with EGFR mutation-positive, advanced NSCLC. Although the combination was tolerable, as predicted by the phase I results, the combination resulted in inferior PFS and response rates compared with erlotinib alone, which led to early termination of the trial.
One possible explanation for the observed reduced efficacy of linsitinib plus erlotinib is an unanticipated PK drug-drug interaction between the agents. However, in our study, as in the earlier phase I trial, steady-state, predose concentrations of erlotinib were similar in both the linsitinib and placebo groups. Furthermore, the plasma concentrations of linsitinib were also similar to those seen in clinical trials of single-agent linsitinib.35,36 As in clinical studies of other IGF-1R—targeted agents, we additionally analyzed plasma levels of IGF-1 in our study as a putative biomarker of IGF-1R inhibition.22,24 The increased and increasing plasma IGF-1 over 5 cycles of treatment in the linsitinib group provides evidence that linsitinib was present in concentrations sufficient to inhibit IGF-1R activity in these patients. These findings suggest that PK or PD drug-drug interactions are unlikely to account for the negative results of this study.
A more likely contributing factor may have been reduced treatment exposure in the linsitinib group compared with the placebo group, with a median 228 days of erlotinib exposure in the linsitinib group compared with 305 days in the placebo group. Whereas the linsitinib plus erlotinib combination was tolerable, it was associated with increased grade 3 toxicity, and this was reflected in higher rates of erlotinib dose interruptions, reductions, and discontinuation of all treatment. Whereas disease progression was the cause for treatment discontinuation in most patients in both arms (75.0% linsitinib; 91.7% placebo; n = 33 vs. 40), drug-related AEs were the cause of treatment discontinuation in 15.6% of patients in the linsitinib group versus none in the placebo group. Whereas reduced erlotinib exposure likely contributed to the negative result, even patients receiving full-dose erlotinib plus linsitinib had inferior outcomes, suggesting additional contributing factors.
The exploratory analysis of PFS performed in the subgroup of patients who received full doses of both study drugs until disease progression or end of study is disconcerting. Although the difference was not statistically significant, median PFS was even poorer for the linsitinib group in this comparison, 2.8 versus 11.0 months (HR, 2.40; P = .10). Although exploratory, this comparison suggests the presence of an unforeseen biologic interaction between the 2 drugs that resulted in a negative impact on the clinical efficacy of the combined drugs.
This is not the first clinical trial of an IGF-1R-targeted agent failing to match the promise suggested by preclinical data. IGF-1R inhibition has long been considered a promising target for cancer treatment, and has been extensively investigated as a treatment for NSCLC, both as monotherapy and in combination with chemotherapy or targeted therapy.19–21 Three clinical trials in addition to the current study have now demonstrated less efficacy when combining IGF-1R targeted drugs with erlotinib compared with erlotinib alone in NSCLC, with all 3 reporting HRs > 1 for PFS.23–25 These negative results further support the hypothesis that as yet unknown mechanistic factors may underlie the decreased clinical activity of EGFR-and IGF-1R-targeted combinations. IGF-1R signaling is inherently complex, involving crosstalk interactions with various feedback, compensatory, and redundant signaling pathways in cancer cells.19–21 For example, in addition to well-described interactions between the IGF-1R and EGFR signaling pathways, a feedback loop involving re-activation of AKT by mammalian target of rapamycin (mTOR) upon IGF-1R inhibition has also been described.45 Given this complexity, it is possible that unanticipated signaling could occur upon dual inhibition of IGF-1R and EGFR in human tumors, resulting in increased activation of alternative pathways, increased tumor cell proliferation, or diminished EGFR inhibition. At this point, however, these hypotheses are speculative.
Conclusion
In summary, our results highlight the complexity of targeting IGF-1R signaling in the treatment of patients with advanced NSCLC. Prior to clinical development of this class of drugs in lung cancer, further understanding of the involved signaling pathways that may yield a robust biomarker that can predict efficacy is needed.
Clinical Practice Points
Combination therapy with linsitinib and erlotinib in advanced NSCLC is supported by the literature. Despite scientific rationale and a positive signal from a phase I study, this randomized study of linsitinib plus erlotinib versus placebo plus erlotinib demonstrated an unfavorable effect that led to reduced efficacy of the linsitinib combination in chemotherapy-naive patients with EGFR mutation-positive, advanced NSCLC.
A likely contributing factor may have been reduced treatment exposure in the linsitinib group compared with the placebo group. However, even patients receiving full-dose erlotinib plus linsitinib had inferior outcomes.
These results further support the hypothesis that as yet unknown mechanistic factors may underlie the decreased clinical activity of EGFR-and insulin-like growth factor-1 receptor (IGF-1R)-targeted combinations.
Given this complexity, it is possible that unanticipated signaling could occur upon dual inhibition of IGF-1R and EGFR in human tumors. Further understanding of the involved signaling pathways that may yield a robust biomarker that can predict efficacy is needed for clinical development of IGF-1R inhibitors in lung cancer.
Supplementary Material
Acknowledgments
Editorial assistance was provided by Scientific Connexions, Inc, Lyndhurst, NJ, an Ashfield Company, part of UDG Healthcare plc, and was funded by Astellas. This study was supported by Astellas.
Footnotes
Disclosure
Drs Chen, Poondru, Singh, and Steinberg are employees of Astellas Pharma Inc, and Sonja Medley was employed with Astellas at the time of the study. Drs Bazhenova and Chow have received honoraria from Astellas Pharma Inc. Dr Bazhenova is currently a paid speaker for Genentech. Dr Chow is also a member of the advisory board for Merck and Novartis, and has received research grants and funding from the following companies: AstraZeneca, Bristol-Myers Squibb, GlaxoSmithKline, Pfizer, Merck, Novartis, and Roche. Dr Eaton received a funding grant from Astellas Pharma, Inc for the conduct of this trial. Dr Chiappori has received honoraria as a member of the speakers’ bureaus at Boehringer Ingelheim, Celgene, Genentech, Novartis, and Pfizer. Dr Rizvi has been a consultant for Bristol Myers Squibb, Genentech/Roche, MedImmune/AstraZeneca, and Merck. Dr Rudin has been a consultant for AbbVie, Boehringer Ingelheim, Celgene, GlaxoSmithKline, and Merck. Drs Juergens and Lopes received institutional funding and compensation for the conduct of this trial via Astellas Pharma; Dr Juergen has also previously developed educational materials for Roche, and Dr Lopes has been a consultant for Roche. Dr Leighl has previously received funding from Novartis for an unrelated clinical trial. Drs Ahn, Dechaphunkul, Gadgeel, and Sunpaweravong state that they have no conflicts of interest.
Supplemental Data
Supplemental tables accompanying this article can be found in the online version at http://dx.doi.org/10.1016/j.cllc.2016.07.007.
References
- 1.World Health Organization. Cancer: Fact Sheet No. 297. World Health Organization Web site; Updated February 2015. Available at: http://www.who.int/mediacentre/factsheets/fs297/en. Accessed: July 27, 2015. [Google Scholar]
- 2.Rosell R, Moran T, Queralt C, et al. Screening for epidermal growth factor receptor mutations in lung cancer. N Engl J Med. 2009;361:958–67. doi: 10.1056/NEJMoa0904554. [DOI] [PubMed] [Google Scholar]
- 3.Han B, Tjulandin S, Hagiwara K, et al. Determining the prevalence of EGFR mutations in Asian and Russian patients with advanced non-small-cell lung cancer of adenocarcinoma (ADC) and non-ADC histology: IGNITE study. ELCC Congress. 2015 doi: 10.1016/j.lungcan.2017.08.021. abstract 96O. [DOI] [PubMed] [Google Scholar]
- 4.Mok TS, Wu Y, Thongprasert S, et al. Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. N Engl J Med. 2009;361:947–57. doi: 10.1056/NEJMoa0810699. [DOI] [PubMed] [Google Scholar]
- 5.Rosell R, Carcereny E, Gervais R, et al. Erlotinib versus standard chemotherapy as first-line treatment for European patients with advanced EGFR mutation-positive non-small-cell lung cancer (EURTAC): a multicentre, open-label, randomised phase 3 trial. Lancet. 2012;13:239–46. doi: 10.1016/S1470-2045(11)70393-X. [DOI] [PubMed] [Google Scholar]
- 6.Sequist LV, Yang JC, Yamamoto N, et al. Phase III study of afatinib or cisplatin plus pemetrexed in patients with metastatic lung adenocarcinoma with EGFR mutations. J Clin Oncol. 2013;31:3327–34. doi: 10.1200/JCO.2012.44.2806. [DOI] [PubMed] [Google Scholar]
- 7.Arcila ME, Oxnard GR, Nafa K, et al. Rebiopsy of lung cancer patients with acquired resistance to EGFR inhibitors and enhanced detection of the T790M mutation using a locked nucleic acid-based assay. Clin Cancer Res. 2011;17:1169–80. doi: 10.1158/1078-0432.CCR-10-2277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sequist LV, Waltman BA, Dias-Santagata D, et al. Genotypic and histological evolution of lung cancers acquiring resistance to EGFR inhibitors. Sci Transl Med. 2011;3:75ra26. doi: 10.1126/scitranslmed.3002003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Engelman JA, Zejnullahu K, Mitsudomi T, et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science. 2007;316:1039–43. doi: 10.1126/science.1141478. [DOI] [PubMed] [Google Scholar]
- 10.Oxnard GR, Arcila ME, Chmielecki J, et al. New strategies in overcoming acquired resistance to epidermal growth factor receptor tyrosine kinase inhibitors in lung cancer. Clin Cancer Res. 2011;17:5530–7. doi: 10.1158/1078-0432.CCR-10-2571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang Z, Lee JC, Lin L, et al. Activation of the AXL kinase causes resistance to EGFR-targeted therapy in lung cancer. Nat Genet. 2012;44:852–60. doi: 10.1038/ng.2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Byers LA, Diao L, Wang J, et al. An epithelial-mesenchymal transition gene signature predicts resistance to EGFR and PI3K inhibitors and identifies Axl as a therapeutic target for overcoming EGFR inhibitor resistance. Clin Cancer Res. 2013;19:279–90. doi: 10.1158/1078-0432.CCR-12-1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Della Corte CM, Bellevicine C, Vicidomini G, et al. SMO amplification and activation of the hedgehog pathway as novel mechanisms of resistance to anti-epidermal growth factor receptor drugs in human lung cancer. Clin Cancer Res. 2015;21:4686–97. doi: 10.1158/1078-0432.CCR-14-3319. [DOI] [PubMed] [Google Scholar]
- 14.Stewart EL, Tan SZ, Liu G, et al. Known and putative mechanisms of resistance to EGFR targeted therapies in NSCLC patients with EGFR mutations-a review. Transl Lung Cancer Res. 2015;4:67–81. doi: 10.3978/j.issn.2218-6751.2014.11.06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jones HE, Goddard L, Gee JM, et al. Insulin-like growth factor-1 receptor signaling and acquired resistance to gefitinib (ZD1839; Iressa) in human breast and prostate cancer cells. Endocr Relat Cancer. 2004;11:793–814. doi: 10.1677/erc.1.00799. [DOI] [PubMed] [Google Scholar]
- 16.Morgillo F, Woo JK, Kim ES, et al. Heterodimerization of insulin-like growth factor receptor/epidermal growth factor receptor and induction of survivin expression counteract the antitumor action of erlotinib. Cancer Res. 2006;66:10100–11. doi: 10.1158/0008-5472.CAN-06-1684. [DOI] [PubMed] [Google Scholar]
- 17.Morgillo F, Kim WY, Kim ES, et al. Implication of the insulin-like growth factor-IR pathway in the resistance of non-small cell lung cancer cells to treatment with gefitinib. Clin Cancer Res. 2007;13:2795–803. doi: 10.1158/1078-0432.CCR-06-2077. [DOI] [PubMed] [Google Scholar]
- 18.Kim JS, Kim ES, Liu D, et al. Prognostic implications of tumoral expression of insulin like growth factors 1 and 2 in patients with non-small-cell lung cancer. Clin LungCancer. 2014;15:213–21. doi: 10.1016/j.cllc.2013.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fidler MJ, Shersher DD, Borgia JA. Targeting the insulin-like growth factor receptor pathway in lung cancer: problems and pitfalls. Ther Adv Med Oncol. 2012;4:51–60. doi: 10.1177/1758834011427576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gao J, Chang YS, Jallal B, et al. Targeting the insulin-like growth factor axis for the development of novel therapeutics in oncology. Cancer Res. 2012;72:3–12. doi: 10.1158/0008-5472.CAN-11-0550. [DOI] [PubMed] [Google Scholar]
- 21.Pollak M. The insulin and insulin-like growth factor receptor family in neoplasia: an update. Nat Rev Cancer. 2012;12:159–69. doi: 10.1038/nrc3215. [DOI] [PubMed] [Google Scholar]
- 22.Karp DD, Paz-Ares LG, Novello S, et al. Phase II study of the anti-insulin-like growth factor type 1 receptor antibody CP-751,871 in combination with paclitaxel and carboplatin in previously untreated, locally advanced, or metastatic non-small-cell lung cancer. J Clin Oncol. 2009;27:2516–22. doi: 10.1200/JCO.2008.19.9331. [DOI] [PubMed] [Google Scholar]
- 23.Ramalingam SS, Spigel DR, Chen D, et al. Randomized phase II study of erlotinib in combination with placebo or R1507, a monoclonal antibody to insulin-like growth factor-1 receptor, for advanced-stage non-small-cell lung cancer. J Clin Oncol. 2011;29:4574–80. doi: 10.1200/JCO.2011.36.6799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Weickhardt A, Doebele R, Oton A, et al. A phase I/II study of erlotinib in combination with the anti-insulin-like growth factor-1 receptor monoclonal antibody IMC-A12 (cixutumumab) in patients with advanced non-small cell lung cancer. J Thorac Oncol. 2012;7:419–26. doi: 10.1097/JTO.0b013e31823c5b11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Scagliotti GV, Bondarenko I, Blackhall F. Randomized, phase III trial of figitumumab in combination with erlotinib versus erlotinib alone in patients with nonadenocarcinoma nonsmall-cell lung cancer. Ann Oncol. 2015;26:497–504. doi: 10.1093/annonc/mdu517. [DOI] [PubMed] [Google Scholar]
- 26.Belfiore A, Frasca F, Pandini G, et al. Insulin receptor isoforms and insulin receptor/insulin-like growth factor receptor hybrids in physiology and disease. Endocr Rev. 2009;30:586–623. doi: 10.1210/er.2008-0047. [DOI] [PubMed] [Google Scholar]
- 27.Buck E, Gokhale PC, Koujak S, et al. Compensatory insulin receptor (IR) activation on inhibition of insulin-like growth factor-1 receptor (IGF-1R): rationale for cotargeting IGF-1R and IR in cancer. Mol Cancer Ther. 2010;9:2652–64. doi: 10.1158/1535-7163.MCT-10-0318. [DOI] [PubMed] [Google Scholar]
- 28.Ulanet DB, Ludwig DL, Kahn CR, et al. Insulin receptor functionally enhances multistage tumor progression and conveys intrinsic resistance to IGF-1R targeted therapy. Proc Natl Acad Sci U S A. 2010;107:10791–8. doi: 10.1073/pnas.0914076107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mulvihill MJ, Cooke A, Rosenfeld-Franklin M, et al. Discovery of OSI-906: a selective and orally efficacious dual inhibitor of the IGF-1 receptor and insulin receptor. Future Med Chem. 2009;1:1153–71. doi: 10.4155/fmc.09.89. [DOI] [PubMed] [Google Scholar]
- 30.Ji QS, Mulvihill MJ, Rosenfeld-Franklin M, et al. A novel, potent, and selective insulin-like growth factor-I receptor kinase inhibitor blocks insulinlike growth factor-I receptor signaling in vitro and inhibits insulin-like growth factor-I receptor dependent tumor growth in vivo. Mol Cancer Ther. 2007;6:2158–67. doi: 10.1158/1535-7163.MCT-07-0070. [DOI] [PubMed] [Google Scholar]
- 31.Buck E, Eyzaquirre A, Rosenfeld-Franklin M, et al. Feedback mechanisms promote cooperativity for small molecule inhibitors of epidermal and insulin-like growth factor receptors. Cancer Res. 2008;68:8322–32. doi: 10.1158/0008-5472.CAN-07-6720. [DOI] [PubMed] [Google Scholar]
- 32.McKinley ET, Bugaj JE, Zhao P, et al. 18FDG-PET predicts pharmacodynamics response to OSI-906, a dual IGF-1R/IR inhibitor, in preclinical mouse models of lung cancer. Clin Cancer Res. 2011;17:3332–40. doi: 10.1158/1078-0432.CCR-10-2274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zinn RL, Gardner EE, Marchionni L, et al. ERK phosphorylation is predictive of resistance to IGF-1R inhibition in small cell lung cancer. Mol Cancer Ther. 2013;12:1131–9. doi: 10.1158/1535-7163.MCT-12-0618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fassnacht M, Berruti A, Baudin E, et al. Linsitinib (OSI-906) versus placebo for patients with locally advanced or metastatic adrenocortical carcinoma: a double-blind, randomized, phase 3 study. Lancet Oncol. 2015;16:426–35. doi: 10.1016/S1470-2045(15)70081-1. [DOI] [PubMed] [Google Scholar]
- 35.Jones R, Kim E, Nava-Parada P, et al. Phase I study of intermittent oral dosing of the insulin-like growth factor-1 and insulin receptors inhibitor OSI-906 in patients with advanced solid tumors. Clin Cancer Res. 2015;21:693–700. doi: 10.1158/1078-0432.CCR-14-0265. [DOI] [PubMed] [Google Scholar]
- 36.Puzanov I, Lindsay CR, Goff L, et al. Phase I dose-escalation study of continuous oral dosing of OSI-906, a dual inhibitor of insulin-like growth factor receptor 1 and insulin receptor tyrosine kinases, in patients with advanced solid tumors. Clin Cancer Res. 2015;21:701–11. doi: 10.1158/1078-0432.CCR-14-0303. [DOI] [PubMed] [Google Scholar]
- 37.Pillai RN, Ramalingam SS. Inhibition of insulin-like growth factor receptor: end of a targeted therapy? Transl Lung Cancer Res. 2013;2:14–22. doi: 10.3978/j.issn.2218-6751.2012.11.05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Macaulay VM, Middleton MR, Eckhardt SG, et al. Phase I dose escalation study of linsitinib (OSI-906) and erlotinib in patients with advanced solid tumors. Clin Cancer Res. 2016;22:2897–907. doi: 10.1158/1078-0432.CCR-15-2218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Eisenhauer EA, Therasse P, Bogaerts J, et al. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1) Eur J Cancer. 2009;45:228–47. doi: 10.1016/j.ejca.2008.10.026. [DOI] [PubMed] [Google Scholar]
- 40.Bedzhou I, Liszewska E, Kanzler B, et al. IGF-1R signaling is indispensable for preimplantation development and is activated via a novel function of e-cadherin. PLoS Genet. 2012;8:e1002609. doi: 10.1371/journal.pgen.1002609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yauch RL, Januario T, Eberhard DA, et al. Epithelial versus mesenchymal phenotype determines in vitro sensitivity and predicts clinical activity of erlotinib in lung cancer patients. Clin Cancer Res. 2005;11:8686–98. doi: 10.1158/1078-0432.CCR-05-1492. [DOI] [PubMed] [Google Scholar]
- 42.Richardson F, Young GD, Sennello R, et al. The evaluation of E-cadherin and vimentin as biomarkers of clinical outcomes among patients with non-small-cell lung cancer treated with erlotinib as second-or third-line therapy. Anticancer Res. 2012;32:537–52. [PubMed] [Google Scholar]
- 43.Hidalgo M, Siu LL, Nemunaitis J, et al. Phase I and pharmacologic study of OSI-774, an epidermal growth factor receptor tyrosine kinase inhibitor, in patients with advanced solid malignancies. J Clin Oncol. 2001;19:3267–79. doi: 10.1200/JCO.2001.19.13.3267. [DOI] [PubMed] [Google Scholar]
- 44.Lu JF, Eppler SM, Wolf J, et al. Clinical pharmacokinetics of erlotinib in patients with solid tumors and exposure-safety relationship in patients with non-small cell lung cancer. Clin Pharmacol Ther. 2006;80:136–45. doi: 10.1016/j.clpt.2006.04.007. [DOI] [PubMed] [Google Scholar]
- 45.Wan X, Harkavy B, Shen N, et al. Rapamycin induces feedback activation of Akt signaling through an IGF-1R-dependent mechanism. Oncogene. 2007;26:1932–40. doi: 10.1038/sj.onc.1209990. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.