

SUMMARY
Stimulation of CD95/Fas drives and maintains cancer stem cells (CSCs). We now report that this involves activation of STAT1, induction of STAT1 regulated genes, and this process is inhibited by active caspases. STAT1 is enriched in CSCs in cancer cell lines, patient-derived human breast cancer, and CD95high expressing glioblastoma neurospheres. CD95 stimulation of cancer cells induced secretion of Type I interferons (IFNs) that bind to Type I IFN receptors, resulting in activation of JAK kinases, activation of STAT1, and induction of a number of STAT1-regulated genes that are part of a gene signature recently linked to therapy resistance in 5 primary human cancers. Consequently, we identified Type I IFNs as drivers of cancer stemness. Knockdown or knock-out of STAT1 resulted in a strongly reduced ability of CD95L or type I IFN to increase cancer stemness. This identifies STAT1 as a key regulator of the CSC-inducing activity of CD95.
Keywords: STAT1, Fas, cancer stem cells, type I interferons, breast cancer, head and neck cancer
INTRODUCTION
CD95 (APO-1/Fas) is an apoptosis-inducing death receptor but also has multiple nonapoptotic and tumor promoting activities (Peter et al., 2015). Stimulation of CD95 increases migration of multiple cancer cells (Barnhart et al., 2004; Kleber et al., 2008). CD95 acts as a general growth promoter for many cancers (Chen et al., 2010) and it is also a driver and maintaining factor for cancer stem cells (CSCs) (Ceppi et al., 2014; Drachsler et al., 2016; Teodorczyk et al., 2014). CD95 couples to multiple signaling pathways, which all contribute to CD95’s nonapoptotic activities. These include MAP kinases, NF-κB, Scr kinases, and PI3K (reviewed in (Peter et al., 2015)). Activation of all these pathways are detectable within a few hours following CD95 stimulation. We recently demonstrated persistent chronic stimulation of CD95 in tumor cells, as it would occur in a tumor in vivo, by CD95 ligand (CD95L) resulted in an increase in cells with stem cell characteristics (Ceppi et al., 2014).
Signal transducer and activator of transcription 1 (STAT1) is an important regulator of the interferon (IFN) pathway (Levy and Darnell, 2002). Upon activation of IFN or certain cytokine receptors, STAT1 is phosphorylated by Janus-activated kinases (JAK) 1, JAK2 and TYK2 resulting in its tyrosine phosphorylation, dimerization and translocation to the nucleus either as a homodimer or heterodimer with STAT2 or STAT3 (Platanias, 2005) where it activates a large number of IFN regulated genes. When stimulated by either IFNα or IFNβ, IFN type I receptors (IFNAR1 and IFNAR2) activate a complex comprised of STAT1/STAT2 heterodimers together with IFN-regulatory factor (IRF) 9 (Platanias, 2005). STAT1 is viewed as a tumor suppressor in breast cancer, which is supported by the observation that STAT1 deficient mice spontaneously develop estrogen receptor α-positive luminal mammary carcinomas (Chan et al., 2012; Schneckenleithner et al., 2011). However, multiple reports also suggest that STAT1 could be tumor promoting in various cancers (Greenwood et al., 2012; Hix et al., 2013; Khodarev et al., 2010; Tymoszuk et al., 2014). STAT1 has also been linked to both radiation resistance and resistance to DNA damaging chemotherapy (Khodarev et al., 2007; Rickardson et al., 2005; Roberts et al., 2005; Tsai et al., 2007).
We have now identified STAT1 as a mediator of cancer stemness induced by CD95. Long-term stimulation of CD95 resulted in production of type I IFNs, which activates STAT1 following binding to IFNAR1 and IFNAR2, resulting in the upregulation of a number of IFN response genes that were recently shown to contribute to therapy resistance in human cancers. Consistently, treating either breast cancer cells or a squamous carcinoma cell line with either CD95L or type I IFNs increased their stemness after causing phosphorylation of STAT1, STAT2 and STAT3. Knockdown or knockout of STAT1 not only blunted the ability of CD95L or type I IFN to mediate cancer stemness, but also completely blocked the phosphorylation of STAT2 and STAT3. We have identified an IFNα/β-STAT1 signaling pathway as a key driver of cancer stemness that is utilized by CD95 to mediate its tumor promoting activities.
RESULTS
SILAC Analysis Identifies Proteins Upregulated after Long-Term Stimulation of CD95 and Downregulated after Treatment with a CD95L-Derived shRNA
Our recent data demonstrated that long-term stimulation of CD95 in cancer cells increases their stemness (Ceppi et al., 2014). This reprogramming process can take up to two weeks. We reasoned that proteins upregulated in cells induced to become CSCs after long-term stimulation of CD95 and downregulated in cells after treatment with a CD95L-derived shRNA (which we previously demonstrated to reduce cancer stemness (Ceppi et al., 2014)) could be candidates to mediate the CSC-inducing activity of CD95. In order to identify such proteins, we performed a stable isotope labeling by amino acids in cell culture (SILAC) analysis. The labeled cells were treated with either isotype matched control Ab or anti-APO-1 for 2 weeks or infected with either pLKO-scrambled or pLKO-shCD95L virus for 12 days. Figure S1A documents that the stimulation of the labeled cells with anti-APO-1 increased the number of CD44hi/CD24lo cells and the treatment with the CD95L-derived shRNA caused a reduction of these cells.
For each treatment set (CD95 stimulated or shCD95L treatment), SILAC labeled cells were subjected to mass spectrometry analysis. About 9000 individual peptides were identified to be differentially regulated in the two treatment groups with 229 peptides being significantly deregulated (FDR 5% and q values <0.05) in opposite directions in the cells stimulated through CD95 or after knockdown of CD95L (Figure 1A). These peptides represented 103 unique proteins. 18 of these proteins were deregulated at least 1.5 fold in each of the two treatment groups in opposite directions (Figure 1B). Interestingly, for 12 of the 18 proteins, we found evidence in the literature of the gene being involved in either CSCs, metastasis, or tumor promotion (proteins in red in Figure 1B). A highly and most significantly upregulated protein in the cells stimulated through CD95 and also most significantly downregulated protein in cells treated with shCD95L was STAT1. This was validated by Western blotting (Figure 1C).
Figure 1. STAT1 Is Phosphorylated and Upregulated after Long-Term Stimulation of CD95.
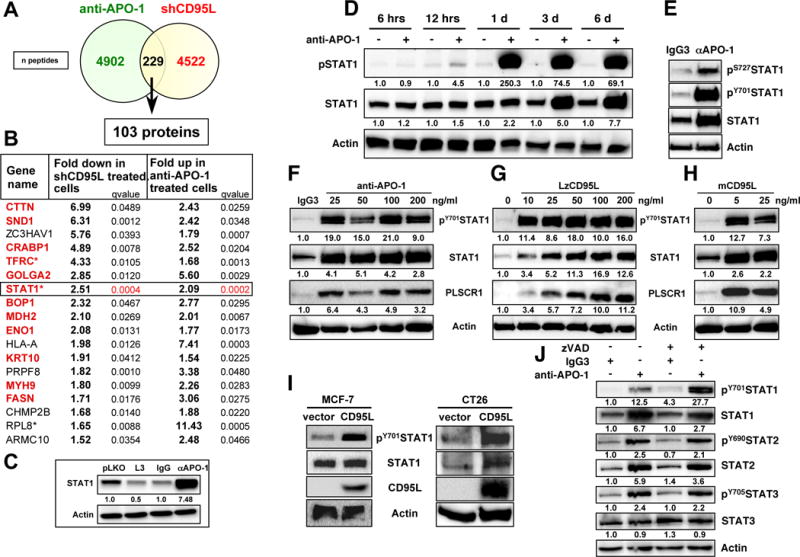
(A) Venn diagram showing the overlap of the number of peptides detected by SILAC to be upregulated in MCF-7 cells stimulated with anti-APO-1 for 14 days or following treatment with the CD95L derived shRNA shL3 for 12 days.
(B) List of all proteins from A that were deregulated at least 1.5 fold in opposite directions in either analysis. Proteins in red have been associated with either CSCs or tumor metastasis (see supplement for references. * These proteins were found to be regulated by STAT1 in a ChIP-Seq analysis (see Table S3).
(C) Western blot validation of STAT1. Quantification of bands normalized to actin is given.
(D) Western blot analysis of MCF-7 cells treated with control IgG3 (−) or anti-APO-1 (+) for the indicated times. Quantification of bands normalized to actin is given.
(E–H) Western blot analysis of MCF-7 cells treated with IgG3, anti-APO-1, LzCD95L, or membrane bound CD95L (mCD95L) for 4 days. For F-H quantification of bands normalized to actin is given.
(I) Western blot analysis of MCF-7 (left) or CT26 (right) cells stably expressing membrane bound human CD95L.
(J) Western blot analysis of MCF-7 cells treated with either IgG3 or anti-APO-1 with or without pretreatment with zVAD-fmk. Quantification of bands normalized to actin is given.
See also Figure S1 and Table S1.
Long-Term Stimulation of Breast Cancer Cells Induces STAT1
We decided to focus on STAT1, not only because STAT1 changes in the SILAC analysis were the most statistically significant, but also because STAT1 has been shown to confer resistance to CD95 mediated apoptosis (Zimmerman et al., 2012). Furthermore, as a transcription factor, it could be involved in regulating some of the other genes on the list. Indeed, binding of STAT1 has been reported using STAT1 specific Chip-Seq analyses for 10 of the 18 genes identified in the SILAC analysis (Table S1). Finally, Western blot analysis showed STAT1 to be the most highly induced protein after CD95 stimulation (Figure 1C and data not shown).
STAT1 is an early response gene in IFNγ stimulated cells (Levy and Darnell, 2002). A kinetic analysis over 6 days revealed that STAT1 upregulation was a fairly late event with no change in protein amount detectable one day after CD95 stimulation (Figure 1D). STAT1 phosphorylation on tyrosine 701 (pY701STAT1) was clearly detectable at day one and a very faint pSTAT1 band was detectable 12 hours after stimulation suggesting that tyrosine phosphorylation of STAT1 preceded its upregulation. In addition to phosphorylation on tyrosine 701, STAT1 also needs to be phosphorylated on serine 727 to achieve full transcriptional activity (Wen et al., 1995). 4 days after CD95 stimulation, both pY701STAT1 and pS727STAT1 were detectable (Figure 1E).
Activation of STAT1 downstream of CD95 was not limited to the stimulation with very small amounts of anti-APO-1 (Figure 1F) but was also observed in cells treated with a low dose of a highly active leucine-zipper tagged (Lz)CD95L (Figure 1G). Lower doses of LzCD95L resulted in a much larger increase in the appearance of pSTAT1 while induction of STAT1 protein expression required higher LzCD95L concentrations. pSTAT1 and STAT1 expression was also induced by treating cells with vesicles enriched in membrane bound CD95L (Figure 1H). Finally, we tested MCF-7 cells stably expressing a form of membrane bound CD95L that resulted in their reprogramming (as evidenced by increased expression of EMT and CSC markers (Ceppi et al., 2014) and an increase in ALDH1 activity (Figure S1B)). STAT1 was more highly phosphorylated in the CD95L-expressing cells when compared to control infected cells (Figure 1I, left panel). The same was found in the mouse colon cancer cell line CT26 stably expressing human membrane bound CD95L (CT26L) (Figure 1I, right panel). We previously showed that CT26L cells form spheres more readily than the parental CT26 cells (Ceppi et al., 2014).
One of the most prominent activities of CD95 is to activate caspases. To test whether caspase activation was required for a CD95-linked pathway that results in activation of STAT1, we treated MCF-7 cells with anti-APO-1 and pretreated the cells with the pancaspase inhibitor zVAD-fmk (zVAD) (Figure 1J). Inhibition of caspases did not impair the ability of CD95 to activate STAT1, suggesting that caspases are not required for CD95 to cause activation of STAT1. In fact, STAT1 phosphorylation was increased in zVAD treated CD95 stimulated cells. In addition, we determined that both STAT2 and STAT3 were also tyrosine phosphorylated, and STAT2 (just like STAT1) showed higher expression after 4 days of CD95 stimulation (Figure 1J). In summary, the data demonstrate that chronic stimulation of CD95 induces STAT1, STAT2 and STAT3 phosphorylation, followed by upregulation of STAT1 and STAT2 proteins.
Caspase-3 Blocks the Ability of CD95 to Activate STAT1
Phosphorylation and upregulation of STAT1 in CD95 stimulated cells was not limited to the ER positive breast cancer cell line MCF-7. It was also observed in the triple negative breast cancer cell line Hs578T (Figure S2A). Other breast cancer cell lines, including SK-BR-3 and HCC70, also responded to CD95 stimulation with activation of STAT1 (Figure 2A). Also, this phenomenon was not restricted to breast cancer cells as it was also found in the melanoma cell line MDA-MB-435 (Figure 2A). Interestingly, no STAT1 phosphorylation or upregulation was detected during CD95 stimulation of the ER positive breast cancer cell line T47D, even for up to 12 days (Figure S2B). However, in all cases (including T47D cells), pretreatment with zVAD allowed for (a more) efficient phosphorylation of STAT1 after CD95 stimulation, which in most cases resulted in an increase in STAT1 protein expression (Figure 2A). This suggested caspase activation is suppressing STAT1 activation downstream of CD95.
Figure 2. Caspase 3 Inhibits Activation of STAT1 Following Stimulation of CD95.
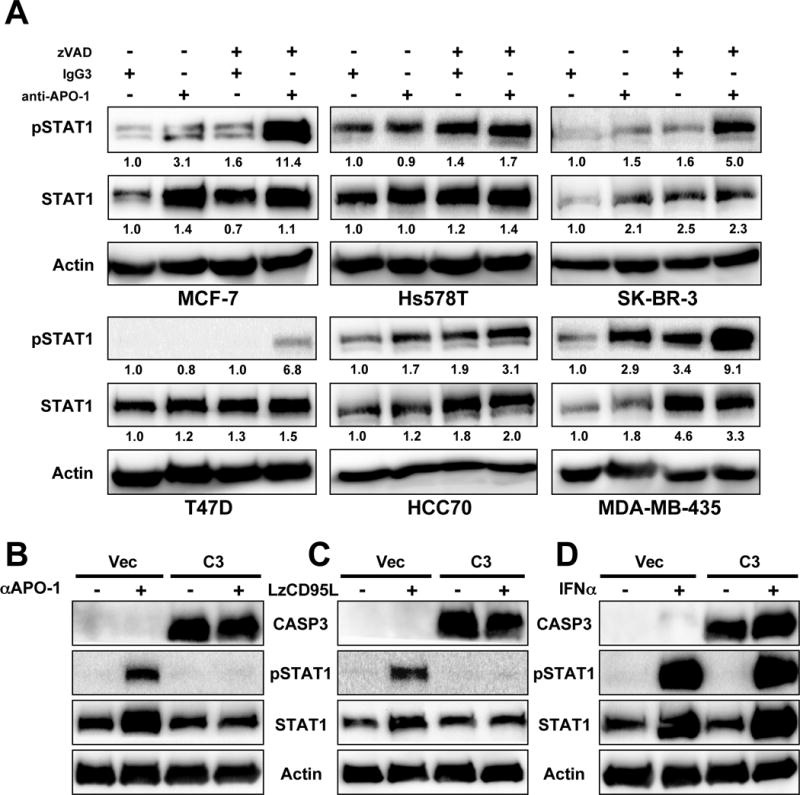
(A) Western blot analysis of 6 cancer cell lines after a 4 day stimulation through CD95 with or without treatment with zVAD. Quantification of bands normalized to actin is given.
(B–D) Western blot analysis of MCF-7 cells reconstituted to express caspase-3 (C3), or control vector (Vec) treated with either anti-APO-1 (+) (compared to IgG3 (-) (B), LzCD95L (C) or IFNα (D).
See also Figure S2.
To test whether lack of caspase-3 shifted the CD95 signaling pathway towards STAT1 activation, we tested MCF-7 cells (which lack caspase-3 (Janicke et al., 1998)) reconstituted to express caspase-3. Strikingly, in these MCF-7(C3) cells, both phosphorylation of STAT1 and STAT1 upregulation were completely blocked following treatment with either anti-APO-1 or LzCD95L (Figure 2B and C). In contrast, when cells were directly treated with interferon α (IFNα) to activate STAT1, caspase-3 expression did not have any effect (Figure 2D). The data suggest that when cancer cells acquire resistance to CD95-mediated apoptosis (i.e. through inhibition of caspases), they may activate STAT1 when chronically stimulated through CD95.
Identification of STAT1-Regulated Genes that Are Induced upon Activation of CD95
In order to identify genes induced in cancer cells upon prolonged stimulation of CD95, we subjected MCF-7 cells stimulated through CD95 for 2 weeks to a gene array analysis (Table S2). 88 (33.2%) of the 265 probes (231 genes) induced at least 1.5 (log2) fold upon CD95 stimulation were type I IFN-stimulated genes (ISG) reported to be upregulated in cells in response to viral infection (Schoggins et al., 2011). Remarkably, of the ten mostly highly upregulated genes, 7 were ISGs (Table S2) consistent with STAT1 being involved in their regulation.
The impression that CD95-induced genes were dominated by ISGs was confirmed when we subjected all genes that were up or downregulated at least 1.0 (log2) fold in MCF-7 cells stimulated through CD95 (Figure S3A) to a Lincscloud analysis (lincscloud.org). A connectivity analysis gave an indication of how many genes were coregulated in the MCF-7 data set between our CD95 stimulated MCF-7 cells and the Lincscloud data sets. Consistent with stimulation of CD95 inducing an IFN response among the top 25 most similar analyses 15 of them (60%) involved treatment of mostly breast cancer cell lines with IFNs (Figure S3B). Multiple gene signatures derived from cell lines representing other cancers treated with IFNs were also detected with slightly lower significance (Figure S3C).
To identify the genes that are directly regulated by STAT1 we performed a STAT1-specific ChIP-Seq analysis of MCF-7 cells stimulated for 10 days with anti-APO-1 (Table S3). This identified 1095 sites that were occupied by STAT1 upon CD95 stimulation (>4 fold enrichment over control treated cells), many of which have been described as STAT1 target genes before.
Strong Correlation between CD95 Induced Genes in MCF-7 cells and Genes Upregulated in the Radioresistant Squamous Cell Carcinoma Cell Line Nu61
Recent data suggest that STAT1 is upregulated in therapy-resistant cancer cells (Khodarev et al., 2007; Pitroda et al., 2009; Rickardson et al., 2005; Tsai et al., 2007; Weichselbaum et al., 2008). STAT1 was found to be upregulated in Nu61 cells, when compared to its parental squamous cell carcinoma cell line SCC61. Nu61 was rendered resistant to gamma radiation through 8 rounds of in vivo radiation in mice (Khodarev et al., 2004). Nu61 cells were reported to express a number of genes in a STAT1-dependent way, which were shown to contribute to their therapy resistance. The most prominent genes were part of an IFN regulated gene signature of 36 IFN-related DNA damage resistance signature (IRDS) that was recently described to be associated with both radiation resistance and general therapy resistance in 5 human cancers (Weichselbaum et al., 2008). Strikingly 19 (52.8%) of the IRDS genes were also upregulated in MCF-7 cells stimulated long-term through CD95 (Figure 3A). Of these genes STAT1, PLSCR1, USP18, and HERC8 were identified as direct STAT1 target genes in our STAT1-specific ChIP-Seq analysis (Figure 3B and C). The upregulation of these genes following CD95 stimulation was confirmed by real-time PCR (Figure 3D) and used in the following as general markers for CD95L and type I IFN induced STAT1 activation. PLSCR1 in particular tracked well with the induction of phosphorylated STAT1 (see Figure 1F–H).
Figure 3. Genes Upregulated in Long-Term Stimulated MCF-7 Cells Largely Overlap with IFN Response Genes Overexpressed in the Radioresistant Variant of SCC61, Nu61.
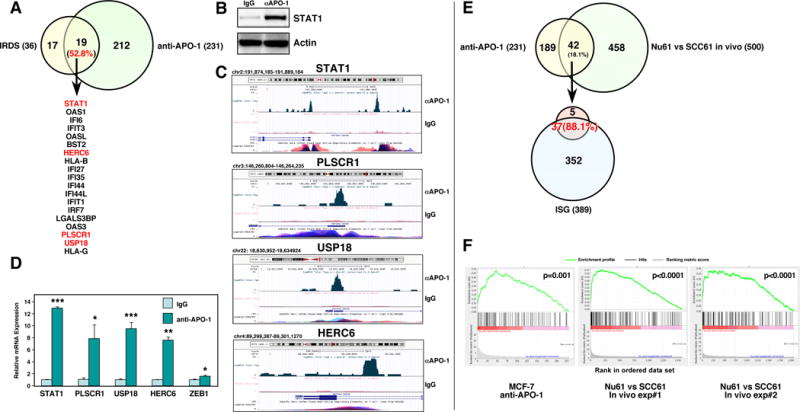
(A) Venn diagram showing the overlap between the IFN-related DNA damage resistance signature (IRDS) (Weichselbaum et al., 2008) and the genes upregulated in MCF-7 cells after 14 days of stimulation with anti-APO-1 (see Table S2). Genes in red were identified as direct targets of STAT1 using ChIP-Seq analysis (see C).
(B) Western blot of the samples (stimulated for 4 days) that were used for the ChIP-Seq analysis. (C) Read distribution and genomic localization around the promotor of three genes that were found to be induced in CD95 stimulated cells and identified as direct STAT1 targets.
(D) Validation of the upregulation of identified STAT1 regulated genes and of ZEB1 by real-time PCR in MCF-7 cells treated with anti-APO-1 for two days.
(E) Top, Venn diagram with the overlap between the CD95 stimulated genes and the genes that were differentially expressed between Nu61 and SCC61 when grown as tumors in mice (two independent experiments). Bottom, Venn diagram showing the overlap between the 42 genes identified above and the ISG genes.
(F) GSEA enrichment plot showing significant enrichment of up-regulated ISG genes in the data set of CD95 induced genes in MCF-7 cells and the two data sets containing the genes differentially expressed in two in vivo experiments between Nu61 and SCC61 tumors. p-value *<0.05, **<0.001; ***<0.0001.
See also Figure S3 and Tables S2 and S3.
Because of the similarity of the set of IFN-induced genes found in both a long-term CD95 stimulated breast cancer cell line and a radioresistant squamous cell carcinoma cell line, we wondered how similar the sets of genes were that were upregulated in these two very different cellular systems. Of the 231 genes upregulated in the CD95 stimulated MCF-7 cells, 42 (18.1%) were also differentially expressed in Nu61 cells grown as tumors in vivo when compared to SCC61 cells (Figure 3E). Of these 42 genes, 37 (88.1%) were part of the ISG signature of 389 genes (Schoggins et al., 2011). A gene set enrichment analysis of the ISG genes revealed that in both CD95 stimulated MCF-7 cells and Nu61 compared to SCC61 cells, ISG genes were some of the most highly upregulated genes (Figure 3F). These data suggest that CD95 may trigger a general STAT1-dependent mechanism that contributes to therapy resistance. Since long-term CD95 stimulation increases cancer stemness, which is a well established mechanism for cancer cells to become less sensitive to therapy (Meacham and Morrison, 2013), we wondered whether STAT1 activation would be involved in increasing CSCs in both these cell systems.
STAT1 Expression and Its Phosphorylation Correlates with Cancer Stemness
To determine whether activation of STAT1 is associated with an increase in cancer stemness upon CD95 stimulation, MCF-7 were treated for 6 days with either IgG3 or anti-APO-1 (Figure 4A). Together with the phosphorylation of STAT1, cortactin (CTTN), another CD95-regulated protein identified by the SILAC analysis (Figure 1B), was upregulated. Interestingly, upregulation of the breast cancer stem cell markers SOX2, and CD44 as well as downregulation of CD24 was also observed (Figure 4A). These data are consistent with a model in which STAT1 contributes to the acquisition of stemness in MCF-7 cells after long-term stimulation of CD95. Consistently, in addition to STAT1, both the STAT1 target PLSCR1 and SOX2 were upregulated in CSC-enriched 3rd generation spheres (Figure 4B). To test whether activation of STAT1 was detected in other established models of increased cancer stemness, we tested MCF-7 cells with inhibited miR-200c expression, which we recently showed to increase cancer stemness (Ceppi et al., 2014). In MCF-7 cells, expressing a miR-200c inhibitor reduced expression of CD24 (Figure 4C), enhanced STAT1 phosphorylation, and upregulated STAT1, PLSCR1 and SOX2 proteins (Figure 4D). STAT1 target genes PLSCR1, USP18 and HERC6, as well as the miR-200 target ZEB1 and SOX2 mRNAs, were upregulated as well (Figure 4E). To determine whether these findings in established cancer cell lines would also apply to primary breast cancer cells, we sorted breast cancer cells from patient-derived tumor xenografted (PDX) mice for CD44 high and CD44 low expression. Supporting the role of STAT1 as a CSC marker, CD44 high cells expressed much higher STAT1, PLSCR1, HERC6, USP18, SOX2, ZEB1, and BMI1 mRNA levels than CD44 low cells (Figure 4F). STAT1 protein was also more abundant in the CD44 high cells (Figure 4G). All these data suggest that STAT1 is activated in breast CSCs. Recently, it was demonstrated that CD95 acts as a marker and driver of stemness in glioblastoma of the mesenchymal subtype (Drachsler et al., 2016). In fact, it was shown that CD95 expression was a better predictor of cancer stemness than the established CSC marker CD133. To test whether CD95 high expressing GBM cells also showed evidence of increased STAT1 signaling, we sorted a human GBM cell line of the mesenchymal subtype grown in neurospheres into CD95 high (CD95hi) and CD95 low (CD95lo) populations and harvested RNA from each. We subjected the RNA to a qPCR analysis (Figure 4H). Compared to the unsorted and CD95lo cells CD95hi cells expressed much higher levels of STAT1, the STAT1 marker genes PLSCR1, HERC6 and USP18, the general stem cell maker BMI1, and ZEB1. These data are consistent with CD95 expression to correlate with cancer stemness in GBM.
Figure 4. STAT1 Is Phosphorylated and Upregulated in Cancer Cells with Enriched Stemness.
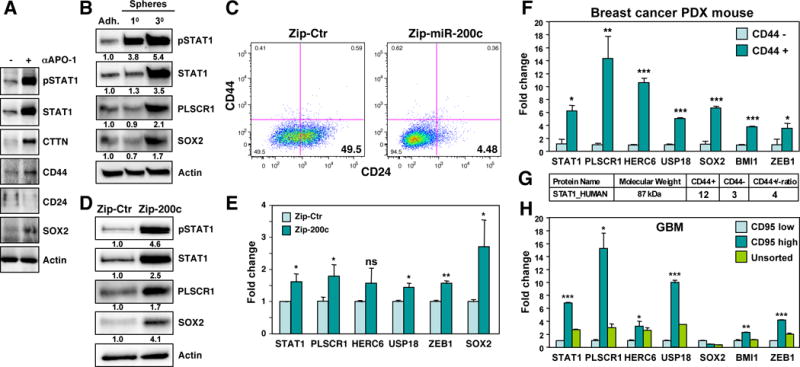
(A) Western blot analysis of MCF-7 cells treated with either IgG3 or anti-APO-1 for 6 days.
(B) Western blot analysis of MCF-7 cells grown under adherence or as first or third generation mammospheres. Quantification of bands normalized to actin is given.
(C) CD44/CD24 surface staining of MCF-7 cells infected with either a miR-Zip control vector (Zip-Ctr) or the miR-Zip-200c inhibitor.
(D) Western blot analysis of the cells in C. Quantification of bands normalized to actin is given.
(E) Real-time PCR analysis of STAT1, STAT1 target genes and the miR-200 targets ZEB1 and SOX2 of the cells in D.
(F) Real time PCR analysis of PDX mouse derived primary human breast cancer cells sorted for high or low CD44 expression.
(G) Identification of STAT1 protein derived peptides by mass spec analysis of the cells in F.
(H) Real time PCR analysis of GSC20 GBM cells sorted for high or low CD95 expression. p-value *<0.05, **<0.001; ***<0.0001; ns, not significant.
See also Figure S4.
We recently showed long-term stimulation through CD95 increases the ability of MCF-7 cells to grow in spheres (Ceppi et al., 2014), and we now show this involves activation of STAT1. The data on increased STAT1 expression suggested that Nu61 cells would also have increased stemness when compared to SCC61 cells. Not only did Nu61 cells have increased expression of STAT1 and its target genes, they also showed increased expression of the stem cell factors SOX2 and BMI1 (Figure S4A). In addition to increased STAT1 and PLCSC1 protein expression and increased STAT1 phosphorylation, they also expressed more SOX2 protein when compared to SCC61 cells (Figure S4B). Interestingly, Nu61 cells formed spheres more readily when compared to SCC61 cells (Figure S4C). Also, similar to MCF-7 cells, growing SCC61 cells in spheres increased the amount of pSTAT1, STAT1, PLSCR1 and SOX2 (Figure S4D). In contrast, growing Nu61 cells under sphere forming conditions did not further increase the expression of these proteins. Finally, we found that also in SCC61 cells long-term stimulation of CD95 induced pSTAT1, STAT1 and PLSCR1 expression, suggesting that STAT1 was activated. Consistent with the sphere forming data this had less of an effect on Nu61 cells (Figure S4E). Finally, CD95 stimulation of SCC61 increased their ability to form spheres (Figure S4F). Again, the CD95-mediated effect was much less pronounced in Nu61 cells. The data suggest that Nu61 cells are enriched in CSCs, that STAT1 may be involved in the generation of CSCs, and that this can be induced by long-term CD95 stimulation.
Type I IFNs Are Required for CD95 to Induce CSCs
Based on our data we hypothesized that CD95 stimulation upregulates IFNs, which leads to STAT1 phosphorylation and subsequent upregulation of ISGs, through binding to IFN receptors and activation of JAK kinases. To test this hypothesis we stimulated MCF-7 cells for 4 days with either anti-APO-1 or LzCD95L and quantified IFNα and IFNγ in the cell lysates and cell supernatants by ELISA (Figure 5A). Both treatments caused a robust production and secretion of IFNα by the cells, whereas IFNγ expression was barely induced. The induction of IFNα was not inhibited by treating MCF-7 cells with the caspase inhibitor zVAD (Figure S5A) and therefore was not the result of dying cells releasing danger signals. Because MCF-7 cells are devoid of caspase-3 this experiments excluded that the activation of any other was required for CD95 driven production of type I IFNs.
Figure 5. CD95 Stimulation Induces Type I IFNs which Have Stem Cell Driving Activities.
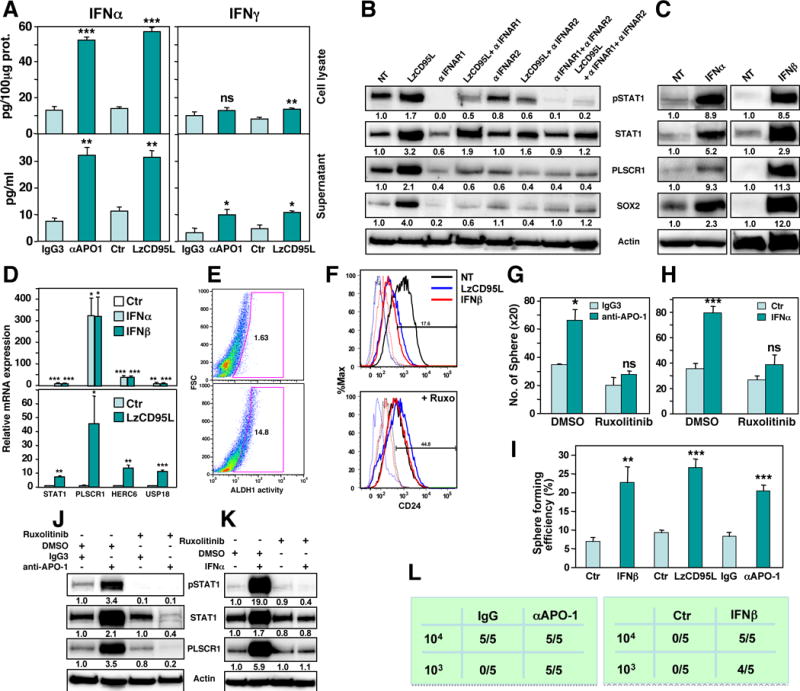
(A) ELISA quantification of IFNα and IFNγ produced by MCF-7 cells treated with either IgG3, anti-APO-1, or LzCD95L for 2 days.
(B) Western blot analysis of MCF-7 cells treated with LzCD95L for 4 days in the presence of neutralizing anti-IFNAR1 and/or IFNAR2 antibodies. Quantification of bands normalized to actin is given.
(C) Western blot analysis of MCF-7 cells control treated or treated with IFNα or IFNβ for 4 days. Quantification of bands normalized to actin is given.
(D) Real-time PCR quantification of mRNAs of STAT1 regulated genes in MCF-7 cells treated with either IFNα, IFNβ (top) or LzCD95L (bottom).
(E) Quantification of ADLH1 activity in MCF-7 cells treated for 6 days with IFNα.
(F) CD24 surface staining of MCF-7 cells treated with IgG3, anti-APO-1, or IFNβ for 6 days in the absence or presence of Ruxolitinib.
(G, H) Sphere formation of MCF-7 cells after treatment with either IgG3 or anti-APO-1 (G) or IFNα (H) for 6 days in the absence or presence of Ruxolitinib.
(I) Single cell sphere formation assay of MCF-7 cells after treatment with either IgG or anti-APO-1 or IFNβ for 6 days.
(J, K) Western blot analysis of MCF-7 cells treated with either IgG3 or anti-APO-1 (J), or IFNα (K) for 4 days and DMSO solvent control or Ruxolitinib.
(L) Tumor initiation frequency in NSG mice injected with MCF-7 cells, treated with either IgG3, anti-APO-1 or IFNβ for 9 days. 103 cells were injected into the left 4th mammary duct or 104 cells the right 4th duct and tumor initiation was monitored by IVIS imaging 7 days after tumor cell injection. p-value *<0.05, **<0.001; ***<0.0001; ns, not significant.
See also Figures S5 and S6.
To determine the contribution of type I IFNs to the STAT1 activating activity of CD95 we pretreated CD95 stimulated cells with antibodies blocking either IFN α/β receptor 1 or 2 (IFNAR1, IFNAR2) or both (Figure 5B). The combination of both antibodies almost completely blocked CD95L-induced phosphorylation of STAT1 and severely reduced upregulation of STAT1 and its target PLSCR1. In addition, it also blocked the induction of SOX2 expression, suggesting that type I IFNs may be mediating the stemness inducing activity of CD95. Interestingly, while we detected a fraction of cells that expressed IFNAR1 or IFNAR2 with little CD95 expression, there were barely any cells that only expressed CD95 (Figure S5B) suggesting that most cells stimulated through CD95 should be receptive to type I IFN.
Type I IFNs Promote Cancer Stemness through Activation of STAT1
Type I IFNs have not been implicated in inducing cancer stemness. However, similar to CD95 stimulation treatment of MCF-7 cells with universal IFNα or IFNβ not only caused phosphorylation of STAT1 and upregulation of STAT1 and PLSCR1 protein, but also induced SOX2 expression (Figure 5C). Interestingly, the relative ratio of mRNA induction of STAT1 and its targets PLSCR1, HERC6 and USP18 was very similar between MCF-7 cells treated with either IFNα/β or CD95L (Figure 5D). Most importantly, Type I IFN treatment increased cancer stemness as monitored by increased ALDH1 activity (Figure 5E). It caused a similar reduction in CD24 expression as in CD95 stimulated cells (Figure 5F) and increased sphere formation (Figure 5G and H). Increased capacity to form spheres was also seen when cells stimulated with either IFNα or anti-APO-1 or LzCD95L were plated at one cell per well (Figure 5I and Figure S5C). This activity of type I IFN and CD95 stimulation was dependent on the canonical JAK/STAT pathway as treatment with the JAK1/JAK2 inhibitor Ruxolitinib blocked the phosphorylation and upregulation of STAT1 and its target PLSCR1 in CD95 and IFNα stimulated cells (Figure 5J and K), the reduction of CD24 expression (Figure 5F) and the ability to form spheres (Figure 5G, H). These data confirmed that CD95 activates STAT1 through the canonical type I IFN pathway resulting in an increase in cancer stemness and pointed at type I IFNs as a driver of cancer stemness. To test whether stimulation through CD95 or treatment with IFNs increase the tumor initiating capacity of the cells, MCF-7 cells were treated for 9 days with either anti-APO-1 or IFNβ and injected into the fat pad of NGS mice at either 103 or 104 cells per injection (Figure 5L). At 103 cells per mouse only the cells treated with either IFNβ or anti-APO-1 formed tumors after a week confirming that either stimulus increased the tumor initiating frequency of the cells.
Many of these results, including the production of IFNα upon CD95 stimulation and the activity of IFNα to drive cancer stemness, were also found with SCC61 (Figure S6). Nu61 already had a constitutively high expression of stem cell markers. These data suggest a type I IFN-driven increase in cancer stemness.
STAT1 Is Part of the Stem Cell-Inducing Activity of CD95 and Contributes to the Stemness-Inducing Activity of Type IFN
STAT1 has not been linked to CSCs before. To test whether STAT1 played a role in the CSC inducing activity of CD95, STAT1 was knocked down using two shRNAs (Figure S7A). Both shRNAs impaired the ability of CD95 stimulation to cause upregulation of CTTN and downregulation of CD24 protein by Western blotting and reduced the number of CD95 induced CD44hi/CD24lo cells (Figure S7B). A similar result was obtained when cells were treated with a siRNA pool targeting STAT1, which efficiently silenced STAT1 expression (Figure S7C). Knockdown of STAT1 significantly reduced the ability of CD95 to induce stemness as shown by CD44/CD24 surface staining (Figure S7D), ALDH1 activity (Figure S7E), and sphere formation (Figure S7F). Interestingly, knockdown of STAT1 alone resulted in a reduction of the ability of cells to form spheres. This was also found for both SCC61 and Nu61 cells (Figure S7G). However, it was more effective in Nu61 cells that express STAT1 the most resulting in reduced SOX2 expression after STAT1 knockdown.
Finally, we deleted two regions in STAT1 using CRISPR/Cas9 gene editing. We either deleted part of the gene in exon 3/4 containing the start codon (E deletion in Figure 6A–C) or a piece of DNA within the region in exon 23/24 that contains the two phosphorylation sites (Y701 and S727, P deletion in Figure 6A–C). Both deletions resulted in cells lacking any detectable STAT1 protein (Figure 6D). Long-term stimulation of CD95 with either LzCD95L (Figure 6E) or anti-APO-1 (data not shown) demonstrated that the STAT1 knockout cells had a severely reduced ability to unable to respond to CD95 stimulation with upregulation of the STAT1 target PLSCR1 protein (Figure 6D) or any of the STAT1 activation marker mRNAs (Figure S8A). This defect was not due to reduced CD95 expression as evidenced by both Western blot analysis (data not shown) and surface staining for CD95 (Figure S8B).
Figure 6. STAT1 Is a Key Factor in the Signaling Pathway that Connects CD95 to Cancer Stemness.
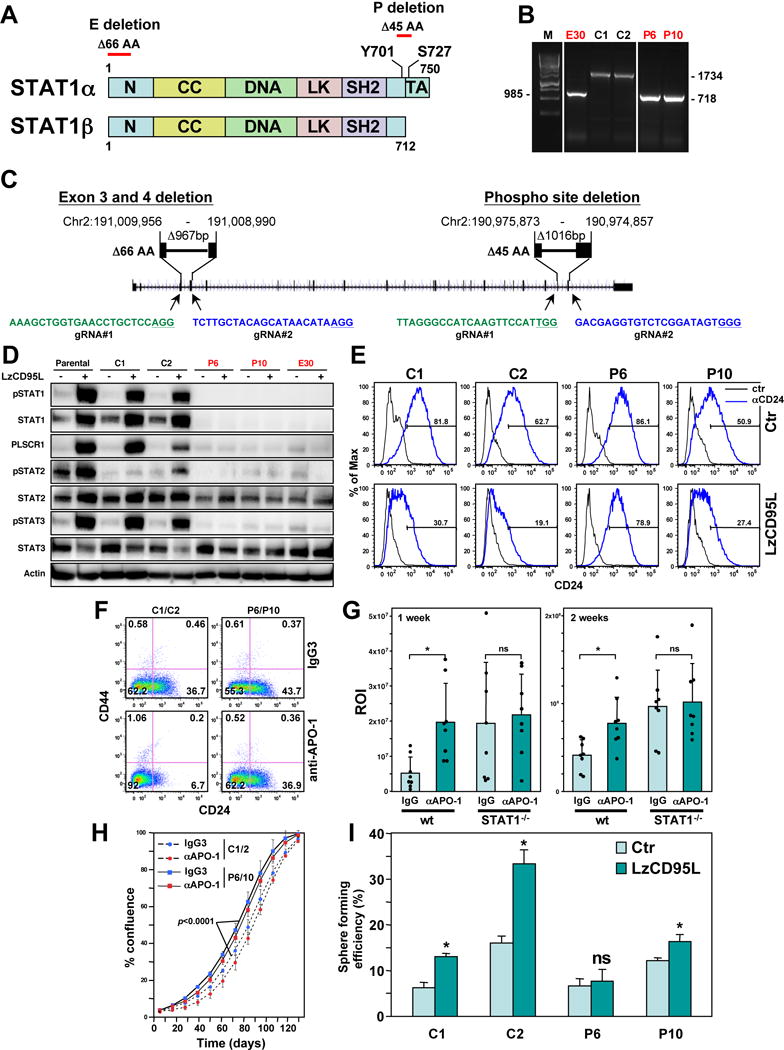
(A) Schematic of the STAT1 protein and the two sites deleted by CRISPR/Cas9 gene editing.
(B) PCR confirmation of homozygous deletions at the two sites in STAT1 in three isolated clones (labeled in red).
(C) Genomic localization of the two CRIPSR/Cas9 deletions in the human STAT1 gene (Human Dec. 2013 (GRCh38/hg38) assembly). The PAM sites in the guide (g)RNAs are underlined. gRNAs in green target the sense and in blue the antisense strand.
(D) Western blot analysis of MCF-7 cells, Cas9 transfected wt clones and STAT1 k.o. clones either control treated or treated with LzCD95L for 4 days.
(E) CD24 surface staining of the clones treated as in D.
(F) CD44/CD24 surface staining of cell pools of clones C1 and C2 and P6 and P10 (now expressing a luciferase plasmid for the in vivo experiment in H) after 7 days of treatment with either IgG3 or anti-APO-1.
(G) Bioluminescence of tumors in NSG mice one week and two weeks after injection of the cells analyzed in F into the fat pad.
(H) Growth of the pooled cells after 9 days of treatment with either IgG3 or anti-APO-1. p-value was calculated using ANOVA.
(I) Percent of wells with spheres after plating MCF-7 cells (treated with LzCD95L for 6 days) at one cell per well in 96-well plates. p-value *<0.05; ns, not significant.
See also Figures S7–S9.
Interestingly, cells lacking STAT1 could also no longer phosphorylate STAT2 or STAT3, suggesting that activation of these STAT proteins required the presence of STAT1 in CD95 stimulated cells. In addition, STAT2 protein was reduced in cells lacking STAT1 as STAT2 was reported to be a STAT1 target gene by ChIP-Seq analysis (Satoh and Tabunoki, 2013). Consistent with STAT1 playing an important role in the stemness-inducing activity of CD95 STAT1 deficient cells stimulated through CD95 were less able to downregulate CD24 (Figure 6E).
To determine the contribution of STAT1 to the tumor promoting activity of CD95 in vivo, we treated two Cas9 expressing wt MCF-7 clones or two STAT1 k.o. P deletion clones with anti-APO-1 for 9 days and injected the pooled cells (C1/C2 or P6/P10) into the fat pad of female NSG mice. We chose to stimulate CD95 with anti-APO-1, which does not crossreact with mouse CD95, to ensure that endogenous CD95 would not be stimulated in the mice after injection of MCF-7 cells likely decorated with the antibody. CD44/CD24 staining of the two cell pools two days before injection demonstrated that the STAT1 k.o. cells were less efficiently reprogrammed by CD95 stimulation when compared to the pool of wt cells (Figure 6F). One week after injection, the mice that received the wt cells, prestimulated through CD95, had tumors about three times the size of control tumors (Figure 6G). This increase was still significant two weeks after injection but less pronounced, presumably due to the waning of the effect of CD95 stimulation. The fact that the viability of the wt cells stimulated through CD95 was lower at the time of injection than the control treated cells (Figure 6H), but the cells gave rise to larger tumors suggests that not general cell viability but increased tumor initiating capacity was the cause for the increased tumor size in the mice harboring CD95 stimulated wt cells. This was in contrast to the STAT1 k.o. cells. Here, CD95 stimulation did not increase tumor growth, although growth of the k.o. cells was higher than that of the wt cells. The increased growth of the k.o. cells was not due to a decrease in CD95 mediated apoptosis sensitivity of the k.o. clones (Figure S8C) but the STAT1−/− cell pool exhibited a significantly higher growth rate in vitro than the pool of wt clones (Figure 6H). This was confirmed by performing TUNEL staining on the tumors. Less than 1% TUNEL positive cells were detected in any of the conditions (data not shown). Although we could not continuously stimulate MCF-7 cells in vivo, MCF-7 control and STAT1 k.o. clones stimulated for 6 days with either anti-APO-1 or LzCD95L showed an increased ability to form spheres from single cells; this activity that was strongly reduced in the STAT1 k.o. cells (Figure 6I). In summary, the data suggest that STAT1 is a factor that contributes to cancer stemness induced by CD95 and that STAT1 may generally have stem cell promoting activities.
Using the STAT1 k.o. cells, we tested whether the activity of type I IFN to increase cancer stemness was also dependent on STAT1. Parental MCF-7 cells, the two wt Cas9 expressing clones C1, C2 and the two STAT1−/− clones P6 and P10 were treated with IFNβ for 6 days and analyzed by Western blotting (Figure S9A). Similar to the stimulation with CD95L the direct stimulation with IFNβ resulted in robust phosphorylation of STAT1, STAT2 and STAT3 and upregulation of STAT1 and STAT2. Again, in the absence of STAT1 neither STAT1 nor STAT3 could be phosphorylated suggesting that in MCF-7 cells activation of both STAT2 and STAT3 are dependent on the presence of STAT1. Importantly, however, downregulation of CD24 and the ability to from spheres from single cells was severely impaired in the STAT1 k.o. cells (Figure S9B and S9C).
To confirm that it was the STAT1 deficiency in the k.o. cells that impaired the ability of the cells to respond to either CD95 stimulation or IFN treatment two of the STAT1−/− clones (P6 and P10) were reconstituted with lentiviral STAT1. These STAT1 reconstituted cells regained the ability to phosphorylate STAT1 and STAT3 and upregulate STAT1 and PLSCR1 in response to either LzCD95L (Figure 7A) or IFNβ (Figure 7B). Both reconstituted clones responded with a more pronounced downregulation of CD24 when stimulated (Figure 7C). The P6 clone which had lost the ability to respond to either CD95L or IFNβ completely was able to activate STAT1 in response to both stimuli (Figure 7D) and these STAT1 reconstituted cells formed spheres more efficiently when plated as single cells per well (Figure 7E) suggesting that they regained the ability to respond to CD95L and IFNs with increased stemness. In summary, our data identify type I IFN as part of a cell autonomous pathway to induce CSCs when cancer cells are stimulated through CD95 and identify type IFNs as novel driver of cancer stemness.
Figure 7. Reconstitution of STAT1 into STAT1 Knock-Out Cells Reconnects CD95 to the IFN/JAK/STAT1 Signaling Pathway.
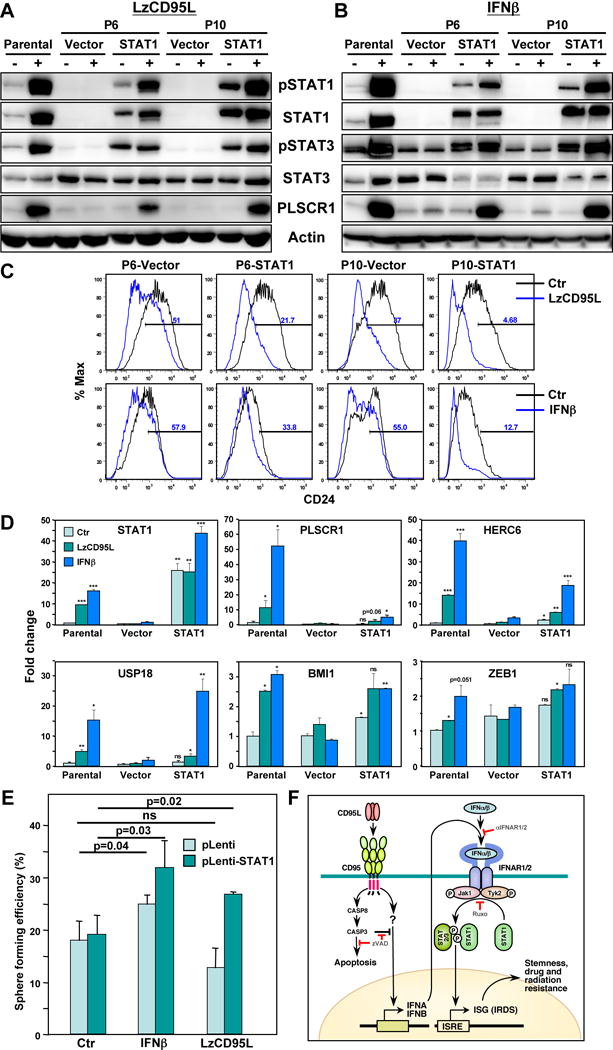
(A,B) Western blot analysis of parental MCF-7 cells, two STAT1 k.o. clones (P6 and P10) infected with either a control virus (Vector) or a STAT1 expression virus cultured without (-) or with (+) LzCD95L (A) or IFNβ (B) for 4 days.
(C) CD24 surface staining of STAT1 k.o. clones infected with pLenti vector or pLenti-STAT1 either untreated and treated with either LzCD95L or IFNβ. for six days.
(D) Real-time PCR analysis of mRNAs in parental MCF-7 cells, clone P6 cells infected with and empty (Vector) or with a STAT1 expressing lentivirus after a six day treatment with either LzCD95 or IFNβ. p-value *<0.05, **<0.001; ***<0.0001; ns, not significant. P-values of parental cells were compared to Ctr. P-values of STAT1 reconstituted cells are compared to Vector expressing cells.
(E) Percent of wells with spheres after plating MCF-7-STAT1 k.o. P6 cells expressing Vector or STAT1 (treated with either IFNβ or LzCD95L for 6 days) at one cell per well in 96-well plates.
(F) Model on the activity of CD95 to mediate cancer stemness through activating a type I IFN/JAK/STAT1 pathway. The JAK/STAT1 signaling pathway and its targeted genes is depicted in a simplified form as it is not known whether all canonical components are involved in the newly described CSC driving activity. zVAD, zVAD-fmk; Ruxo, Ruxolinitib. ISG, IFN sensitive genes; IRDS, interferon-related DNA damage resistance signature. For details see text.
DISCUSSION
CD95 possesses tumor promoting activities (Peter et al., 2015). We recently demonstrated that chronic stimulation of CD95 increases and maintains cancer stemness (Ceppi et al., 2014). We now demonstrate that this activity greatly depends on the activation of a STAT1-dependent type I IFN pathway and identify type I IFNs as drivers of cancer stemness.
Short-term stimulation (8 hours) of CD95 expressing MCF-7 cells, rendered apoptosis-resistant by overexpressing Bcl-xL, previously resulted in the identification of a gene signature that was consistent with activation of the transcription factor NF-κB (Barnhart et al., 2004). In fact, a re-analysis of these data revealed that of the 56 gene probes that showed a signal change of >1.5 fold in two independent analyses, 33 (58.9%) were known NF-κB regulated genes (Table S4). This analysis also identified 5 known IFN regulated genes, including TNFAIP3 (A20), IRF1, CEBPD, BCL3, and JUNB. These data suggest that early after CD95 stimulation NF-κB is activated. However, upon long-term stimulation CD95, the NF-kB signature of induced genes switches to an IFN signature of genes. Experiments are ongoing to determine whether NF-κB activation is involved in the induction of ISGs downstream of CD95.
By testing STAT1 k.o. MCF-7 cells, we found that in cells treated with either CD95L or type I IFN, STAT1 not only drives its own expression but also the expression of STAT2; this is consistent with STAT2 being a STAT1 regulated gene (Satoh and Tabunoki, 2013). Both STAT2 and STAT3 were phosphorylated in CD95L or IFNβ treated cells. Remarkably, in complete STAT1 MCF-7 k.o. cells, neither CD95L nor IFNβ could cause any detectable phosphorylation of STAT3. This activity was fully restored upon reexpression of STAT1. These observations can be explained by assuming that both STAT2 and STAT3 need to heterodimerize with STAT1 to undergo phosphorylation.
Based on our data, we propose the following model to explain some of the tumor promoting activities of CD95 (Figure 7F). Upon short-term exposure to CD95L, cancer cells have at least two ways to respond: If they are sensitive to CD95-mediated apoptosis, they will undergo canonical caspase-mediated apoptosis. However, most cancer cells are resistant to CD95-mediated apoptosis. When these cells are stimulated, they activate nonapoptotic signaling pathways including NF-κB, which increases motility and invasiveness (Barnhart et al., 2004). Upon chronic stimulation, as one would expect in a physiologically relevant situation of slowly growing tumors with infiltrating immune cells, and especially under conditions of reduced caspase activity, type I IFNs are secreted and bind to type I IFN receptors, activating JAK kinases and causing activation of STAT1, either as a heterodimer with STAT2 or alternatively with STAT3. This in turn results in the induction of a number of ISGs, including members of the ISRE signature of genes that contribute to the increase in therapy resistance of cancer presumably through an increase in stemness.
It was recently suggested that cells produce IFNβ when undergoing caspase-inhibited apoptosis (White et al., 2014). CD95 mediated apoptosis is dependent on activation of caspases (Peter and Krammer, 1998). Our findings that in all tested cells (including the caspase-3 deficient MCF-7 cells), stimulation of CD95 resulted in a more profound activation of STAT1 when caspases were inhibited combined with our observation that under such conditions cells do not show any signs of cell death now suggests that cell death is not required for CD95 to engage a Type I IFN pathway. The response of cells to IFN downstream of CD95 is not due to dying cells. Interestingly, however, while inhibiting caspases allowed for a more efficient activation of STAT1 (see Figures 1J and 2A) it did not increase the amount of IFN produced (see Figure S5A). This suggests that a caspase other than caspase-3 inhibits activation of STAT1 independent of the release of IFNs (not shown in Figure 7F).
In addition to STAT1, there were three genes significantly upregulated at the mRNA and protein level in CD95-stimulated MCF-7 cells that were found to be regulated by STAT1 in the ChIP-Seq analysis and also upregulated in Nu61 versus SCC61: 1) Phospholipid scramblase 1 (PLSCR1) is a member of a family of membrane proteins that has been proposed to contribute to the reorganization of plasma membrane phospholipids (Zhou et al., 1997). It functionally contributes to cytokine-regulated cell proliferation and differentiation, and it is required for normal myelopoiesis. Stem cell factor and G-CSF were reported to induce marked increases in PLSCR1 levels (Zhou et al., 2002) and blocking PLSCR1 impairs tumorigenesis of colorectal carcinomas (Fan et al., 2012). 2) USP18/UBP43 is an ubiquitin-specific protease that removes ISG15 from ISGylated IFN target proteins, and mice lacking USP18 are hypersensitive to IFN (Malakhova et al., 2003), suggesting that USP18 acts to dampen the IFN response. It has been shown to serve as a negative prognostic marker for bladder cancer (Kim et al., 2014). 3) HERC6 belongs to the HERC family of ubiquitin ligases. These three genes were also more highly expressed in cells with increased stemness, either in MCF-7 cells after repressing miR-200c, SKBR3 or SCC61 cells when grown under sphere forming conditions, in CD44 high primary PDX mouse-derived breast cancer cells, and in glioblastoma cells of the mesenchymal subtype grown as neurospheres and sorted for CD95. Finally, they were highly upregulated in MCF-7 and SCC61 cells treated with type I IFN. We have used these three genes throughout our work as markers of STAT1 activation. It remains to be tested which STAT1 regulated genes are required for the stemness-inducing activity of CD95 and IFN signaling.
While acute IFNα treatment promotes the proliferation of dormant hematopoietic stem cells in vivo (Essers et al., 2009), neither STAT1 nor any of the three target genes have been directly connected to cancer stemness. STAT1 has been described as a cell autonomous tumor suppressor of mouse mammary gland tumorigenesis, and globally STAT1 deficient mice spontaneously develop ER positive luminal breast cancer (Chan et al., 2012; Klover et al., 2010; Schneckenleithner et al., 2011). In contrast to these reports, recent studies have identified STAT1 as a tumor promoter for a number of human cancers and it serves as an adverse prognostic marker (Greenwood et al., 2012; Hix et al., 2013; Khodarev et al., 2010; Kovacic et al., 2006; Tymoszuk et al., 2014). In addition, a link between STAT1 and therapy resistance has been uncovered (Khodarev et al., 2004; Khodarev et al., 2007; Rickardson et al., 2005; Weichselbaum et al., 2008). Interestingly, one report demonstrated that STAT1 signaling contributes to radioresistance in breast cancer-initiating cells (Zhan et al., 2011).
The findings reported here are unlikely limited to breast cancer or squamous cell carcinoma. CD95 was recently described as a marker and driver of cancer stemness in GBM (Drachsler et al., 2016). Our data on the increased expression of STAT1 and STAT1 targets and the stem cell marker BMI1 in CD95hi GBM cells when compared to CD95lo cells suggests that STAT1 may also be involved in maintaining stemness of mesenchymal subtype of glioma cancer cells.
We acknowledge that the study of cancer stem cells is complex (Medema, 2013) and it is not entirely clear what role CD95 and type I IFN play in promoting all aspects of CSCs. However, the combination of monitoring proper surface markers, expressed CSC driving genes, single cell sphere formation assays and in vivo tumor initiation assays used in this study all point toward a strong and novel cell autonomous function of both CD95 and type I IFNs in increasing cancer stemness. This explains some interesting earlier reports that connected STAT1 with therapy resistance in human cancers. One remarkable finding is that we found an 88% overlap in gene expression between the 42 shared genes expressed in a breast cancer cell line stimulated in vitro through CD95 and a squamous carcinoma cell line rendered radioresistant in vivo, and the 389 genes that were originally identified as antiviral ISGs (Schoggins et al., 2011). Furthermore, the expression of a subgroup of only 36 of these genes (the IRDS) distinguishes 5 different human cancers (head & neck, lung, breast, prostate cancer, and high grade glioma) patients into groups of poor and favorable prognosis (Weichselbaum et al., 2008). Most remarkably, for breast cancer the IRDS was found to serve as a therapy (both radiation and adjuvant chemotherapy) predictive marker for relapse while the expression of the IRDS in patients without any treatment was not predictive of outcome. Our data now suggest that the IRDS could be the result of chronic stimulation of CD95 on the cancer cells.
EXPERIMENTAL PROCEDURES
General Methods
Long-term stimulation of cancer cells through CD95, knock down of CD95L using shRNAs, sphere forming assays, CD44/CD24 surface staining, Western blot analyses, quantitative real time PCR were done as previously described (Ceppi et al., 2014).
SILAC Analysis
For SILAC labeling, MCF-7 cells were grown in media with 13C6-L-lysine and 13C6-L-arginine (heavy) in the treatment group or with regular lysine and arginine (light) in the control group. All reagents were from Pierce® (SILAC Protein Quantitation Kit, Fisher Scientific). Cells were labeled with the heavy amino acids for 5 doublings until more than 98% of amino acids in the cells had been replaced by heavy amino acids as assessed by mass spec analysis (data not shown). The light labeled and heavy labeled cells were treated with either anti-APO-1 or isotype control antibody for 12 days or infected with pLKO-scrambled or pLKO-shCD95L virus for 10 days, respectively. After treatment (six biological replicates per condition), 2×106 labeled cells were lysed with 8M urea, 20mM HEPES, pH8, and sonicated three times (30 second per cycle with 30 second of resting time) on ice. Lysates were centrifuged at 12,000g for 15 min, and the samples were diluted to 4M urea by adding 20mM HEPES, pH8. After protein quantification, lysates were kept frozen at -80C until mass-spec analysis.
CRISPR/Cas9 Deletion of STAT1
Two regions in STAT1, one covering parts of exon 3 and 4 and one parts of exon 23 and 24 were deleted in MCF7 cells using two guideRNAs. Deletions in single cell clones were verified by genomic PCR using external primer pairs and by DNA sequencing.
Supplementary Material
Acknowledgments
We are grateful to Denise Scholten for performing the GSEA analysis and to Drs. Platanias and Horvath for helpful discussions. We would like to thank Dr. Youjia Hua for analyzing the Illumina gene array data and Dr. Yingming Zhao for assessing the labeling efficiency of the cells used in the SILAC experiment, and Angel Alvarez for providing the GSC20 cells. This work was funded by a DOD postdoctoral fellowship W81XWH-13-1-0301 (to P.C.), R00CA160638 and ACS127951-RSG-15-025-01-CS (to H.L.), a Northwestern Memorial Foundation-Lynn Sage Cancer Research Foundation grant, R01CA149356 and R35CA197450 (to M.E.P.).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information includes Supplemental Experimental Procedures, 9 figures and five tables, can be found with this article online at
ACCESSION NUMBERS
The accession number for the ChIP-Seq and expression data reported in this paper is GEO: GSE81860.
AUTHOR CONTRIBUTIONS
P.C. planned the study and performed experiments. A.S.Q, C.L., A.E.M., W.P., and S.B. performed experiments. N.N.K. and R.R.W. provided the squamous carcinoma cells and gene array data of these cells grown in mice. L.M. helped with the orthotopic injection of tumor cells. J.K. and J.Y. assisted with the ChIP-Seq analysis, J.C.Z. performed computational analyses. W.C., X.L provided the sorted PDX mouse cells and X.L. performed mass spec analysis of these cells. Z.C. and A.R.S performed the SILAC mass spec analysis, and M.E.P. directed the study and wrote the manuscript.
References
- Barnhart BC, Legembre P, Pietras E, Bubici C, Franzoso G, Peter ME. CD95 ligand induces motility and invasiveness of apoptosis-resistant tumor cells. EMBO J. 2004;23:3175–3185. doi: 10.1038/sj.emboj.7600325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ceppi P, Hadji A, Kohlhapp F, Pattanayak A, Hau A, Xia L, Liu H, Murmann AE, Peter ME. CD95 and CD95L promote and protect cancer stem cells. Nature Commun. 2014;5:5238. doi: 10.1038/ncomms6238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan SR, Vermi W, Luo J, Lucini L, Rickert C, Fowler AM, Lonardi S, Arthur C, Young LJ, Levy DE, et al. STAT1-deficient mice spontaneously develop estrogen receptor alpha-positive luminal mammary carcinomas. Breast Cancer Res. 2012;14:R16. doi: 10.1186/bcr3100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Park SM, Tumanov AV, Hau A, Sawada K, Feig C, Turner JR, Fu YX, Romero IL, Lengyel E, et al. CD95 promotes tumour growth. Nature. 2010;465:492–496. doi: 10.1038/nature09075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drachsler M, Kleber S, Mateos A, Volk K, Mohr N, Chen S, Cirovic B, Tuttenberg J, Gieffers C, Sykora J, et al. CD95 maintains stem cell-like and non-classical EMT programs in primary human glioblastoma cells. Cell Death Dis. 2016;7:e2209. doi: 10.1038/cddis.2016.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Essers MA, Offner S, Blanco-Bose WE, Waibler Z, Kalinke U, Duchosal MA, Trumpp A. IFNalpha activates dormant haematopoietic stem cells in vivo. Nature. 2009;458:904–908. doi: 10.1038/nature07815. [DOI] [PubMed] [Google Scholar]
- Fan CW, Chen CY, Chen KT, Shen CR, Kuo YB, Chen YS, Chou YP, Wei WS, Chan EC. Blockade of phospholipid scramblase 1 with its N-terminal domain antibody reduces tumorigenesis of colorectal carcinomas in vitro and in vivo. J Transl Med. 2012;10:254. doi: 10.1186/1479-5876-10-254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenwood C, Metodieva G, Al-Janabi K, Lausen B, Alldridge L, Leng L, Bucala R, Fernandez N, Metodiev MV. Stat1 and CD74 overexpression is co-dependent and linked to increased invasion and lymph node metastasis in triple-negative breast cancer. J Proteomics. 2012;75:3031–3040. doi: 10.1016/j.jprot.2011.11.033. [DOI] [PubMed] [Google Scholar]
- Hix LM, Karavitis J, Khan MW, Shi YH, Khazaie K, Zhang M. Tumor STAT1 transcription factor activity enhances breast tumor growth and immune suppression mediated by myeloid-derived suppressor cells. J Biol Chem. 2013;288:11676–11688. doi: 10.1074/jbc.M112.441402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janicke RU, Sprengart ML, Wati MR, Porter AG. Caspase-3 is required for DNA fragmentation and morphological changes associated with apoptosis. J Biol Chem. 1998;273:9357–9360. doi: 10.1074/jbc.273.16.9357. [DOI] [PubMed] [Google Scholar]
- Khodarev N, Ahmad R, Rajabi H, Pitroda S, Kufe T, McClary C, Joshi MD, MacDermed D, Weichselbaum R, Kufe D. Cooperativity of the MUC1 oncoprotein and STAT1 pathway in poor prognosis human breast cancer. Oncogene. 2010;29:920–929. doi: 10.1038/onc.2009.391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khodarev NN, Beckett M, Labay E, Darga T, Roizman B, Weichselbaum RR. STAT1 is overexpressed in tumors selected for radioresistance and confers protection from radiation in transduced sensitive cells. Proc Natl Acad Sci U S A. 2004;101:1714–1719. doi: 10.1073/pnas.0308102100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khodarev NN, Minn AJ, Efimova EV, Darga TE, Labay E, Beckett M, Mauceri HJ, Roizman B, Weichselbaum RR. Signal transducer and activator of transcription 1 regulates both cytotoxic and prosurvival functions in tumor cells. Cancer Res. 2007;67:9214–9220. doi: 10.1158/0008-5472.CAN-07-1019. [DOI] [PubMed] [Google Scholar]
- Kim YH, Kim WT, Jeong P, Ha YS, Kang HW, Yun SJ, Moon SK, Choi YH, Kim IY, Kim WJ. Novel combination markers for predicting survival in patients with muscle invasive bladder cancer: USP18 and DGCR2. J Korean Med Sci. 2014;29:351–356. doi: 10.3346/jkms.2014.29.3.351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleber S, Sancho-Martinez I, Wiestler B, Beisel A, Gieffers C, Hill O, Thiemann M, Mueller W, Sykora J, Kuhn A, et al. Yes and PI3K Bind CD95 to Signal Invasion of Glioblastoma. Cancer Cell. 2008;13:235–248. doi: 10.1016/j.ccr.2008.02.003. [DOI] [PubMed] [Google Scholar]
- Klover PJ, Muller WJ, Robinson GW, Pfeiffer RM, Yamaji D, Hennighausen L. Loss of STAT1 from mouse mammary epithelium results in an increased Neu-induced tumor burden. Neoplasia. 2010;12:899–905. doi: 10.1593/neo.10716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovacic B, Stoiber D, Moriggl R, Weisz E, Ott RG, Kreibich R, Levy DE, Beug H, Freissmuth M, Sexl V. STAT1 acts as a tumor promoter for leukemia development. Cancer Cell. 2006;10:77–87. doi: 10.1016/j.ccr.2006.05.025. [DOI] [PubMed] [Google Scholar]
- Levy DE, Darnell JE., Jr Stats: transcriptional control and biological impact. Nat Rev Mol Cell Biol. 2002;3:651–662. doi: 10.1038/nrm909. [DOI] [PubMed] [Google Scholar]
- Malakhova OA, Yan M, Malakhov MP, Yuan Y, Ritchie KJ, Kim KI, Peterson LF, Shuai K, Zhang DE. Protein ISGylation modulates the JAK-STAT signaling pathway. Genes Dev. 2003;17:455–460. doi: 10.1101/gad.1056303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meacham CE, Morrison SJ. Tumour heterogeneity and cancer cell plasticity. Nature. 2013;501:328–337. doi: 10.1038/nature12624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medema JP. Cancer stem cells: the challenges ahead. Nat Cell Biol. 2013;15:338–344. doi: 10.1038/ncb2717. [DOI] [PubMed] [Google Scholar]
- Peter ME, Hadji A, Murmann AE, Brockway S, Putzbach W, Pattanayak A, Ceppi P. The role of CD95 and CD95 ligand in cancer. Cell Death Differ. 2015;22:885–886. doi: 10.1038/cdd.2015.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peter ME, Krammer PH. Mechanisms of CD95 (APO-1/Fas)-mediated apoptosis. Curr Opin Immunol. 1998;10:545–551. doi: 10.1016/s0952-7915(98)80222-7. [DOI] [PubMed] [Google Scholar]
- Pitroda SP, Wakim BT, Sood RF, Beveridge MG, Beckett MA, MacDermed DM, Weichselbaum RR, Khodarev NN. STAT1-dependent expression of energy metabolic pathways links tumour growth and radioresistance to the Warburg effect. BMC Med. 2009;7:68. doi: 10.1186/1741-7015-7-68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Platanias LC. Mechanisms of type-I- and type-II-interferon-mediated signalling. Nat Rev Immunol. 2005;5:375–386. doi: 10.1038/nri1604. [DOI] [PubMed] [Google Scholar]
- Rickardson L, Fryknas M, Dhar S, Lovborg H, Gullbo J, Rydaker M, Nygren P, Gustafsson MG, Larsson R, Isaksson A. Identification of molecular mechanisms for cellular drug resistance by combining drug activity and gene expression profiles. Br J Cancer. 2005;93:483–492. doi: 10.1038/sj.bjc.6602699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts D, Schick J, Conway S, Biade S, Laub PB, Stevenson JP, Hamilton TC, O’Dwyer PJ, Johnson SW. Identification of genes associated with platinum drug sensitivity and resistance in human ovarian cancer cells. Br J Cancer. 2005;92:1149–1158. doi: 10.1038/sj.bjc.6602447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satoh J, Tabunoki H. A Comprehensive Profile of ChIP-Seq-Based STAT1 Target Genes Suggests the Complexity of STAT1-Mediated Gene Regulatory Mechanisms. Gene Regul Syst Bio. 2013;7:41–56. doi: 10.4137/GRSB.S11433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneckenleithner C, Bago-Horvath Z, Dolznig H, Neugebauer N, Kollmann K, Kolbe T, Decker T, Kerjaschki D, Wagner KU, Muller M, et al. Putting the brakes on mammary tumorigenesis: loss of STAT1 predisposes to intraepithelial neoplasias. Oncotarget. 2011;2:1043–1054. doi: 10.18632/oncotarget.371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoggins JW, Wilson SJ, Panis M, Murphy MY, Jones CT, Bieniasz P, Rice CM. A diverse range of gene products are effectors of the type I interferon antiviral response. Nature. 2011;472:481–485. doi: 10.1038/nature09907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teodorczyk M, Kleber S, Wollny D, Sefrin JP, Aykut B, Mateos A, Herhaus P, Sancho-Martinez I, Hill O, Gieffers C, et al. CD95 promotes metastatic spread via Sck in pancreatic ductal adenocarcinoma. Cell Death Differ. 2014;22:1192–1202. doi: 10.1038/cdd.2014.217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai MH, Cook JA, Chandramouli GV, DeGraff W, Yan H, Zhao S, Coleman CN, Mitchell JB, Chuang EY. Gene expression profiling of breast, prostate, and glioma cells following single versus fractionated doses of radiation. Cancer Res. 2007;67:3845–3852. doi: 10.1158/0008-5472.CAN-06-4250. [DOI] [PubMed] [Google Scholar]
- Tymoszuk P, Charoentong P, Hackl H, Spilka R, Muller-Holzner E, Trajanoski Z, Obrist P, Revillion F, Peyrat JP, Fiegl H, et al. High STAT1 mRNA levels but not its tyrosine phosphorylation are associated with macrophage infiltration and bad prognosis in breast cancer. BMC Cancer. 2014;14:257. doi: 10.1186/1471-2407-14-257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weichselbaum RR, Ishwaran H, Yoon T, Nuyten DS, Baker SW, Khodarev N, Su AW, Shaikh AY, Roach P, Kreike B, et al. An interferon-related gene signature for DNA damage resistance is a predictive marker for chemotherapy and radiation for breast cancer. Proc Natl Acad Sci U S A. 2008;105:18490–18495. doi: 10.1073/pnas.0809242105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen Z, Zhong Z, Darnell JE., Jr Maximal activation of transcription by Stat1 and Stat3 requires both tyrosine and serine phosphorylation. Cell. 1995;82:241–250. doi: 10.1016/0092-8674(95)90311-9. [DOI] [PubMed] [Google Scholar]
- White MJ, McArthur K, Metcalf D, Lane RM, Cambier JC, Herold MJ, van Delft MF, Bedoui S, Lessene G, Ritchie ME, et al. Apoptotic caspases suppress mtDNA-induced STING-mediated type I IFN production. Cell. 2014;159:1549–1562. doi: 10.1016/j.cell.2014.11.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhan JF, Chen LH, Yuan YW, Xie GZ, Sun AM, Liu Y, Chen ZX. STAT1 promotes radioresistance of CD44(+)/CD24(-/low) cells in breast cancer. Exp Biol Med (Maywood) 2011;236:418–422. doi: 10.1258/ebm.2011.010287. [DOI] [PubMed] [Google Scholar]
- Zhou Q, Zhao J, Stout JG, Luhm RA, Wiedmer T, Sims PJ. Molecular cloning of human plasma membrane phospholipid scramblase. A protein mediating transbilayer movement of plasma membrane phospholipids. J Biol Chem. 1997;272:18240–18244. doi: 10.1074/jbc.272.29.18240. [DOI] [PubMed] [Google Scholar]
- Zhou Q, Zhao J, Wiedmer T, Sims PJ. Normal hemostasis but defective hematopoietic response to growth factors in mice deficient in phospholipid scramblase 1. Blood. 2002;99:4030–4038. doi: 10.1182/blood-2001-12-0271. [DOI] [PubMed] [Google Scholar]
- Zimmerman MA, Rahman NT, Yang D, Lahat G, Lazar AJ, Pollock RE, Lev D, Liu K. Unphosphorylated STAT1 promotes sarcoma development through repressing expression of Fas and bad and conferring apoptotic resistance. Cancer Res. 2012;72:4724–4732. doi: 10.1158/0008-5472.CAN-12-1347. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.