

Abstract
Uridine 5′-monophosphate-5-fluoro-galactopyranose (UDP-5F-Galp, 7) was synthesized and its effect on UDP-Galp mutase (UGM) was investigated. UGM facilitated the hydrolysis of 7 to yield UDP and 5-oxo-galactose (24), but no 11 was detected. 19F-NMR and trapping experiments demonstrated that the reaction involves initial formation of a substrate-cofactor adduct followed by decomposition of the resulting C5 gem-fluorohydrin to generate a 5-oxo-intermediate (10). The results support the current mechanistic proposal for UGM and suggest new directions for designing mechanism-based inhibitors.
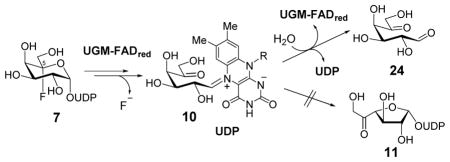
Graphical Abstract
Uridine 5′-monophosphate (UDP)-galactopyranose mutase (UGM) is a flav0enzyme that catalyzes the redox-neutral interconversion of UDP-galactopyranose (UDP-Galp, 1) and UDP-galactofuranose (UDP-Galf, 2).1 This is an important enzyme for many pathogenic bacteria, including Mycobacterium tuberculosis, the causative agent of tuberculosis, since UDP-Galf (2) is the precursor of Galf residues found in their cell surfaces.2 The emergence of multidrug-resistant strains of M. tuberculosis has prompted the search for new biomedical approaches to combat this life-threatening disease.3 The absence of UGM in mammalian cells has made inhibition of UGM to disrupt this biosynthetic pathway a promising target in the development of new antimicrobial agents. Indeed, the inhibition of UGM has been demonstrated to adversely affect mycobacterial cell growth.4
In addition to its therapeutic potential, the unique catalytic mechanism of UGM has also attracted much attention. It had been shown that UGM is catalytically active only under reducing conditions where its flavin adenine dinucleotide (FADred, 3) coenzyme remains reduced throughout this overall redox neutral reaction.5 The reduced FAD (3) acts as a nucleophile to displace the UDP moiety from UDP-Galp (1) or UDP-Galf (2) to form a covalent linkage between N5 of FAD and C1 of Galp (1 → 4) or Galf (2 → 6).6 Subsequent scission of the C1–O5 or C1–O4 bond is assisted by the lone-pair on N5 of FAD to yield an acyclic iminium ion intermediate 5, which was firstly detected by trapping with hydride reagent,7 and was observed in a recent crystal structure of UGM mutant.8 Recyclization of 5 produces the furanosyl ring of Galf (5 → 6) or the pyranosyl ring of Galp (5 → 4). This recyclization reaction is followed by the elimination of reduced FAD, which may occur concurrently with nucleophilic attack at C1 by UDP, leading to the formation of UDP-Galf (6 → 2) or UDP-Galp (4 → 1) as the products (Scheme 1). At equilibrium, the ratio of UDP-Galp to UDP-Galf is approximately 10 to 1.5 A series of FAD analogues were used to verify the role of FADred in this isomerization reaction. The data supported a chemical mechanism for UGM involving an SN2-type displacement of UDP from UDP-Galp/Galf by N5 of FADred.6c,6i
Scheme 1.

Current mechanistic model of UGM catalysis.
In an effort to learn more about the catalytic properties of UGM and to develop new mechanistic-based inhibitors targeting UGM, UDP-5F-Galp (7) was recognized as a promising core structure. It is expected that 7 would react with UGM to form a cofactor-substrate adduct 8. The subsequent ring opening of 8 to form the iminium ion intermediate would result in a gem-fluorohydrin moiety at C5 (9) that should undergo rapid dehydrofluorination9 to afford 10 (Scheme 2). The absence of C5–OH in 10 would prevent cyclization of 10 to regenerate the pyranosyl ring, but would still allow C1-O4 bond formation to yield UDP-5-oxo-Galf (11). The 5-oxo group in 11 may react with a nucleophilic residue in the active site to form a covalent adduct and thus inhibit the enzyme. In addition, further modification of the C6 hydroxyl group to a better leaving group in 7 could enhance the nucleophilic susceptibility at C6 in 10 or 11 and promote enzyme modification and inactivation.
Scheme 2.

Predicted reaction of UGM with UDP-5F-Galp (7) based on the working mechanistic model.
To test these premises, we have prepared the targeted compounds and investigated their effects on the activity of UGM. Reported herein are the chemical syntheses of 7 along with its C6-fluoro derivative (26), characterization of their reactions with UGM, and the mechanistic implications of the incubation out-comes.
The epoxide fluoridolysis strategy developed by Coward et al.10 was applied to synthesize UDP-5F-Galp (7). As depicted in Scheme 3, the reaction was initiated by derivatization of the C6 hydroxyl group of methyl α-D-galacto-pyranoside (12) with triphenylmethyl chloride (12 → 13). Benzyl protection of the remaining hydroxyl groups followed by acid hydrolysis selectively exposed the C6 hydroxyl (13 → 15),11 which was then phenylselenylated via bromination and substitution (15 → 17). Deprotection of the anomeric hydroxyl group of 17 and subsequent reaction with freshly-prepared dibenzyl phosphorochloridate gave the α-phosphate 19 exclusively. Oxidation of 19 and thermal decomposition of the resulting selenoxide produced the exo-olefin 20.12 Epoxidation using dimethyldioxirane (DMDO) generated in-situ13 and subsequent ring-opening using hydrogen fluoride (HF)10 gave the desired fluorohydrin (21) as the major product along with its L-isomer (Section S2). Global benzyl deprotection and coupling with uridine 5′-monophosphate (UMP)14 provided UDP-5F-Galp (7).
Scheme 3.

Synthesis of UDP-5F-Galp (7).
Incubation of 7 (200 μM) with UGM (less than 1 μM) was carried out at 37 °C for 5 min in 50 μL 100 mM potassium phosphate (KPi) buffer (pH 7.5) in the presence of 20 mM Na2S2O4.5 No consumption of 7 was apparent as monitored by high-performance liquid chromatography (HPLC, see Supporting information for HPLC methods). However, depletion of 7 (200 μM) was ob-served when the enzyme concentration and reaction time were increased to 20 μM and 1 h, respectively. Meanwhile, the appearance of two new peaks in the HPLC traces of the reaction work-ups, one at 24.1 min and the other at 30.5 min, was also noted (Figure 1, trace c). The species responsible for these new peaks were determined to be UDP and FAD based on co-elution with standards and the mass of each species verified by mass spectrometry (Section S3). While UDP was derived from 7, FAD was detected due to its dissociation from UGM during the work-up. In the absence of enzyme, formation of UDP and consumption of 7 was also observable, but only over extended period of time (24 h, Figure 1, trace d). No reaction product consistent with 11 was detected under all HPLC conditions tested.
Figure 1.

HPLC traces (Method A, see Supporting Information) of the incubation of UDP-5F-Galp (7) with UGM. Trace a: standards of related uridine-containing species; b: synthetic 7; and reaction of 200 μM 7 with c: 20 μM UGM for 1 h and d: buffer for 24 h.
To assess whether the hydrolysis of UDP-5F-Galp to release UDP is catalyzed by UGM, UGM with varied concentrations (0.0, 0.8, 2, and 5 μM) was incubated with 200 μM UDP-5F-Galp (7) anaerobically at 37 °C. The consumption of 7 and formation of UDP were followed up to 24 h as shown in Figure S5. Except UDP, no other uridine-containing product was detected in the reaction (section S4). The apparent first-order hydrolysis rate of 7 increased from 0.112 ± 0.003 h−1 in the absence of UGM to 1.108 ± 0.008 h−1 with 5 μM UGM. It is thus clear that UGM can accelerate the hydrolysis of 7. A comparison was also made using assay mixtures containing 7 and apo-UGM, or apo-UGM reconstituted with either FAD or 5-deaza-FAD. Only incubation with the FAD-reconstituted UGM showed significant hydrolysis activity as compared to the no-enzyme control (section S5). A reductant such as Na2S2O4 is also required for the hydrolysis (section S5). These results demonstrated that reduced FAD (3) plays a direct role in UGM-catalyzed hydrolysis of 7.
To study the catalytic function of the reduced FAD (3), the reaction mixture was treated with NaBH3CN in order to trap the putative Schiff base adduct formed between the reduced FAD and 7.7 Two new species were indeed detected by liquid chromatography-electrospray ionization-mass spectrometry (LC-ESI-MS) analysis with m/z ratios of 948.2 and 473.6 consistent with 22 (948.2 for [M – H] − and 473.6 for [M – 2H]2−) as well as m/z ratios of 950.2 and 474.6 consistent with 23 (950.2 for [M – H] − and 474.6 for [M – 2H]2−, Figure 2A and section S6). Hence, the reduced FAD (3) acts as a nucleophile to displace UDP of UDP-5F-Galp (7) as it does during the catalysis of the UDP-Galp/UDP-Galf isomerization reaction. When the reaction of 7 and UGM in KPi buffer (in D2O) was monitored using 19F NMR, a time-dependent reduction of the 5-F triplet signal of 7 (at −119 ppm) was observed, while a new singlet signal appeared at −122 ppm (Figure 2B and section S7). The chemical shift of the latter peak is consistent with the reported value of free fluoride.15 These results suggest that the reaction between 7 and UGM proceeds at least up to 9, followed by its decomposition to 10 as shown in Scheme 2. However, the reaction ensues no further than 10 since UDP-5-oxo-Galf (11) predicted as the product of the reaction of 7 with UGM was not detected under the HPLC conditions examined. The fact that UGM does not lose activity during incubation with 7 (data not shown) indicates that the reduced FAD could somehow be regenerated from 10.
Figure 2.

(A) ESI-MS (negative ion mode) of the adduct 22 and 23 trapped from reactions of UGM with UDP-5F-Galp (7) in the presence of NaBH3CN. (B) 19F NMR spectra of the reaction of 7 with UGM acquired every 15 min for 12 h. Here showed the spectra for the first 135 min.
To further characterize the turnover product from the reaction of 7 with UGM, the reaction mixture after lyophilization was incubated with O-(2,3,4,5,6-pentafluoro-benzyl)hydroxylamine (PFBHA) in pyridine, followed by the treatment with acetic anhydride.16 LC-ESI-MS analysis of the work-up revealed the occurrence of two new species with m/z ratios of 564.1 and 759.1, consistent with mono- and di-O-pentafluorobenzyl oxime acetates of 5-oxo-D-galactose (24), respectively (section S8). The identification of 24 as the turnover product in this experiment suggested that 10 was hydrolyzed to regenerate the reduced FAD and the active enzyme (Scheme 4).
Scheme 4.

Proposed hydrolysis of 7 by UGM through the intermediacy of 10.
The reason that 10 cannot cyclize to form the furanosyl ring via the C4–OH is not clear. One possible scenario is that the C5 carbonyl group of 10 may be involved in a hydrogen-bonding network that hinders the proper alignment of the C4 hydroxyl group to reach C1 of 10 in the active site. The extended lifetime of 10 would result in its hydrolysis. In fact, we did observe that UGM could catalyze the hydrolysis of UDP-Galp (1) under high enzyme concentration and extended incubation time (section S9). It is thus likely that hydrolysis of Schiff base intermediate (5 or 10) is an inherent side-activity of UGM but is typically suppressed by minimizing the lifetime of the intermediate.
The formation of the C5-oxo-bearing intermediate 10 during the enzymatic reaction prompted the design and synthesis of UDP-[5,6-F2]-Galp (26), which is expected to react with UGM similarly to generate 6-deoxy-6-fluoro-10 whose α-fluoro carbonyl functionality might be susceptible to modification by an active-site residue. As shown in Scheme 5, the hydroxyl group at C6 of 21 was subjected to fluorination using di-ethylaminosulfur trifluoride (DAST) reagent to generate 25. The resulting product was hydrogenated to remove the benzyl protecting groups followed by coupling with UMP to give 26. Although facilitated hydrolysis of 26 at C1 to yield UDP and release of fluoride were noted in the presence of UGM as observed for 7, no apparent decrease in activity of UGM was observed when 2 μM of the enzyme was pre-incubated with 200 μM of 26 up to 24 h (data not shown).
Scheme 5.

Synthesis of UDP-[5,6-F2]-Galp.
In summary, the C5-fluorinated substrate analog 7 was prepared and its reaction with UGM was fully characterized. Release of UDP from 7 is UGM-dependent and compound 24 was identified as the turnover product. Our results clearly revealed the intermediacy of 5 (or 9/10) in the catalytic mechanism of UGM and lend further credence to the currently accepted mechanism of UGM. They also suggest a more associative mechanism (SN2-like) during substrate-FAD adduct formation. In addition, the inherent hydrolytic activity of UGM was also unraveled. These findings, in conjunction with the observation that a C5-oxo intermediate is generated from the C5-F substrate analogue during turnover, may be of use in the de-sign of mechanism-based inhibitors for UGM. Although our first attempt (26) was not successful, exploration of the chemical space at C6 of 7 is nevertheless a promising direction for future research.
Supplementary Material
Acknowledgments
This work is supported by grants from the National Institutes of Health (GM035906) and Welch Foundation (F-1511).
Footnotes
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The Supporting Information is available free of charge on the ACS Publications website. Experimental procedures regarding chemical synthesis and enzymatic assays along with their results. Complete spectroscopic characterization of all new compounds are also included. (PDF)
References
- 1.Tanner JJ, Boechi L, McCammon JA, Sobrado P. Arch Biochem Biophys. 2014;544:128–141. doi: 10.1016/j.abb.2013.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.(a) Peltier P, Euzen R, Daniellou R, Nugier-Chauvin C, Ferrières V. Carbohydr Res. 2008;343:1892–1923. doi: 10.1016/j.carres.2008.02.010. [DOI] [PubMed] [Google Scholar]; (b) Richards MR, Lowary TL. Chem Biochem. 2009;10:1920–1938. doi: 10.1002/cbic.200900208. [DOI] [PubMed] [Google Scholar]; (c) de Lederkremer RM, Agusti R. Adv Carbohydr Chem Biochem. 2009;62:311–366. doi: 10.1016/S0065-2318(09)00007-9. [DOI] [PubMed] [Google Scholar]; (d) Oppenheimer M, Valenciano AL, Sobrado P. Enzyme Res. 2011:415976. doi: 10.4061/2011/415976. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Tefsen B, Ram AF, van Die I, Routier FH. Glycobiology. 2012;22:456–469. doi: 10.1093/glycob/cwr144. [DOI] [PubMed] [Google Scholar]
- 3.(a) Koul A, Arnoult E, Lounis N, Guillemont J, Andries K. Nature. 2011;469:483–490. doi: 10.1038/nature09657. [DOI] [PubMed] [Google Scholar]; (b) Leung CC, Lange C, Zhang Y. Respirology. 2013;18:1047–1055. doi: 10.1111/resp.12156. [DOI] [PubMed] [Google Scholar]; (c) Ramazanzadeh R, Roshani D, Shakib P, Rouhi S. J Res Med Sci. 2015;20:78–88. [PMC free article] [PubMed] [Google Scholar]
- 4.(a) Dykhuizen EC, May JF, Tongpenyai A, Kiessling LL. J Am Chem Soc. 2008;130:6706–6707. doi: 10.1021/ja8018687. [DOI] [PubMed] [Google Scholar]; (b) Borrelli S, Zandberg WF, Mohan S, Ko M, Martinez-Gutierrez F, Partha SK, Sanders DAR, Av-Gay Y, Pinto BM. Int J Antimicrob Agents. 2010;36:364–368. doi: 10.1016/j.ijantimicag.2010.06.030. [DOI] [PubMed] [Google Scholar]
- 5.Zhang Q, Liu H-w. J Am Chem Soc. 2000;122:9065–9070. [Google Scholar]
- 6.(a) Barlow JN, Girvin ME, Blanchard JS. J Am Chem Soc. 1999;121:6968–6969. [Google Scholar]; (b) Zhang Q, Liu H-w. J Am Chem Soc. 2001;123:6756–6766. doi: 10.1021/ja010473l. [DOI] [PubMed] [Google Scholar]; (c) Huang Z, Zhang Q, Liu H-w. Bioorg Chem. 2003;31:494–502. doi: 10.1016/j.bioorg.2003.08.002. [DOI] [PubMed] [Google Scholar]; (d) Fullerton SWB, Daff S, Sanders DAR, Ingledaw WJ, Whitfield C, Chapman SK, Naismisth JH. Biochemistry. 2003;42:2104–2109. doi: 10.1021/bi027077f. [DOI] [PubMed] [Google Scholar]; (e) Caravano A, Sinaÿ P, Vincent SP. Bioorg Med Chem Lett. 2006;16:1123–1125. doi: 10.1016/j.bmcl.2005.11.106. [DOI] [PubMed] [Google Scholar]; (f) Itoh K, Huang Z, Liu H-w. Org Lett. 2007;9:879–882. doi: 10.1021/ol0631408. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Yuan Y, Bleile DW, Wen X, Sanders DAR, Itoh K, Liu H-w, Pinto M. J Am Chem Soc. 2008;130:3157–3168. doi: 10.1021/ja7104152. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Sadeghi-Khomami A, Forcada TJ, Wilson C, Sanders DAR, Thomas NR. Org Biomol Chem. 2010;8:1596–1602. doi: 10.1039/b917409e. [DOI] [PubMed] [Google Scholar]; (i) Sun HG, Ruszczycky MW, Chang W-c, Thibodeaux CJ, Liu H-w. J Biol Chem. 2012;287:4602–4608. doi: 10.1074/jbc.M111.312538. [DOI] [PMC free article] [PubMed] [Google Scholar]; (j) Huang W, Gauld JW. J Phys Chem B. 2012;116:14040–14050. doi: 10.1021/jp310952c. [DOI] [PubMed] [Google Scholar]
- 7.(a) Soltero-Higgin M, Carlson EE, Gruber TD, Kiessling LL. Nat Struct Mol Biol. 2004;11:539–543. doi: 10.1038/nsmb772. [DOI] [PubMed] [Google Scholar]; (b) Gruber TD, Westler WM, Kiessling LL, Forest KT. Biochemistry. 2009;48:9171–9173. doi: 10.1021/bi901437v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mehra-Chaudhary R, Dai Y, Sobrado P, Tanner JJ. Biochemistry. 2016;55:833–836. doi: 10.1021/acs.biochem.6b00035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Some reported examples in which decomposition of gem-fluorohydrin into carbonyl was implied. Marcotte PA, Robinson CH. Biochemistry. 1982;21:2773–2778. doi: 10.1021/bi00540a030.Haufe G, Pietz S, Wölker R. Eur J Org Chem. 2003;21:2166–2175.Lermontov SA, Ushakova LL, Kuryleva NV. J Fluorine Chem. 2008;129:332–334.Purkayastha N, Shendage DM, Fröhlich R, Haufe G. J Org Chem. 2010;75:222–225. doi: 10.1021/jo901872a.
- 10.(a) Hartman MCT, Coward JK. J Am Chem Soc. 2002;124:10036–10053. doi: 10.1021/ja0127234. [DOI] [PubMed] [Google Scholar]; (b) Hagena TL, Coward JK. Tetrahedron: Asymme-try. 2009;20:781–794. [Google Scholar]
- 11.Bernotas RC, Pezzone MA, Ganem B. Carbohydr Res. 1987;167:305–311. doi: 10.1016/0008-6215(87)80289-6. [DOI] [PubMed] [Google Scholar]
- 12.(a) Inage M, Chaki H, Kusumoto S, Shiba T. Chem Lett. 1982:1281–1284. [Google Scholar]; (b) Endo T, Kajihara Y, Kodama H, Hashimoto H. Bioorg Med Chem. 1996;4:1939–1948. doi: 10.1016/s0968-0896(96)00176-9. [DOI] [PubMed] [Google Scholar]
- 13.Yang D, Wong MK, Yip YC. J Org Chem. 1995;60:3887–3889. [Google Scholar]
- 14.Wittmann V, Wong CH. J Org Chem. 1997;62:2144–2147. doi: 10.1021/jo9620066. [DOI] [PubMed] [Google Scholar]
- 15.Silverstein RM, Webster FX, Kiemle DJ. Spectrometric Identification of Organic Compounds. 7. John Wiley & Sons; Hoboken: 2005. [Google Scholar]
- 16.Biondi PA, Manca F, Nergi A, Secchi C. J Chromatogr. 1987;411:275. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.