

Abstract
Mouse models are great tools to study the mechanisms of disease development. Theiler’s murine encephalomyelitis virus is used in two distinct viral infection mouse models to study the human diseases multiple sclerosis (MS) and epilepsy. Intracerebral (i.c.) infection of the SJL/J mouse strain results in persistent viral infection of the central nervous system and a MS-like disease, while i.c. infection of the C57BL/6J mouse strain results in acute seizures and epilepsy. Our understanding of how the immune system contributes to the development of two disparate diseases caused by the same virus is presented.
Keywords: Theiler’s murine encephalomyelitis virus (TMEV), Multiple Sclerosis (MS), Epilepsy, Seizures, Mouse model, Immune response
1. Introduction
Theiler’s murine encephalomyelitis virus (TMEV) is a naturally occurring enteric pathogen of the mouse (Lipton, 1975; Theiler, 1937). This virus is used as a mouse model for two distinct diseases of humans: multiple sclerosis (MS) and epilepsy. These mouse models are a powerful tool to study the mechanisms of how the immune response to viral infection can contribute to MS and epilepsy and the current knowledge in the field will be reviewed herein.
TMEV was first described by Max Theiler in 1937, when he isolated the Theiler’s original (TO) strain from the central nervous system (CNS) of mice having flaccid hind leg paralysis (Lipton, 1975; Theiler, 1937). In 1952 Daniels et al isolated another TMEV strain, called Daniels (DA), from spontaneously paralyzed mice that was shown to induce spinal cord demyelination (Daniels et al., 1952). It was not until 1975, when Lipton described an experimental mouse model of TMEV-DA-induced demyelination, that this infection model became a commonly used animal model to study MS (Lipton, 1975).
TMEV is a non-enveloped, single stranded RNA virus from the cardiovirus genus, picornavirus family (Clatch et al., 1986; Ohara et al., 1988). TMEV normally infects mouse gut without causing symptoms, however TMEV can, rarely, naturally infect the CNS. Based on neurovirulence in the CNS of susceptible mouse strains, TMEV can be divided into two different subgroups: 1) GDVII, comprised of the strains GDVII and FA, is extremely virulent to mice and mice die within 1–2 weeks of infection; and 2) TO, that includes the DA and BeAn strains, causes encephalitis during the acute stage of infection and demyelinating disease during the chronic stage of infection (Libbey et al., 2008; Lipton, 1975; Tsunoda and Fujinami, 2010). In this review the focus will be on the DA strain.
Disease development following TMEV infection depends on the mouse strain (Figure 1). The SJL/J mouse strain, when intracerebrally (i.c.) infected with TMEV, develops a MS-like disease termed TMEV-induced demyelinating disease (TMEV-IDD) (Figure 1A) (Dal Canto et al., 1996; Lipton, 1975; Lipton and Jelachich, 1997), while the C57BL/6J mouse strain, when i.c. infected with TMEV, develops acute seizures, which progress to epilepsy (Figure 1C) (Libbey et al., 2008). The difference between the mouse strains seems to be partially due to the strong antiviral innate immune response, such as type I interferon (IFN), observed in C57BL/6J mice (Rodriguez et al., 1995).
Figure 1. Schematic representation of two distinct mouse models using TMEV infection.
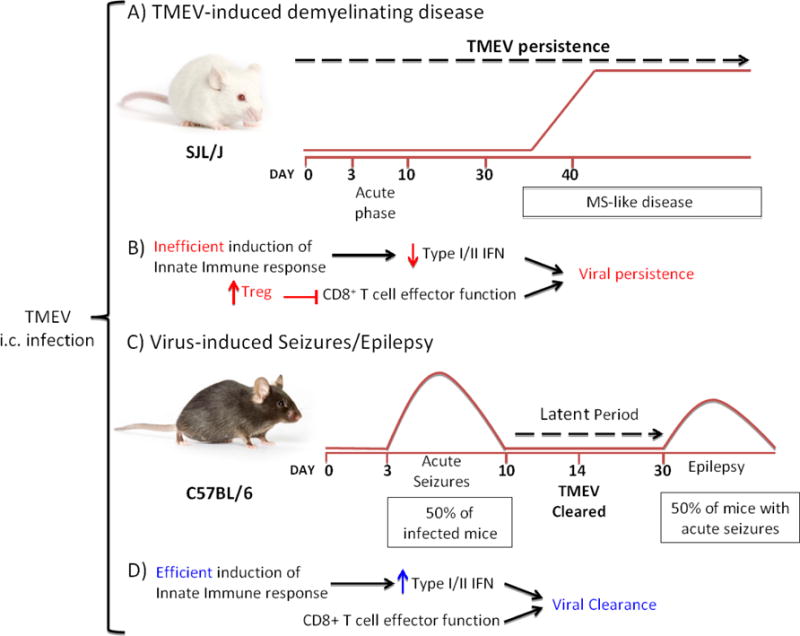
A) TMEV-IDD: I.c infection of SJL/J mice with TMEV results in persistent viral infection that lasts throughout the lifespan of the mouse. This results in a biphasic disease. An acute phase occurs between 3-10 days post infection. Viral persistence in the CNS results in chronic neuroinflammation leading to a MS-like phase, which appears after 30 days post infection. In this phase weakness of the hind limbs and an ataxic paralysis are observed. This model mimics some of the pathological and clinical features observed in MS patients with the progressive form of the disease. B) TMEV infection of the susceptible SJL/J mouse strain induces low type I (IFN-α and IFN-β) and type II (IFN-γ) IFN responses during the acute phase. Additionally, CD8+ T cell effector function is suppressed by the elevated induction of Tregs in this mouse strain, resulting in viral persistence and demyelination. C) Virus-induced seizures/epilepsy: I.c infection of C57BL/6J mice with TMEV results in acute seizures in 50% of the infected mice between 3–10 days post infection. After viral clearance, a variable latent period occurs where no behavioral seizures are observed. After the latent period, approximately 50% of the mice that have experienced acute seizures develop epilepsy. D) TMEV infection of the C57BL/6 mouse strain results in an efficient induction of antiviral immune response by type I and type II IFNs. Also, the lower number of Tregs induced in this mouse strain after TMEV infection allows for viral clearance by CD8+ T effector cells.
2. Multiple sclerosis
MS is the most common inflammatory demyelinating disease of the CNS affecting over 2.5 million people world-wide, and with approximately 400,000 cases in North America (Mazrouei et al., 2016). It is a chronic and progressive autoimmune disease and it is the leading cause of non-traumatic neurological disability in young adults (Compston and Coles, 2008). As an autoimmune disease, the host immune system attacks its own tissue. In the case of MS, the myelin proteins (myelin sheath) are attacked by the patient’s own immune system (Figure 2). Axons projecting from the neurons are usually wrapped in a material called myelin, which “insulates” the nerve cell, and gaps in the myelin called Ranvier nodes allow the saltatory impulse conduction to occur (Figure 2A). In MS patients, due to the myelin destruction, the saltatory conduction is impaired resulting in inefficient (slow) or blocked impulse conduction (Figure 2B). Signs and symptoms of the disease include cognitive and motor impairment (including paralysis), vertigo, tremor, loss of vision, weakness and dementia (Compston and Coles, 2008; Miller et al., 2012).
Figure 2. Demyelination as a result of TMEV infection of the CNS.
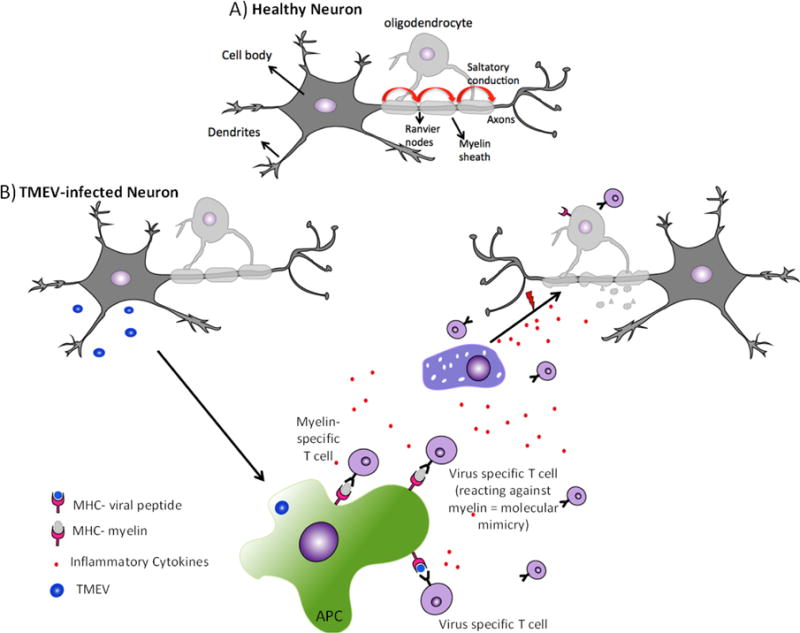
A) Healthy neurons have an intact myelin sheath, produced by oligodendrocytes. Nodes of Ranvier between myelin sheaths facilitate the saltatory conduction mechanism (curved red arrows). B) Immune cell infiltration during TMEV-IDD: myelin-specific and/or viral-specific T cells, together with other inflammatory immune cells, infiltrate the CNS through the blood-brain barrier. In response to viral infection, antigen-presenting cells (APCs) process and present viral peptides to T cells, which become activated resulting in the secretion of pro-inflammatory cytokines leading to the recruitment of other immune cells such as macrophages, resulting in tissue damage and release of self-antigens. Self-antigens can also be released through direct lysis of virus-infected neurons and oligodendrocytes. Self-antigens are processed and presented by APCS to T cells, a process called epitope spreading. The structural and sequence homology shared between viral-peptides and self-peptides can also result in the activation of virus-specific T cell in the periphery. These T cells when migrate into the CNS, react against self-antigens (molecular mimicry), leading to tissue damage. Prolonged inflammation in the CNS results in immune-mediated tissue damage, where myelin is targeted, leading to demyelination. In the absence of myelin, impulse conduction by neurons is impaired resulting in clinical signs of the disease.
MS is characterized by a multifocal inflammatory demyelination, resulting in the formation of chronic multifocal sclerotic plaques (Compston and Coles, 2008). Neuronal degeneration and axonal loss also occur during the progression of the disease and are related to permanent neurological deficit (Bjartmar et al., 2000). The clinical course of the disease consists of an early stage that is characterized by the first attack (Clinically Isolated Syndrome), which involves inflammation and some characteristics of demyelination. The disease progresses to the relapsing-remitting phase, which is characterized by attacks separated by periods of no clinical symptoms. This phase can last for decades, and it is characterized by inflammatory demyelination that results in the formation of demyelinating plaques in the white matter (Lassmann, 2011; Lassmann and Bradl, 2016; Libbey et al., 2014). Increased demyelination and axonal loss are found when disease involves a progressive clinical course (primary progressive or secondary progressive) and just a few options for treatment are available at this stage of the disease. Active demyelinating lesions in the brain and spinal cord are the pathologic hallmark of MS (Frohman et al., 2006). Lesions are comprised predominantly of infiltrating immune cells, such as macrophages and T cells, and resident CNS microglia, and are accompanied by the disruption of the blood-brain barrier (BBB) [(Compston and Coles, 2008); reviewed in (Perez-Cerda et al., 2016)].
The etiology of MS is largely unknown, and it is widely accepted that MS is a multifactorial disease. The contribution of genetic factors is well established in MS susceptibility, and close to 10% of MS patients have first or second degree relatives with the disease (Ebers, 1983). Although genetic factors specifically associated with the major histocompatibility complex (MHC) class II play a role in MS development, studies with monozygotic twins demonstrated a disease concordance rate of only 30 to 35%, indicating that genetic factors alone may not trigger the disease (Baranzini et al., 2010). Although specific infectious agents have not been linked to MS, epidemiological studies suggest that herpes simplex virus type-1 (HSV-1), human herpes virus type-6 (HHV-6), T cell leukemia virus type-1, Epstein-Barr virus, influenza virus, and human picornaviruses, such as rhinovirus, may be associated with MS [reviewed in (Libbey and Fujinami, 2010) (Kriesel et al., 2004)], suggesting that environmental factors such as viral infections may be related to the development and/or exacerbation of MS in genetically susceptible individuals. In fact, viral antigens and virus-specific antibodies were detected in subsets of MS patients [reviewed in (Virtanen and Jacobson, 2012)]. Actually, antigens from HHV-6 were found in the MS plaques (Cermelli et al., 2003; Challoner et al., 1995; Soldan et al., 1997; Virtanen et al., 2011). The development of experimental viral infection models, such as TMEV-IDD, made possible the study of the contribution of the inflammatory immune response, induced by viral infection, to the development of neuroinflammation and demyelination, allowing for the advancement of our knowledge in the MS field.
2.1. Immune response and immunological tolerance
After T cell receptor (TCR) rearrangement and during T cell maturation, T cells undergo processes called positive and negative selection within the thymus. In the cortex of the thymus, epithelial cells express MHC molecules; positive selection occurs when T cells with TCR that recognize and bind, in a relatively weak manner, self-peptide loaded on MHC molecules are selected to “survive”; T cells that fail to recognize self-peptide-MHC molecules (death by neglect), or recognize it too strongly (death by negative selection), die by apoptosis (Benoist and Mathis, 1989, 1999; Sebzda et al., 1999; Sprent, 1993; Sprent and Webb, 1987). T cells that recognize MHC-II become CD4+ T cells while T cells that bind to MHC-I become CD8+ T cells. In order to not initiate an immune response against self, CD4+ and CD8+ T cells are subjected to a mechanism called negative selection (central tolerance). In the thymic medulla, TCRs that recognize self-peptide, displayed by thymic antigen presenting cells (APCs), with high avidity in the context of the MHCs are deleted [reviewed in (Sprent and Kishimoto, 2001)]. However, T cells with low or weak affinity recognition of MHC-peptides are selected, generating a functional and self-tolerant T cell repertoire. These processes occur to ensure that T cells are only going to react to non-self-peptides presented by APCs in the context of MHC molecules. However, although several mechanisms exist in order to ensure central tolerance, T cells can still escape from these “checkpoints” resulting in the generation of autoreactive T cells, which migrate to the periphery [reviewed elsewhere (Goverman, 2011; Sprent and Kishimoto, 2001)]. Peripheral T cell tolerance is another checkpoint mechanism to prevent activation of autoreactive T cells that escaped central tolerance. In the absence of pathogens, APCs are kept in a non-activated state (quiescent), expressing low levels of costimulatory molecules. Engagement of TCR by self-peptide loaded on MHC at the cell surface of a non-activated APC can result in T cell clonal deletion by apoptosis (Klein et al., 2014; Sprent and Webb, 1987). Another mechanism for peripheral tolerance occurs when antigen recognition happens in the absence of costimulatory signals, resulting in unresponsive T cells (anergy), or these cells can be suppressed by regulatory T cells (Tregs), which secrete immunosuppressive cytokines, blocking T cell activation and effector function (Klein et al., 2014; Sprent and Webb, 1987). Despite all the mechanisms to prevent the generation of autoreactive T cells, these cells can still escape and may be capable of inducing autoimmunity.
Tight junctions of the BBB can limit naïve T cell infiltration into the CNS (Ransohoff and Brown, 2012; Schwartz and Deczkowska, 2016). However, once these T cells become activated, upregulation and expression of cell surface molecules allows these cells to penetrate the BBB (Ransohoff and Brown, 2012; Schwartz and Deczkowska, 2016). Escape from central and peripheral tolerance leads to the activation of myelin-specific T cells in the periphery, potentially through virus infection, allowing these cells to invade the CNS and cause disease. Once inside the CNS, these activated T cells recognize myelin presented by local APCs, eliciting the T cells’ effector functions, resulting in cytokine and chemokine secretion that leads to increased lymphocyte and macrophage infiltration and, consequently, inflammation and demyelination. It is believed that in genetically susceptible individuals, environmental factors such as viral infections may induce loss of tolerance and activation of T cells that are specific for myelin.
2.2. TMEV-IDD: A mouse model to study MS
I.c. infection of the SJL/J mouse strain with the DA strain of TMEV results in persistent viral infection of the CNS, leading to the development of a chronic progressive biphasic disease called TMEV-IDD (Figure 1A, B), which is a model for progressive forms of MS. This mouse model develops clinical and histopathological features that are similar to those observed in MS patients. TMEV-IDD in SJL/J mice is characterized by an acute phase, that occurs 1-week post infection, and a chronic phase, which begins within 1 month after TMEV inoculation (Tsunoda and Fujinami, 1996, 2010). During the chronic phase of the disease, TMEV-infected SJL/J mice develop weakness of the hind limbs, which advances to a severe spastic paralysis and no recovery is observed, similar to what is observed in patients with the primary progressive form of MS. Each mouse is scored based on the impairment severity: asymptomatic (0), mild gait abnormalities (1), severe gait abnormalities (2), mild spastic paralysis (3), hind limb paralysis (4), moribund (5) (McCarthy et al., 2012).
In this disease model, demyelination observed in the CNS is associated with a sustained inflammatory immune response due to viral persistence (Figure 3). In contrast, i.c. infection of the C57BL/6J mouse strain does not result in an inflammatory demyelinating disease since TMEV is cleared. The ability of TMEV to persist in the SJL/J mouse strain throughout the animal’s lifespan seems to be in part related to the innate immune response. Cellular pattern recognition receptors such as Toll Like Receptors (TLR) recognize pathogens through pathogen-associated molecular patterns (PAMPs) resulting in the induction of the innate immune response. Once the innate immune response is triggered, secretion of pro-inflammatory chemokines and cytokines contributes to the induction of the adaptive immune response, by recruiting immune cells to the site of infection. The major cytokines induced by viral infection are type I IFN, such as IFN-α and IFN-β, and the type II IFN-γ. Activation of the type I IFN pathway leads to increased levels of expression of interferon-stimulated genes (ISGs), which can exert an antiviral effect (Figure 1D). It was demonstrated that, in contrast to C57BL/6J, the SJL/J mouse strain induces low ISG protein expression, which could contribute to viral persistence (Figure 1B) (Li et al., 2015). Also an early study conducted by Murray et al demonstrated the inverse relationship between IFN-γ production and viral persistence in TMEV-infected mice (Murray et al., 2002). It was also reported that TMEV infection of a C57BL/6J mouse strain lacking IFN-γ, and in which virus persisted, developed demyelination (Bowen and Olson, 2013).
Figure 3. Infection of SJL/J mice with TMEV leads to demyelination.
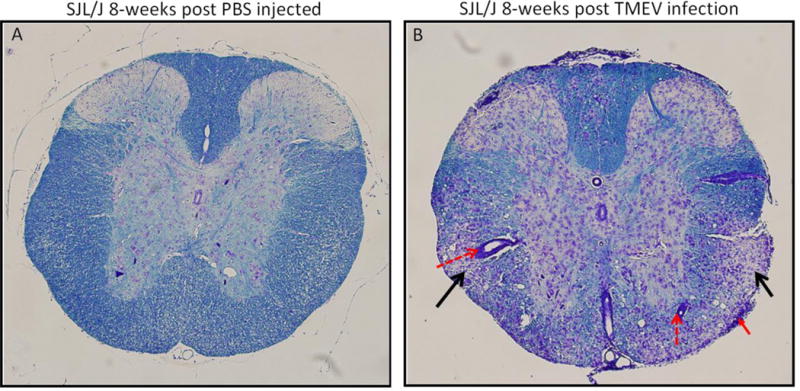
Spinal cord sections obtained from a PBS injected mouse (A) and a TMEV infection mouse (B), both at 8-weeks post injection/infection, and stained with Luxol fast blue. Infiltration of inflammatory cells into the CNS is observed. Perivascular cuffing (dashed red arrows) in the white matter; meningitis (solid red arrow) and demyelination (black arrows) are indicated.
While Tregs play an important role in protecting against autoimmunity, these cells have also been implicated in the decreased anti-viral CD8+ T cell response and the decreased viral clearance associated with chronic viral infections. Tregs are known to modulate the immune response to viral infection, and numerous infection models have demonstrated that Treg depletion increases viral clearance and IFN-γ production by CD8+ T cells (Dittmer et al., 2004; Eggena et al., 2005; Groux et al., 1997; He et al., 2004). Richards et al demonstrated that viral persistence in TMEV-infected SJL/J mice may in part be related to the high induction of Tregs during the acute stage of infection (Richards et al., 2011). Infection with TMEV resulted in an increase in the number of Tregs. The C57BL/6J mouse strain had a ratio of 1:1.2 (CD8+ Teffector:Treg), while the SJL/J mouse strain had a 1:4 ratio during the acute phase, and these Tregs were capable of suppressing the CD8+ T cells effector function, decreasing viral clearance and allowing for viral persistence.
TMEV was demonstrated to initially infect predominantly neurons (Liu et al., 1967). During the acute phase of the disease, viral antigens and viral genome are found in neurons and in apoptotic neurons in the grey matter, such as the hippocampus and the cerebral cortex (Tsunoda et al., 1997). Increased infiltration of CD4+ and CD8+ T cells, monocytes and a few B cells and plasma cells are observed in the grey matter of the brain, characteristic of CNS inflammation (encephalitis) (Oleszak et al., 2004; Schlitt et al., 2003; Tsunoda and Fujinami, 2010). After the acute phase, the viral load decreases but the immune system fails to completely clear the viral infection, leading to progression to the chronic phase. In the chronic phase of the disease the inflammatory cells persist in the white matter while demyelination in the spinal cord and axonal damage are also observed (Tsunoda and Fujinami, 2002). Unlike the acute phase, TMEV is not found in neurons but persists in oligodendrocytes, astrocytes and microglia/macrophages, as demonstrated by immunohistochemistry and in situ hybridization (Clatch et al., 1990; Girard et al., 2002; Jelachich et al., 1995; Misra et al., 2008; Oleszak et al., 2004; Rodriguez et al., 1983). Infiltrating immune cells, at this stage of the disease, are predominantly CD4+ and CD8+ T cells and to a lesser extent macrophages, B cells and plasma cells. Therefore, while TMEV persistence is important to induce demyelination, the trigger mechanism for demyelination is inflammation and the induction of autoimmunity.
2.2.1. Immune cells and demyelination in TMEV-IDD
2.2.1.1. Microglia/macrophages
The role of microglia/macrophages in TMEV-IDD is correlated in part with the ability of the virus to persist in these cells. Also, the presence of macrophages in or close to the demyelinating lesion suggests an active role of these immune cells in MS pathology. The presence of myelin-containing macrophages (foamy macrophages) has been documented in MS patients and in TMEV-infected mice (Oleszak et al., 2004; Oleszak et al., 1997). Macrophages and microglia (the last is defined as the resident macrophages within the CNS) are the first line of defense during TMEV infection; however, due to their intense secretion of pro-inflammatory cytokines during the chronic phase, microglial/macrophage activation may result in local tissue damage/immunopathology. In agreement with the negative impact of microglial/macrophage activation during the chronic phase is the observation that these cells can degrade myelin basic protein (MBP) by secreting proteolytic factors, cytokines, nitric oxide metabolites and reactive oxygen species (Langrish et al., 2005; Liuzzi et al., 1995; Oleszak et al., 2004). Furthermore, production of tumor necrosis factor (TNF)-α by macrophages in the CNS of TMEV-infected mice was associated with myelin loss. One direct contribution of macrophages to autoimmunity was demonstrated by Katz-Levy et al: macrophages isolated from TMEV-infected mice were able to present not only viral-specific antigens but also self-antigens to T cells, which may result in activation of autoreactive T cells and could initiate autoimmune disease (Katz-Levy et al., 2000).
2.2.1.2. T cells
2.2.1.2.1. CD4+ T cells
Genome-wide association studies have demonstrated a correlation between human leukocyte antigens (HLA)-DRB1*-1501 and -DQ0601 and MS. These alleles encode MHC-II molecules that present antigen to CD4+ T cells. The initial concept that CD4+ T cells were involved in the development of MS originated due to results obtained using the experimental autoimmune encephalomyelitis (EAE) model [extensively reviewed elsewhere (Fletcher et al., 2010)]. In this disease model, EAE-induction using myelin-derived peptides results in these antigens being presented by MHC-II, eliciting induction of CD4+ T cell, but not CD8+ T cell, effector functions. Importantly, adoptive transfer of activated myelin-specific CD4+ T cells into a naïve recipient animal was sufficient to trigger disease, supporting the idea that MS is a T cell-mediated autoimmune disease. While the EAE model is the most common model used to study MS, the induction of the myelin-specific immune response is somewhat contrived and does not correlate with the development of spontaneous disease. Although this model mimics many of the pathological and clinical symptoms of MS, it does not recapitulate all of them.
In TMEV-IDD, a strong induction of a virus-specific CD4+ T cell response (Th1 type) is observed. As disease progresses to the chronic phase, epitope-spreading (discussed below) occurs, generating immune responses against the myelin proteolipid protein (PLP)139–151 peptide, resulting in CD4+ T cells that recognize and attack myelin proteins, causing demyelination (Katz-Levy et al., 2000; Miller et al., 1997). Furthermore, secretion of pro-inflammatory cytokines by viral- and myelin-specific CD4+ T cells results in increased infiltration of macrophages into the CNS, leading to myelin destruction (Piccio et al., 2002). Support for the role of CD4+ T cells in TMEV-IDD was demonstrated when less severe demyelination was observed in TMEV-infected SJL/J mice treated with anti-CD4 antibody (Montero et al., 2004). The importance of CD4+ T cells was also highlighted by the ability of astrocytes to express MHC-II in response to inflammatory stimuli, such as IFN-γ, and present antigens to CD4+ T cells. Astrocytes are known to maintain the integrity of the BBB; therefore, the observation that astrocytes, involved in presenting antigens to CD4+ T cells, died by apoptosis, suggests that this could contribute to deregulation of BBB homeostasis, resulting in neuroinflammation, cellular infiltration and demyelination (Palma et al., 1999).
2.2.1.2.2. CD8+ T cells
Viral infections have been implicated in the generation and exacerbation of MS, possibly through the production of autoreactive CD8+ T cells. Although MS has been viewed as a CD4+ T cell-mediated disease, in part because of its association with MHC-II, several groups have shown that specific alleles of the MHC class I, such as HLA-A3, have strong associations with MS susceptibility (Friese et al., 2008; Naito et al., 1972). Importantly, analysis of inflammatory infiltrate in CNS lesions from MS patients showed enhanced numbers of CD8+ T cells compared to CD4+ T cells (ratio 10:1) and these CD8+ T cells were shown to be clonally expanded within the site of active demyelination (Babbe et al., 2000; Neumann et al., 2002). Furthermore, CD8+ T cells were found to be preferentially clonally expanded in the cerebrospinal fluid (CSF) and in the circulation of MS patients compared to CD4+ T cells (Babbe et al., 2000; Skulina et al., 2004). In vitro studies comparing MS patients and healthy individuals showed that the frequency of CD8+ T cells specific for CNS antigens was higher than the frequency of CD4+ T cells specific for CNS antigens in MS patients (Crawford et al., 2004). Additionally, MHC-I, which normally has low expression on astrocytes, neurons and oligodendrocytes, has increased expression on these cells in MS lesions, and CD8+ T cells were found interacting with APCs in active lesions (Hoftberger et al., 2004).
The role of CD8+ T cells in MS remains controversial regarding its protective or destructive function. In the SJL/J mouse strain, CD8+ T cells were shown to play a role in viral clearance during the acute phase, but these cells caused tissue damage during the chronic phase by recognizing MHC-I expressed on axons and inducing immune responses against these cells. Initial studies demonstrated that TMEV-infected SJL/J mice lacking CD8+ T cells developed early onset and more severe disease compared to the control group (Begolka et al., 2001; Borrow et al., 1992; Murray et al., 1998), suggesting that these cells have an immunomodulatory function. However, several lines of evidence point to a pathogenic rather than regulatory function of CD8+ T cells. As mentioned above, one possible mechanism that is regulating CD8+ T cell function is induction of Tregs in TMEV-infected SJL/J mice. Tregs induced following TMEV infection lead to decreased CD8+ T cell effector function, allowing for viral persistence. Treg depletion using monoclonal antibody resulted in increased viral clearance, increased adaptive antiviral immune response and increased antibody response, demonstrating an important role of Tregs in regulating CD8+ T cell function and its contribution to viral clearance and induction of an autoimmune disease (Richards et al., 2011).
In order to study the role of CD8+ T cells in demyelination, CD8+ T cells isolated from healthy individuals and MS patients were stimulated with peptides derived from human myelin proteins. These CD8+ T cell clones obtained from MS patients were specific for MBP, PLP and myelin associated glycoprotein and these cells produced IFN-γ (Honma et al., 1997; Tsuchida et al., 1994; Zang et al., 2004). This suggests that autoreactive CD8+ T cells can recognize myelin protein. Interestingly, these CD8+ T cell clones reacted with and lysed targets that were coated with peptides derived from Saccharomyces cerevisiae, suggesting that these autoreactive CD8+ T cells can recognize microbe- and self-peptides, possibly by CD8+ T cells having dual TCRs or through molecular mimicry (discussed below). In TMEV-IDD, some autoreactive CD8+ T cell clones have been found to be dual-reactive, reacting with both a self-peptide and a microorganism, such as a virus or bacterium, suggesting the possibility of dual TCRs. We and others have shown that adoptive transfer of autoreactive CD8+ T cells was sufficient to induce an EAE-like disease (Huseby et al., 2001; Libbey et al., 2012; Sun et al., 2001), suggesting that CD8+ T cells are likely playing a role in the establishment and progression of MS.
However, while depletion of CD4+ T cells in MS patients has no significant effect on symptoms of the disease (van Oosten et al., 1997), MS patients treated with alemtuzumab, a drug that depletes both CD4+ and CD8+ T cells, showed significant disease improvement (Coles et al., 2006). This implies that more than one cell type is likely responsible for the disease development and/or exacerbation in MS, and that this disease results from a combination of events involving different immune cells.
2.2.1.3. B cells
B cells are essential for both the innate and adaptive immune responses. Although very few B cells are found in the CNS of TMEV-infected SJL/J mice, anti-TMEV neutralizing antibodies are found in the serum 1 week post infection and high levels of neutralizing antibodies are found in persistent infection (Tsunoda, 1996). Both in MS patients and in animal models of MS, B cells and plasma cells are observed at active lesion sites. Also, the presence of antibodies has been documented in areas of demyelination (Baranzini et al., 2010; Kabat et al., 1942; Krumbholz and Meinl, 2014; Pachner et al., 2011; Yamada et al., 1990).
Due to the development of autoantibodies in MS patients, B cell depletion therapy is considered a treatment for this disease (Barr et al., 2012; Hauser et al., 2008). In order to determine the consequences of B cell depletion, Gilli et al administered anti-CD20 monoclonal antibody to TMEV-infected mice (Gilli et al., 2015). B cell depletion was confirmed by measuring the numbers of B cells in the peripheral blood. They found that upon B cell depletion, increased viral load in the CNS, accompanied by microglial activation, T cell infiltration, demyelination and axonal damage, was observed, resulting in a faster progression and disease exacerbation. Similar results were also obtained with B cell depletion using the EAE model (Matsushita et al., 2008; Weber et al., 2010).
However, contrary to the results obtained in mouse models, treatment of MS patients with anti-CD20 antibody resulted in disease improvement, where patients experienced fewer attacks. However, one important consideration is whether B cell depletion treatment would be beneficial and safe for patients at high risk of viral infections. As the result obtained using the TMEV-IDD mouse model suggests, patients without B cells may experience worse disease manifestation during a viral infection due to failure to induce a strong antibody-immune response and/or the inability to control inflammation, since regulatory B cells are also depleted.
2.3. How immune response to viral infection can lead to MS
As discussed, lymphocytes that escape from central and peripheral tolerance are autoreactive, and can trigger autoimmune responses. Several mechanisms for how autoreactive T cells can be activated after infection have been proposed. Three different ways in which viral infection may trigger an autoimmune response resulting in MS include epitope spreading, molecular mimicry and dual TCR:
Epitope spreading: It occurs following persistent viral infection of the target organ causing recruitment and activation of viral-specific T cells. These T cells will mediate an immune response leading to inflammation, which results in tissue damage and release of self-antigens. These self-antigens are engulfed, processed and presented by APCs to T cells, resulting in T cells reactive against self-antigens. If tissue damage continues, other self-peptides are released, leading to the spread of self-epitopes, thus inducing autoimmunity (Lehmann et al., 1992; Lehmann et al., 1993). As the identification of the first epitope used to induce an immune response is very challenging, it is very difficult, although not impossible, to prove epitope spreading in human patients. The existence of this mechanism in MS was first demonstrated using the EAE model. In this model, disease was induced by priming the immune system with a defined CNS antigen (such as PLP or MBP), resulting in an antigen-specific T cell response. Interestingly it was observed that after disease initiation and tissue damage, antigen specificity changed, confirming epitope spreading as a mechanism to induce autoimmunity.
Molecular mimicry: It was first suggested by Fujinami et al in 1983 and is the most accepted hypothesis to explain how viruses can induce autoimmune disease (Fujinami et al., 1983). It occurs when self and virus share sequence or structural homology. Infection outside the CNS would result in activation of a virus-specific T cell response that, upon migration into the CNS, would target self-antigens, through cross-reactive epitopes, initiating the disease. It has been suggested that viruses such as Epstein-Barr share amino acid sequences with CNS structures (Wandinger et al., 2000). Furthermore, it was also demonstrated that a viral protein from HHV-6, known as U24, and MBP share identical amino acid sequences, and T cells obtained from MS patients were able to cross-react with both MBP and HHV-6 viral protein (Tejada-Simon et al., 2003). The sharing of sequences may contribute to the induction of MS in infected, susceptible individuals.
Dual TCR: During T cell development V-D-J recombination occurs, giving rise to TCR α and β chains which are expressed on T cells. Although it is believed that T cells express only one TCR, recent studies in humans and mice suggest that T cells carrying more than one TCR exist. One possible mechanism for how dual TCR escape tolerance was described by Blichfeldt et al [(Blichfeldt et al., 1996; Cusick et al., 2013a)]. One potential dual TCR scenario is where one combination of TCR Vα/Vβ chains is specific for self-antigen while another combination of TCR Vα/Vβ chains is specific for viral antigen, and which, once activated, would react against both virus and self [reviewed in (Cusick et al., 2013a)]. Supporting this dual TCR hypothesis is the finding using the MBP transgenic TCR model (Ji et al., 2010). In this model CD8+ T cells specific for MBP encode multiple TCRs and infection of these mice, with vaccinia virus, induced EAE. This result suggests a role for dual TCR, where one receptor recognizes viral peptide while the other TCR recognizes MBP. Following vaccinia virus infection, these cells became activated, in order to overcome the viral infection, and infiltrated into the CNS, resulting in disease development due to its ability to recognize MBP as non-self, inducing demyelination. Our lab demonstrated that following peripheral TMEV infection, autoreactive CD8+ T cells containing more than one TCR are induced. Hybridomas were generated using these CD8+ T cell clones and adoptive transfer of these cells into naïve mice induced disease, suggesting that T cells containing dual TCRs may play a role in the induction of autoimmune disease in vivo (Libbey et al., 2012).
2.4. Conclusions
MS is a neurological, progressive disease that results in severe disability. It is an immune mediated disease characterized by immune cells attacking the myelin sheath. The etiology of MS is unknown, and evidence suggests that it is a multifactorial disease where environmental factors, such as viral infection, of genetically susceptible individuals seem to play a role in disease establishment and exacerbation. Decades of extensive work in neuroimmunology have clearly demonstrated a link between inflammatory response and disease development. Importantly, the development of mouse models such as TMEV-IDD to study the progressive form of the disease allow for significant advances in understanding important immunological and virological aspects of this disease. However, despite these advances, in order to identify and develop potential therapeutic targets to treat or even cure MS, future studies are needed to better understand the mechanism for how the immune response against myelin is developed.
3. Seizures and epilepsy
Epilepsy is a serious chronic neurological disorder characterized by recurrent seizures (Vezzani et al., 2016). It is estimated that around 70 million people suffer from epilepsy worldwide (Ngugi et al., 2010), and that in the USA alone more than 5 million people have been diagnosed with epilepsy (Thurman et al., 2016). The annual cost to treat and care for patients with epilepsy in the USA is estimated to be $15.5 billion (England et al., 2012). Although treatments to prevent seizures are available, they are mainly anticonvulsants and around 30% of the patients do not respond to the medications, and numerous side effects related to the drugs have been reported (reviewed in (Laxer et al., 2014).
Seizures develop as a result of imbalances between excitatory and inhibitory inputs within the brain, with these inputs shifting toward excitation. Changes in excitability may result from alterations in neuronal cell surface receptor expression and phosphorylation status. Glutamate is the most common excitatory neurotransmitter in the CNS and clearance of glutamate from the synaptic cleft is essential to maintain CNS homeostasis (Hu et al., 2000; Tilleux and Hermans, 2008). Thus, increased expression and function of glutamate can contribute to the development of seizures (Nadler, 2012). Ionotropic glutamate receptors are divided into three subfamilies: AMPA (α-amino-3-hydroxy-5-methyl-4-isoxazolepropionate), NMDA (N- methyl-D-aspartate) and KA (Kainate) receptors (Dingledine, 2012; Noebels et al., 2010). These receptors are found to be expressed on neurons, astrocytes, oligodendrocytes and microglia. While excess extracellular glutamate is removed by astrocytes in order to prevent hyperexcitation and excitotoxicity, it has been demonstrated that pro-inflammatory cytokines can inhibit glutamate uptake into astrocytes (Hu S et al, 2000), suggesting a mechanistic role of inflammation in the development of seizures (discussed below).
Approximately 50% of seizures are idiopathic and about 50% of seizures develop as a result of CNS injuries, such as traumatic brain injury (TBI), brain tumors and CNS infection (Annegers et al., 1988; Delorenzo et al., 2005). In these latter cases, inflammation of the brain parenchyma (encephalitis) is the major risk factor for the development of seizures and epilepsy (Misra et al., 2008). There are over 200 different viruses that are known to cause disease in humans, and at least 100 different neurotropic viruses can cause encephalitis (Bale, 2015; Griffin, 2003; Libbey and Fujinami, 2011). Viral encephalitic patients have a 20% increased risk for developing seizures than the general population (Getts et al., 2008; Libbey and Fujinami, 2011; Vezzani et al., 2016). Additionally, patients who survive viral encephalitis are 16 times more likely to develop epilepsy (Annegers et al., 1988; Getts et al., 2008; Misra et al., 2008). Several viruses such as influenza virus, West Nile virus (WNV), herpes viruses and non-polio picornaviruses have been associated with seizure development and epilepsy (Bonello et al., 2015; Libbey and Fujinami, 2011). An important distinction that needs to be made is related to seizures that occur after infection (1–2 weeks post infection), called acute seizures, and seizures that appear later (months to years after infection), that are unprovoked recurrent seizures defined as epilepsy (Berg et al., 2010; Lowenstein, 2009). Although not everyone who experiences acute seizure will develop epilepsy, a relationship exists, and acute seizures are a risk factor for epilepsy (Berg et al., 2010; Lowenstein, 2009).
3.1. TMEV-induced acute seizures and epilepsy
Examination of the mechanism(s) of viral encephalitis-driven epilepsy and the development of new therapeutic agents has been limited in part due to the absence of an appropriate and relevant animal model to study this disorder. Over the last several years, a number of models have attempted to reproduce seizures and epilepsy in mice following viral encephalitis. Viruses that were frequently used in these studies were WNV, measles virus and HSV-1; however, a common limitation was a high mortality rate during the acute infection, making it impossible to study long-term effects such as epilepsy (Getts et al., 2007; Lehrmann et al., 2008; Wu et al., 2003).
In order to study the contribution of viral encephalitis to the development of acute seizures and epilepsy, our lab has developed an experimental mouse model of virus-induced seizures/epilepsy, which has now been used by several groups and similar results have been obtained (Broer et al., 2016; Libbey et al., 2008; Stewart et al., 2010b). In this model (Figure 1C, D), i.c. infection of C57BL/6J mice with the DA strain of TMEV results in acute behavioral seizures, accompanied by weight loss, in 50% of the infected mice between 3 and 10 days post infection. Typically, day 3 is the first day that seizures are observed, while day 6 is the peak of seizure activity, and by day 10 acute seizures have resolved. Mice are monitored for acute behavioral seizures daily from days 2 to 10, for 2 hours, twice a day (Libbey et al., 2008). Seizures were shown to last approximately 1–2 minutes and are scored following the Racine scale (stage 3-forelimb clonus, 4-rearing with forelimb clonus, 5-rearing and falling with forelimb clonus). However, continuous video-electroencephalogram (video-EEG) monitoring demonstrated seizure activity in 75% instead of 50% of mice, suggesting some seizures may be missed when observed for only a small portion of the day (Stewart et al., 2010a). Within 2 weeks post infection, virus is cleared from the CNS, probably through activation of antiviral CD8+ T cells and antibodies. After seizures resolve, mice progress to a latent period, which is characterized by an asymptomatic phase, where seizures are not observed (Monteyne et al., 1997). However, approximately 50% of the infected mice that experienced acute behavioral seizures will subsequently develop spontaneous recurrent seizures (epilepsy) (Libbey et al., 2008; Stewart et al., 2010b). Therefore, this mouse model, where mice display an initial insult followed by a latent period and then epilepsy, resembles features observed in patients with temporal lobe epilepsy (TLE), which is the most prevalent form of epilepsy affecting humans (Al Sufiani and Ang, 2012).
In TLE more than 50% of patients have detectable levels of HHV-6 genomic DNA and protein, and the hippocampal sclerosis is characterized by gliosis, neuronal loss and glial scarring (Al Sufiani and Ang, 2012; Crespel et al., 2002; Donati et al., 2003). Analysis of the histopathological features in TMEV-infected mice that experienced acute seizures demonstrated a profound loss of CA1 to CA2 pyramidal neurons in the hippocampus, likely due to apoptosis, microglial activation and astrogliosis, which mimic features found in epileptic foci in the brains of human patients (Kirkman et al., 2010; Libbey et al., 2011b; Loewen et al., 2016). Additionally, by analyzing the brains of TMEV-infected mice experiencing seizures, a correlation was demonstrated between increased inflammation in the hippocampus and acute seizures (Kirkman et al., 2010; Libbey et al., 2011b). Because mice begin to experience acute seizures at day 3 post infection, prior to the development of the adaptive immune response, one plausible hypothesis is that seizures are induced as a consequence of the activation of the innate immune response (Kirkman et al., 2010; Libbey et al., 2008; Libbey et al., 2010).
3.2. Innate immune response to viral infection of the CNS and its contribution to the development of acute seizures
While observed CNS infections are relatively rare, they can be extremely destructive. The BBB works to prevent circulating peripheral cells, proteins and pathogens from entering the CNS. Activation of the innate immune response occurs hours after viral infection and relies on: 1) innate immune cells, such as macrophages, natural killer (NK) cells and neutrophils; 2) CNS resident cells, microglia and astrocytes; 3) proteins secreted by these cells, such as pro-inflammatory cytokines; and 4) the complement system (Chakraborty et al., 2010; Libbey et al., 2010). Viruses can gain access to the CNS using different routes, such as by infecting peripheral cells that penetrate into the CNS, or by infecting peripheral nerves and disseminating into the CNS via axonal or retrograde transport (Eeg-Olofsson, 2003). Viruses that have a tropism for neurons (neurotropic viruses), after gaining access to the CNS, can spread from neuron to neuron using connecting synapses (Ehrengruber et al., 2002; Young and Rall, 2009). Direct viral infection of neurons can result in neuronal death leading to the release of danger signals, comprised of cellular products, and pro-inflammatory cytokines, which can lead to the activation and recruitment of innate immune cells to the CNS, resulting in inflammation [Reviewed in (Vezzani et al., 2016)]. Additionally, upon viral infection of neurons, these cells signal astrocytes and microglia through the production of cytokines, such as type I IFN, and chemokines, such as CX3CL1 (fractalkine). Microglia and macrophages express the fractalkine receptor (CX3CR1) and become activated upon CX3CL1 secretion. Activated microglia and macrophages produce pro-inflammatory cytokines, such as interleukin (IL)-6, IL-1 and TNF-α, resulting in additional microglial activation. Also, secretion of chemokines results in peripheral leukocyte recruitment to the CNS, leading to an increase in inflammation [Reviewed in (Vezzani et al., 2016)].
3.2.1. Microglia/macrophages
Brain sections from TMEV-infected mice stained with Ricinus communis agglutinin (RCA)-I lectin, which recognizes macrophages and activated microglia, revealed significantly greater numbers of RCA-I positive cells in mice with seizures, compared with mice without seizures. Cusick et al clearly demonstrated that infiltration of macrophages into the brain occurs as early as 3 days post infection, further confirming the importance of macrophages in seizure development (Cusick et al., 2013b). Although there is not a specific marker to distinguish macrophages from microglia, the use of flow cytometry to analyze the level of expression of CD45 and CD11b on the cell surface made it possible to separate these two cell populations: microglia are characterized by CD45low/int CD11bhi expression and macrophages are characterized by CD45hi CD11bhi expression (Cusick et al., 2013b).
The role of macrophages in this animal model was explored further through the use of wogonin and minocycline. Wogonin is a natural product extracted from the root of the plant Scutellaria baicalensis (Baikal Skullcap) which has antioxidant and anti-inflammatory activities (Fas et al., 2006; Shao et al., 1999). Minocycline is an antibiotic with an anti-inflammatory property that was reported to decrease excitability within the CNS (Giuliani et al., 2005; Tikka and Koistinaho, 2001; Yrjanheikki et al., 1998). Fewer TMEV-infected mice developed seizures when treated with wogonin or minocycline (Cusick et al., 2013b). Importantly, analysis of the cellular infiltration into the brain demonstrated that wogonin-treated mice had decreased infiltration of macrophages, supporting the role of macrophage infiltration in seizure development. Furthermore, a study by Howe et al has shown that while depletion of myeloid cells, including neutrophils, decreased neuron damage, depletion of only neutrophils had no effect on disease (Howe et al., 2012).
Macrophages are a highly heterogeneous population of myeloid cells characterized by their plasticity (Mantovani et al., 2013). Macrophages can respond to a variety of stimuli leading to functional changes. In reponse to TLR4, through lipopolysaccharide (LPS) stimulation, and IFN-γ, macrophages undergo classical (M1) activation, while alternative (M2) activation is induced by IL-4 and IL-13 (Davis et al., 2013). Since M1 activated macrophages produce pro-inflammatory cytokines and M2 activated macrophages secrete anti-inflammatory cytokines, the presence of M1 macrophages are normally associated with inflammation while M2 macrophages are related to resolution or control of the inflammatory response. Inflammatory macrophages can secrete pro-inflammatory cytokines such as IL-6 and TNF-α. Interestingly, after viral infection of the CNS, increased levels of IL-6, IL-1β and TNF-α were observed [reviewed in (Chakraborty et al., 2010)]. Additionally, mRNA expression of IL-6 and TNF-α was found to be increased in TMEV-infected mice (Kirkman et al., 2010; Theil et al., 2000). To assess the requirement of these cytokines in the development of acute seizures, Kirkman et al infected C57BL/6J knockout (KO) mouse strains for IL-1 receptor 1 (IL-1R1), IL-6, TNF-R1 or myeloid differentiation primary response gene 88 (MyD88) with TMEV. While no differences in seizure frequencies were observed in IL-1R1 and MyD88 deficient mice compared to control, in the absence of IL-6 and TNF-R1, fewer mice experienced seizures, suggesting these cytokines are contributing to the development of acute seizures (Kirkman et al., 2010).
While infiltration of macrophages following TMEV infection was associated with seizure development, it was unclear whether secretion of IL-6 and TNF-α was linked to macrophages or activated microglia. Using green fluorescence protein (GFP) chimeric mice, where microglia are GFP negative and macrophages are GFP positive, Cusick et al demonstrated that IL-6 secretion was associated with infiltrated macrophages while microglia were the major producer of TNF-α in the CNS (Cusick et al., 2013b). In support of the role of IL-6 in the induction of acute seizures, it was demonstrated that transgenic mice overexpressing IL-6 within the CNS, under the control of the astrocyte promoter glial fibrillary acidic protein (GFAP), developed spontaneous seizures (Campbell et al., 1993). Additionally, increased CSF and serum levels of IL-6 have been observed in several conditions associated with seizures, such as TBI (Yang et al., 2013). Therefore, these results confirm an inflammatory role for the infiltrating macrophages in the development of acute seizures.
3.2.2. NK cells
NK cells are part of the innate immune system and are implicated in a variety of functions ranging from dendritic cell maturation to T cell maturation and the killing of lymphoid and glial cells (Lunemann et al., 2009). The function of NK cells in the CNS is not well established and opposite functions such as neuroprotection and neurotoxicity have been attributed to these cells (Hao et al., 2010; Winkler-Pickett et al., 2008). However, NK cells are known to play an important antiviral role, as demonstrated by the increased susceptibility to herpes virus infection of patients lacking NK cells [reviewed in (Poli et al., 2013)].
NK cells can be recruited to the CNS by the secretion of chemokines such as CCL2, CXCL10, CXCL9, CXCL11 and XCR1, and are one of the first effector cells at an inflammation site (Poli et al., 2013). Analysis of mRNA expression demonstrated that at 2 days post infection, C57BL/6J mice infected with TMEV had levels of CXCL9 and CXCL11 which were highly elevated, but this elevation was not sustained at 6 days post infection which represents the peak of seizure activity (unpublished). Additionally, although NK cells have been shown to infiltrate into the CNS after TMEV infection of C57BL/6J mice (Cusick et al., 2013b), depletion of NK cells in TMEV-infected mice using anti-NK1.1 antibody had no significant effect in the frequency of acute seizures, suggesting that this innate immune cell is not playing a role in the induction and/or establishment of acute behavioral seizures (Libbey et al., 2011a).
3.3. How inflammation within the CNS can result in acute seizures
The balance between excitatory and inhibitory inputs is essential for maintaining homeostasis in the CNS. A disruption in this balance, such as an increase in excitatory or a decrease in inhibitory inputs, can lead to hyperexcitability and, in turn, seizures (Getts et al., 2008). One way by which this disruption can take place is through changes in receptor expression and function on post-synaptic neurons. For example, a decrease in the main inhibitory transmitter γ-aminobutyric acid (GABA), or its receptor, can lead to an increase in neuronal excitation due to a loss of inhibitory tone (Ben-Ari and Dudek, 2010). Alternatively, an increase in signaling mediated by glutamate can result in hyperexcitability and eventually excitotoxicity. In fact, a number of changes in the glutamate signaling system have previously been shown to lead to an increase in excitability. These changes include, but are not limited to, an increase in the amount of glutamate released into the synaptic cleft, disruptions in glutamate clearance from the synapse, changes in neuronal receptor expression or function, and how efficiently a neurotransmitter is degraded (Coulter and Steinhauser, 2015; Dietrich et al., 2002; Prince et al., 1995). The three main subfamilies of ionotropic glutamate receptors, AMPA, NMDA, and KA, have a variety of different subunit combinations that can influence how glutamate signals through them (Meldrum, 2000; Sanchez et al., 2001). In addition to ionotropic glutamate receptors, there are several classes of metabotropic glutamate receptors (mGluRs) that modulate neuronal excitability and synaptic transmission through regulation of various membrane ion channels and intracellular second-messenger systems (Notenboom et al., 2006). Disruption in the expression or function of mGluRs can disrupt the excitatory/inhibitory balance and lead to hyperexcitable states (Dietrich et al., 2002; Merlin et al., 1995; Merlin and Wong, 1997).
During viral infection of the CNS, four main acute phase cytokines are produced. These cytokines include: IL-6, TNF-α, IL-1β and type 1 IFNs (Choi et al., 2009; Koj, 1996). The acute phase cytokines IL-1β and TNF-α have been shown to lead to neurotoxicity in human fetal brain cell and glial co-culture, an effect that was significantly reduced by NMDA receptor antagonists (55% reduction) or nitric oxide synthase inhibition (55%) (Chao et al., 1995). In a later study, it was demonstrated that IL-1β and TNF-α inhibit glutamate uptake into cultured primary human fetal astrocytes (Hu et al., 2000). These experiments suggest that IL-1β and TNF-α can lead to increased exposure to, and extended availability of, glutamate in the synapse, which in turn can result in increased excitation and excitotoxicity (Campbell and Hablitz, 2008). The potential role of TNF-α and IL-1β on the development of seizures and epilepsy has been reviewed elsewhere [reviewed further in (Rijkers et al., 2009; Vezzani and Viviani, 2015)].
IL-6 has been reported to be increased in the plasma and CSF of epilepsy patients with generalized motor seizures (Ishikawa et al., 2015; Lehtimaki et al., 2007; Peltola et al., 2000). Furthermore, studies have shown that IL-6 alone can induce epileptiform activity in both in vivo and ex vivo models. In a study by Levy et al, mice treated with intraventricular injections of IL-6 were monitored for cortical seizure activity and they displayed significantly more seizure-like activity when compared to control mice (Levy et al., 2015). Single cell recordings performed in cortical rat slice preparations have shown that IL-6 application causes a decrease in evoked inhibitory post synaptic current amplitude without changing evoked excitatory post synaptic current amplitudes (Garcia-Oscos et al., 2012); suggesting IL-6 can drive neurons into a more excitable state. Moreover, transgenic mice with increased astrocytic production of IL-6 driven by GFAP (GFAP-IL6) have been found to be significantly more sensitive to NMDA- and KA-induced seizures (Campbell et al., 1993; Samland et al., 2003). The brains from these mice have neurodegeneration in the CA1 consisting of a reduction in dendritic branching (Campbell et al., 1993), and a loss of parvalbumin-positive hippocampal interneurons, a specific subset of GABAergic inhibitory neurons (Heyser et al., 1997). However, there was no significant difference in the number of general GABAergic neurons (Samland et al., 2003). Furthermore, GFAP-IL6 mice have enhanced dendritic post synaptic potentials and somatic population spikes in the CA1 region of the hippocampus as compared to controls, suggesting that increased IL-6 in the CNS can affect neuronal excitability (Nelson et al., 2012). Other disease models have also given insight to the role of IL-6 in hyperexcitability. Use of trigeminal ganglia cell cultures has demonstrated that an application of IL-6 can lead to an increased number of action potentials in response to a stimulus along with a decreased latency to the first spike as compared to controls, due to downstream modulation of sodium ion channels (Yan et al., 2012). These results underscore the ability of IL-6 to signal through neurons in a way that results in channel modifications leading to increased excitability.
The inflammatory action of IL-6 also appears to effect neuronal glutamate receptor expression and function during development. Two examples of receptor proteins that are observed to change in response to exposure to IL-6 are the group-II metabotropic glutamate receptors (mGluR2/3), and the AMPA receptor subunit 2 (GluA2). mGluR2/3 are mainly presynaptic receptors that negatively regulate glutamate release, and a loss of these receptors could increase the levels of glutamate released into the synapse (Dietrich et al., 2002; Kim et al., 2008; Ure et al., 2006). Moreover, GluA2 is an AMPA receptor subunit that renders the receptor impermeable to calcium if expressed (Isaac et al., 2007). It has been shown that a decrease in GluA2 leads to expression of more calcium permeable AMPA receptors and, in turn, increased excitability (Prince et al., 1995; Sanchez et al., 2001). IL-6 treatment of cultured hippocampal neurons during the first 2 weeks in vitro, the main period of physiological development, showed a significant decrease in expression mGluR2/3, and GluA2 at 14 days in vitro (Vereyken et al., 2007). This same study also found that GFAP-IL6 mice also displayed a decrease in mGluR2/3 expression. This study demonstrates the downregulation of several proteins, which could result in increased neuronal excitability. Furthermore, research utilizing cultured cerebellar granule neurons demonstrated that IL-6 treatments increased membrane and current responses to NMDA receptor stimulation and increased calcium influx through voltage-sensitive calcium channels in developing neurons (Qiu et al., 1998).
Previously described research has clearly demonstrated there is an association between inflammatory responses and hyperexcitability in the CNS. Furthermore, pro-inflammatory cytokines such as IL-6 are not only increased in patients after seizures, but have been shown to influence neuronal excitability by affecting glutamate clearance, changing expression of glutamate receptors and receptor subunits, and by decreasing inhibitory tone.
3.4. Conclusion
Seizures/epilepsy are neurological disorders highly associated with viral infection of the CNS. Available treatments are mainly anticonvulsants and 1/3 of the patients are refractory to current treatment. Therefore, the discovery and characterization of new therapeutics to prevent, ameliorate and inhibit seizures/epilepsy are in urgent need.
With development of the TMEV-induced seizures/epilepsy model, the first viral-induced epilepsy model, it has become clear that the immune response plays a striking role in the development of acute seizures. Although adaptive immune cells, such as T and B cells, do not seem to play a role (not discussed in the review), the innate immune response is implicated in this disorder. Using this model, it was demonstrated that following viral infection, increased infiltration of IL-6-producing macrophages within the CNS is associated with seizure development. In agreement with this, in IL-6 KO mice infected with TMEV fewer mice experienced seizures compared to the control group. Similar effects are observed in mice treated with anti-inflammatory compounds, suggesting that new effective therapeutics may need to target the innate immune response, by modulating cellular infiltration into the CNS or altering its inflammatory phenotype and effector functions.
Future studies are still required to understand at the cellular and molecular levels the mechanism of how viral encephalitis induces seizures/epilepsy. One extremely important question is to determine how IL-6 functions in the CNS, timing of IL-6 production and cell types that are affected by this cytokine. Also, the majority of the studies are focused on acute seizures and not on the spontaneous recurrent seizures. It will be important to use this model to study and demonstrate the mechanism of epileptogenesis and to understand the reason why patients are refractory to treatment.
Highlights.
TMEV-IDD is a mouse model of the progressive form of multiple sclerosis
The adaptive immune response plays a major role in the development of TMEV-IDD
The TMEV-induced seizure model replicates many aspects of acute seizures & epilepsy
Infiltration of immune cells into the CNS contributes to seizure development
Viral infection models allow for the study of the immune response and disease link
Acknowledgments
Funding: This work was supported by the National Institutes of Health [grant numbers: 5R01NS065714 & 5R01NS082102].
Abbreviations
- AMPA
α-amino-3-hydroxy-5-methyl-4-isoxazolepropionate
- APCs
antigen presenting cells
- BBB
blood-brain barrier
- CNS
central nervous system
- CSF
cerebrospinal fluid
- DA
Daniels
- EAE
experimental autoimmune encephalomyelitis
- GABA
γ-aminobutyric acid
- GFAP
glial fibrillary acidic protein
- GFP
green fluorescence protein
- HHV-6
human herpes virus type-6
- HSV-1
herpes simplex virus type-1
- i.c.
intracerebrally
- IFN
interferon
- IL
interleukin
- IL-1R1
IL-1 receptor 1
- ISGs
interferon-stimulated genes
- KA
Kainate
- KO
knockout
- LPS
lipopolysaccharide
- MBP
myelin basic protein
- mGluRs
metabotropic glutamate receptors
- MHC
major histocompatibility complex
- MS
multiple sclerosis
- MyD88
myeloid differentiation primary response gene 88
- NK
natural killer
- NMDA
N-methyl-D-aspartate
- PAMPs
pathogen-associated molecular patterns
- PLP
myelin proteolipid protein
- RCA
Ricinus communis agglutinin
- TBI
traumatic brain injury
- TCR
T cell receptor
- TLE
temporal lobe epilepsy
- TLR
Toll Like Receptors
- TMEV
Theiler’s murine encephalomyelitis virus
- TMEV-IDD
TMEV-induced demyelinating disease
- TNF
tumor necrosis factor
- TO
Theiler’s original
- Tregs
regulatory T cells
- video-EEG
video-electroencephalogram
- WNV
West Nile virus
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Al Sufiani F, Ang LC. Neuropathology of temporal lobe epilepsy. Epilepsy Res Treat. 2012;2012:624519. doi: 10.1155/2012/624519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Annegers JF, Hauser WA, Beghi E, Nicolosi A, Kurland LT. The risk of unprovoked seizures after encephalitis and meningitis. Neurology. 1988;38:1407–1410. doi: 10.1212/wnl.38.9.1407. [DOI] [PubMed] [Google Scholar]
- Babbe H, Roers A, Waisman A, Lassmann H, Goebels N, Hohlfeld R, Friese M, Schroder R, Deckert M, Schmidt S, Ravid R, Rajewsky K. Clonal expansions of CD8(+) T cells dominate the T cell infiltrate in active multiple sclerosis lesions as shown by micromanipulation and single cell polymerase chain reaction. The Journal of experimental medicine. 2000;192:393–404. doi: 10.1084/jem.192.3.393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bale JF., Jr Virus and Immune-Mediated Encephalitides: Epidemiology, Diagnosis, Treatment, and Prevention. Pediatr Neurol. 2015;53:3–12. doi: 10.1016/j.pediatrneurol.2015.03.013. [DOI] [PubMed] [Google Scholar]
- Baranzini SE, Mudge J, van Velkinburgh JC, Khankhanian P, Khrebtukova I, Miller NA, Zhang L, Farmer AD, Bell CJ, Kim RW, May GD, Woodward JE, Caillier SJ, McElroy JP, Gomez R, Pando MJ, Clendenen LE, Ganusova EE, Schilkey FD, Ramaraj T, Khan OA, Huntley JJ, Luo S, Kwok PY, Wu TD, Schroth GP, Oksenberg JR, Hauser SL, Kingsmore SF. Genome, epigenome and RNA sequences of monozygotic twins discordant for multiple sclerosis. Nature. 2010;464:1351–1356. doi: 10.1038/nature08990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barr TA, Shen P, Brown S, Lampropoulou V, Roch T, Lawrie S, Fan B, O’Connor RA, Anderton SM, Bar-Or A, Fillatreau S, Gray D. B cell depletion therapy ameliorates autoimmune disease through ablation of IL-6-producing B cells. The Journal of experimental medicine. 2012;209:1001–1010. doi: 10.1084/jem.20111675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Begolka WS, Haynes LM, Olson JK, Padilla J, Neville KL, Dal Canto M, Palma J, Kim BS, Miller SD. CD8-deficient SJL mice display enhanced susceptibility to Theiler’s virus infection and increased demyelinating pathology. Journal of neurovirology. 2001;7:409–420. doi: 10.1080/135502801753170264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Ari Y, Dudek FE. Primary and secondary mechanisms of epileptogenesis in the temporal lobe: there is a before and an after. Epilepsy currents. 2010;10:118–125. doi: 10.1111/j.1535-7511.2010.01376.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benoist C, Mathis D. Positive and negative selection of the T cell repertoire in MHC class II transgenic mice. Semin Immunol. 1989;1:117–124. [PubMed] [Google Scholar]
- Benoist C, Mathis D. T-cell development: a new marker of differentiation state. Curr Biol. 1999;9:R59–61. doi: 10.1016/s0960-9822(99)80011-6. [DOI] [PubMed] [Google Scholar]
- Berg AT, Berkovic SF, Brodie MJ, Buchhalter J, Cross JH, van Emde Boas W, Engel J, French J, Glauser TA, Mathern GW, Moshe SL, Nordli D, Plouin P, Scheffer IE. Revised terminology and concepts for organization of seizures and epilepsies: report of the ILAE Commission on Classification and Terminology, 2005–2009. Epilepsia. 2010;51:676–685. doi: 10.1111/j.1528-1167.2010.02522.x. [DOI] [PubMed] [Google Scholar]
- Bjartmar C, Kidd G, Mork S, Rudick R, Trapp BD. Neurological disability correlates with spinal cord axonal loss and reduced N-acetyl aspartate in chronic multiple sclerosis patients. Annals of neurology. 2000;48:893–901. [PubMed] [Google Scholar]
- Blichfeldt E, Munthe LA, Rotnes JS, Bogen B. Dual T cell receptor T cells have a decreased sensitivity to physiological ligands due to reduced density of each T cell receptor. Eur J Immunol. 1996;26:2876–2884. doi: 10.1002/eji.1830261211. [DOI] [PubMed] [Google Scholar]
- Bonello M, Michael BD, Solomon T. Infective Causes of Epilepsy. Semin Neurol. 2015;35:235–244. doi: 10.1055/s-0035-1552619. [DOI] [PubMed] [Google Scholar]
- Borrow P, Tonks P, Welsh CJ, Nash AA. The role of CD8+T cells in the acute and chronic phases of Theiler’s murine encephalomyelitis virus-induced disease in mice. J Gen Virol. 1992;73(Pt 7):1861–1865. doi: 10.1099/0022-1317-73-7-1861. [DOI] [PubMed] [Google Scholar]
- Bowen JL, Olson JK. IFNgamma influences type I interferon response and susceptibility to Theiler’s virus-induced demyelinating disease. Viral Immunol. 2013;26:223–238. doi: 10.1089/vim.2013.0004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broer S, Kaufer C, Haist V, Li L, Gerhauser I, Anjum M, Bankstahl M, Baumgartner W, Loscher W. Brain inflammation, neurodegeneration and seizure development following picornavirus infection markedly differ among virus and mouse strains and substrains. Exp Neurol. 2016;279:57–74. doi: 10.1016/j.expneurol.2016.02.011. [DOI] [PubMed] [Google Scholar]
- Campbell IL, Abraham CR, Masliah E, Kemper P, Inglis JD, Oldstone MB, Mucke L. Neurologic disease induced in transgenic mice by cerebral overexpression of interleukin 6. Proceedings of the National Academy of Sciences of the United States of America. 1993;90:10061–10065. doi: 10.1073/pnas.90.21.10061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell SL, Hablitz JJ. Decreased glutamate transport enhances excitability in a rat model of cortical dysplasia. Neurobiol Dis. 2008;32:254–261. doi: 10.1016/j.nbd.2008.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cermelli C, Berti R, Soldan SS, Mayne M, D’Ambrosia JM, Ludwin SK, Jacobson S. High frequency of human herpesvirus 6 DNA in multiple sclerosis plaques isolated by laser microdissection. J Infect Dis. 2003;187:1377–1387. doi: 10.1086/368166. [DOI] [PubMed] [Google Scholar]
- Chakraborty S, Nazmi A, Dutta K, Basu A. Neurons under viral attack: victims or warriors? Neurochem Int. 2010;56:727–735. doi: 10.1016/j.neuint.2010.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Challoner PB, Smith KT, Parker JD, MacLeod DL, Coulter SN, Rose TM, Schultz ER, Bennett JL, Garber RL, Chang M, et al. Plaque-associated expression of human herpesvirus 6 in multiple sclerosis. Proceedings of the National Academy of Sciences of the United States of America. 1995;92:7440–7444. doi: 10.1073/pnas.92.16.7440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chao CC, Hu S, Ehrlich L, Peterson PK. Interleukin-1 and tumor necrosis factor-alpha synergistically mediate neurotoxicity: involvement of nitric oxide and of N-methyl-D-aspartate receptors. Brain Behav Immun. 1995;9:355–365. doi: 10.1006/brbi.1995.1033. [DOI] [PubMed] [Google Scholar]
- Choi J, Nordli DR, Jr, Alden TD, DiPatri A, Jr, Laux L, Kelley K, Rosenow J, Schuele SU, Rajaram V, Koh S. Cellular injury and neuroinflammation in children with chronic intractable epilepsy. Journal of neuroinflammation. 2009;6:38. doi: 10.1186/1742-2094-6-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clatch RJ, Lipton HL, Miller SD. Characterization of Theiler’s murine encephalomyelitis virus (TMEV)-specific delayed-type hypersensitivity responses in TMEV-induced demyelinating disease: correlation with clinical signs. Journal of immunology. 1986;136:920–927. [PubMed] [Google Scholar]
- Clatch RJ, Miller SD, Metzner R, Dal Canto MC, Lipton HL. Monocytes/macrophages isolated from the mouse central nervous system contain infectious Theiler’s murine encephalomyelitis virus (TMEV) Virology. 1990;176:244–254. doi: 10.1016/0042-6822(90)90249-q. [DOI] [PubMed] [Google Scholar]
- Coles AJ, Cox A, Le Page E, Jones J, Trip SA, Deans J, Seaman S, Miller DH, Hale G, Waldmann H, Compston DA. The window of therapeutic opportunity in multiple sclerosis: evidence from monoclonal antibody therapy. J Neurol. 2006;253:98–108. doi: 10.1007/s00415-005-0934-5. [DOI] [PubMed] [Google Scholar]
- Compston A, Coles A. Multiple sclerosis. Lancet. 2008;372:1502–1517. doi: 10.1016/S0140-6736(08)61620-7. [DOI] [PubMed] [Google Scholar]
- Coulter DA, Steinhauser C. Role of astrocytes in epilepsy. Cold Spring Harb Perspect Med. 2015;5:a022434. doi: 10.1101/cshperspect.a022434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crawford MP, Yan SX, Ortega SB, Mehta RS, Hewitt RE, Price DA, Stastny P, Douek DC, Koup RA, Racke MK, Karandikar NJ. High prevalence of autoreactive, neuroantigen-specific CD8+ T cells in multiple sclerosis revealed by novel flow cytometric assay. Blood. 2004;103:4222–4231. doi: 10.1182/blood-2003-11-4025. [DOI] [PubMed] [Google Scholar]
- Crespel A, Coubes P, Rousset MC, Brana C, Rougier A, Rondouin G, Bockaert J, Baldy-Moulinier M, Lerner-Natoli M. Inflammatory reactions in human medial temporal lobe epilepsy with hippocampal sclerosis. Brain Res. 2002;952:159–169. doi: 10.1016/s0006-8993(02)03050-0. [DOI] [PubMed] [Google Scholar]
- Cusick MF, Libbey JE, Fujinami RS. Multiple sclerosis: autoimmunity and viruses. Current opinion in rheumatology. 2013a;25:496–501. doi: 10.1097/BOR.0b013e328362004d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cusick MF, Libbey JE, Patel DC, Doty DJ, Fujinami RS. Infiltrating macrophages are key to the development of seizures following virus infection. Journal of virology. 2013b;87:1849–1860. doi: 10.1128/JVI.02747-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dal Canto MC, Kim BS, Miller SD, Melvold RW. Theiler’s Murine Encephalomyelitis Virus (TMEV)-Induced Demyelination: A Model for Human Multiple Sclerosis. Methods. 1996;10:453–461. doi: 10.1006/meth.1996.0123. [DOI] [PubMed] [Google Scholar]
- Daniels JB, Pappenheimer AM, Richardson S. Observations on encephalomyelitis of mice (DA strain) The Journal of experimental medicine. 1952;96:517–530. doi: 10.1084/jem.96.6.517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis MJ, Tsang TM, Qiu Y, Dayrit JK, Freij JB, Huffnagle GB, Olszewski MA. Macrophage M1/M2 polarization dynamically adapts to changes in cytokine microenvironments in Cryptococcus neoformans infection. MBio. 2013;4:e00264–00213. doi: 10.1128/mBio.00264-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delorenzo RJ, Sun DA, Deshpande LS. Cellular mechanisms underlying acquired epilepsy: the calcium hypothesis of the induction and maintainance of epilepsy. Pharmacol Ther. 2005;105:229–266. doi: 10.1016/j.pharmthera.2004.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dietrich D, Kral T, Clusmann H, Friedl M, Schramm J. Presynaptic group II metabotropic glutamate receptors reduce stimulated and spontaneous transmitter release in human dentate gyrus. Neuropharmacology. 2002;42:297–305. doi: 10.1016/s0028-3908(01)00193-9. [DOI] [PubMed] [Google Scholar]
- Dingledine R. Glutamatergic Mechanisms Related to Epilepsy: Ionotropic Receptors. In: Noebels JL, Avoli M, Rogawski MA, Olsen RW, Delgado-Escueta AV, editors. Jasper’s Basic Mechanisms of the Epilepsies. 4. Bethesda (MD): 2012. [PubMed] [Google Scholar]
- Dittmer U, He H, Messer RJ, Schimmer S, Olbrich AR, Ohlen C, Greenberg PD, Stromnes IM, Iwashiro M, Sakaguchi S, Evans LH, Peterson KE, Yang G, Hasenkrug KJ. Functional impairment of CD8(+) T cells by regulatory T cells during persistent retroviral infection. Immunity. 2004;20:293–303. doi: 10.1016/s1074-7613(04)00054-8. [DOI] [PubMed] [Google Scholar]
- Donati D, Akhyani N, Fogdell-Hahn A, Cermelli C, Cassiani-Ingoni R, Vortmeyer A, Heiss JD, Cogen P, Gaillard WD, Sato S, Theodore WH, Jacobson S. Detection of human herpesvirus-6 in mesial temporal lobe epilepsy surgical brain resections. Neurology. 2003;61:1405–1411. doi: 10.1212/01.wnl.0000094357.10782.f9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebers GC. Genetic factors in multiple sclerosis. Neurol Clin. 1983;1:645–654. [PubMed] [Google Scholar]
- Eeg-Olofsson O. Virological and immunological aspects of seizure disorders. Brain Dev. 2003;25:9–13. doi: 10.1016/s0387-7604(02)00162-6. [DOI] [PubMed] [Google Scholar]
- Eggena MP, Barugahare B, Jones N, Okello M, Mutalya S, Kityo C, Mugyenyi P, Cao H. Depletion of regulatory T cells in HIV infection is associated with immune activation. Journal of immunology. 2005;174:4407–4414. doi: 10.4049/jimmunol.174.7.4407. [DOI] [PubMed] [Google Scholar]
- Ehrengruber MU, Ehler E, Billeter MA, Naim HY. Measles virus spreads in rat hippocampal neurons by cell-to-cell contact and in a polarized fashion. Journal of virology. 2002;76:5720–5728. doi: 10.1128/JVI.76.11.5720-5728.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- England MJ, Liverman CT, Schultz AM, Strawbridge LM. Epilepsy across the spectrum: promoting health and understanding. A summary of the Institute of Medicine report. Epilepsy Behav. 2012;25:266–276. doi: 10.1016/j.yebeh.2012.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fas SC, Baumann S, Zhu JY, Giaisi M, Treiber MK, Mahlknecht U, Krammer PH, Li-Weber M. Wogonin sensitizes resistant malignant cells to TNFalpha- and TRAIL-induced apoptosis. Blood. 2006;108:3700–3706. doi: 10.1182/blood-2006-03-011973. [DOI] [PubMed] [Google Scholar]
- Fletcher JM, Lalor SJ, Sweeney CM, Tubridy N, Mills KH. T cells in multiple sclerosis and experimental autoimmune encephalomyelitis. Clin Exp Immunol. 2010;162:1–11. doi: 10.1111/j.1365-2249.2010.04143.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friese MA, Jakobsen KB, Friis L, Etzensperger R, Craner MJ, McMahon RM, Jensen LT, Huygelen V, Jones EY, Bell JI, Fugger L. Opposing effects of HLA class I molecules in tuning autoreactive CD8+ T cells in multiple sclerosis. Nat Med. 2008;14:1227–1235. doi: 10.1038/nm.1881. [DOI] [PubMed] [Google Scholar]
- Frohman EM, Racke MK, Raine CS. Multiple sclerosis–the plaque and its pathogenesis. The New England journal of medicine. 2006;354:942–955. doi: 10.1056/NEJMra052130. [DOI] [PubMed] [Google Scholar]
- Fujinami RS, Oldstone MB, Wroblewska Z, Frankel ME, Koprowski H. Molecular mimicry in virus infection: crossreaction of measles virus phosphoprotein or of herpes simplex virus protein with human intermediate filaments. Proceedings of the National Academy of Sciences of the United States of America. 1983;80:2346–2350. doi: 10.1073/pnas.80.8.2346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Oscos F, Salgado H, Hall S, Thomas F, Farmer GE, Bermeo J, Galindo LC, Ramirez RD, D’Mello S, Rose-John S, Atzori M. The stress-induced cytokine interleukin-6 decreases the inhibition/excitation ratio in the rat temporal cortex via trans-signaling. Biol Psychiatry. 2012;71:574–582. doi: 10.1016/j.biopsych.2011.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Getts DR, Balcar VJ, Matsumoto I, Muller M, King NJ. Viruses and the immune system: their roles in seizure cascade development. Journal of neurochemistry. 2008;104:1167–1176. doi: 10.1111/j.1471-4159.2007.05171.x. [DOI] [PubMed] [Google Scholar]
- Getts DR, Matsumoto I, Muller M, Getts MT, Radford J, Shrestha B, Campbell IL, King NJ. Role of IFN-gamma in an experimental murine model of West Nile virus-induced seizures. J Neurochem. 2007;103:1019–1030. doi: 10.1111/j.1471-4159.2007.04798.x. [DOI] [PubMed] [Google Scholar]
- Gilli F, Li L, Campbell SJ, Anthony DC, Pachner AR. The effect of B-cell depletion in the Theiler’s model of multiple sclerosis. J Neurol Sci. 2015;359:40–47. doi: 10.1016/j.jns.2015.10.012. [DOI] [PubMed] [Google Scholar]
- Girard S, Gosselin AS, Pelletier I, Colbere-Garapin F, Couderc T, Blondel B. Restriction of poliovirus RNA replication in persistently infected nerve cells. J Gen Virol. 2002;83:1087–1093. doi: 10.1099/0022-1317-83-5-1087. [DOI] [PubMed] [Google Scholar]
- Giuliani F, Hader W, Yong VW. Minocycline attenuates T cell and microglia activity to impair cytokine production in T cell-microglia interaction. J Leukoc Biol. 2005;78:135–143. doi: 10.1189/jlb.0804477. [DOI] [PubMed] [Google Scholar]
- Goverman JM. Immune tolerance in multiple sclerosis. Immunol Rev. 2011;241:228–240. doi: 10.1111/j.1600-065X.2011.01016.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffin DE. Immune responses to RNA-virus infections of the CNS. Nat Rev Immunol. 2003;3:493–502. doi: 10.1038/nri1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groux H, O’Garra A, Bigler M, Rouleau M, Antonenko S, de Vries JE, Roncarolo MG. A CD4+ T-cell subset inhibits antigen-specific T-cell responses and prevents colitis. Nature. 1997;389:737–742. doi: 10.1038/39614. [DOI] [PubMed] [Google Scholar]
- Hao J, Liu R, Piao W, Zhou Q, Vollmer TL, Campagnolo DI, Xiang R, La Cava A, Van Kaer L, Shi FD. Central nervous system (CNS)-resident natural killer cells suppress Th17 responses and CNS autoimmune pathology. The Journal of experimental medicine. 2010;207:1907–1921. doi: 10.1084/jem.20092749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauser SL, Waubant E, Arnold DL, Vollmer T, Antel J, Fox RJ, Bar-Or A, Panzara M, Sarkar N, Agarwal S, Langer-Gould A, Smith CH, Group, H.T. B-cell depletion with rituximab in relapsing-remitting multiple sclerosis. The New England journal of medicine. 2008;358:676–688. doi: 10.1056/NEJMoa0706383. [DOI] [PubMed] [Google Scholar]
- He H, Messer RJ, Sakaguchi S, Yang G, Robertson SJ, Hasenkrug KJ. Reduction of retrovirus-induced immunosuppression by in vivo modulation of T cells during acute infection. J Virol. 2004;78:11641–11647. doi: 10.1128/JVI.78.21.11641-11647.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heyser CJ, Masliah E, Samimi A, Campbell IL, Gold LH. Progressive decline in avoidance learning paralleled by inflammatory neurodegeneration in transgenic mice expressing interleukin 6 in the brain. Proceedings of the National Academy of Sciences of the United States of America. 1997;94:1500–1505. doi: 10.1073/pnas.94.4.1500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoftberger R, Aboul-Enein F, Brueck W, Lucchinetti C, Rodriguez M, Schmidbauer M, Jellinger K, Lassmann H. Expression of major histocompatibility complex class I molecules on the different cell types in multiple sclerosis lesions. Brain pathology. 2004;14:43–50. doi: 10.1111/j.1750-3639.2004.tb00496.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honma K, Parker KC, Becker KG, McFarland HF, Coligan JE, Biddison WE. Identification of an epitope derived from human proteolipid protein that can induce autoreactive CD8+ cytotoxic T lymphocytes restricted by HLA-A3: evidence for cross-reactivity with an environmental microorganism. Journal of neuroimmunology. 1997;73:7–14. doi: 10.1016/s0165-5728(96)00161-0. [DOI] [PubMed] [Google Scholar]
- Howe CL, Lafrance-Corey RG, Sundsbak RS, Lafrance SJ. Inflammatory monocytes damage the hippocampus during acute picornavirus infection of the brain. J Neuroinflammation. 2012;9:50. doi: 10.1186/1742-2094-9-50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu S, Sheng WS, Ehrlich LC, Peterson PK, Chao CC. Cytokine effects on glutamate uptake by human astrocytes. Neuroimmunomodulation. 2000;7:153–159. doi: 10.1159/000026433. [DOI] [PubMed] [Google Scholar]
- Huseby ES, Liggitt D, Brabb T, Schnabel B, Ohlen C, Goverman J. A pathogenic role for myelin-specific CD8(+) T cells in a model for multiple sclerosis. The Journal of experimental medicine. 2001;194:669–676. doi: 10.1084/jem.194.5.669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isaac JT, Ashby MC, McBain CJ. The role of the GluR2 subunit in AMPA receptor function and synaptic plasticity. Neuron. 2007;54:859–871. doi: 10.1016/j.neuron.2007.06.001. [DOI] [PubMed] [Google Scholar]
- Ishikawa N, Kobayashi Y, Fujii Y, Kobayashi M. Increased interleukin-6 and high-sensitivity C-reactive protein levels in pediatric epilepsy patients with frequent, refractory generalized motor seizures. Seizure. 2015;25:136–140. doi: 10.1016/j.seizure.2014.10.007. [DOI] [PubMed] [Google Scholar]
- Jelachich ML, Bandyopadhyay P, Blum K, Lipton HL. Theiler’s virus growth in murine macrophage cell lines depends on the state of differentiation. Virology. 1995;209:437–444. doi: 10.1006/viro.1995.1276. [DOI] [PubMed] [Google Scholar]
- Ji Q, Perchellet A, Goverman JM. Viral infection triggers central nervous system autoimmunity via activation of CD8+ T cells expressing dual TCRs. Nat Immunol. 2010;11:628–634. doi: 10.1038/ni.1888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabat EA, Moore DH, Landow H. An Electrophoretic Study of the Protein Components in Cerebrospinal Fluid and Their Relationship to the Serum Proteins. J Clin Invest. 1942;21:571–577. doi: 10.1172/JCI101335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katz-Levy Y, Neville KL, Padilla J, Rahbe S, Begolka WS, Girvin AM, Olson JK, Vanderlugt CL, Miller SD. Temporal development of autoreactive Th1 responses and endogenous presentation of self myelin epitopes by central nervous system-resident APCs in Theiler’s virus-infected mice. Journal of immunology. 2000;165:5304–5314. doi: 10.4049/jimmunol.165.9.5304. [DOI] [PubMed] [Google Scholar]
- Kim CH, Lee J, Lee JY, Roche KW. Metabotropic glutamate receptors: phosphorylation and receptor signaling. J Neurosci Res. 2008;86:1–10. doi: 10.1002/jnr.21437. [DOI] [PubMed] [Google Scholar]
- Kirkman NJ, Libbey JE, Wilcox KS, White HS, Fujinami RS. Innate but not adaptive immune responses contribute to behavioral seizures following viral infection. Epilepsia. 2010;51:454–464. doi: 10.1111/j.1528-1167.2009.02390.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein L, Kyewski B, Allen PM, Hogquist KA. Positive and negative selection of the T cell repertoire: what thymocytes see (and don’t see) Nat Rev Immunol. 2014;14:377–391. doi: 10.1038/nri3667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koj A. Initiation of acute phase response and synthesis of cytokines. Biochim Biophys Acta. 1996;1317:84–94. doi: 10.1016/s0925-4439(96)00048-8. [DOI] [PubMed] [Google Scholar]
- Kriesel JD, White A, Hayden FG, Spruance SL, Petajan J. Multiple sclerosis attacks are associated with picornavirus infections. Mult Scler. 2004;10:145–148. doi: 10.1191/1352458504ms1005oa. [DOI] [PubMed] [Google Scholar]
- Krumbholz M, Meinl E. B cells in MS and NMO: pathogenesis and therapy. Semin Immunopathol. 2014;36:339–350. doi: 10.1007/s00281-014-0424-x. [DOI] [PubMed] [Google Scholar]
- Langrish CL, Chen Y, Blumenschein WM, Mattson J, Basham B, Sedgwick JD, McClanahan T, Kastelein RA, Cua DJ. IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. The Journal of experimental medicine. 2005;201:233–240. doi: 10.1084/jem.20041257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lassmann H. Pathophysiology of inflammation and tissue injury in multiple sclerosis: what are the targets for therapy. J Neurol Sci. 2011;306:167–169. doi: 10.1016/j.jns.2010.07.023. [DOI] [PubMed] [Google Scholar]
- Lassmann H, Bradl M. Multiple sclerosis: experimental models and reality. Acta Neuropathol. 2016 doi: 10.1007/s00401-016-1631-4. Epub ahead of print Oct 20, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laxer KD, Trinka E, Hirsch LJ, Cendes F, Langfitt J, Delanty N, Resnick T, Benbadis SR. The consequences of refractory epilepsy and its treatment. Epilepsy Behav. 2014;37:59–70. doi: 10.1016/j.yebeh.2014.05.031. [DOI] [PubMed] [Google Scholar]
- Lehmann PV, Forsthuber T, Miller A, Sercarz EE. Spreading of T-cell autoimmunity to cryptic determinants of an autoantigen. Nature. 1992;358:155–157. doi: 10.1038/358155a0. [DOI] [PubMed] [Google Scholar]
- Lehmann PV, Sercarz EE, Forsthuber T, Dayan CM, Gammon G. Determinant spreading and the dynamics of the autoimmune T-cell repertoire. Immunol Today. 1993;14:203–208. doi: 10.1016/0167-5699(93)90163-F. [DOI] [PubMed] [Google Scholar]
- Lehrmann E, Guidetti P, Love A, Williamson J, Bertram EH, Schwarcz R. Glial activation precedes seizures and hippocampal neurodegeneration in measles virus-infected mice. Epilepsia. 2008;49(Suppl 2):13–23. doi: 10.1111/j.1528-1167.2008.01489.x. [DOI] [PubMed] [Google Scholar]
- Lehtimaki KA, Keranen T, Palmio J, Makinen R, Hurme M, Honkaniemi J, Peltola J. Increased plasma levels of cytokines after seizures in localization-related epilepsy. Acta Neurol Scand. 2007;116:226–230. doi: 10.1111/j.1600-0404.2007.00882.x. [DOI] [PubMed] [Google Scholar]
- Levy N, Milikovsky DZ, Baranauskas G, Vinogradov E, David Y, Ketzef M, Abutbul S, Weissberg I, Kamintsky L, Fleidervish I, Friedman A, Monsonego A. Differential TGF-beta Signaling in Glial Subsets Underlies IL-6-Mediated Epileptogenesis in Mice. Journal of immunology. 2015;195:1713–1722. doi: 10.4049/jimmunol.1401446. [DOI] [PubMed] [Google Scholar]
- Li L, Ulrich R, Baumgartner W, Gerhauser I. Interferon-stimulated genes-essential antiviral effectors implicated in resistance to Theiler’s virus-induced demyelinating disease. Journal of neuroinflammation. 2015;12:242. doi: 10.1186/s12974-015-0462-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Libbey JE, Cusick MF, Tsunoda I, Fujinami RS. Antiviral CD8(+) T cells cause an experimental autoimmune encephalomyelitis-like disease in naive mice. Journal of neurovirology. 2012;18:45–54. doi: 10.1007/s13365-012-0077-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Libbey JE, Fujinami RS. Potential triggers of MS. Results and problems in cell differentiation. 2010;51:21–42. doi: 10.1007/400_2008_12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Libbey JE, Fujinami RS. Neurotropic viral infections leading to epilepsy: focus on Theiler’s murine encephalomyelitis virus. Future virology. 2011;6:1339–1350. doi: 10.2217/fvl.11.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Libbey JE, Kennett NJ, Wilcox KS, White HS, Fujinami RS. Interleukin-6, produced by resident cells of the central nervous system and infiltrating cells, contributes to the development of seizures following viral infection. Journal of virology. 2011a;85:6913–6922. doi: 10.1128/JVI.00458-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Libbey JE, Kennett NJ, Wilcox KS, White HS, Fujinami RS. Lack of correlation of central nervous system inflammation and neuropathology with the development of seizures following acute virus infection. Journal of virology. 2011b;85:8149–8157. doi: 10.1128/JVI.00730-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Libbey JE, Kirkman NJ, Smith MC, Tanaka T, Wilcox KS, White HS, Fujinami RS. Seizures following picornavirus infection. Epilepsia. 2008;49:1066–1074. doi: 10.1111/j.1528-1167.2008.01535.x. [DOI] [PubMed] [Google Scholar]
- Libbey JE, Kirkman NJ, Wilcox KS, White HS, Fujinami RS. Role for complement in the development of seizures following acute viral infection. Journal of virology. 2010;84:6452–6460. doi: 10.1128/JVI.00422-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Libbey JE, Lane TE, Fujinami RS. Axonal pathology and demyelination in viral models of multiple sclerosis. Discov Med. 2014;18:79–89. [PMC free article] [PubMed] [Google Scholar]
- Lipton HL. Theiler’s virus infection in mice: an unusual biphasic disease process leading to demyelination. Infection and immunity. 1975;11:1147–1155. doi: 10.1128/iai.11.5.1147-1155.1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipton HL, Jelachich ML. Molecular pathogenesis of Theiler’s murine encephalomyelitis virus-induced demyelinating disease in mice. Intervirology. 1997;40:143–152. doi: 10.1159/000150541. [DOI] [PubMed] [Google Scholar]
- Liu C, Collins J, Sharp E. The pathogenesis of Theiler’s GD VII encephalomyelitis virus infection in mice as studied by immunofluorescent technique and infectivity titrations. Journal of immunology. 1967;98:46–55. [PubMed] [Google Scholar]
- Liuzzi GM, Riccio P, Dal Canto MC. Release of myelin basic protein-degrading proteolytic activity from microglia and macrophages after infection with Theiler’s murine encephalomyelitis virus: comparison between susceptible and resistant mice. J Neuroimmunol. 1995;62:91–102. doi: 10.1016/0165-5728(95)00110-n. [DOI] [PubMed] [Google Scholar]
- Loewen JL, Barker-Haliski ML, Dahle EJ, White HS, Wilcox KS. Neuronal Injury, Gliosis, and Glial Proliferation in Two Models of Temporal Lobe Epilepsy. J Neuropathol Exp Neurol. 2016;75:366–378. doi: 10.1093/jnen/nlw008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowenstein DH. Epilepsy after head injury: an overview. Epilepsia. 2009;50(Suppl 2):4–9. doi: 10.1111/j.1528-1167.2008.02004.x. [DOI] [PubMed] [Google Scholar]
- Lunemann A, Lunemann JD, Munz C. Regulatory NK-cell functions in inflammation and autoimmunity. Mol Med. 2009;15:352–358. doi: 10.2119/molmed.2009.00035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mantovani A, Biswas SK, Galdiero MR, Sica A, Locati M. Macrophage plasticity and polarization in tissue repair and remodelling. J Pathol. 2013;229:176–185. doi: 10.1002/path.4133. [DOI] [PubMed] [Google Scholar]
- Matsushita T, Yanaba K, Bouaziz JD, Fujimoto M, Tedder TF. Regulatory B cells inhibit EAE initiation in mice while other B cells promote disease progression. J Clin Invest. 2008;118:3420–3430. doi: 10.1172/JCI36030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazrouei F, Ganjalikhani-Hakemi M, Salehi R, Alesahebfosoul F, Etemadifar M, Pouladian M, Meshkat R, Nekoueian S, Zarkesh-Esfahani H, Ziyaee-Ghahnaviyeh M. Association of TIM-1 5383-5397ins/del and TIM-3 -1541C>T polymorphisms with multiple sclerosis in Isfahan population. International journal of immunogenetics. 2016;43:131–134. doi: 10.1111/iji.12264. [DOI] [PubMed] [Google Scholar]
- McCarthy DP, Richards MH, Miller SD. Mouse models of multiple sclerosis: experimental autoimmune encephalomyelitis and Theiler’s virus-induced demyelinating disease. Methods Mol Biol. 2012;900:381–401. doi: 10.1007/978-1-60761-720-4_19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meldrum BS. Glutamate as a neurotransmitter in the brain: review of physiology and pathology. J Nutr. 2000;130:1007S–1015S. doi: 10.1093/jn/130.4.1007S. [DOI] [PubMed] [Google Scholar]
- Merlin LR, Taylor GW, Wong RK. Role of metabotropic glutamate receptor subtypes in the patterning of epileptiform activities in vitro. J Neurophysiol. 1995;74:896–900. doi: 10.1152/jn.1995.74.2.896. [DOI] [PubMed] [Google Scholar]
- Merlin LR, Wong RK. Role of group I metabotropic glutamate receptors in the patterning of epileptiform activities in vitro. J Neurophysiol. 1997;78:539–544. doi: 10.1152/jn.1997.78.1.539. [DOI] [PubMed] [Google Scholar]
- Miller DH, Chard DT, Ciccarelli O. Clinically isolated syndromes. Lancet Neurol. 2012;11:157–169. doi: 10.1016/S1474-4422(11)70274-5. [DOI] [PubMed] [Google Scholar]
- Miller SD, Vanderlugt CL, Begolka WS, Pao W, Yauch RL, Neville KL, Katz-Levy Y, Carrizosa A, Kim BS. Persistent infection with Theiler’s virus leads to CNS autoimmunity via epitope spreading. Nat Med. 1997;3:1133–1136. doi: 10.1038/nm1097-1133. [DOI] [PubMed] [Google Scholar]
- Misra UK, Tan CT, Kalita J. Viral encephalitis and epilepsy. Epilepsia. 2008;49(Suppl 6):13–18. doi: 10.1111/j.1528-1167.2008.01751.x. [DOI] [PubMed] [Google Scholar]
- Montero E, Nussbaum G, Kaye JF, Perez R, Lage A, Ben-Nun A, Cohen IR. Regulation of experimental autoimmune encephalomyelitis by CD4+, CD25+ and CD8+ T cells: analysis using depleting antibodies. J Autoimmun. 2004;23:1–7. doi: 10.1016/j.jaut.2004.05.001. [DOI] [PubMed] [Google Scholar]
- Monteyne P, Bureau JF, Brahic M. The infection of mouse by Theiler’s virus: from genetics to immunology. Immunol Rev. 1997;159:163–176. doi: 10.1111/j.1600-065x.1997.tb01014.x. [DOI] [PubMed] [Google Scholar]
- Murray PD, McGavern DB, Pease LR, Rodriguez M. Cellular sources and targets of IFN-gamma-mediated protection against viral demyelination and neurological deficits. Eur J Immunol. 2002;32:606–615. doi: 10.1002/1521-4141(200203)32:3<606::aid-immu606>3.0.co;2-d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray PD, Pavelko KD, Leibowitz J, Lin X, Rodriguez M. CD4(+) and CD8(+) T cells make discrete contributions to demyelination and neurologic disease in a viral model of multiple sclerosis. J Virol. 1998;72:7320–7329. doi: 10.1128/jvi.72.9.7320-7329.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nadler JV. Plasticity of Glutamate Synaptic Mechanisms. In: Noebels JL, Avoli M, Rogawski MA, Olsen RW, Delgado-Escueta AV, editors. Jasper’s Basic Mechanisms of the Epilepsies. 4. Bethesda (MD): 2012. [PubMed] [Google Scholar]
- Naito S, Namerow N, Mickey MR, Terasaki PI. Multiple sclerosis: association with HL-A3. Tissue Antigens. 1972;2:1–4. doi: 10.1111/j.1399-0039.1972.tb00111.x. [DOI] [PubMed] [Google Scholar]
- Nelson TE, Olde Engberink A, Hernandez R, Puro A, Huitron-Resendiz S, Hao C, De Graan PN, Gruol DL. Altered synaptic transmission in the hippocampus of transgenic mice with enhanced central nervous systems expression of interleukin-6. Brain Behav Immun. 2012;26:959–971. doi: 10.1016/j.bbi.2012.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann H, Medana IM, Bauer J, Lassmann H. Cytotoxic T lymphocytes in autoimmune and degenerative CNS diseases. Trends in neurosciences. 2002;25:313–319. doi: 10.1016/s0166-2236(02)02154-9. [DOI] [PubMed] [Google Scholar]
- Ngugi AK, Bottomley C, Kleinschmidt I, Sander JW, Newton CR. Estimation of the burden of active and life-time epilepsy: a meta-analytic approach. Epilepsia. 2010;51:883–890. doi: 10.1111/j.1528-1167.2009.02481.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noebels JL, Avoli M, Rogawski M, Olsen R, Delgado-Escueta AV. “Jasper’s Basic Mechanisms of the Epilepsies” Workshop. Epilepsia. 2010;51(Suppl 5):1–5. doi: 10.1111/j.1528-1167.2010.02792.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Notenboom RG, Hampson DR, Jansen GH, van Rijen PC, van Veelen CW, van Nieuwenhuizen O, de Graan PN. Up-regulation of hippocampal metabotropic glutamate receptor 5 in temporal lobe epilepsy patients. Brain. 2006;129:96–107. doi: 10.1093/brain/awh673. [DOI] [PubMed] [Google Scholar]
- Ohara Y, Stein S, Fu JL, Stillman L, Klaman L, Roos RP. Molecular cloning and sequence determination of DA strain of Theiler’s murine encephalomyelitis viruses. Virology. 1988;164:245–255. doi: 10.1016/0042-6822(88)90642-3. [DOI] [PubMed] [Google Scholar]
- Oleszak EL, Chang JR, Friedman H, Katsetos CD, Platsoucas CD. Theiler’s virus infection: a model for multiple sclerosis. Clin Microbiol Rev. 2004;17:174–207. doi: 10.1128/CMR.17.1.174-207.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oleszak EL, Katsetos CD, Kuzmak J, Varadhachary A. Inducible nitric oxide synthase in Theiler’s murine encephalomyelitis virus infection. J Virol. 1997;71:3228–3235. doi: 10.1128/jvi.71.4.3228-3235.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pachner AR, Li L, Lagunoff D. Plasma cells in the central nervous system in the Theiler’s virus model of multiple sclerosis. J Neuroimmunol. 2011;232:35–40. doi: 10.1016/j.jneuroim.2010.09.026. [DOI] [PubMed] [Google Scholar]
- Palma JP, Yauch RL, Lang S, Kim BS. Potential role of CD4+ T cell-mediated apoptosis of activated astrocytes in Theiler’s virus-induced demyelination. Journal of immunology. 1999;162:6543–6551. [PubMed] [Google Scholar]
- Peltola J, Palmio J, Korhonen L, Suhonen J, Miettinen A, Hurme M, Lindholm D, Keranen T. Interleukin-6 and interleukin-1 receptor antagonist in cerebrospinal fluid from patients with recent tonic-clonic seizures. Epilepsy Res. 2000;41:205–211. doi: 10.1016/s0920-1211(00)00140-6. [DOI] [PubMed] [Google Scholar]
- Perez-Cerda F, Sanchez-Gomes MV, Matute C. The link of inflammation and neurodegeneration in progressive multiple sclerosis. Multiple Sclerosis and Demyelinating Disorders. 2016;1:9. [Google Scholar]
- Piccio L, Rossi B, Scarpini E, Laudanna C, Giagulli C, Issekutz AC, Vestweber D, Butcher EC, Constantin G. Molecular mechanisms involved in lymphocyte recruitment in inflamed brain microvessels: critical roles for P-selectin glycoprotein ligand-1 and heterotrimeric G(i)-linked receptors. Journal of immunology. 2002;168:1940–1949. doi: 10.4049/jimmunol.168.4.1940. [DOI] [PubMed] [Google Scholar]
- Poli A, Kmiecik J, Domingues O, Hentges F, Blery M, Chekenya M, Boucraut J, Zimmer J. NK cells in central nervous system disorders. Journal of immunology. 2013;190:5355–5362. doi: 10.4049/jimmunol.1203401. [DOI] [PubMed] [Google Scholar]
- Prince HK, Conn PJ, Blackstone CD, Huganir RL, Levey AI. Down-regulation of AMPA receptor subunit GluR2 in amygdaloid kindling. J Neurochem. 1995;64:462–465. doi: 10.1046/j.1471-4159.1995.64010462.x. [DOI] [PubMed] [Google Scholar]
- Qiu Z, Sweeney DD, Netzeband JG, Gruol DL. Chronic interleukin-6 alters NMDA receptor-mediated membrane responses and enhances neurotoxicity in developing CNS neurons. J Neurosci. 1998;18:10445–10456. doi: 10.1523/JNEUROSCI.18-24-10445.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ransohoff RM, Brown MA. Innate immunity in the central nervous system. J Clin Invest. 2012;122:1164–1171. doi: 10.1172/JCI58644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards MH, Getts MT, Podojil JR, Jin YH, Kim BS, Miller SD. Virus expanded regulatory T cells control disease severity in the Theiler’s virus mouse model of MS. J Autoimmun. 2011;36:142–154. doi: 10.1016/j.jaut.2010.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rijkers K, Majoie HJ, Hoogland G, Kenis G, De Baets M, Vles JS. The role of interleukin-1 in seizures and epilepsy: a critical review. Exp Neurol. 2009;216:258–271. doi: 10.1016/j.expneurol.2008.12.014. [DOI] [PubMed] [Google Scholar]
- Rodriguez M, Leibowitz JL, Lampert PW. Persistent infection of oligodendrocytes in Theiler’s virus-induced encephalomyelitis. Annals of neurology. 1983;13:426–433. doi: 10.1002/ana.410130409. [DOI] [PubMed] [Google Scholar]
- Rodriguez M, Pavelko K, Coffman RL. Gamma interferon is critical for resistance to Theiler’s virus-induced demyelination. J Virol. 1995;69:7286–7290. doi: 10.1128/jvi.69.11.7286-7290.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samland H, Huitron-Resendiz S, Masliah E, Criado J, Henriksen SJ, Campbell IL. Profound increase in sensitivity to glutamatergic- but not cholinergic agonist-induced seizures in transgenic mice with astrocyte production of IL-6. J Neurosci Res. 2003;73:176–187. doi: 10.1002/jnr.10635. [DOI] [PubMed] [Google Scholar]
- Sanchez RM, Koh S, Rio C, Wang C, Lamperti ED, Sharma D, Corfas G, Jensen FE. Decreased glutamate receptor 2 expression and enhanced epileptogenesis in immature rat hippocampus after perinatal hypoxia-induced seizures. J Neurosci. 2001;21:8154–8163. doi: 10.1523/JNEUROSCI.21-20-08154.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlitt BP, Felrice M, Jelachich ML, Lipton HL. Apoptotic cells, including macrophages, are prominent in Theiler’s virus-induced inflammatory, demyelinating lesions. J Virol. 2003;77:4383–4388. doi: 10.1128/JVI.77.7.4383-4388.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz M, Deczkowska A. Neurological Disease as a Failure of Brain-Immune Crosstalk: The Multiple Faces of Neuroinflammation. Trends Immunol. 2016;37:668–679. doi: 10.1016/j.it.2016.08.001. [DOI] [PubMed] [Google Scholar]
- Sebzda E, Mariathasan S, Ohteki T, Jones R, Bachmann MF, Ohashi PS. Selection of the T cell repertoire. Annu Rev Immunol. 1999;17:829–874. doi: 10.1146/annurev.immunol.17.1.829. [DOI] [PubMed] [Google Scholar]
- Shao ZH, Li CQ, Vanden Hoek TL, Becker LB, Schumacker PT, Wu JA, Attele AS, Yuan CS. Extract from Scutellaria baicalensis Georgi attenuates oxidant stress in cardiomyocytes. J Mol Cell Cardiol. 1999;31:1885–1895. doi: 10.1006/jmcc.1999.1021. [DOI] [PubMed] [Google Scholar]
- Skulina C, Schmidt S, Dornmair K, Babbe H, Roers A, Rajewsky K, Wekerle H, Hohlfeld R, Goebels N. Multiple sclerosis: brain-infiltrating CD8+ T cells persist as clonal expansions in the cerebrospinal fluid and blood. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:2428–2433. doi: 10.1073/pnas.0308689100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soldan SS, Berti R, Salem N, Secchiero P, Flamand L, Calabresi PA, Brennan MB, Maloni HW, McFarland HF, Lin HC, Patnaik M, Jacobson S. Association of human herpes virus 6 (HHV-6) with multiple sclerosis: increased IgM response to HHV-6 early antigen and detection of serum HHV-6 DNA. Nat Med. 1997;3:1394–1397. doi: 10.1038/nm1297-1394. [DOI] [PubMed] [Google Scholar]
- Sprent J. The thymus and T-cell tolerance. Ann N Y Acad Sci. 1993;681:5–15. doi: 10.1111/j.1749-6632.1993.tb22863.x. [DOI] [PubMed] [Google Scholar]
- Sprent J, Kishimoto H. The thymus and central tolerance. Transplantation. 2001;72:S25–28. [PubMed] [Google Scholar]
- Sprent J, Webb SR. Function and specificity of T cell subsets in the mouse. Adv Immunol. 1987;41:39–133. doi: 10.1016/s0065-2776(08)60030-9. [DOI] [PubMed] [Google Scholar]
- Stewart KA, Wilcox KS, Fujinami RS, White HS. Development of postinfection epilepsy after Theiler’s virus infection of C57BL/6 mice. Journal of neuropathology and experimental neurology. 2010a;69:1210–1219. doi: 10.1097/NEN.0b013e3181ffc420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart KA, Wilcox KS, Fujinami RS, White HS. Theiler’s virus infection chronically alters seizure susceptibility. Epilepsia. 2010b;51:1418–1428. doi: 10.1111/j.1528-1167.2009.02405.x. [DOI] [PubMed] [Google Scholar]
- Sun D, Whitaker JN, Huang Z, Liu D, Coleclough C, Wekerle H, Raine CS. Myelin antigen-specific CD8+ T cells are encephalitogenic and produce severe disease in C57BL/6 mice. Journal of immunology. 2001;166:7579–7587. doi: 10.4049/jimmunol.166.12.7579. [DOI] [PubMed] [Google Scholar]
- Tejada-Simon MV, Zang YC, Hong J, Rivera VM, Zhang JZ. Cross-reactivity with myelin basic protein and human herpesvirus-6 in multiple sclerosis. Annals of neurology. 2003;53:189–197. doi: 10.1002/ana.10425. [DOI] [PubMed] [Google Scholar]
- Theil DJ, Tsunoda I, Libbey JE, Derfuss TJ, Fujinami RS. Alterations in cytokine but not chemokine mRNA expression during three distinct Theiler’s virus infections. J Neuroimmunol. 2000;104:22–30. doi: 10.1016/s0165-5728(99)00251-9. [DOI] [PubMed] [Google Scholar]
- Theiler M. Spontaneous Encephalomyelitis of Mice, a New Virus Disease. The Journal of experimental medicine. 1937;65:705–719. doi: 10.1084/jem.65.5.705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thurman DJ, Kobau R, Luo YH, Helmers SL, Zack MM. Health-care access among adults with epilepsy: The U.S. National Health Interview Survey, 2010 and 2013. Epilepsy Behav. 2016;55:184–188. doi: 10.1016/j.yebeh.2015.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tikka TM, Koistinaho JE. Minocycline provides neuroprotection against N-methyl-D-aspartate neurotoxicity by inhibiting microglia. Journal of immunology. 2001;166:7527–7533. doi: 10.4049/jimmunol.166.12.7527. [DOI] [PubMed] [Google Scholar]
- Tilleux S, Hermans E. Down-regulation of astrocytic GLAST by microglia-related inflammation is abrogated in dibutyryl cAMP-differentiated cultures. J Neurochem. 2008;105:2224–2236. doi: 10.1111/j.1471-4159.2008.05305.x. [DOI] [PubMed] [Google Scholar]
- Tsuchida T, Parker KC, Turner RV, McFarland HF, Coligan JE, Biddison WE. Autoreactive CD8+ T-cell responses to human myelin protein-derived peptides. Proceedings of the National Academy of Sciences of the United States of America. 1994;91:10859–10863. doi: 10.1073/pnas.91.23.10859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsunoda I, Fujinami RS. Two models for multiple sclerosis: experimental allergic encephalomyelitis and Theiler’s murine encephalomyelitis virus. J Neuropathol Exp Neurol. 1996;55:673–686. doi: 10.1097/00005072-199606000-00001. [DOI] [PubMed] [Google Scholar]
- Tsunoda I, Fujinami RS. Inside-Out versus Outside-In models for virus induced demyelination: axonal damage triggering demyelination. Springer Semin Immunopathol. 2002;24:105–125. doi: 10.1007/s00281-002-0105-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsunoda I, Fujinami RS. Neuropathogenesis of Theiler’s murine encephalomyelitis virus infection, an animal model for multiple sclerosis. J Neuroimmune Pharmacol. 2010;5:355–369. doi: 10.1007/s11481-009-9179-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsunoda I, Kurtz CI, Fujinami RS. Apoptosis in acute and chronic central nervous system disease induced by Theiler’s murine encephalomyelitis virus. Virology. 1997;228:388–393. doi: 10.1006/viro.1996.8382. [DOI] [PubMed] [Google Scholar]
- Ure J, Baudry M, Perassolo M. Metabotropic glutamate receptors and epilepsy. J Neurol Sci. 2006;247:1–9. doi: 10.1016/j.jns.2006.03.018. [DOI] [PubMed] [Google Scholar]
- van Oosten BW, Lai M, Hodgkinson S, Barkhof F, Miller DH, Moseley IF, Thompson AJ, Rudge P, McDougall A, McLeod JG, Ader HJ, Polman CH. Treatment of multiple sclerosis with the monoclonal anti-CD4 antibody cM-T412: results of a randomized, double-blind, placebo-controlled, MR-monitored phase II trial. Neurology. 1997;49:351–357. doi: 10.1212/wnl.49.2.351. [DOI] [PubMed] [Google Scholar]
- Vereyken EJ, Bajova H, Chow S, de Graan PN, Gruol DL. Chronic interleukin-6 alters the level of synaptic proteins in hippocampus in culture and in vivo. Eur J Neurosci. 2007;25:3605–3616. doi: 10.1111/j.1460-9568.2007.05615.x. [DOI] [PubMed] [Google Scholar]
- Vezzani A, Fujinami RS, White HS, Preux PM, Blumcke I, Sander JW, Loscher W. Infections, inflammation and epilepsy. Acta Neuropathol. 2016;131:211–234. doi: 10.1007/s00401-015-1481-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vezzani A, Viviani B. Neuromodulatory properties of inflammatory cytokines and their impact on neuronal excitability. Neuropharmacology. 2015;96:70–82. doi: 10.1016/j.neuropharm.2014.10.027. [DOI] [PubMed] [Google Scholar]
- Virtanen JO, Jacobson S. Viruses and multiple sclerosis. CNS & neurological disorders drug targets. 2012;11:528–544. doi: 10.2174/187152712801661220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Virtanen JO, Pietilainen-Nicklen J, Uotila L, Farkkila M, Vaheri A, Koskiniemi M. Intrathecal human herpesvirus 6 antibodies in multiple sclerosis and other demyelinating diseases presenting as oligoclonal bands in cerebrospinal fluid. J Neuroimmunol. 2011;237:93–97. doi: 10.1016/j.jneuroim.2011.06.012. [DOI] [PubMed] [Google Scholar]
- Wandinger K, Jabs W, Siekhaus A, Bubel S, Trillenberg P, Wagner H, Wessel K, Kirchner H, Hennig H. Association between clinical disease activity and Epstein-Barr virus reactivation in MS. Neurology. 2000;55:178–184. doi: 10.1212/wnl.55.2.178. [DOI] [PubMed] [Google Scholar]
- Weber MS, Prod’homme T, Patarroyo JC, Molnarfi N, Karnezis T, Lehmann-Horn K, Danilenko DM, Eastham-Anderson J, Slavin AJ, Linington C, Bernard CC, Martin F, Zamvil SS. B-cell activation influences T-cell polarization and outcome of anti-CD20 B-cell depletion in central nervous system autoimmunity. Annals of neurology. 2010;68:369–383. doi: 10.1002/ana.22081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winkler-Pickett R, Young HA, Cherry JM, Diehl J, Wine J, Back T, Bere WE, Mason AT, Ortaldo JR. In vivo regulation of experimental autoimmune encephalomyelitis by NK cells: alteration of primary adaptive responses. Journal of immunology. 2008;180:4495–4506. doi: 10.4049/jimmunol.180.7.4495. [DOI] [PubMed] [Google Scholar]
- Wu HM, Huang CC, Chen SH, Liang YC, Tsai JJ, Hsieh CL, Hsu KS. Herpes simplex virus type 1 inoculation enhances hippocampal excitability and seizure susceptibility in mice. Eur J Neurosci. 2003;18:3294–3304. doi: 10.1111/j.1460-9568.2003.03075.x. [DOI] [PubMed] [Google Scholar]
- Yamada M, Zurbriggen A, Fujinami RS. Monoclonal antibody to Theiler’s murine encephalomyelitis virus defines a determinant on myelin and oligodendrocytes, and augments demyelination in experimental allergic encephalomyelitis. The Journal of experimental medicine. 1990;171:1893–1907. doi: 10.1084/jem.171.6.1893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan J, Melemedjian OK, Price TJ, Dussor G. Sensitization of dural afferents underlies migraine-related behavior following meningeal application of interleukin-6 (IL-6) Mol Pain. 2012;8:6. doi: 10.1186/1744-8069-8-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang SH, Gustafson J, Gangidine M, Stepien D, Schuster R, Pritts TA, Goodman MD, Remick DG, Lentsch AB. A murine model of mild traumatic brain injury exhibiting cognitive and motor deficits. J Surg Res. 2013;184:981–988. doi: 10.1016/j.jss.2013.03.075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young VA, Rall GF. Making it to the synapse: measles virus spread in and among neurons. Current topics in microbiology and immunology. 2009;330:3–30. doi: 10.1007/978-3-540-70617-5_1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yrjanheikki J, Keinanen R, Pellikka M, Hokfelt T, Koistinaho J. Tetracyclines inhibit microglial activation and are neuroprotective in global brain ischemia. Proceedings of the National Academy of Sciences of the United States of America. 1998;95:15769–15774. doi: 10.1073/pnas.95.26.15769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zang YC, Li S, Rivera VM, Hong J, Robinson RR, Breitbach WT, Killian J, Zhang JZ. Increased CD8+ cytotoxic T cell responses to myelin basic protein in multiple sclerosis. J Immunol. 2004;172:5120–5127. doi: 10.4049/jimmunol.172.8.5120. [DOI] [PubMed] [Google Scholar]