

Abstract
The esophagus is derived from the anterior portion of the developmental intermediate foregut, a structure that also gives rise to other organs including the trachea, lung, and stomach. Genetic studies have shown that multiple signaling pathways (e.g. Bmp) and transcription factors (e.g. SOX2) are required for the separation of the esophagus from the neighboring respiratory system. Notably, some of these signaling pathways and transcription factors continue to play essential roles in the subsequent morphogenesis of the esophageal epithelium which undergoes a simple columnar-to-stratified squamous conversion. Reactivation of the relevant signaling pathways has also been associated with pathogenesis of esophageal diseases that affect the epithelium and its stem cells in adults. In this review we will summarize these findings. We will also discuss new data regarding the cell-of-origin for the striated and smooth muscles surrounding the esophagus and how they are differentiated from the mesenchyme during development.
Keywords: foregut, tracheoesophageal separation, EA, esophageal atresia/tracheoesophageal fistula, basal cells, epithelial morphogenesis
1. Introduction
The esophagus serves as a channel to transport food from the mouth to the stomach. Fitting the need, the esophagus is ensheathed by multiple layers of muscles that are essential to generate peristalsis to move food. Within the lumen a thick stratified squamous epithelium is required to sustain the passing of the abrasive raw food, which is facilitated by secretions of the esophageal submucosal glands. During embryonic development, the esophagus and trachea initially share a single-lumen tube at the anterior region of the foregut. As organogenesis proceeds, the anterior foregut separates and generates the trachea ventrally and the esophagus dorsally. This tracheal-esophageal separation occurs at around E9.5-E11.5 in mice and approximately 4-6 weeks of gestation in humans. Although the exact cellular and molecular mechanisms remain elusive, recent studies of mouse genetic models suggest that this separation involves dorsal-ventral patterning of signaling molecules (e.g. Bmps, Wnts) and transcription factors (e.g. SOX2, NKX2.1). Disruption of this expression pattern leads to various anomalies such as esophageal atresia with or without tracheoesophageal fistula (EA/TEF) where the esophagus is closed as a blind end sac proximally, and in a worse scenario the foregut remains as a single-lumen tube [1]. Some of the signaling molecules and transcription factors continue to play essential roles in the subsequent development of the esophagus, especially for the morphogenesis of the epithelium which involves a transition from simple columnar to stratified squamous. These molecules are also required for the maintenance of the epithelium in the adult esophagus. Abnormal activities of relevant signaling pathways or abnormal expression of the transcription factors have been associated with the pathogenesis of several common esophageal diseases including eosinophilic esophagitis, Barrett's esophagus, and even cancer. This review will summarize these findings with a focus on how signaling pathways and transcription factors regulate tracheal-esophageal separation and morphogenesis of the esophageal epithelium. We will also discuss new findings regarding the development of the mesenchyme, especially the formation of the muscle layers in the esophagus.
The anterior foregut exhibits dorsal-ventral differences in terms of gene expression prior to the start of tracheal-esophageal separation. For example, the transcription factor SOX2 is abundantly expressed in the dorsal epithelium [2]. By contrast, NKX2.1 (also known as TTF1) is enriched in the epithelium at the ventral side [3-5]. As discussed in more details below this dorsal-ventral expression of signaling molecules and transcription factors is essential for the specification of the early foregut endoderm into different territories, i.e. respiratory epithelium and esophageal epithelium (Fig. 1). When the esophagus is completely separated from the respiratory system, the epithelium lining the nascent organ is simple columnar. The epithelium promptly starts to stratify and differentiate to form a multiple-layered squamous epithelium that consists of basal and suprabasal cells (Fig. 2). The squamous epithelium is about 4-6 cell thick with a single layer of basal cells in the adult mouse esophagus, while in the human esophagus the squamous epithelium contains 20-30 layer of cells with several layers of basal cells at the bottom [6]. In addition, an acellular layer of keratin is found on the top of the squamous epithelium in rodent esophagus similar to the skin, but this keratin layer is absent in the human esophagus (Fig. 2). On the other hand, human but not rodent esophagus contains abundant submucosal glands that are responsible for producing mucins and bicarbonate etc. [7]. The mechanism leading to the formation of these glands remains unknown due to the lack of animal models.
Fig. 1. Establishment of the esophagus from the anterior foregut.
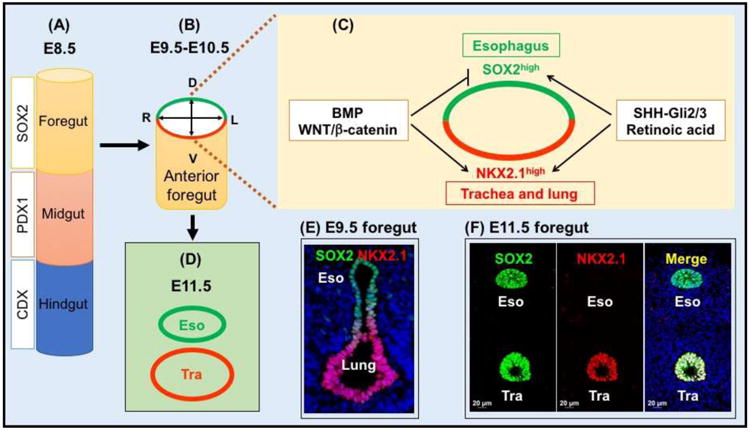
(A) The gut endoderm is specified into the foregut (SOX2+), midgut (PDX1+) and hindgut (CDX+) after gastrulation. (B-D) Dorsal-ventral patterning of the transcription factors and signaling molecules is required for the generation of the esophagus from the anterior foregut. The transcription factor SOX2 is highly expressed in the epithelial precursors of the dorsal foregut (future esophagus) at E9.5, while NKX2.1 is enriched in the ventral foregut (future trachea and lung). The activities of the Bmp and canonical Wnt signaling pathways promote lung lineage differentiation while repressing the esophageal fate. SHH-Gli2/3 and Retinoic acid are required for the growth of the esophagus and lung. (E) Dorsal-ventral expression of SOX2 and NKX2.1 in the E9.5 foregut (cross section). (F) High levels of SOX2 are maintained in the esophagus and also present in the trachea following tracheal-esophageal separation. However, NKX2.1 is only present in the trachea. Abbreviation: Tra, trachea.
Fig. 2. The mouse and human esophagus (hematoxylin and eosin staining).
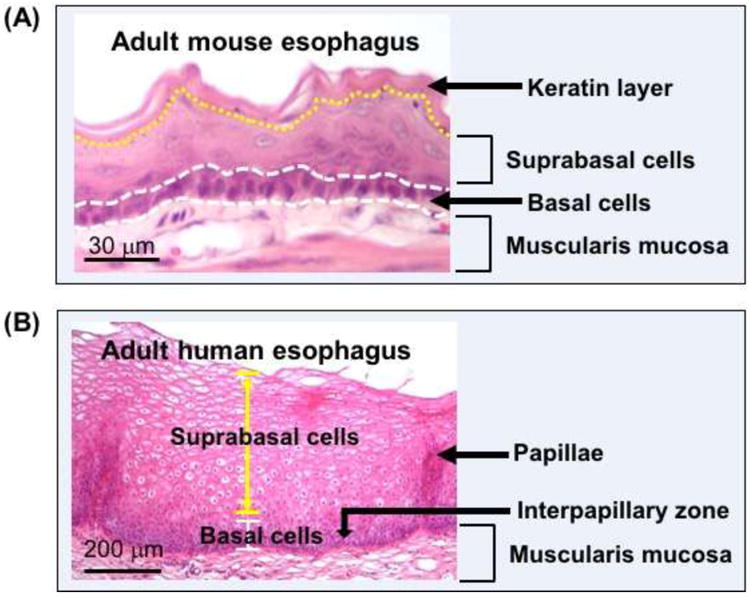
(A) The stratified squamous epithelium in the adult esophagus. (B) The stratified squamous epithelium in the adult esophagus. The epithelium consists of basal cells (undifferentiated) and suprabasal cells (differentiated) in both mouse and human esophagus. The average thickness of the epithelial is 4-6 and 30-40 cells in the mouse and human esophagus, respectively. Note that the keratin layer is absent in the human esophagus which contains several layers of basal cells as compared to a single layer in the mouse esophagus.
The mesenchyme enclosing the epithelium is seemingly homogeneous when the esophagus is established from the mouse foregut at E11.5. Similar to the epithelium the mesenchyme is also highly proliferative at this stage. Previous genetic studies have shown that several transcription factors including FOXP1 and FOXP2 are important for the differentiation of the mesenchyme into layers of muscle cells (see review by Jacobs et al. [8]). Many questions remain unanswered, however, including determining the cell-of-origin for the surrounding striated muscle. Recent lineage tracing experiments provide some unexpected results. These findings will be discussed below.
2. Establishment of the Esophagus from the Early Foregut
Previously three models have been proposed to describe how the esophagus is separated from the trachea, including: (1) The outgrowth model in which the trachea extends with the lung primordium from the early foregut, (2) the watershed model in which a yet-to-be identified mesenchymal condensation serves as a wedge to split the anterior foregut into the trachea and esophagus, and (3) the septation model where epithelial cells at the dorsal–ventral midline make contact across the midline of the lumen and fuse to form a septum [1]. Recent evidence, however, has suggested that none of these models fully accounts for tracheal-esophageal separation. For example, the outgrowth model predicts that the common tube above the lung budding site will be specified as one part of the esophagus. Nevertheless, characterization studies showed that the expression of the respiratory marker NKX2.1 is present in the ventral common foregut throughout tracheal-esophageal separation [2, 9]. According to the watershed model, the common tube above the mesenchymal condensation would remain undivided. Contrary to this prediction, the length of the undivided foregut becomes shorter as separation proceeds [10]. Regarding the septation model, the predicted septum has never been detected by scanning electron microscopy during chicken foregut separation. Instead the authors observed an epithelial saddle at the site where the lung buds initiate, which is continuously present at the location where the trachea and esophagus are splitting [11].
We recently developed a mouse foregut culture system to visualize the separation process and proposed an alternative model termed “splitting and extension model”. We used a SOX2-GFP knockin allele to monitor the movement of the foregut epithelium and observed an epithelial saddle that is initiated at the lung-esophageal boundary in the E9.5 foregut [1]. The saddle subsequently moves in a caudal-cranial direction to split the trachea and esophagus, while both nascent organs extend caudally (see movie in [1]). The epithelial saddle is composed of cells from the lung and future esophagus (Fig. 3), raising the possibility that abnormal lung development (e.g. branching defects) is associated with abnormal separation of the esophagus from the trachea. It is worth mentioning that up to 72% of surviving adolescents and adults with treated EA/TEF continue to suffer from respiratory problems throughout their lifetime [12-14]. Consistently, lung lobe fusion (horseshoe), agenesis, or hypoplasia with abnormal epithelial differentiation in the airways has been reported in patients with EA/TEF [15]. The etiology and mechanism of EA/TEF formation remains largely unknown. Nevertheless, recent studies with animal models are beginning to provide insight into the dysmorphogenetic processes. Several signaling pathways (e.g. Bmp, Wnt) and transcription factors (e.g. SOX2) have been shown to play important roles in the regulation of tracheal-esophageal separation [2, 3, 16]. Intriguingly, in these animal models EA/TEF is always accompanied by disturbances in lung development most commonly characterized by lobulation and branching defects [8]. These observations support the hypothesis that tracheal-esophageal separation and lung development are closely linked. It remains unclear however how these developmental processes are connected and which underlying common mechanisms exist. A combination of live imaging, lineage tracing, and genetic manipulation will be instrumental in addressing these issues.
Fig. 3. Tracheal–esophageal separation: Splitting and extension model.
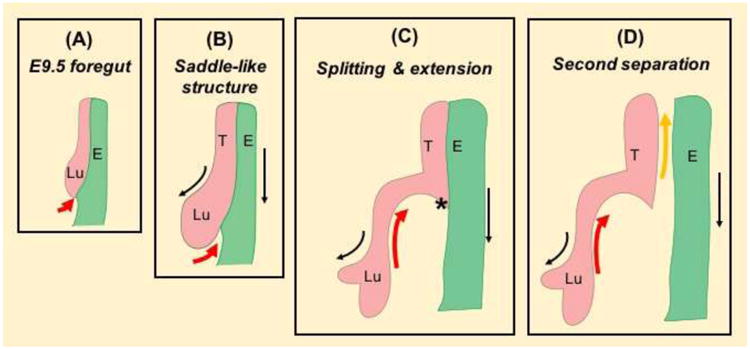
(A) A saddle-like structure starts to form at the distal end of the anterior foregut at E9.5 (19-somite stage). (B, C) The saddle-like structure moves in a bottom-up manner (red arrow) and splits the lung from the esophagus as the lung and esophagus grow rostrally (black arrow). (D) A second wave of bottom-up movement occurs to complete the separation process (yellow arrow). The lung and esophagus are highlighted by pink and green, respectively. Asterisk labels constriction site where the first wave of separation ends. Abbreviation: Lu, lung; T, trachea; E, esophagus.
2.1 Transcription factors controlling tracheal-esophageal separation
SOX2 and NKX2.1 in the epithelium
SOX2 is a key family member of SRY-related transcription factors which are critical for organ development, stem cell proliferation, and differentiation [17, 18]. Abnormal levels of SOX2 have been associated with pathogenesis of multiple diseases such as anophthalmia-esophageal-genital (AEG) syndrome [19] and even cancer malignancy [18]. For a comprehensive overview of SOX2 function in development and disease we refer to a recently published book (Sox2: Biology and Role in Development and Disease [20]). Abnormal levels of SOX2 are also found in patients with EA/TEF [19]. We showed that significant downregulation of SOX2 in the early foregut leads to EA/TEF in SOX2GFP/COND hypomorphic mouse mutants. Notably, these mutants present abnormal lung branching morphogenesis with elongated main bronchi and shortened trachea [2], further supporting a connection between abnormal lung development and EA/TEF etiology. Furthermore, TEFs connecting the trachea and the stomach contain respiratory epithelial cells expressing NKX2.1 and Scgb1a1.
The expression of SOX2 is enriched in the dorsal epithelium in contrast to the NKX2.1-enriched ventral epithelium prior to the initiation of the tracheal-esophageal separation. This unique dorsal-ventral expression pattern of the two transcription factors is important for the separation [2, 21]. It is possible that downregulation of SOX2 promotes the expansion of NKX2.1+ respiratory cells into the dorsal domain, thereby affecting the formation of the epithelial saddle at the very beginning of the separation process. This possibility can be tested with careful characterization of the saddle in SOX2GFP/COND hypomorphic mutants. Live imaging of tracheal-esophageal separation in these mutants should provide additional information. EA/TEF also develops in mutants lacking the Nkx2.1 gene, which is essential for early lung morphogenesis; deletion of NKX2.1 leads to severely hypoplastic lungs [21]. In this case it is reasonable to postulate that Nkx2.1 deletion directly impacts the formation of the saddle which fails to move anteriorly to separate the esophagus from the trachea. Again characterization of the saddle at the stage of tracheal-esophageal separation will facilitate to address the possibility. Many questions remain, however, regarding the underlying cellular and molecular mechanisms. For example, both SOX2 and NKX2.1 are transcription factors regulating an array of downstream targets. Do they share common downstream targets that directly regulate the movement of the saddle in addition to their roles for the specification of the foregut endoderm? It was previously shown that inhibition of SOX2 function leads to altered migration of neural progenitor cells in the chick neural tube [22]. Moreover, NKX2.1 is also involved in cell movement through transcriptional regulation of myosin binding protein H (MYBPH) which physically interacts with and inhibits Rho kinase 1 (ROCK1). Downregulation of MYBPH with siRNAs leads to increased motility of lung adenocarcinoma cells [23]. Along these lines examining cytoskeletal organization in the epithelial saddle may reveal surprising results in SOX2 or NKX2.1 mutants.
FOXF1 and BARX1 in the mesenchyme
The role of mesenchymal cells in tracheal-esophageal separation remains obscure. Deletion of the Bmp antagonist Noggin leads to ectopic cartilage nodules in the TEF connecting the trachea and stomach [9], but whether/how mesenchymal specification contributes to tracheal-esophageal separation remains unknown. In our foregut culture the mesenchyme is highly proliferative which is consistent with in vivo characterization of the separating foregut [1, 10]. Whether the proliferating mesenchyme is the driving force for foregut separation, however, is subject to debate. We observed extensive migration of the expanding mesenchyme in a direction opposite to where the saddle moves (see movie in [1]). Nevertheless, heterozygous deletion of the transcription factor FOXF1 which is specifically expressed in the mesenchyme leads to reduced growth of the foregut and EA/TEF formation accompanied by fusion of right lung lobes on CD1 genetic background [24]. The phenotypes are similar to what were observed in mutants having defects in sonic hedgehog (Shh) signaling, suggesting that FOXF1 is a downstream target of the pathway. Consistently, the transcript levels of Foxf1 are reduced in the foregut of Shh null mutants [24]. These findings inform a regulatory axis where the epithelium signal (i.e. Shh) modulates mesenchymal morphogenesis. Interestingly, a recent study showed that GLI2 and GLI3, downstream targets of Shh pathway, regulate the expression of NKX2.1 in the ventral foregut [25], indicating a feedback loop during the separation of the esophagus from the trachea. It will be interesting to determine whether GLI2 or GLI3 or both are required for FOXF1 expression in the mesenchyme.
Findings in frog and mouse genetic models suggest that GLI2 and GLI3 are both required for the transcription of two important Wnt signaling ligands WNT2 and 2b in the mesenchyme of the ventral foregut [25]. WNT2/2b mediated canonical Wnt signaling is essential for the separation of the esophagus from the trachea (as discussed further below). Interestingly, the activities of the canonical Wnt signaling pathway are limited to the ventral side of the foregut prior to separation [16, 26]. Conversely, the transcription factor BARX1 is enriched in the dorsal foregut mesenchyme and mesenchyme between the nascent esophagus and trachea. Genetic studies suggest that BARX1 is required for restricting Wnt activation to the ventral foregut [27]. Deletion of the Barx1 gene leads to expansion of canonical Wnt signaling into the dorsal foregut and the mutants display EA/TEF. Further analysis revealed that BARX1 regulates the expression of secreted Frizzled-related proteins (sFRPs) which serve to antagonize the activity of the Wnt pathway. In the absence of BARX1 the transcripts of sFRP1 and sFRP2 were not detected in the foregut mesenchyme. Notably, it seems that loss of these Wnt antagonists occurs specifically in the foregut of the Barx1 null mutants [27]. As mentioned above, BARX1 expression is limited to the mesenchyme between the nascent esophagus and trachea where the saddle is located. It will be tempting to test whether this transcription factor has additional roles in modulating the formation and/or movement of the saddle besides restricting Wnt signaling activities.
2.2 Signaling pathways controlling tracheal-esophageal separation
Establishment of the esophagus from the anterior foregut requires intimate interaction of the mesenchyme and epithelium. The crosstalk is partly mediated by signaling molecules that are able to freely move between the two compartments. Consistently, disruption of the essential components of multiple signaling pathways e.g. Bmp, Wnt and Shh results in abnormal separation of the esophagus from the trachea.
Bmp and canonical Wnt signaling
The saddle consists of both lung and esophageal cells while it moves anteriorly to separate the respiratory system from the esophagus. Previous studies have shown that activation of Bmp and Wnt/ß-catenin signaling is critical for promoting respiratory while suppressing esophageal cell fate in the ventral side of the foregut (Fig. 1). It is therefore expected that disruption of these two pathways will result in the abnormal formation of the saddle, leading to defective tracheal-esophageal separation. Indeed deletion of multiple components of the Bmp or Wnt pathways results in tracheal agenesis or EA/TEF [3, 16, 26]. For example, EA/TEF develops in mutants lacking the Bmp antagonist Noggin (Noggin-/-) or Bmp receptors (Shh-Cre; Bmpr1aloxp/-; Bmpr1b-/-), while loss of the Bmp ligand BMP4 results in transient expression of NKX2.1 in the tracheal field at E9.25-E10.5 and tracheal agenesis [28]. Notably, lung branching morphogenesis is also affected in all of these mutants. Moreover, loss of one copy of Bmp4 allele [9] or deletion of Bmp7 [29] rescues the separation defects in Noggin null background, further supporting that balanced Bmp signaling activities are required for the formation of an intact esophagus. Of interest is that Bmp signaling is active in the ventral foregut which serves as precursors of respiratory cells. Removal of Bmpr1a and 1b leads to increased expression of the dorsal markers SOX2 and p63 at the expense of NKX2.1 in the E9.75 foregut of Shh-Cre; Bmpr1aloxp/-; Bmpr1b-/- mutants [3]. Further analysis showed that Bmp signaling represses the transcription of the Sox2 gene. Consistent with these findings, Sox2 inactivation is able to restore NKX2.1 expression and normal tracheal-esophageal separation in Shh-Cre; Bmpr1aloxp/-; Bmpr1b-/-; Sox2loxp/loxp compounds [3].
Similar to Bmp, the activities of canonical Wnt signaling are also limited to the ventral side of the anterior foregut. The Wnt ligands WNT2 and WNT2b are expressed specifically in the mesenchyme surrounding the ventral foregut. This expression pattern is correlated with Wnt signaling activation as demonstrated by the Wnt signaling reporter allele BAT-GAL [26]. Both Wnt2/2b double knockouts (DKO) and Shh-Cre; β-cateninloxp/loxp mutants display lung and tracheal agenesis. The expression of NKX2.1 is lost in the mutants and the anterior foregut ends as a single-lumen tube lined by SOX2hi and p63hi esophageal precursors [30]. On the contrary, ß-catenin gain-of-function driven by Shh-Cre leads to a switch of cell fate in the esophagus and proximal stomach as evidenced by ectopic expression of NKX2.1 [16, 26]. Ectopic NKX2.1 expression was also detected in the TEFs of Barx1-/- mutants which show ectopic Wnt signaling activities in the dorsal foregut as described above [27]. It is noteworthy that ß-catenin gain-of-function is unable to rescue NKX2.1 expression in the foregut of Shh-Cre; Bmpr1aloxp/-; Bmpr1b-/- mutants. In addition, Bmp signaling activities are maintained in the foregut of Shh-Cre; β-cateninloxp/loxp mutants, suggesting that the canonical Wnt and Bmp signaling pathways act in parallel to promote respiratory cell fate and tracheal-esophageal separation [3].
Shh and Retinoic acid signaling
The activation of Shh signaling is dynamic during the development of the early foregut endoderm. Shh protein is initially detected in the ventral foregut endoderm where the lung and trachea arise, and it can also be detected in the ventral side of the esophagus [9, 16]. The expression pattern is maintained until E11.5 when the esophagus is completely separated from the trachea. The expression then shifts to the epithelium of the dorsal side of the esophagus, while the expression in the trachea and ventral esophagus becomes attenuated [31]. Deletion of Shh or its downstream effectors GLI2 and GLI3 (GLI2-/-; GLI3+/-) leads to severely hypoplastic lungs and EA/TEF [32, 33], consistent with the important roles for this pathway in foregut development. The levels of NKX2.1 are significantly reduced in the anterior foregut of GLI2-/-; GLI3+/- mutants accompanied by decreased expression of WNT2/2b and BMP4 [25], supporting a crosstalk of multiple signaling pathways during lung morphogenesis and tracheal-esophageal separation.
Retinoic Acid (RA) signaling has recently been shown to modulate the expression of Shh in the anterior foregut endoderm prior to lung bud specification [25]. The authors treated E7.5 whole embryos in culture with the RA inhibitors DEAB or BMS493 for two days. They found that both Shh and Indian Hedgehog (Ihh) are not expressed in the foregut epithelium accompanied by the loss of NKX2.1. Of note is that NKX2.1 expression remains unaltered in the brain and thyroid, suggesting a tissue specific function for the RA/Shh regulatory axis. These findings are consistent with previous reports that RA signaling is crucial for lung development and tracheal-esophageal separation. Rat fetuses fed with food deficient for the RA precursor vitamin A exhibits lung agenesis [34], while mouse mutants lacking the RA-synthesizing enzyme retinaldehyde dehydrogenase 2 (Raldh2) also present lung agenesis and EA/TEF [35]. Interestingly, in organ culture blocking TGFß signaling is able to rescue lung defects induced by RA inhibition [36], suggesting that RA promotes lung bud induction through the inhibition of TGFß signaling.
3. Epithelial Morphogenesis and Stem/Progenitor Cells of the Esophagus
The conversion of simple columnar into stratified squamous epithelium during esophageal development involves dynamic changes in the expression of cytokeratin. KRT8 and its binding partner KRT18 are enriched in the columnar epithelium lining the nascent esophagus. As stratification begins, the bottom layer (basal layer) initiates the expression of KRT5 and its partner KRT14 while the levels of KRT8/18 are reduced (Fig. 4, 5) [31, 37]. However, the top a few layers (suprabasal layer) of the stratified epithelium maintain low levels of KRT8/18 until postnatal (P)3 in mice [37]. Meanwhile these suprabasal cells also start to express KRT1/10, KRT4/13, and gradually lose the proliferating capability [37] (Fig. 4, 5). As the cells further differentiate and move up to the top layers they express Loricrin and Involucrin. Finally in mouse but not human esophagus the epithelial cells undergo enucleating process and lose nuclei while keratin proteins deposit on the top, similar to skin morphogenesis [38, 39]. Recent studies suggested that transcription factors and signaling pathways that are important for the establishment of the esophagus from the foregut also play critical roles in the morphogenesis and maintenance of the stratified squamous epithelium. Some of these transcription factors (e.g. p63) and signaling pathways (e.g. Bmp, Wnt) are also important for skin morphogenesis [38-41].
Fig. 4. Conversion of simple columnar to stratified squamous epithelium in the developing esophagus.
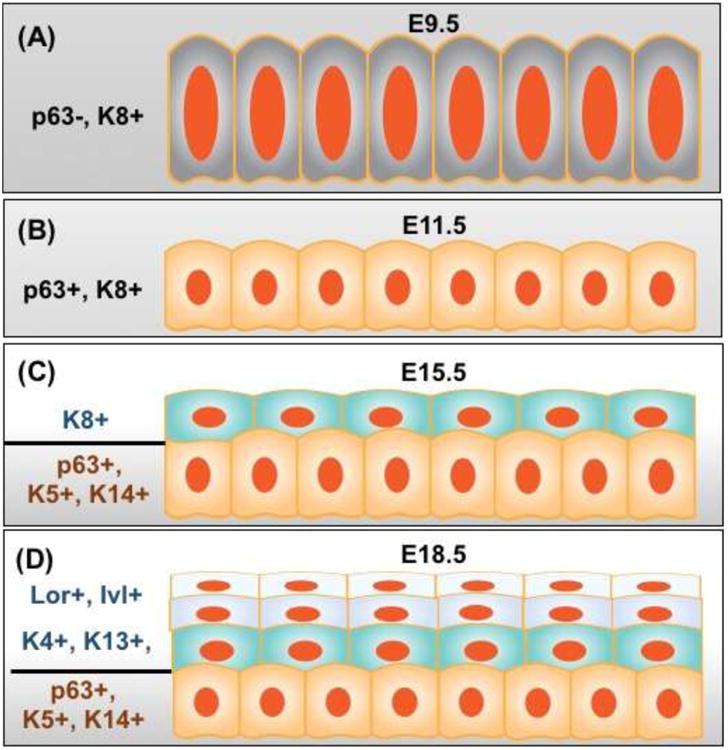
(A) Prior to tracheal-esophageal separation, the esophageal epithelial precursors lining the dorsal foregut are simple columnar (KRT8+). (B) The epithelium lining the nascent esophagus remains columnar (p63+, KRT8+) at E11.5. (C) The stratified epithelium expresses KRT5 and KRT14 in the basal layer and KRT8 in the suprabasal layers at E15.5. (D) Suprabasal cells undergo terminal differentiation and express KRT4, KRT13, Loricrin and Involucrin. Abbreviation: Lor, Loricrin; Inv, Involucrin.
Fig. 5. Gene expression in the mouse esophageal epithelium at E18.5 and adult stages.
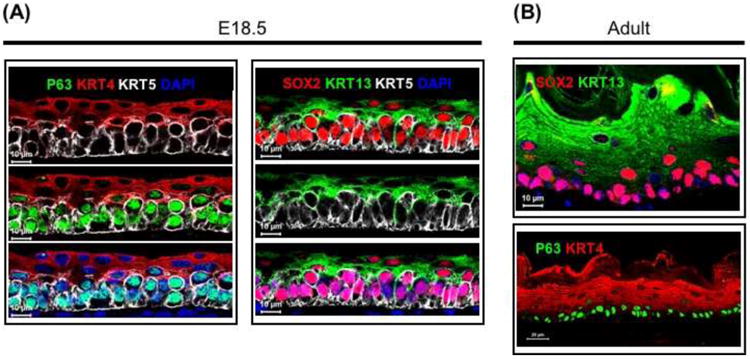
(A) p63, SOX2 and KRT5 are enriched in the basal cells, while KRT4 and KRT13 are expressed in differentiating suprabasal cells of the esophagus at E18.5. (B) The expression of p63 and SOX2 is maintained in the adult esophagus.
3.1 Epithelial morphogenesis in the developing esophagus
Regulation of epithelial stratification and differentiation by P63 and SOX2
P63 is thus far the most potent regulator of the conversion of simple columnar into stratified squamous epithelium [42]. Similar to the skin the epithelium remains simple columnar in the esophagus of p63-/- mutants, expressing high levels of KRT8/18 but not KRT5/14 [40-43] (Fig 6). A significant portion of p63-/- epithelial cells in the esophagus contain multi-cilia [42]. It was reported that ciliated cells are transiently present in the developing esophagus [1, 37, 42], suggesting that the epithelial differentiation is arrested in p63 null mutant. However, it is also possible that transdifferentiation occurs following p63 deletion given that ciliated cells are one of the major cell lineages in the lung. Transcripts of several genes (e.g. Villin) marking intestinal cells were also detected in the simple columnar epithelium of p63-/- mutants [44]. Transcription of the p63 gene results in two major isoforms, TAp63 and ΔNp63 which further generate α, ß, and γ forms depending on splicing [45, 46]. Each isoform has distinct functions in organ development and stem cell maintenance [45]. A detailed characterization study is lacking for the isoforms expressed in the developing esophagus. ΔNp63 is the major isoform in the skin [47]. Conditional deletion of ΔNp63 isoform recapitulates the stratification defects in both skin and the esophagus [48, 49].
Fig. 6. p63 ablation leads to failed stratification of the esophageal epithelium.
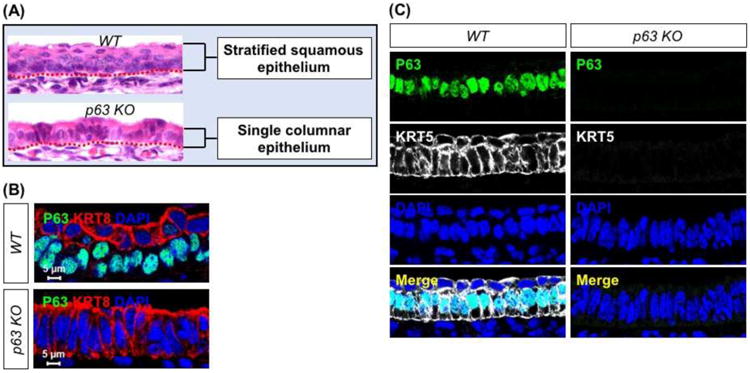
(A) p63 deletion results in a single layer of columnar epithelium at E18.5. (B) The epithelium of the esophagus remains simple columnar (KRT8+) in the E14.5 p63-/- esophagus. Note that KRT8 is present in the suprabasal cells of wild type esophagus at E14.5. (C) p63 deletion leads to the loss of KRT5 expression in the esophageal epithelium at E14.5. Abbreviation: KO, knockout.
SOX2 is expressed highly in the epithelium of the developing esophagus while its expression in the skin is limited to the mesenchyme dermal papilla [50, 51]. SOX2 not only plays important roles in the initial establishment of the esophagus from the anterior foregut, it is also required for generating the organized structure of the stratified squamous epithelium. Significant downregulation of SOX2 protein levels leads to a disorganized epithelium in hypomorphic Sox2GFP/COND mutants that have separated esophagus (∼60% of total mutants) [2]. In addition, SOX2 regulates the commitment of the epithelium to squamous cell fate, and the epithelial cells produce a large amount of mucins (Alcian blue+) in the esophagus of Sox2GFP/COND mutants [2]. The expression of p63 is maintained although the levels are reduced. This is consistent with the finding that the epithelium remains stratified in the esophagus [2].
Regulation of epithelial differentiation by Bmp signaling
Bmp signaling is not activated in the mouse esophageal epithelium until E14.5 when the expression of Noggin is lost [31]. Through analysis with the BRE-lacZ reporter mouse line, we showed that the activity of the Bmp signaling pathway is limited to the differentiating suprabasal cells at E14.5, and the activity is further enhanced at E15.5 and adult stage [31, 52]. When Bmp signaling is inactivated the epithelium fails to differentiate, although the stratification proceeds normally in the esophagus of Shh-Cre; Bmpr1aloxp/loxp mice. All of the epithelial cells express the basal cell proteins p63 and SOX2, and proliferating cells are present in the suprabasal layers [31]. Inhibition of Bmp signaling is also required for the self-renewal of basal cells in the adult esophagus. For example, treatment with Noggin promotes the self-renewal of basal progenitor cells in a 3-D organoid culture [53]. Conversely, treatment with Bmp4 reduces proliferation and promotes squamous differentiation of basal progenitor cells, leading to the expression of the differentiation markers Loricrin and Involucrin [52].
Regulation of epithelial differentiation by Notch signaling
Studies in the skin suggest that p63 controls the epithelial stratification and differentiation through Notch signaling. Activation of the Notch pathway is dependent on the binding of the transmembrane ligands Jagged (Jag1, 2) and Delta-like (Dll 1, 3, 4) proteins provided by the adjacent cells to Notch receptors. The receptors are then cleaved by protease metalloprotease and γ-secretase, thereby producing Notch intracellular domain (NICD). Subsequently, NICD translocates into the nucleus where it directly induces gene transcription by forming a complex with cofactors CSL (CBF1/RBPj/Su(H)/Lag-1) and mastermind-like (MAML1-3) proteins. Downstream targets of the Notch pathway include HES, HEY and ESR [54]. In the embryonic skin deletion of ΔNp63 results in decreased levels of Jag1, Notch1 and Hes1 while the levels of Notch2, Notch3 and RBPjκ are increased [48]. In the adults Notch activation is correlated with decreased expression of KRT5 and KRT14 in skin basal cells [55]. While it remains to be determined whether deletion of p63 in the esophagus invokes similar changes in the expression of Notch pathway components, a recent study demonstrated that Notch3 knockdown blocks squamous differentiation of cultured esophageal epithelium [56]. Moreover, inhibition of Notch signaling by γ-secretase inhibitors or ectopic expression of dominant negative MAML1 also reduces the expression of Involucrin. Consistent with these in vitro findings, overexpression of dominant negative MAML1 impairs the squamous differentiation in the esophagus of adult mice [56].
3.2 Stem/progenitor cells in the adult esophagus
Are basal cells heterogeneous or homogeneous?
Quick turnover of the stratified squamous epithelium requires constant self-renewal of basal cells in the adult esophagus. We previously used a KRT5-CreER transgenic mouse line to show that basal cells proliferate and differentiate to replenish the epithelium during daily tear-and-wear [57]. However, it remains unknown whether every single basal cells possess equal potential, and the findings thus far do not agree with each other (Fig 7).
Fig. 7. Heterogeneous vs. homogeneous basal cell population in the adult esophagus.
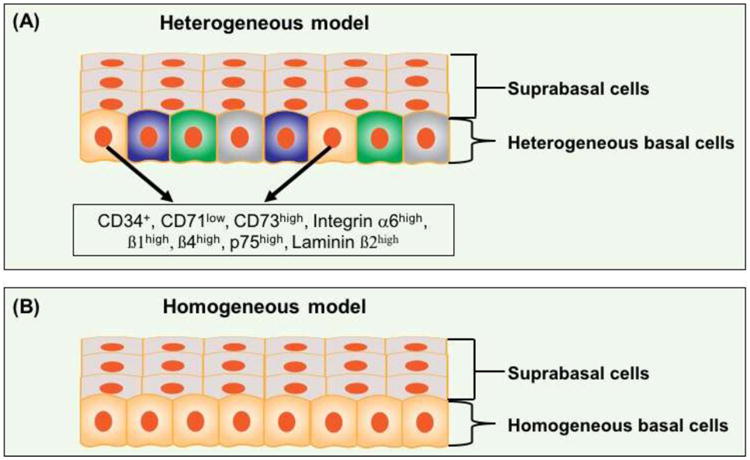
(A) Heterogeneous model. Multiple subpopulations of basal cells were reported to present stem cell capabilities and these subpopulations were indicated as (Laminin ß2high; Integrin ß1high), (CD34+), (Integrin α6high, CD71low) and (Integrin α6high, ß4high CD73high). Whether the subpopulations express overlapping cell surface markers has not been determined. (B) Homogeneous model. Basal cells are considered as a homogeneous population through lineage tracing and mathematical modeling. See text for details.
Seery et al. reported that basal cells in the human esophagus are heterogeneous [58]. They proposed that basal cells at the interpapillary zone contain stem cells characterized by low expression of Integrin ß1 and high levels of the ß2 laminin chain. These stem cells are less proliferative (Ki67+) and tend to go through asymmetric division to generate one stem cell and one differentiated daughter cell in response to cues from the underlying basement membrane [58]. By contrast, a side population (CD34+) in the basal layer of the mouse esophagus was considered as stem cells [59]. The population was initially identified by the Hoechst dye exclusion assay. The group found that these cells express CD34 and maintain proliferative capability after multiple passages. When cultured in a 3D system, CD34+ cells are able to reconstitute the stratified squamous epithelium [59]. Furthermore, the purified basal cell subpopulation (CD34+) is able to repair the esophageal epithelium following scratch-induced injury in a mouse model [59]. Interestingly, according to Croagh et al. the esophageal basal cells are divided into three subpopulations based on the expression levels of Integrin α6 and CD71. These three groups include quiescent stem cells (Itgα6highCD71low), transit-amplifying cells (Itgα6highCD71high) and cells destined to differentiate (Itgα6low CD71high) [60]. On the other hand, a recent report demonstrated that basal cells express differential levels of Integrin α6, ß1 and ß4 [53]. Similar to our findings [61], the authors found that SOX2+ basal cells express high levels of Itgß1 and p75, and they readily form 3-D organoids when cultured in the presence of growth factors including Egf [53, 61]. The authors further found that basal cells expressing high levels of Itgα6, ß4, and CD73 are more proliferative and less-differentiated, and retinoic acid treatment promotes self-renewal of this subpopulation [53]. It remains to be determined whether the basal cell subpopulations identified by different groups exhibit overlapping gene expression. For example, CD34+ subpopulation is also Itgα6highCD71low. We recently showed that Bmp4 is expressed specifically in a subpopulation of basal cells [52]. However, whether the expression pattern is stochastic or indeed labels a unique subpopulation requires further analysis. Genetic lineage labelling combined with in vitro live imaging will help address the issue.
On the other hand, a recent report by Frede et al. suggested that the normal esophageal epithelium is maintained by a single population of progenitor cells, arguing against the heterogeneous hypothesis [62]. This finding is consistent with what the group has previously reported through the use of lineage tracing combined with mathematics modelling [63]. The authors proposed that every basal cell possesses equal potential of self-renewal and differentiation. At homeostasis, cell production and cell loss are balanced as proliferating basal cells generate equal proportions of dividing and non-dividing cells [64]. By contrast, during wound healing basal cells in the injured site generate more proliferating cells until the epithelium is repaired. This potential seems to be shared by all of the basal cells lining the esophagus [63].
4. A New Paradigm for Muscle Development
Generation of peristalsis to move food along the esophagus requires coordinated action of multiple layers of muscle [65]. In adult mice, the cervical and most thoracic regions are composed of striated muscle, while lower thoracic and distal parts consist of smooth muscle [8, 66]. In humans the upper part of the esophagus is also surrounded by two layers of striated muscle. However, the muscle layers in the middle 1/3 is composed of a mixture of striated and smooth muscle, and the bottom 1/3 is surrounded entirely by two layers of smooth muscle [8, 67, 68]. The smooth muscle in the lower esophageal sphincter (LES) is important to maintain basal tonic contraction to prevent reflux of food back into the esophagus from the stomach [69, 70]. Weakening muscle function is believed to promote gastroesophageal reflux disease [69, 71]. Conversely, impaired relaxation of the LES and perturbed peristalsis are the main characteristics of achalasia, a motility disorder leading to accumulation of ingesta in the lower part of the esophagus (i.e. megaesophagus) [72].
When the esophagus is established from the anterior foregut, the mesenchyme surrounding the epithelium is uncommitted. Differentiation of the mesenchyme into the smooth muscle starts at approximately E11.0 in mice equivalent to 4-5 weeks of gestation in humans [73, 74]. Initially the entire esophagus is enclosed by two layers (outer longitudinal and inner circular) of smooth muscle. The precursors of striated muscle are first present in the proximal esophagus at E13.0, and the smooth muscle is replaced by striated muscle in a proximal-to-distal manner over the next three weeks until the adult pattern is achieved [66, 75]. Muscle formation occurs similarly in human esophageal development but the final striated–smooth muscle boundary is more proximal than in mice as described above.
Studies of the cell of origin for the striated muscle in the developing esophagus have been controversial. Earlier studies suggested that the striated muscle is derived from somite, a common source for striated muscles in the body trunk [76]. It was also proposed that transdifferentiation of smooth muscle cells into skeletal muscle cells occurs during esophageal development [77]. However, transdifferentiation was not observed when lineage tracing was used in genetic studies [77, 78]. Interestingly, a recent lineage tracing study demonstrated that the striated muscle is indeed originated from the cranial mesoderm [79]. The authors utilized Pax3Cre; R26mTmG mice to show that somite-derived myogenic Pax3-GFP+ cells do not contribute to esophageal striated muscles, although Pax3Cre-labeled cells contribute extensively to the diaphragm and trunk muscles. By contrast, lineage tracing experiments with Mesoderm progenitor 1 (Mesp1)-Cre and Isl1-Cre showed that cranial mesodermal cells generate the striated muscle layers in the mouse esophagus [79]. It was previously known that cranial mesoderm gives rise to the head muscles and derivatives of the second heart field [80], in contrast to the findings that all body muscles and part of the tongue musculature are established from somites [81]. This new finding now adds the esophageal striated muscle as the third derivative of the cranial mesoderm [79]. Intriguingly, the esophageal striated muscle develops through intercalating into the smooth muscle layer. It seems that the initial smooth muscles in the nascent esophagus form a scaffold to allow the migration of striated muscle progenitors from the cranial arch [79]. In addition, the transcription factor TBX1 is required for the downward migration of the muscle progenitors. Deletion of Tbx1 leads to the loss of striated muscles in the esophagus while the smooth muscles remain throughout the esophagus [79]. This new model of muscle development opens a new window into the possibility of dissecting the molecular mechanisms controlling striated muscle differentiation.
Several genes have been shown to regulate muscle development in the esophagus. For example, the cell surface receptor Cdo (also called Cdon) is required for setting up the striated-smooth muscle boundary. Deletion of Cdo leads to aberrantly proximal location of the boundary, achalasia and megaesophagus [82]. In addition, the bHLH transcription factor Myf5 is required for striated muscle differentiation [66]. Deletion of the Wnt signaling receptor Fz4 also affects the formation of the striated muscle, leading to esophageal distension [83]. Moreover, Pax7-/- mice develop megaesophagus due to the disrupted differentiation of striated muscle and abnormal orientation of smooth muscles [84]. Foxp1+/–; Foxp2-/- mice also display abnormal smooth muscles, and the striated muscle is absent in the esophagus of the compound mutants [85]. Many questions remain regarding the functions of these genes in muscle development. For example, do they regulate the migration and/or differentiation of the muscle precursors? Are they involved in the loss of the smooth muscle while replacing with the striated muscle? With the identification of the cell of origin for the striated muscle it is now possible to perform cell specific deletion/overexpression through Cre/loxP system, thereby providing a new dimension of insights into the muscle differentiation program.
5. Concluding Remarks
The development of the esophagus requires interaction between multiple transcription factors and cellular signaling pathways that mediate the epithelial-mesenchymal crosstalk. Through genetic analysis and in vitro modelling we have begun to gain insights into the mechanism regulating the initial establishment of the esophagus from the foregut and subsequent epithelial morphogenesis. We are also capable of identifying the cell of origin for different components of the mesenchymal derivatives by using lineage tracing tools. These novel findings provide us with new opportunities to study esophageal development and subsequent diseases which have seen significant increase over the past three decades. Some of the signaling molecules and transcription factors from development are reused during disease initiation and progression. For example, we have shown that suppressed Bmp signaling promotes basal cell hyperplasia in EoE [52]; high levels of SOX2 drive the malignant transformation of basal cells, leading to squamous cell carcinoma [61]. How these embryonic signaling molecules are reactivated during disease pathogenesis is an intriguing issue. Understanding these mechanisms will help us devise new tools to prevent or even reverse disease progression. That said, probing these mechanisms will need an integrated application of multiple technologies including genetic manipulation and live imaging. Moreover, we have fairly limited knowledge for the development of the esophageal submucosal glands which are lacking in rodents. With the advances of genome editing tools like CRISPR/Cas9 we now can consider generating genetic models to study these glands in animals such as porcine. We can also develop experimental protocols to directly study human submucosal glands through differentiation of human pluripotent stem cells.
Table 1.
Transcription factors and signaling molecules involved in the development of the esophageal epithelium and mesenchyme.
| Mouse mutants | Genetic function change | Epithelium or Muscle | Phenotypes and pathological disorders | References |
|---|---|---|---|---|
| Sox2EGFP/COND | Hypomorphism | Epithelium | Disrupted esophageal epithelium stratification and the epithelium secrets mucin. | [2] |
| p63-/- | Loss of function | Epithelium | Deficient in basal cells. Esophageal epithelium remains as simple columnar and fails to stratify. | [40-42] |
| Noggin-/- | Loss of function | Epithelium | Multiple abnormal gland-like pits lined with simple columnar epithelium producing Alcian Blue stained mucus. | [31] |
| Shh-Cre; Bmpr1aCA | Ectopic expression of constitutively active Bmpr1a | Epithelium | Multiple clusters of clusters of simple columnar epithelial cells along the esophagus. | [31] |
| Raldh2−/− | Loss of function | Epithelium | Formation of esophageal atresia. | [35] |
| Tbx1−/− | Loss of function | Muscle | Lack of skeletal muscle. | [79] |
| Cdo−/− | Loss of function | Muscle | Proximal move of skeletal-smooth muscle boundary, achalasia and megaesophagus. | [84] |
| Myf5-/- | Loss of function | Muscle | Failed to undergo skeletal myogenesis. | [66] |
| Pax7−/− | Loss of function | Muscle | Megaesophagus due to the disrupted differentiation of striated muscle and abnormal orientation of smooth muscles. | [84] |
| Foxp1+/-; Foxp2-/- | Loss of function | Muscle | Absence of esophageal skeletal muscle. | [85] |
Acknowledgments
We thank members of Que laboratory, Ms. Maria R. Stupnikov and Dr. Wellington V. Cardoso at the Columbia Center for Human Development for critical reading of the manuscript and helpful discussion. The work in Que laboratory is partly supported by NIH/NIDDK R01DK100342, NYSTEM C029555 and March of Dimes Research Grant 1-FY14-528. The work in Dr. Kuancan Liu's laboratory is supported by the National Natural Science Foundation of China (No.81302068), the National High Technology Research and Development Program of China (863 Program, No. 2014AA020541), and the Program for the Top Young Innovative Talents of Fujian Province.
Abbreviations
- EA/TEF
esophageal atresia/tracheoesophageal fistula
- LES
lower esophageal sphincter
- Bmp
bone morphogenetic protein
- Shh
sonic hedgehog
- Ihh
Indian hedgehog
- MYBPH
myosin binding protein H
- ROCK1
Rho kinase 1
- sFRPs
secreted Frizzled-related proteins
- RA
retinoic acid
- Raldh2
retinaldehyde dehydrogenase 2
Footnotes
Conflict of Interest: All authors have no conflict of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Que JM. The initial establishment and epithelial morphogenesis of the esophagus: a new model of tracheal-esophageal separation and transition of simple columnar into stratified squamous epithelium in the developing esophagus. Wiley Interdiscip Rev Dev Biol. 2015;4(4):419–30. doi: 10.1002/wdev.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Que J, Okubo T, Goldenring JR, Nam KT, Kurotani R, Morrisey EE, Taranova O, Pevny LH, Hogan BL. Multiple dose-dependent roles for Sox2 in the patterning and differentiation of anterior foregut endoderm. Development. 2007;134(13):2521–31. doi: 10.1242/dev.003855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Domyan ET, Ferretti E, Throckmorton K, Mishina Y, Nicolis SK, Sun X. Signaling through BMP receptors promotes respiratory identity in the foregut via repression of Sox2. Development. 2011;138(5):971–81. doi: 10.1242/dev.053694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lazzaro D, Price M, Defelice M, Dilauro R. The Transcription Factor-Ttf-1 Is Expressed at the Onset of Thyroid and Lung Morphogenesis and in Restricted Regions of the Fetal Brain. Development. 1991;113(4):1093–&. doi: 10.1242/dev.113.4.1093. [DOI] [PubMed] [Google Scholar]
- 5.Serls AE, Doherty S, Parvatiyar P, Wells JM, Deutsch GH. Different thresholds of fibroblast growth factors pattern the ventral foregut into liver and lung. Development. 2005;132(1):35–47. doi: 10.1242/dev.01570. [DOI] [PubMed] [Google Scholar]
- 6.Treuting PM. Comparative anatomy and histology : a mouse and human atlas. Elsevier/Academic Press; Amsterdam: 2012. [Google Scholar]
- 7.Long JD, Orlando RC. Esophageal submucosal glands: structure and function. Am J Gastroenterol. 1999;94(10):2818–24. doi: 10.1111/j.1572-0241.1999.1422_b.x. [DOI] [PubMed] [Google Scholar]
- 8.Jacobs IJ, Ku WY, Que J. Genetic and cellular mechanisms regulating anterior foregut and esophageal development. Dev Biol. 2012;369(1):54–64. doi: 10.1016/j.ydbio.2012.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Que J, Choi M, Ziel JW, Klingensmith J, Hogan BL. Morphogenesis of the trachea and esophagus: current players and new roles for noggin and Bmps. Differentiation. 2006;74(7):422–37. doi: 10.1111/j.1432-0436.2006.00096.x. [DOI] [PubMed] [Google Scholar]
- 10.Ioannides AS, Massa V, Ferraro E, Cecconi F, Spitz L, Henderson DJ, Copp AJ. Foregut separation and tracheo-oesophageal malformations: the role of tracheal outgrowth, dorso-ventral patterning and programmed cell death. Dev Biol. 2010;337(2):351–62. doi: 10.1016/j.ydbio.2009.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Metzger R, Wachowiak R, Kluth D. Embryology of the early foregut. Seminars in pediatric surgery. 2011;20(3):136–44. doi: 10.1053/j.sempedsurg.2011.03.004. [DOI] [PubMed] [Google Scholar]
- 12.Malmstrom K, Lohi J, Lindahl H, Pelkonen A, Kajosaari M, Sarna S, Malmberg LP, Makela MJ. Longitudinal follow-up of bronchial inflammation, respiratory symptoms, and pulmonary function in adolescents after repair of esophageal atresia with tracheoesophageal fistula. J Pediatr. 2008;153(3):396–401. doi: 10.1016/j.jpeds.2008.03.034. [DOI] [PubMed] [Google Scholar]
- 13.Robertson DF, Mobaireek K, Davis GM, Coates AL. Late pulmonary function following repair of tracheoesophageal fistula or esophageal atresia. Pediatr Pulmonol. 1995;20(1):21–6. doi: 10.1002/ppul.1950200105. [DOI] [PubMed] [Google Scholar]
- 14.Sistonen S, Malmberg P, Malmstrom K, Haahtela T, Sarna S, Rintala RJ, Pakarinen MP. Repaired oesophageal atresia: respiratory morbidity and pulmonary function in adults. Eur Respir J. 2010;36(5):1106–12. doi: 10.1183/09031936.00153209. [DOI] [PubMed] [Google Scholar]
- 15.Fragoso AC, Tovar JA. The multifactorial origin of respiratory morbidity in patients surviving neonatal repair of esophageal atresia. Front Pediatr. 2014;2:39. doi: 10.3389/fped.2014.00039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Harris-Johnson KS, Domyan ET, Vezina CM, Sun X. beta-Catenin promotes respiratory progenitor identity in mouse foregut. Proc Natl Acad Sci U S A. 2009;106(38):16287–92. doi: 10.1073/pnas.0902274106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Avilion AA, Nicolis SK, Pevny LH, Perez L, Vivian N, Lovell-Badge R. Multipotent cell lineages in early mouse development depend on SOX2 function. Genes Dev. 2003;17(1):126–40. doi: 10.1101/gad.224503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu K, Lin B, Zhao M, Yang X, Chen M, Gao A, Liu F, Que J, Lan X. The multiple roles for Sox2 in stem cell maintenance and tumorigenesis. Cell Signal. 2013;25(5):1264–71. doi: 10.1016/j.cellsig.2013.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Williamson KA, Hever AM, Rainger J, Rogers RC, Magee A, Fiedler Z, Keng WT, Sharkey FH, McGill N, Hill CJ, Schneider A, Messina M, Turnpenny PD, Fantes JA, van Heyningen V, FitzPatrick DR. Mutations in SOX2 cause anophthalmia-esophageal-genital (AEG) syndrome. Hum Mol Genet. 2006;15(9):1413–22. doi: 10.1093/hmg/ddl064. [DOI] [PubMed] [Google Scholar]
- 20.Kondoh H, Lovell-Badge R. Sox2: biology and role in development and disease [Google Scholar]
- 21.Minoo P, Su G, Drum H, Bringas P, Kimura S. Defects in tracheoesophageal and lung morphogenesis in Nkx2.1(-/-) mouse embryos. Dev Biol. 1999;209(1):60–71. doi: 10.1006/dbio.1999.9234. [DOI] [PubMed] [Google Scholar]
- 22.Graham V, Khudyakov J, Ellis P, Pevny L. SOX2 functions to maintain neural progenitor identity. Neuron. 2003;39(5):749–65. doi: 10.1016/s0896-6273(03)00497-5. [DOI] [PubMed] [Google Scholar]
- 23.Hosono Y, Yamaguchi T, Mizutani E, Yanagisawa K, Arima C, Tomida S, Shimada Y, Hiraoka M, Kato S, Yokoi K, Suzuki M, Takahashi T. MYBPH, a transcriptional target of TTF-1, inhibits ROCK1, and reduces cell motility and metastasis. EMBO J. 2012;31(2):481–93. doi: 10.1038/emboj.2011.416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mahlapuu M, Enerback S, Carlsson P. Haploinsufficiency of the forkhead gene Foxf1, a target for sonic hedgehog signaling, causes lung and foregut malformations. Development. 2001;128(12):2397–406. doi: 10.1242/dev.128.12.2397. [DOI] [PubMed] [Google Scholar]
- 25.Rankin SA, Han L, McCracken KW, Kenny AP, Anglin CT, Grigg EA, Crawford CM, Wells JM, Shannon JM, Zorn AM. A Retinoic Acid-Hedgehog Cascade Coordinates Mesoderm-Inducing Signals and Endoderm Competence during Lung Specification. Cell Rep. 2016;16(1):66–78. doi: 10.1016/j.celrep.2016.05.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Goss AM, Tian Y, Tsukiyama T, Cohen ED, Zhou D, Lu MM, Yamaguchi TP, Morrisey EE. Wnt2/2b and beta-catenin signaling are necessary and sufficient to specify lung progenitors in the foregut. Dev Cell. 2009;17(2):290–8. doi: 10.1016/j.devcel.2009.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Woo J, Miletich I, Kim BM, Sharpe PT, Shivdasani RA. Barx1-mediated inhibition of Wnt signaling in the mouse thoracic foregut controls tracheo-esophageal septation and epithelial differentiation. PLoS One. 2011;6(7):e22493. doi: 10.1371/journal.pone.0022493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li Y, Gordon J, Manley NR, Litingtung Y, Chiang C. Bmp4 is required for tracheal formation: a novel mouse model for tracheal agenesis. Dev Biol. 2008;322(1):145–55. doi: 10.1016/j.ydbio.2008.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li Y, Litingtung Y, Ten Dijke P, Chiang C. Aberrant Bmp signaling and notochord delamination in the pathogenesis of esophageal atresia. Dev Dyn. 2007;236(3):746–54. doi: 10.1002/dvdy.21075. [DOI] [PubMed] [Google Scholar]
- 30.Shu W, Jiang YQ, Lu MM, Morrisey EE. Wnt7b regulates mesenchymal proliferation and vascular development in the lung. Development. 2002;129(20):4831–42. doi: 10.1242/dev.129.20.4831. [DOI] [PubMed] [Google Scholar]
- 31.Rodriguez P, Da Silva S, Oxburgh L, Wang F, Hogan BL, Que J. BMP signaling in the development of the mouse esophagus and forestomach. Development. 2010;137(24):4171–6. doi: 10.1242/dev.056077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Litingtung Y, Lei L, Westphal H, Chiang C. Sonic hedgehog is essential to foregut development. Nat Genet. 1998;20(1):58–61. doi: 10.1038/1717. [DOI] [PubMed] [Google Scholar]
- 33.Motoyama J, Liu J, Mo R, Ding Q, Post M, Hui CC. Essential function of Gli2 and Gli3 in the formation of lung, trachea and oesophagus. Nat Genet. 1998;20(1):54–7. doi: 10.1038/1711. [DOI] [PubMed] [Google Scholar]
- 34.Wilson JG, Roth CB, Warkany J. An analysis of the syndrome of malformations induced by maternal vitamin A deficienc. Effects of restoration of vitamin A at various times during gestation. Am J Anat. 1953;92(2):189–217. doi: 10.1002/aja.1000920202. [DOI] [PubMed] [Google Scholar]
- 35.Wang Z, Dolle P, Cardoso WV, Niederreither K. Retinoic acid regulates morphogenesis and patterning of posterior foregut derivatives. Dev Biol. 2006;297(2):433–45. doi: 10.1016/j.ydbio.2006.05.019. [DOI] [PubMed] [Google Scholar]
- 36.Chen F, Desai TJ, Qian J, Niederreither K, Lu JN, Cardoso WV. Inhibition of Tgf beta signaling by endogenous retinoic acid is essential for primary lung bud induction. Development. 2007;134(16):2969–2979. doi: 10.1242/dev.006221. [DOI] [PubMed] [Google Scholar]
- 37.Yu WY, Slack JM, Tosh D. Conversion of columnar to stratified squamous epithelium in the developing mouse oesophagus. Dev Biol. 2005;284(1):157–70. doi: 10.1016/j.ydbio.2005.04.042. [DOI] [PubMed] [Google Scholar]
- 38.Fuchs E, Raghavan S. Getting under the skin of epidermal morphogenesis. Nat Rev Genet. 2002;3(3):199–209. doi: 10.1038/nrg758. [DOI] [PubMed] [Google Scholar]
- 39.Fuchs E. Scratching the surface of skin development. Nature. 2007;445(7130):834–42. doi: 10.1038/nature05659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mills AA, Zheng B, Wang XJ, Vogel H, Roop DR, Bradley A. p63 is a p53 homologue required for limb and epidermal morphogenesis. Nature. 1999;398(6729):708–13. doi: 10.1038/19531. [DOI] [PubMed] [Google Scholar]
- 41.Yang A, Schweitzer R, Sun D, Kaghad M, Walker N, Bronson RT, Tabin C, Sharpe A, Caput D, Crum C, McKeon F. p63 is essential for regenerative proliferation in limb, craniofacial and epithelial development. Nature. 1999;398(6729):714–8. doi: 10.1038/19539. [DOI] [PubMed] [Google Scholar]
- 42.Daniely Y, Liao G, Dixon D, Linnoila RI, Lori A, Randell SH, Oren M, Jetten AM. Critical role of p63 in the development of a normal esophageal and tracheobronchial epithelium. Am J Physiol Cell Physiol. 2004;287(1):C171–81. doi: 10.1152/ajpcell.00226.2003. [DOI] [PubMed] [Google Scholar]
- 43.Koster MI, Kim S, Mills AA, DeMayo FJ, Roop DR. p63 is the molecular switch for initiation of an epithelial stratification program. Genes Dev. 2004;18(2):126–31. doi: 10.1101/gad.1165104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang X, Ouyang H, Yamamoto Y, Kumar PA, Wei TS, Dagher R, Vincent M, Lu X, Bellizzi AM, Ho KY, Crum CP, Xian W, McKeon F. Residual embryonic cells as precursors of a Barrett's-like metaplasia. Cell. 2011;145(7):1023–35. doi: 10.1016/j.cell.2011.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Crum CP, McKeon FD. p63 in epithelial survival, germ cell surveillance and neoplasia. Annu Rev Pathol. 2010;5:349–71. doi: 10.1146/annurev-pathol-121808-102117. [DOI] [PubMed] [Google Scholar]
- 46.McKeon F. p63 and the epithelial stem cell: more than status quo? Genes Dev. 2004;18(5):465–9. doi: 10.1101/gad.1190504. [DOI] [PubMed] [Google Scholar]
- 47.Romano RA, Ortt K, Birkaya B, Smalley K, Sinha S. An active role of the DeltaN isoform of p63 in regulating basal keratin genes K5 and K14 and directing epidermal cell fate. PLoS One. 2009;4(5):e5623. doi: 10.1371/journal.pone.0005623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Romano RA, Smalley K, Magraw C, Serna VA, Kurita T, Raghavan S, Sinha S. DeltaNp63 knockout mice reveal its indispensable role as a master regulator of epithelial development and differentiation. Development. 2012;139(4):772–82. doi: 10.1242/dev.071191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pignon JC, Grisanzio C, Geng Y, Song J, Shivdasani RA, Signoretti S. p63-expressing cells are the stem cells of developing prostate, bladder, and colorectal epithelia. Proc Natl Acad Sci U S A. 2013;110(20):8105–10. doi: 10.1073/pnas.1221216110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Driskell RR, Giangreco A, Jensen KB, Mulder KW, Watt FM. Sox2-positive dermal papilla cells specify hair follicle type in mammalian epidermis. Development. 2009;136(16):2815–23. doi: 10.1242/dev.038620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rendl M, Lewis L, Fuchs E. Molecular dissection of mesenchymal-epithelial interactions in the hair follicle. PLoS Biol. 2005;3(11):e331. doi: 10.1371/journal.pbio.0030331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Jiang M, Ku WY, Zhou Z, Dellon ES, Falk GW, Nakagawa H, Wang ML, Liu K, Wang J, Katzka DA, Peters JH, Lan X, Que J. BMP-driven NRF2 activation in esophageal basal cell differentiation and eosinophilic esophagitis. J Clin Invest. 2015;125(4):1557–68. doi: 10.1172/JCI78850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.DeWard AD, Cramer J, Lagasse E. Cellular heterogeneity in the mouse esophagus implicates the presence of a nonquiescent epithelial stem cell population. Cell Rep. 2014;9(2):701–11. doi: 10.1016/j.celrep.2014.09.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kopan R, Ilagan MX. The canonical Notch signaling pathway: unfolding the activation mechanism. Cell. 2009;137(2):216–33. doi: 10.1016/j.cell.2009.03.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Blanpain C, Lowry WE, Pasolli HA, Fuchs E. Canonical Notch signaling functions as a commitment switch in the epidermal lineage. Gene Dev. 2006;20(21):3022–3035. doi: 10.1101/gad.1477606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ohashi S, Natsuizaka M, Yashiro-Ohtani Y, Kalman RA, Nakagawa M, Wu L, Klein-Szanto AJ, Herlyn M, Diehl JA, Katz JP, Pear WS, Seykora JT, Nakagawa H. NOTCH1 and NOTCH3 coordinate esophageal squamous differentiation through a CSL-dependent transcriptional network. Gastroenterology. 2010;139(6):2113–23. doi: 10.1053/j.gastro.2010.08.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Jovov B, Que J, Tobey NA, Djukic Z, Hogan BL, Orlando RC. Role of E-cadherin in the pathogenesis of gastroesophageal reflux disease. Am J Gastroenterol. 2011;106(6):1039–47. doi: 10.1038/ajg.2011.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Seery JP, Watt FM. Asymmetric stem-cell divisions define the architecture of human oesophageal epithelium. Curr Biol. 2000;10(22):1447–50. doi: 10.1016/s0960-9822(00)00803-4. [DOI] [PubMed] [Google Scholar]
- 59.Kalabis J, Oyama K, Okawa T, Nakagawa H, Michaylira CZ, Stairs DB, Figueiredo JL, Mahmood U, Diehl JA, Herlyn M, Rustgi AK. A subpopulation of mouse esophageal basal cells has properties of stem cells with the capacity for self-renewal and lineage specification. Journal of Clinical Investigation. 2008;118(12):3860–3869. doi: 10.1172/JCI35012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Croagh D, Phillips WA, Redvers R, Thomas RJS, Kaur P. Identification of candidate murine esophageal stem cells using a combination of cell kinetic studies and cell surface markers. Stem Cells. 2007;25(2):313–318. doi: 10.1634/stemcells.2006-0421. [DOI] [PubMed] [Google Scholar]
- 61.Liu K, Jiang M, Lu Y, Chen H, Sun J, Wu S, Ku WY, Nakagawa H, Kita Y, Natsugoe S, Peters JH, Rustgi A, Onaitis MW, Kiernan A, Chen X, Que J. Sox2 cooperates with inflammation-mediated Stat3 activation in the malignant transformation of foregut basal progenitor cells. Cell Stem Cell. 2013;12(3):304–15. doi: 10.1016/j.stem.2013.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Frede J, Greulich P, Nagy T, Simons BD, Jones PH. A single dividing cell population with imbalanced fate drives oesophageal tumour growth. Nat Cell Biol. 2016;18(9):967–78. doi: 10.1038/ncb3400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Doupe DP, Alcolea MP, Roshan A, Zhang G, Klein AM, Simons BD, Jones PH. A Single Progenitor Population Switches Behavior to Maintain and Repair Esophageal Epithelium. Science. 2012;337(6098):1091–1093. doi: 10.1126/science.1218835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Alcolea MP, Greulich P, Wabik A, Frede J, Simons BD, Jones PH. Differentiation imbalance in single oesophageal progenitor cells causes clonal immortalization and field change. Nat Cell Biol. 2014;16(6):615–22. doi: 10.1038/ncb2963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Meyer GW, Austin RM, Brady CE, 3rd, Castell DO. Muscle anatomy of the human esophagus. J Clin Gastroenterol. 1986;8(2):131–4. doi: 10.1097/00004836-198604000-00005. [DOI] [PubMed] [Google Scholar]
- 66.Kablar B, Tajbakhsh S, Rudnicki MA. Transdifferentiation of esophageal smooth to skeletal muscle is myogenic bHLH factor-dependent. Development. 2000;127(8):1627–39. doi: 10.1242/dev.127.8.1627. [DOI] [PubMed] [Google Scholar]
- 67.Yazaki E, Sifrim D. Anatomy and physiology of the esophageal body. Dis Esophagus. 2012;25(4):292–8. doi: 10.1111/j.1442-2050.2011.01180.x. [DOI] [PubMed] [Google Scholar]
- 68.Vegesna AK, Chuang KY, Besetty R, Phillips SJ, Braverman AS, Barbe MF, Ruggieri MR, Miller LS. Circular smooth muscle contributes to esophageal shortening during peristalsis. World J Gastroenterol. 2012;18(32):4317–22. doi: 10.3748/wjg.v18.i32.4317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Boeckxstaens GE. The lower oesophageal sphincter. Neurogastroenterol Motil. 2005;17(1):13–21. doi: 10.1111/j.1365-2982.2005.00661.x. [DOI] [PubMed] [Google Scholar]
- 70.Goyal RK, Rattan S. Genesis of basal sphincter pressure: effect of tetrodotoxin on lower esophageal sphincter pressure in opossum in vivo. Gastroenterology. 1976;71(1):62–7. [PubMed] [Google Scholar]
- 71.Babaei A, Bhargava V, Korsapati H, Zheng WH, Mittal RK. A unique longitudinal muscle contraction pattern associated with transient lower esophageal sphincter relaxation. Gastroenterology. 2008;134(5):1322–31. doi: 10.1053/j.gastro.2008.02.031. [DOI] [PubMed] [Google Scholar]
- 72.Park W, Vaezi MF. Etiology and pathogenesis of achalasia: the current understanding. Am J Gastroenterol. 2005;100(6):1404–14. doi: 10.1111/j.1572-0241.2005.41775.x. [DOI] [PubMed] [Google Scholar]
- 73.McHugh KM. Molecular analysis of gastrointestinal smooth muscle development. J Pediatr Gastroenterol Nutr. 1996;23(4):379–94. doi: 10.1097/00005176-199611000-00001. [DOI] [PubMed] [Google Scholar]
- 74.McHugh KM. Molecular analysis of smooth muscle development in the mouse. Dev Dyn. 1995;204(3):278–90. doi: 10.1002/aja.1002040306. [DOI] [PubMed] [Google Scholar]
- 75.Sang Q, Young HM. Development of nicotinic receptor clusters and innervation accompanying the change in muscle phenotype in the mouse esophagus. J Comp Neurol. 1997;386(1):119–36. doi: 10.1002/(sici)1096-9861(19970915)386:1<119::aid-cne11>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- 76.Christ B, Ordahl CP. Early stages of chick somite development. Anat Embryol (Berl) 1995;191(5):381–96. doi: 10.1007/BF00304424. [DOI] [PubMed] [Google Scholar]
- 77.Patapoutian A, Wold BJ, Wagner RA. Evidence for developmentally programmed transdifferentiation in mouse esophageal muscle. Science. 1995;270(5243):1818–21. doi: 10.1126/science.270.5243.1818. [DOI] [PubMed] [Google Scholar]
- 78.Rishniw M, Xin HB, Deng KY, Kotlikoff MI. Skeletal myogenesis in the mouse esophagus does not occur through transdifferentiation. Genesis. 2003;36(2):81–2. doi: 10.1002/gene.10198. [DOI] [PubMed] [Google Scholar]
- 79.Gopalakrishnan S, Comai G, Sambasivan R, Francou A, Kelly RG, Tajbakhsh S. A Cranial Mesoderm Origin for Esophagus Striated Muscles. Dev Cell. 2015;34(6):694–704. doi: 10.1016/j.devcel.2015.07.003. [DOI] [PubMed] [Google Scholar]
- 80.Nathan E, Monovich A, Tirosh-Finkel L, Harrelson Z, Rousso T, Rinon A, Harel I, Evans SM, Tzahor E. The contribution of Islet1-expressing splanchnic mesoderm cells to distinct branchiomeric muscles reveals significant heterogeneity in head muscle development. Development. 2008;135(4):647–57. doi: 10.1242/dev.007989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Relaix F, Rocancourt D, Mansouri A, Buckingham M. A Pax3/Pax7-dependent population of skeletal muscle progenitor cells. Nature. 2005;435(7044):948–53. doi: 10.1038/nature03594. [DOI] [PubMed] [Google Scholar]
- 82.Romer AI, Singh J, Rattan S, Krauss RS. Smooth muscle fascicular reorientation is required for esophageal morphogenesis and dependent on Cdo. Journal of Cell Biology. 2013;201(2):309–323. doi: 10.1083/jcb.201301005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Wang Y, Huso D, Cahill H, Ryugo D, Nathans J. Progressive cerebellar, auditory, and esophageal dysfunction caused by targeted disruption of the frizzled-4 gene. J Neurosci. 2001;21(13):4761–71. doi: 10.1523/JNEUROSCI.21-13-04761.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Chihara D, Romer AI, Bentzinger CF, Rudnicki MA, Krauss RS. PAX7 is required for patterning the esophageal musculature. Skelet Muscle. 2015;5 doi: 10.1186/s13395-015-0068-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Shu W, Lu MM, Zhang Y, Tucker PW, Zhou D, Morrisey EE. Foxp2 and Foxp1 cooperatively regulate lung and esophagus development. Development. 2007;134(10):1991–2000. doi: 10.1242/dev.02846. [DOI] [PubMed] [Google Scholar]