

Abstract
A series of shikonin analogs have been synthesized in a one-pot reaction of quinizarin with β, γ-unsaturated aldehydes in MeOH under mild conditions and investigated for their cytotoxicity against four cancer cell lines and one normal cell line. The synthesized compounds were found to be cytotoxic against HeLa cells with no apparent toxicity against normal cell line. Further modification led to the discovery of a novel tetracyclic anthraquinone (4b/4b′) with potent cytotoxic activities against cervical, breast and pancreatic cancer cell lines with no significant effect on the growth of the control mammary epithelial cell line MCF-10. The good cytotoxicity and selectivity of compound 4b/4b′ suggest that it could be a promising lead for further optimization.
Keywords: Shikonin analogs, Anthraquinone, Synthesis, Antitumor activity
Natural products have long served as a powerful inspiration in human’s quest for new anticancer drugs and continue to be a rich source for novel lead compounds.1 Shikonin is a potent antitumor agent isolated from the roots of Lithospermum erythrorhizon.2 It was shown to display potent cytotoxicity against a wide range of tumor cell lines.3–6 Many of the structural modifications of shikonin have occurred at the hydroxyl groups of the naphthazarin ring and the side chain. Accordingly, much research on the synthesis and antitumor activities of hydroxyl-masked shikonin derivatives has been reported over the past few years (Fig. 1).7–14 However, these derivatives, while displaying varied antitumor activities, have failed to reach their full potential because none has been developed as a clinical candidate so far. This suggests that modifications to the hydroxyl groups may not be a viable strategy to overcome drawbacks in the structure of shikonin itself. Moreover, limited options that exist in shikonin modifications will inevitably hamper the establishment of a robust structure–activity relationship (SAR).
Figure 1.
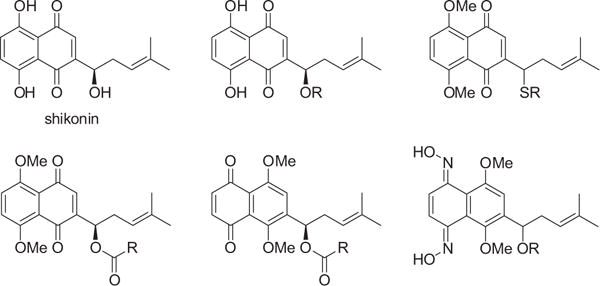
Structures of shikonin and its derivatives from modifications of the hydroxyl groups.
The shikonin molecule consists of two pharmacophores. One is the naphthazarin ring with hydroxyl groups that can produce the reactive oxygen species via the redox cycling and bio-reductive and Michael addition alkylation processes.15–17 The other is the hydroxyl side chain which has two identical methyl substituents at the terminal olefin. We assumed that the free hydroxyl groups at the naphthazarin skeleton of shikonin was important for retaining its drug-like property. This is apparent from the fact that almost all known anthracycline antitumor antibiotics and mitoxantrone contain a quinizarin skeleton with free hydroxyl groups, which may play a key role both in the DNA binding and cell or tissue bioavailability (Fig. 2).18–20 Therefore, the presence of polar hydroxyl substituents on the shikonin skeleton may be essential to maintaining its high therapeutic index.
Figure 2.
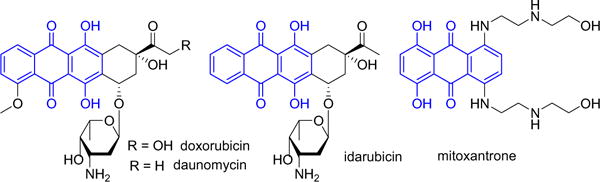
Structures of representative anthracyclines.
Since extensive SAR has been conducted around the hydroxyl groups of shikonin, it was decided that a more radical alteration of the structure might be beneficial. Literature survey indicates that the cinnamyl moiety represents an important pharmacophore in numerous naturally occurring or synthetically derived antitumor agents (Fig. 3).21–24 Hence, we performed the transformation of the terminal methyl substituents of the olefin of shikonin to a phenyl ring and characterized the biological activities resulting from a change of the main skeleton. Based on the biological significance of quinizarin and cinnamyl moieties an ongoing research program in our laboratory has focused on developing bioactive anthraquinones.25–29 We envisioned that the combination of these moieties into one molecule to mimic the structure of shikonin might create a new class of natural product-like molecules with improved therapeutic index compared to shikonin. The resulting molecules may confer desirable biological activities. To this end, development of efficient synthetic approaches to these molecules would be of great value. Herein, we wish to report our recent findings on the assembly of such shikonin analogs in a simple manner and their associated antitumor activities.
Figure 3.
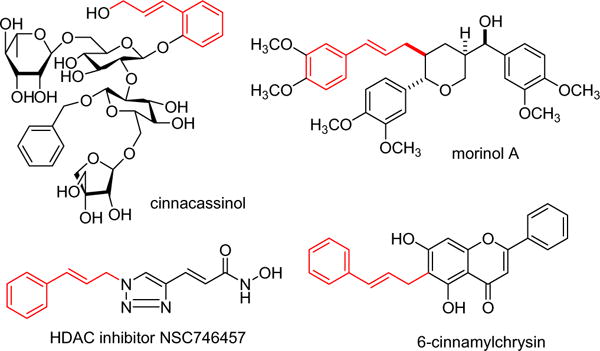
Representative molecules containing cinnamyl moieties.
Because of the hydroxy functional groups in the quinone ring system, the preparation of shikonin or anthraquinone derivatives often involve a multistep reaction sequence (mainly protection and deprotection procedures)7–14,30–32, imposing extra efforts and cost for the synthesis of target compounds. Given the value of shikonin derivatives as potential pharmaceuticals, simple and efficient methods have been highly sought-after tools to access shikonin analogs. In 2013, we reported a simple method to derivatize 1, 4-dihydroxyanthraquinone that avoids tedious protection–deprotection sequences (Scheme 1).26
Scheme 1.
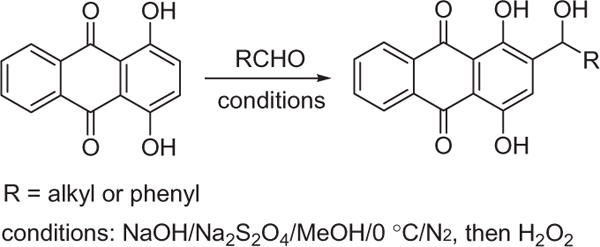
The preparation of hydroxyalkylanthraquinones using aldehyde as the reactant. Conditions: NaOH/Na2S2O4/MeOH/0 °C/N2, then H2O2.
Having optimized reaction conditions for the hydroxyalkylation reaction between quinizarin and aldehydes shown in Scheme 1, we considered whether these reaction conditions could be employed for the addition of a quinizarin to β, γ-unsaturated aldehyde. The presence of a double bond in β, γ-unsaturated aldehyde was first envisioned as a potential difficulty because we recently found that under the similar reaction conditions α, β-unsaturated aldehydes could cyclize with quinizarin to afford the cyclopentanoids.28 However, the reaction of quinizarin 1 and β, γ-unsaturated aldehyde 2a was found to proceed smoothly, furnishing hydroxyalkyl product 3a in good yield (81%) (Scheme 2). By carefully optimizing the conditions, the reaction could afford the desired 3a in 80% yield with the molar ratio of the reagent aldehyde/NaOH/Na2S2O4 being 2.0/5.0/2.0.
Scheme 2.
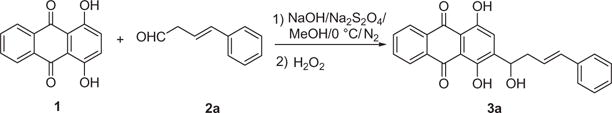
Synthesis of 3a.
Subsequently, the reactions of various β, γ-unsaturated aldehydes 2b–p with quinizarin 1 were explored under the improved reaction conditions, and the results are shown in Table 1. It was found that β, γ-unsaturated aldehydes bearing electron-donating groups (Me and MeO, entries 1–6) or electron-withdrawing groups (F, Cl, and Br, entries 7–15) on the phenyl ring were compatible with the reaction conditions. Meanwhile, the different positions of the substituents have an effect on the reaction yields. Generally, the aldehydes with a substituent at the ortho position of the phenyl ring gave relatively lower yields (entries 1, 4, 7, 10, and 13). Higher yields were observed for substrates bearing a meta substituent on the phenyl ring, as observed for the reactions of 2c, 2f, 2i, 2l, and 2o (entries 2, 5, 8, 11, and 14). In all cases, the reactions proceeded efficiently under mild conditions to afford the corresponding products 3b–p in moderate to excellent yields. All of the new compounds were fully analyzed by 1H and 13C NMR, and high-resolution mass spectrometry (HRMS). Thus, this simple, one-step protocol could be extended as a practical and efficient method to prepare various potentially bioactive shikonin analogs or anthraquinone derivatives. To the best of our knowledge, no description of such a procedure has yet been reported for the synthesis of the shikonin analogs.
Table 1.
Structures and yields of compounds 3b–p
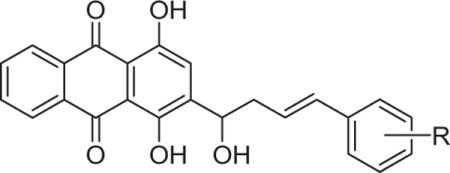
| ||||
|---|---|---|---|---|
|
| ||||
| Entry | Compound | β, γ-Unsaturated aldehyde | R | Yielda (%) |
| 1 | 3b | 2b | 2-CH3 | 51 |
| 2 | 3c | 2c | 3-CH3 | 90 |
| 3 | 3d | 2d | 4-CH3 | 70 |
| 4 | 3e | 2e | 2-CH3O | 49 |
| 5 | 3f | 2f | 3-CH3O | 68 |
| 6 | 3g | 2g | 4-CH3O | 60 |
| 7 | 3h | 2h | 2-F | 49 |
| 8 | 3i | 2i | 3-F | 79 |
| 9 | 3j | 2j | 4-F | 67 |
| 10 | 3k | 2k | 2-Cl | 60 |
| 11 | 3l | 2l | 3-Cl | 90 |
| 12 | 3m | 2m | 4-Cl | 85 |
| 13 | 3n | 2n | 2-Br | 65 |
| 14 | 3o | 2o | 3-Br | 85 |
| 15 | 3p | 2p | 4-Br | 74 |
Isolated yield.
Compounds 3a–p were subjected to antiproliferative assays against four human cancer cell lines: HeLa (cervical cancer), MDA-MB-231 (triple negative breast cancer), MiaPaca-2 (pancreatic adenocarcinoma), MCF-7 (ER+ human breast cancer) and one normal mammary epithelial cell line MCF-10. Shikonin was used as a positive control to evaluate the antitumor activity of the designed compounds. The IC50 (the concentration at which a compound inhibits 50% cell growth) values of the test compounds on each cell line were determined by plotting the inhibition of cells at different concentrations of 33.3, 11.1, 3.7, 1.23, 0.41, 0.137, 0.0457, and 0.0152 μM, as presented in Table 2. Results indicate that HeLa cells appear to be the most susceptible cell line. Apart from some exceptions, inhibition of the other three cancer cell lines required higher concentrations of the tested compounds. While less potent than shikonin overall, the various substitutions did yield subtle structural effects on potency. For example, if one examines the IC50 values of all tested compounds against HeLa cell line, compound 3a, with no substituent at the phenyl ring, was more potent than the rest of the compounds (3b–p), suggesting that the presence of a substituent group at the phenyl ring of the side chain on the synthesized compound is unfavorable to increasing the cytotoxicity.
Table 2.
In vitro cytotoxicity of compounds 3a–p
| Compound | IC50a (μM)
|
||||
|---|---|---|---|---|---|
| HeLa | MDA-MB-231 | MiaPaca-2 | MCF-7 | MCF-10 | |
| 3a | 18.64 | IAb | IA | IA | IA |
| 3b | 28.31 | IA | 26.12 | 32.81 | IA |
| 3c | 27.19 | IA | 28.47 | 32.81 | IA |
| 3d | 29.00 | IA | IA | IA | IA |
| 3e | 29.00 | IA | IA | IA | IA |
| 3f | 28.01 | IA | 27.08 | IA | IA |
| 3g | 24.12 | 26.90 | 26.29 | 32.81 | IA |
| 3h | 28.65 | 29.16 | 27.32 | IA | IA |
| 3i | 25.15 | 26.50 | 26.33 | 32.13 | IA |
| 3j | 32.87 | 28.00 | 23.19 | 28.48 | IA |
| 3k | 27.20 | IA | IA | 31.49 | IA |
| 3l | 25.79 | 31.21 | 26.38 | 32.19 | IA |
| 3m | 27.36 | 28.54 | 27.44 | 32.29 | IA |
| 3n | 25.60 | 30.71 | 27.10 | 31.10 | IA |
| 3o | 29.12 | IA | 27.55 | 31.73 | IA |
| 3p | IA | IA | IA | IA | IA |
| Shikonin | 6.42 | 3.94 | 0.04 | 5.49 | 4.56 |
IC50 values were calculated from three independent experiments.
IA = inactivity. Inactivity is defined as having greater than 50% cell at the highest concentration (33.3 μM).
Another encouraging observation was that the synthetic shikonin analogs exhibited no apparent cytotoxicity in MCF-10, a normal human mammary epithelial cell line. This observation is significant since shikonin does not show such selectivity and the development of anticancer drugs that selectively target abnormal tumor cell with minimal impact on normal cells is a fundamental objective of cancer research. The selectivity of our shikonin analogs suggests that the two terminal methyl groups of the side chain at shikonin scaffold could be linked to the indiscriminating high toxicity and the removal of the methyl groups at the side chain might improve the safety profile of shikonin.
This finding prompted us to further investigate the cytotoxicity of other shikonin analogs without the methyl groups at the terminal olefin. The strategy was to modify the side chain in compound 3 to form a five-membered carbocyclic ring. Our rationale in designing such compounds was that most of the anthracycline antitumor antibiotics contain a tetracyclic carbon skeleton which comprises one quinizarin moiety and one carbocyclic ring. Despite the fact that >2000 analogs of doxorubicin have been synthesized and tested, so far no clinical candidates or drugs have emerged with substantially enhanced efficacy.33 Most of the synthetic tetracyclic anthraquinones contain a fused six-membered ring, but analogs made by changing the six-membered to a five-membered one are particularly rare. Thus, preparation of tetracyclic anthraquinones containing a fused five-membered ring were attempted, followed by an evaluation of cell toxicities (Table 3). Compounds 4 were prepared according to the procedure described previously by our group.28 Reaction of 1,4-dihydroxyanthraquinone with cinnamaldehyde under previous conditions smoothly provided the desired product 4a. However, when R1 ≠ H (e.g., R1 = bromo), there were two possible positions for the cyclization to occur, thus a mixture of regioisomers (4b/4b′ or 4c/4c′) was obtained when the cyclization reaction was treated with 6-bromo-1,4-dihydroxyanthraquinone and cinnamaldehyde or 3-m-tolylacrylaldehyde. Because we were unable to successfully purify 4b and 4b′ or 4c and 4c′ from the regioisomeric mixture, the activity of 4b/4b′ and 4c/4c′ was tested using the regioisomeric mixture. To our delight, a significant improvement in the activity against tested cancer cell lines was observed of the tetracyclic anthraquinones that contain a fused five-membered ring. For example, compounds 4b/4b′ caused significant cytotoxicity at lower concentration (IC50 = 6.22 μM) which were comparable with the shikonin-induced cytotoxicity (IC50 = 6.42 μM) in HeLa cells. The improvement of potency against various cancer cell lines of compounds 4b/4b′ relative to compounds 3 has been ascribed mainly to the presence of a bromo substituent in the anthraquinone ring since compound 4a did not show significant improvement in cytotoxic activity despite its structural similarity with 4b/4b′. It is also interesting to note that 4b/4b′ showed low cytotoxicity to control mammary epithelial cells (IC50 = 28.14 μM), suggesting it might serve as a lead compound with potentially little side effects for the treatment of cancer. In addition, compounds 4c/4c′ showed weak activity compared with 4b/4b′, indicating that the presence of a substituent on the phenyl ring was unfavorable for the increase in cytotoxicity and the result was consistent with the SAR of compound 3 against HeLa cell line (Table 1, 3a vs. 3b–p).
Table 3.
In vitro cytotoxicity of compounds 4a–c
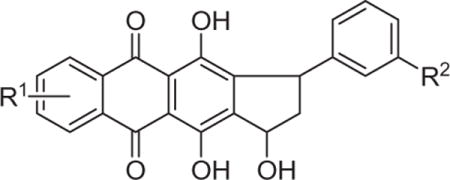
| |||||||
|---|---|---|---|---|---|---|---|
|
| |||||||
| Compound | R1 | R2 | IC50a (μM)
|
||||
| HeLa | MDA-MB-231 | MiaPaca-2 | MCF-7 | MCF-10 | |||
| 4a | H | H | 29.19 | 28.77 | 27.65 | IA | IA |
| 4b/4b′ | 7-Br/8-Br | H | 6.22 | 6.25 | 6.55 | 6.57 | 28.14 |
| 4c/4c′ | 7-Br/8-Br | CH3 | 25.95 | 26.28 | IAb | 28.74 | IA |
| Shikonin | 6.42 | 3.94 | 0.04 | 5.49 | 4.56 | ||
| Doxorubicin | 0.03 | 0.16 | 0.02 | 1.17 | 0.16 | ||
IC50 values were calculated from three independent experiments.
IA = inactivity. Inactivity is defined as having greater than 50% cell at the highest concentration (33.3 μM).
Previous pharmaceutical studies on shikonin derivatives mainly focused on the modification of the hydroxyl substituents. In the present study, we report for the first time the modification to the terminal methyl groups at the side chain of shikonin and the resulting cytotoxicity towards neoplastic as well as normal cells. The chemical optimization efforts afforded a class of novel shikonin analogs, along with a much simplified synthetic scheme (the shikonin derivatives are normally difficult to prepare in a single step). These compounds were evaluated in vitro for cytotoxicity against four human cancer cell lines and one normal cell line and showed some selectivity towards cancer cell lines over the normal cell line. The selectivity might be due to the removal of methyl groups on the side chain of shikonin. Further modifications were done to produce several tetracyclic anthraquinones. Among them, compounds 4b/4b′ with potent activity to all tested cell lines but weak toxicity to normal cell line may serve as the lead compound for relevant cancer therapy. The discoveries reported herein provide a platform for the development of more potent and selective antiproliferative agents based on the shikonin skeleton.
Supplementary Material
Acknowledgments
We are thankful for the financial support from Major Program of Natural Science Foundation (12KJA150005) and PAPD of Jiangsu Higher Education Institutions (L.-M. Zhao), NIGMS grant number 1U54GM104940 (G. Wang), the Natural Science Foundation of Jiangsu Province (BK2012576) (L.-M. Zhao), and NIMHD grant number 2G12MD007595 (G. Wang).
Footnotes
Supplementary data
Supplementary data associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.bmcl.2016.04.006.
References and notes
- 1.Newman DJ, Cragg GM. J Nat Prod. 2012;75:311. doi: 10.1021/np200906s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Papageorgiou VP, Assimopoulou AN, Couladouros EA, Hepworth D, Nicolaou KC. Angew Chem Int Ed. 1999;38:271. doi: 10.1002/(SICI)1521-3773(19990201)38:3<270::AID-ANIE270>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- 3.Wang R, Yin R, Zhou W, Xu D, Li S. Expert Opin Ther Pat. 2012;22:977. doi: 10.1517/13543776.2012.709237. [DOI] [PubMed] [Google Scholar]
- 4.Lu J-J, Bao J-L, Wu G-S, Xu W-S, Huang M-Q, Chen X-P, Wang Y-T. Anticancer Agents Med Chem. 2013;13:456. [PubMed] [Google Scholar]
- 5.Andujar I, Recio MC, Giner RM, Rios JL. Cur Med Chem. 2013;20:2892. doi: 10.2174/09298673113209990008. [DOI] [PubMed] [Google Scholar]
- 6.Chen C, Shanmugasundaram K, Rigby AC, Kung AL. Eur J Pharm Sci. 2013;49:18. doi: 10.1016/j.ejps.2013.02.003. [DOI] [PubMed] [Google Scholar]
- 7.Ahn B-Z, Baik K-U, Kweon G-R, Lim K, Hwang B-D. J Med Chem. 1995;38:1044. doi: 10.1021/jm00006a025. [DOI] [PubMed] [Google Scholar]
- 8.Zhao L-M, Xie T-P, He Y-Q, Xu D-F, Li S-S. Eur J Med Chem. 2009;44:1410. doi: 10.1016/j.ejmech.2008.09.039. [DOI] [PubMed] [Google Scholar]
- 9.Su Y, Xie J, Wang Y, Hu X, Lin X. Eur J Med Chem. 2010;45:2713. doi: 10.1016/j.ejmech.2010.02.002. [DOI] [PubMed] [Google Scholar]
- 10.Zhou W, Peng Y, Li S-S. Eur J Med Chem. 2010;45:6005. doi: 10.1016/j.ejmech.2010.09.068. [DOI] [PubMed] [Google Scholar]
- 11.Zhou W, Zhang X, Xiao L, Ding J, Liu Q-H, Li S-S. Eur J Med Chem. 2011;46:3420. doi: 10.1016/j.ejmech.2011.05.006. [DOI] [PubMed] [Google Scholar]
- 12.Rao Z, Liu X, Zhou W, Yi J, Li S-S. Eur J Med Chem. 2011;46:3934. doi: 10.1016/j.ejmech.2011.05.065. [DOI] [PubMed] [Google Scholar]
- 13.He H, Bai L-P, Jiang Z-H. Bioorg Med Chem Lett. 2012;22:1582. doi: 10.1016/j.bmcl.2011.12.143. [DOI] [PubMed] [Google Scholar]
- 14.Wang R, Zhang X, Song H, Zhou S, Li S. Bioorg Med Chem Lett. 2014;24:4304. doi: 10.1016/j.bmcl.2014.07.012. [DOI] [PubMed] [Google Scholar]
- 15.Duan D, Zhang B, Yao J, Liu Y, Fang J. Free Radical Biol Med. 2014;182 doi: 10.1016/j.freeradbiomed.2014.02.016. [DOI] [PubMed] [Google Scholar]
- 16.Yang J-T, Li Z-L, Wu J-Y, Lu F-J, Chen C-H. PLoS One. 2014;9:e94180. doi: 10.1371/journal.pone.0094180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mao X, Yu CR, Li WH, Li WX. Cell Res. 2008;18:879. doi: 10.1038/cr.2008.86. [DOI] [PubMed] [Google Scholar]
- 18.Traganos F, Evenson DP, Staiano-Coico L, Darzynkiewicz Z, Melamed MR. Cancer Res. 1980;40:671. [PubMed] [Google Scholar]
- 19.Patterson LH. Cancer Metastasis Rev. 1993;12:119. doi: 10.1007/BF00689805. [DOI] [PubMed] [Google Scholar]
- 20.Gholivand MB, Kashanian S, Peyman H, Roshanfekr H. Eur J Med Chem. 2011;46:2630. doi: 10.1016/j.ejmech.2011.03.034. [DOI] [PubMed] [Google Scholar]
- 21.Usia T, Banskota AH, Tezuka Y, Midorikawa K, Matsushige K, Kadota S. J Nat Prod. 2002;65:673. doi: 10.1021/np010486c. [DOI] [PubMed] [Google Scholar]
- 22.Ngoc TM, Lee IS, Ha DT, Kim HJ, Min BS, Bae KH. J Nat Prod. 2009;72:1205. doi: 10.1021/np900031q. [DOI] [PubMed] [Google Scholar]
- 23.Hou J, Feng C, Li Z, Fang Q, Wang H, Gu G, Shi Y, Liu P, Xu F, Yin Z. Eur J Med Chem. 2011;46:3190. doi: 10.1016/j.ejmech.2011.04.027. [DOI] [PubMed] [Google Scholar]
- 24.Yamauchi S, Kawahara S, Wukirsari T, Nishiwaki H, Nishi K, Sugahara T, Akiyama K, Kishida T. Bioorg Med Chem Lett. 2013;23:4923. doi: 10.1016/j.bmcl.2013.06.067. [DOI] [PubMed] [Google Scholar]
- 25.Zhao L-M, Zhang L-M, Liu J-J, Wan L-J, Cheng Y-Q, Zhang S-Q, Yan Z-W, Jiang J-H. Eur J Med Chem. 2012;47:255. doi: 10.1016/j.ejmech.2011.10.050. [DOI] [PubMed] [Google Scholar]
- 26.Zhao LM, Ma FY, Jin HS, Ma J, Wang H, Fu CZ. Eur J Org Chem. 2013;7193 [Google Scholar]
- 27.Zhao L-M, Zhang L-M, Ma F-Y, Wang X-S, Jin H-S. Tetrahedron Lett. 2013;54:2802. [Google Scholar]
- 28.Cao F-X, Zhao L-M. Tetrahedron Lett. 2015;56:2724. [Google Scholar]
- 29.Zhao L-M, Ma F-Y, Jin H-S, Zheng S, Zhong Q, Wang G. Eur J Med Chem. 2015;102:303. doi: 10.1016/j.ejmech.2015.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nicolaou KC, Hepworth D. Angew Chem Int Ed. 1998;37:839. doi: 10.1002/(SICI)1521-3773(19980403)37:6<839::AID-ANIE839>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- 31.Jin GZ, Song GY, Zheng XG, Kim Y, Sok DE, Ahn BZ. Arch Pharm Res. 1998;21:198. doi: 10.1007/BF02974028. [DOI] [PubMed] [Google Scholar]
- 32.Tam MN, Nam NH, Jin GZ, Song GY, Ahn BZ. Arch Pharm. 2000;333:189. doi: 10.1002/1521-4184(20006)333:6<189::aid-ardp189>3.0.co;2-t. [DOI] [PubMed] [Google Scholar]
- 33.Weiss RB. Semin Oncol. 1992;19:670. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.