

Abstract
Lafora disease (LD, OMIM 254780) is a rare fatal neurodegenerative disorder that usually occurs during childhood with generalized tonic-clonic seizures, myoclonus, absences, drop attacks or visual seizures. Unfortunately, at present, available treatments are only palliatives and no curative drugs are available yet. The hallmark of the disease is the accumulation of insoluble polyglucosan inclusions, called Lafora bodies (LBs), within the neurons but also in heart, muscle and liver cells. Mouse models lacking functional EPM2A or EPM2B genes (the two major loci related to the disease) recapitulate the Lafora disease phenotype: they accumulate polyglucosan inclusions, show signs of neurodegeneration and have a dysregulation of protein clearance and endoplasmic reticulum stress response. In this study, we have subjected a mouse model of LD (Epm2b−/−) to different pharmacological interventions aimed to alleviate protein clearance and endoplasmic reticulum stress. We have used two chemical chaperones, trehalose and 4-phenylbutyric acid. In addition, we have used metformin, an activator of AMP-activated protein kinase (AMPK), as it has a recognized neuroprotective role in other neurodegenerative diseases. Here, we show that treatment with 4-phenylbutyric acid or metformin decreases the accumulation of Lafora bodies and polyubiquitin protein aggregates in the brain of treated animals. 4-Phenylbutyric acid and metformin also diminish neurodegeneration (measured in terms of neuronal loss and reactive gliosis) and ameliorate neuropsychological tests of Epm2b−/− mice. As these compounds have good safety records and are already approved for clinical uses on different neurological pathologies, we think that the translation of our results to the clinical practice could be straightforward.
Keywords: Lafora disease, polyglucosan accumulation, metformin, 4-phenylbutyric acid, trehalose, pharmacological intervention
INTRODUCTION
Lafora disease (LD, OMIM 254780) is a rare fatal neurodegenerative disorder that usually occurs during childhood with generalized tonic-clonic seizures, myoclonus, absences, drop attacks or visual seizures ([1], [2]). Unfortunately, at present, available treatments are only palliatives and no curative drugs are available yet. The hallmark of the disease is the accumulation of insoluble polyglucosan inclusions, called Lafora bodies (LBs), within the neurons but also in heart, muscle and liver cells. Recessive mutations in two main loci, EPM2A and EPM2B, are known to produce LD ([3], [4], [5]). EPM2A gene encodes the glucan phosphatase laforin and EPM2B gene encodes the E3-ubiquitin ligase malin. Although it is known that both proteins form a functional complex ([6], [7], [8]), the underlying molecular mechanisms of this pathology are not fully understood. In this sense, although laforin and malin have been involved in the regulation of glycogen metabolism, recent reports indicate that they also play a role in alternative physiological pathways such as endoplasmic reticulum stress response ([9], [10], [11]) and protein clearance ([10], [12], [13], [14]).
Protein homeostasis (proteostasis) is achieved by the coordinated action of an efficient folding and transport of newly synthesized proteins and an active quality control with degradative mechanisms to reduce the load of unfolded/misfolded proteins thereby to prevent abnormal protein aggregation [15]. When the folding capacity is saturated, unfolded/misfolded proteins are usually tagged with ubiquitin moieties to target them for proteasome and/or lysosomal degradation. If these protein clearance mechanisms are not efficient then unfolded/misfolded proteins accumulate and tend to form oligomeric structures. Thus, the presence of protein aggregates is an indication of an altered proteostasis [15]. LBs contain, in addition to insoluble glycogen-like polysaccharides (polyglucosans), ubiquitinated proteins, advanced glycation-end products, chaperones, autophagy components and proteasome subunits ([12], [16]). Therefore LD can be considered as another example of proteostasis dysfunction in which as indicated above, protein clearance and endoplasmic reticulum stress are defective. So, we reasoned that the use of compounds that could alleviate proteostasis dysfuction could be beneficial in the treatment of LD. In this sense, in this work we have used 4-phenylbutyric acid (4-PBA), a chemical chaperone that reverses misfolding and aggregation of proteins associated with several human neurodegenerative diseases ([17], [18], [19]) and trehalose, a non-reducing disaccharide that also acts as a chemical chaperone preventing protein denaturation and protecting cell integrity against several stresses ([20], [21]).
AMP-activated protein kinase (AMPK) is a heterotrimeric enzyme consisting in one catalytic AMPKα, one scaffold AMPKβ and one regulatory AMPKγ subunit, that senses energy status in cells and responds to drops in ATP by inhibiting catabolic and activating anabolic pathways, to restore ATP homeostasis ([22] [23]). AMPK also plays a role in promoting autophagy by inhibiting mTOR kinase or by phosphorylating ULK1 [23]. As it has been reported that AMPK activation plays a neuroprotective role in different neurodegenerative diseases ([24], [25], [26], [27]), we checked whether metformin, an activator of AMPK widely used in the treatment of type2 diabetes, could have a beneficial effect on LD.
Our results indicate that among the drugs mentioned above, 4-PBA and metformin have a clear beneficial effect on all the assayed neurophatological symptoms of Epm2b−/− mice.
MATERIAL AND METHODS
Ethic statement, animal care, mice and husbandry
This study was carried out in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the Consejo Superior de Investigaciones Cientificas (CSIC, Spain). All mouse procedures were approved by the animal committee of the Instituto de Biomedicina de Valencia-CSIC [Permit Number: INTRA12 (IBV-4)]. All efforts were made to minimize animal suffering. To minimize the effect of differences in the genetic background of the animals, and since LD is a recessive autosomal disorder, we crossed Epm2b−/− mice (with a mixed background 129sv:C57BL/6) previously described [14] with control C57BL/6JRccHsd mice obtained from Harlan laboratories (Barcelona, Spain) to obtain heterozygous Epm2b+/− mice. These were crossed among them to obtain heterozygous Epm2b+/− (used as control) and homozygous Epm2b−/− littermates, that were used in this study. Mice were maintained in the IBV-CSIC facility on a 12/12 light/dark cycle under constant temperature (23°C) with food and water provided ad libitum.
Pharmacological interventions
Trehalose, metformin and 4-phenylbutyric acid (4-PBA), all these compounds having good safety records and already approved for clinical uses on different neurological pathologies, were purchased from Sigma (Madrid, Spain). Trehalose (2%), metformin (12 mM) and 4-PBA (20 mM) were administered dissolved in the drinking water. Epm2b+/− (control) and Epm2b−/− (KO) male mice (groups of 8–12 animals) at three months of age (we chose this age because at this time the Emp2b−/− mice clearly show the presence of Lafora bodies in the brain and they are at the beginning of their neurological impairment) were treated or not with trehalose, 4-PBA or metformin. Treatments were applied for one month, and mice were subjected to the behavioral tests described below. Then, treatment was continued for an extra month. After this period, mice were subjected again to the same behavioral tests and sacrificed by cervical dislocation. Brain was recovered, cut in two equal pieces, one part conserved at −80°C for processing for western blot analyses, and the other part fixed in 4% paraformaldehyde in phosphate buffer saline (PBS) and dehydrated for posterior histological analyses.
Histological and immunohistofluorescence analyses
Dehydrated tissues were embedded in paraffin and sectioned at 4 μm. For periodic acid-Schiff (PAS) staining, sections were deparaffined, rehydrated and incubated with 1% periodic acid for 7 min, followed by additional 21 min incubation with Schiff reagent (Sigma, Madrid, Spain). Alternatively, for immunohistochemistry analyses (IHC), sections were deparaffined, rehydrated, and warmed at 95°C for 30 min in 10 mM citrate buffer for antigen retrieval. Sections were blocked in blocking buffer (3% BSA — 3% fat-free milk proteins, in PBS) and incubated overnight at 4°C with primary antibody diluted at 1/200 in blocking buffer [anti-NeuN, (Millipore, Madrid, Spain)]. After three washes of 10 min in PBS, sections were incubated for one hour at room temperature with the biotin-conjugated rabbit secondary antibody diluted at 1/250 in blocking buffer, washed three times with PBS for 5 min, incubated with the immunopure ABC kit (Fisher scientific, Madrid, Spain) for 30 min in darkness, washed three times in PBS for 5 min and developed with using metal enhanced dab method (Fisher scientific). Both PAS staining than IHC sections were finally counterstained with haematoxylin (Sigma, Madrid, Spain), dehydrated and mounted in DPX (Merck, Germany). For immunohistofluoresence analyses (IHF), sections were deparaffined, rehydrated, incubated for 20 min in sodium borohydrate (1 mg/ml) to lower background and warmed at 95°C for 30 min in 10 mM citrate buffer for antigen retrieval. Sections were blocked in blocking buffer (3% BSA — 3% fat-free milk proteins, in PBS) and incubated overnight at 4°C with primary antibody diluted at 1/250 in blocking buffer [anti-polyubiquitin, (Enzo Life Sciences, Madrid, Spain) or anti-GFAP (Sigma, Madrid, Spain)]. After three washes of 10 min in PBS, sections were incubated for one hour at room temperature with the appropriate secondary antibody diluted at 1/200 in blocking buffer, washed once with PBS, incubated with DAPI (Sigma, Madrid, Spain), washed twice with PBS and mounted in FluoMount (Polysciences Inc., USA). Images were acquired with a DM6000 Leica Microscope and fluorescence was quantified using Image J software (NIH, USA).
Western blot analyses
Mouse brain homogenates were lysed in RIPA buffer [20 mM Tris-HCl, pH 7.5; 150 mM NaCl; 1 mM EDTA; 1 mM EGTA; 1% NP-40; 1% sodium deoxycholate; 1 mM Na3VO4 and complete protease inhibitor cocktail (Roche, Spain)] for 20 min at 4°C and centrifuged at 10,000 g for 15 min. The supernatants were collected and 25 μg of total protein subjected to SDS-PAGE, transferred into PVDF membrane and revealed with the appropriated antibodies, using Li-Cor protocol and the Odissey software (LI-COR Corporate, USA). The antibodies used in this study were anti-BiP/GRP78, anti-pThr172-AMPKalpha anti-AMPKbeta1/2, anti-pS641-glycogen synthase, anti-glycogen synthase kinase 3b, anti-pSer9-glycogen synthase kinase 3b, from Cell Signaling Technology Inc. (Barcelona, Spain); anti-LC3B and anti-actin from Sigma (Madrid, Spain); anti-p62 from Abcam (Cambridge, UK); anti-glycogen synthase (Epitomics, Burlingame, USA).
Behavioral tests
Mouse neuropsychological behavior was examined using a selection of standard tests. Briefly, the tests were conducted as follow:
Tail Suspension Test (TST): It was performed as indicated in [28]. Mice were suspended at 15 cm height by the tail for 30s and the foot-clasping posture was scored as follows: 0, with both extended feet and 1, with one clasped foot, alternative foot-clasping or with both clasped feet.
Vertical Pole Test: It was performed as indicated in [29]. Mice were placed head upward near the top of a vertical rough-surfaced pole (diameter 10 mm, height 51 cm). The time taken for the mice to turn completely downward (time to turn) and the total time taken to reach the floor (locomotion activity time) were recorded, with a cut-off limit of 120s for the combined movements.
Open Field Test: It was performed as indicated in [30]. Mice were placed in the left-bottom corner of a dark square open field (40cm × 40cm × 40cm) and exploration was tracked for 20 min. Distance, immobility, spent time and number of entries into 3 concentric zones were measured with the Smart software (PanLab, Barcelona, Spain).
Beam-balance Test: It was performed as indicated in [31]. The mouse’s home-cage was placed at one end of an 80 cm long beam of 26, 12 or 5 mm width and the start point was the other wider end of the beam. The tapered beam had ledges (2 cm wide) that provide “safety” when the mouse’s foot slipped off of the beam. The mouse was given a single trial to learn to walk from the start of the beam to its home-cage. Once the mouse reached its home-cage, it was permitted to remain in the cage to reinforce the target location. The mouse was given three additional trials, each separated by the 60s reinforcement. Time to cross the beam was recorded with a cut-off limit of 120s.
Statistical analysis
The data were expressed as mean ± standard error of the mean (SEM). Statistical significance was determined as indicated in each case by Kruskall-Wallis test, followed by Conover-Inman’s multiple comparison test, by Fisher exact test or by Wilcoxon-Mann-Whitney test using R software [32].
RESULTS
The different treatments affect their corresponding molecular targets
In order to find a beneficial treatment for LD, we used different strategies aimed either to alleviate proteostasis, which is impaired in the disease, or to activate AMPK, since as indicated above, activation of this kinase plays a neuroprotective role in different neurodegenerative diseases. Three-month old male heterozygous Epm2b+/− (control) and homozygous Epm2b−/− (KO) mice were subjected to different pharmacological interventions [trehalose and 4-phenylbutiric acid (4-PBA) to alleviate proteostasis dysfunction, and metformin, to activate AMPK], and after the treatment period, mice were subjected to different behavioral tests, sacrificed by cervical dislocation and brain was recovered for biochemical and histological analyses.
We first checked by western blot analyses of total brain extracts the efficiency of the different drugs in affecting their corresponding anticipated molecular targets. Trehalose and 4-PBA may both act as chemical chaperones to relieve proteostasis dysfucntion. In agreement with this assumption, we found that after two months of treatment with any of these compounds, there was an increase in the levels of the chaperone BiP/GRP78, in both control (Epm2b+/−, p=0.006 w/o TTT vs trehalose and p=0.03 w/o TTT vs 4-PBA) and KO malin (Epm2b−/−, p=0.002 w/o TTT vs trehalose and p=0.007 w/o TTT vs 4-PBA) mice (Fig. 1A). No significant differences were observed between the untreated samples of control (Epm2b+/−) and KO (Epm2b−/−) mice at this age, in agreement with previous results [10]. On the other hand, treatment with metformin for two months induced the activation of AMPK in both control (p=0.03) and KO malin mice (p=0.03), as indicated by higher levels of the phosphorylated form of the catalytic subunit of the AMPK complex (pThr172-AMPKalpha) (Fig. 1B). In this case we use the levels of AMPKβ subunits as loading control, since this antibody gives better results than the anti-AMPKα antibody when analyzing endogenous levels of the trimeric AMPK complex.
Figure 1.
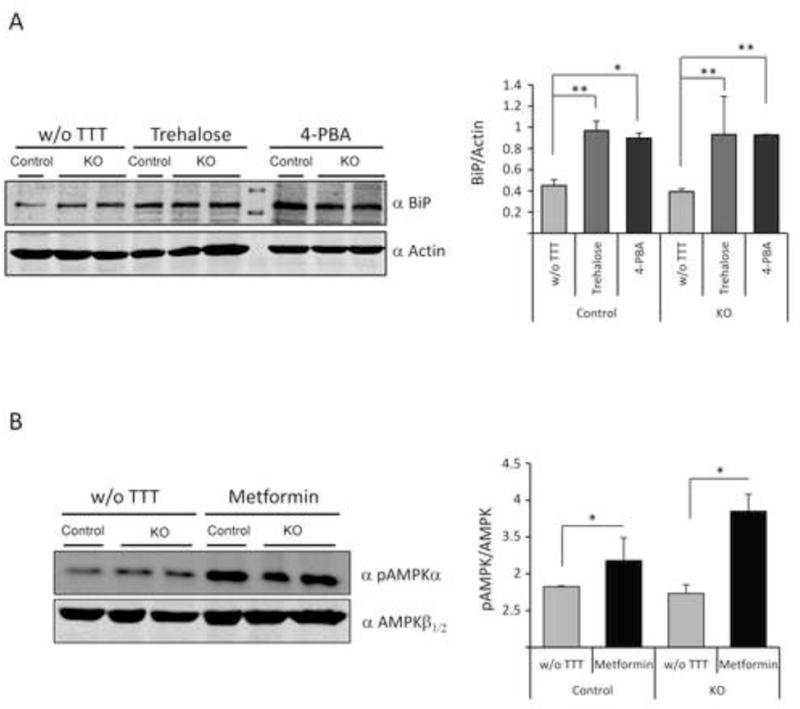
Drugs used in this study affect their corresponding targets in mice brain. Western blot analyses of whole brain extracts using the indicated antibodies and densitometric quantification of the corresponding blots were carried out as described in Material and Methods. A representative blot is presented. (A) anti-BiP/GRP78 (BiP) and anti-actin (loading control) from Epm2b+/− (control) and Epm2b−/− (KO) mice untreated (w/o TTT) or treated with trehalose (2%) or 4-PBA (20 mM) for two months. *p<0.05, **p<0.01 (n: 3 [control] or n:4 [KO]) comparing the indicated groups with the basal condition according to Kruskal-Wallis non-parametric test followed by Conover-Inman post-hoc test. (B) anti-pThr172-AMPKalpha (pAMPKα) and anti-AMPKβ1/2 (loading control) from Epm2b+/− control and Epm2b−/− (KO) mice untreated (w/o TTT) or treated with 12 mM metformin for two months. Bars indicate mean values of the relative intensity of each protein of at least three independent samples ± SEM. Asterisks denote significant differences *p<0.05, (n: 3 [control] or n:4 [KO]) comparing the indicated groups with the basal condition according to the Wilcoxon-Mann-Whitney test.
4-PBA or metformin treatments decrease the accumulation of polyglucosan inclusions and polyubiquitinated protein aggregates
Next, we investigated the effects of all drugs on the formation of polyglucosan inclusions (that stained positive with the periodic acid-Schiff protocol; PAS+), the hallmark of LD, in different brain areas: cerebellum, pontine, hippocampus, olfactory bulb, cortex and midbrain. As observed in Fig. 2, no PAS+ inclusions were detected in the brain areas of control (Epm2b+/−) mice. However, untreated Epm2b−/− mice accumulated polyglucosan inclusions in different areas with different extent, with higher levels in cerebellum, pontine and hippocampus and lower levels in olfactory bulb, cortex and midbrain (Fig. 2B, white bars). The treatment with trehalose did not modify the accumulation of PAS+ granules (Fig. 2A and 2B). However, treatment with 4-PBA or metformin clearly reduced the number of PAS+ aggregates in the cerebellum (p<0.001 Epm2b−/− w/o TTT vs 4-PBA and p<0.001 w/o TTT vs metformin), pontine (p=0.011 Epm2b−/− w/o TTT vs 4-PBA and p<0.04 w/o TTT vs metformin) and hippocampus (p<0.001 Epm2b−/− w/o TTT vs 4-PBA and p<0.001 w/o TTT vs metformin) (Fig. 2A and 2B).
Figure 2.
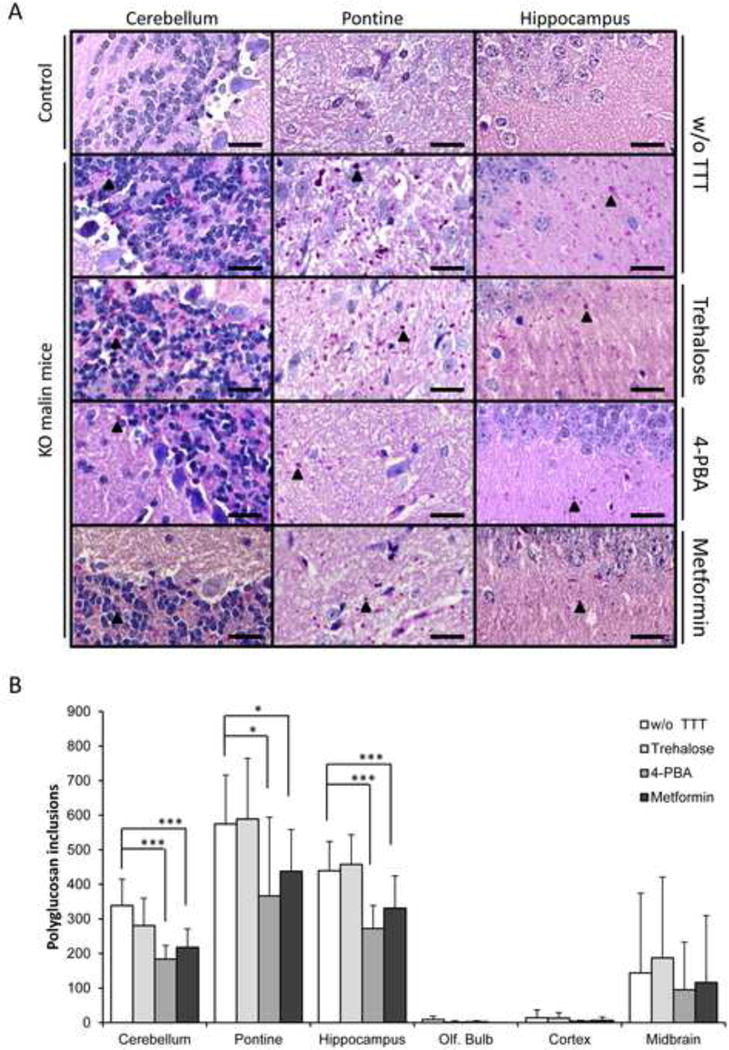
Histological analyses of PAS+ inclusions in brain of Epm2b+/− (control) or Epm2b−/− (KO) mice untreated (w/o TTT) or treated with trehalose, 4-PBA or metformin (two months each). (A) Representative frontal sections of the cerebellum, pontine and hippocampus from Epm2b+/− control or Epm2b−/− (KO) mice untreated (w/o TTT) or treated as indicated in the text, were stained with periodic acid-Schiff (PAS) and counterstained with haematoxylin. Black arrow heads point to Lafora bodies. Scale bars: 25 μm. (B) PAS+ inclusions were counted by eye in three independent samples of cerebellum, pontine, hippocampus, olfactive bulb, cerebral cortex and midbrain of seven mice of each group, and mean quantification of the number of polyglucosan inclusions in an area of 0.09 mm2 was represented. Bars indicate mean of values corresponding to seven mice ± SEM. Asterisks denote significant differences *p< 0.05, **p< 0.01, ***p<0.001 comparing the indicated groups with the basal condition (Epm2b−/− w/o TTT) according to Kruskal-Wallis non-parametric test followed by Conover-Inman post-hoc test.
It is known that polyglucosan inclusions that accumulate in LD are also enriched in polyubiquitinated proteins ([1], [2]). We confirmed this fact by immunohistofluorescence. As it is shown in Fig. 3, untreated Epm2b−/− mice presented high levels of polyubiquitinated protein aggregates in cerebellum, pontine and hippocampus, whereas Epm2b+/− control mice did not present any of these aggregates. Then, we checked whether the treatment with the different drugs reduced the levels of polyubiquitinated protein aggregates present in Epm2b−/− mice. Consistent with the results described above, trehalose did not modify or slightly increased (pontine: p=0.02; hippocampus: p=0.02) the levels of polyubiquitinated protein aggregates present in the different studied brain areas of the treated animals (Fig. 3). However, treatment with 4-PBA or metformin clearly decreased the levels of polyubiquitinated protein aggregates in all the assayed areas of the brain (cerebellum: p=0.002 [4-PBA], p<0.001 [metformin]; pontine: p=0.004 [4-PBA], p<0.001 [metformin]; hippocampus: p<0.001 [4-PBA], p<0.001 [metformin]; cortex: p=0.003 [4-PBA], p<0.001 [metformin]) (Fig. 3A and 3B).
Figure 3.
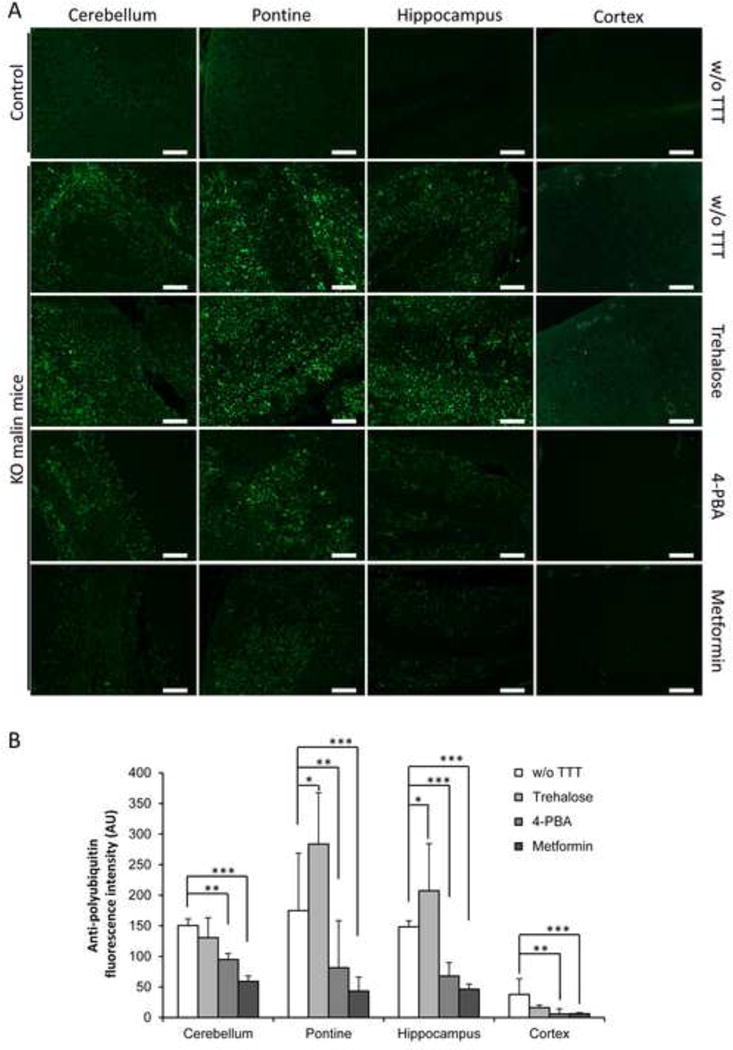
Immunohistofluorescence analyses of polyubiquitinated protein aggregates in brain of Epm2b+/− (control) or Epm2b−/− (KO) mice untreated (w/o TTT) or treated with trehalose, 4-PBA or metformin (two months). (A) Representative frontal sections of the cerebellum, pontine, hippocampus and cortex from Epm2b+/− control or Epm2b−/− KO mice untreated (w/o TTT) or treated as indicated, were incubated with anti-polyubiquitin antibody and AlexaFluor488 conjugated anti-rabbit antibody. Scale bars: 100 μm. (B) Polyubiquitin-associated fluorescence was quantified with ImageJ software in two independent pictures of cerebellum, pontine, hippocampus and cerebral cortex from five mice per group, and mean fluorescence in an area of 0.4 mm2 was represented. Bars indicate mean of fluorescence values corresponding to five mice ± SEM. Asterisks denote significant differences *p< 0.05, **p< 0.01, ***p<0.001 comparing the indicated groups with the basal condition (Epm2b−/− w/o TTT) according to Kruskal-Wallis non-parametric test followed by Conover-Inman post-hoc test.
Neurodegeneration and hippocampal gliosis is reduced by 4-PBA or metformin treatments
Lafora disease is characterized by neuronal loss and the appearance of the concomitant gliosis ([2], [33], [34]). We confirmed these facts in the untreated Epm2b−/− mice where we observed that selected areas of the hippocampus [dentate gyrus (DG) and cornus ammonis 1 (CA1) regions] had reduced NeuN (Neuronal Nuclei) staining, a neuron-specific marker (DG p=0.016 and CA1 p=0.006 in untreated control vs Epm2b−/−) (Fig. 4A and 4B). Untreated Epm2b−/− mice presented a less compacted staining (see black arrow heads) suggesting the loss of neurons in these areas (Fig. 4A). Trehalose treatment did not prevent neuronal loss in DG or CA1 areas of the hippocampus (Fig. 4A and 4B). However, when compared with untreated Epm2b−/− mice, treatment with 4-PBA or metformin presented a significant higher NeuN staining in both analyzed hippocampus regions (DG: p=0.005 [4-PBA], p=0.016 [metformin]; CA1: p=0.005 [4-PBA], p=0.008 [metformin]), suggesting that these treatments prevented neuronal loss.
Figure 4.
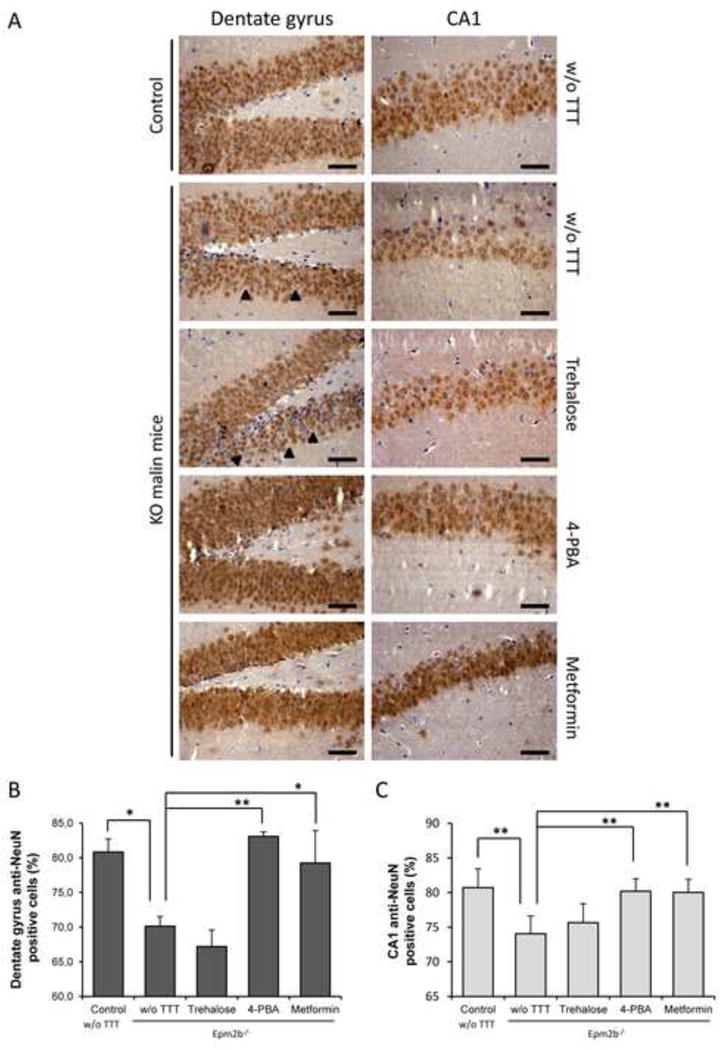
Immunohistochemistry analyses of neurodegeneration in hippocampus of Epm2b+/− (control) and Epm2b−/− (KO) mice untreated (w/o TTT) or treated with trehalose, 4-PBA or metformin (two months). (A) Representative frontal sections of the dentate gyrus (DG) and the cornus ammonis 1 (CA1) of the hippocampus from Epm2b+/− control or Epm2b−/− KO mice untreated (w/o TTT) or treated as indicated, were incubated with anti-NeuN (Neuronal nuclei) antibody. Black arrow heads point to empty space between neurons in DG. Scale bars 25 μm. NeuN-positive and negative cells were counted in DG (B) and CA1 (C) sections from three independent mice and percentage of NeuN-postive cells in an area of 0.09 mm2 was represented. Bars indicate mean values corresponding to three independent mice ± SEM. Asterisks denote significant differences *p< 0.05 **p<0.01 comparing the indicated groups with the basal condition (Epm2b−/− w/o TTT) according to Kruskal-Wallis non-parametric test followed by Conover-Inman post-hoc test.
Similar areas of the hippocampus were also stained with an antibody that recognized GFAP (glial fibrillary acidic protein), an astrocyte marker (Fig. 5). This staining was higher in preparations from untreated Epm2b−/− in comparison to control mice (DG: p=0.006, and CA1: p=0.03 in control vs Epm2b−/−) (Fig. 5), confirming the presence of reactive gliosis. Treatment of Epm2b−/− with 4-PBA significantly decreased around 20% (p=0.02) and 40% (p<0.001) the gliosis in DG and CA1, respectively (Fig. 5). Similarly, metformin treatment reduced gliosis around 40% (p<0.001) and 60% (p<0.001) in DG and CA1, respectively (Fig. 5). Surprisingly, treatment with trehalose also significantly diminished around 40% the astrogliosis in both regions (p<0.001 in both cases) (Fig. 5).
Figure 5.
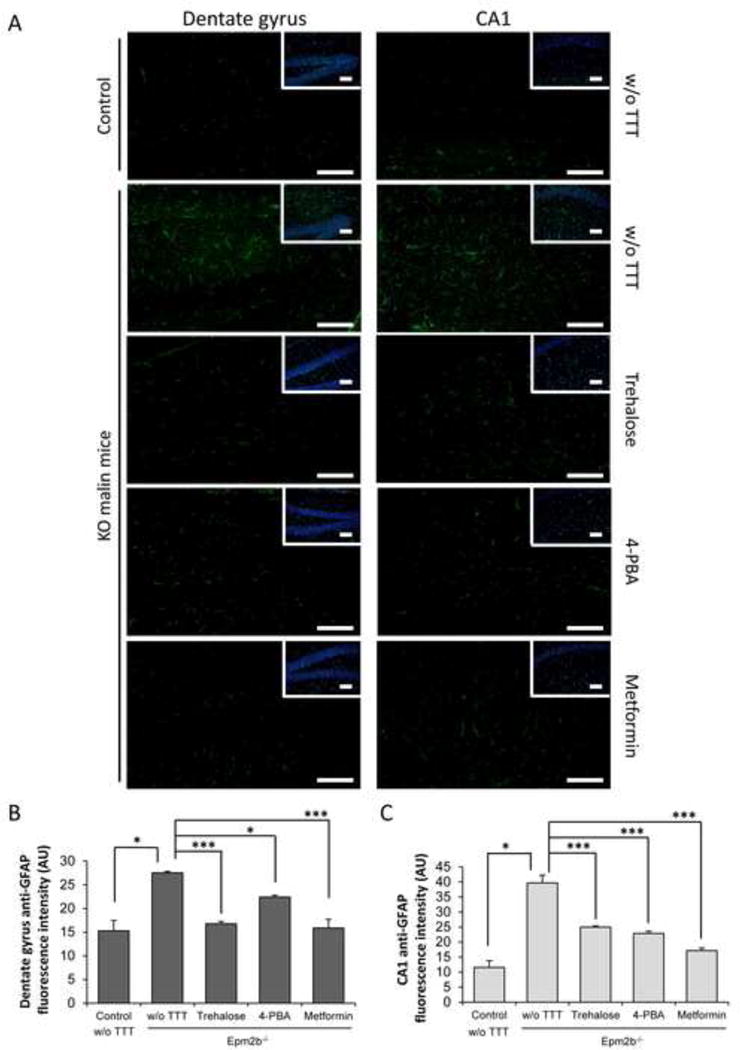
Immunohistofluorescence analyses of gliosis in hippocampus of Epm2b+/− (control) or Epm2b−/− (KO) mice untreated (w/o TTT) or treated with trehalose, 4-PBA or metformin (two months). (A) Representative frontal sections of the dentate gyrus (DG) and the cornus ammonis 1 (CA1) of the hippocampus from Epm2b+/− control or Epm2b−/− KO mice untreated (w/o TTT) or treated as indicated, were incubated with anti-GFAP (glial fibrillary acidic protein) antibody and AlexaFluor488 conjugated anti-rabbit antibody and counterstained with DAPI to show the area where the analysis was performed (insets). Scale bars 100 μm. GFAP-associated fluorescence was quantified with ImageJ software in two pictures from DG (B) and CA1 (C) sections from five mice per group, and mean fluorescence in an area of 0.26 mm2 was represented. Bars indicate mean of fluorescence values corresponding to five mice ± SEM. Asterisks denote significant differences *p< 0.05 ***p<0.001 comparing the indicated groups with the basal condition (Epm2b−/− w/o TTT) according to Kruskal-Wallis non-parametric test followed by Conover-Inman post-hoc test.
Trehalose, 4-PBA or metformin ameliorate some Epm2b−/− neuropsychological dysfunctions
In addition to the above described molecular and histological analyses, each group of mice was subjected to a battery of neuropsychological tests after one month of treatment and also at the end of the period of two months of treatment. One of the tests was the tail suspension test (TST). Originally designed to screen antidepressant drugs [28], it is based on the fact that animals subjected to the short-term, inescapable stress of being suspended by their tail, develop an immobile posture with both extended feet. Clasping of the feet is recorded as a sign of neurodegeneration. As observed in Fig. 6, untreated Epm2b−/− mice presented a significant worse performance than untreated control mice (p=0.002 and p=0.007, after one and two months of treatment, respectively). Treatment with 4-PBA or metformin showed a tendency to improve the performance after one month of treatment but the differences became statistically significant after two months of treatment (p=0.045 [4-PBA] and p=0.017 [metformin]) (Fig. 6). Trehalose treatment did not improve the performance of Epm2b−/− mice in any case.
Figure 6.
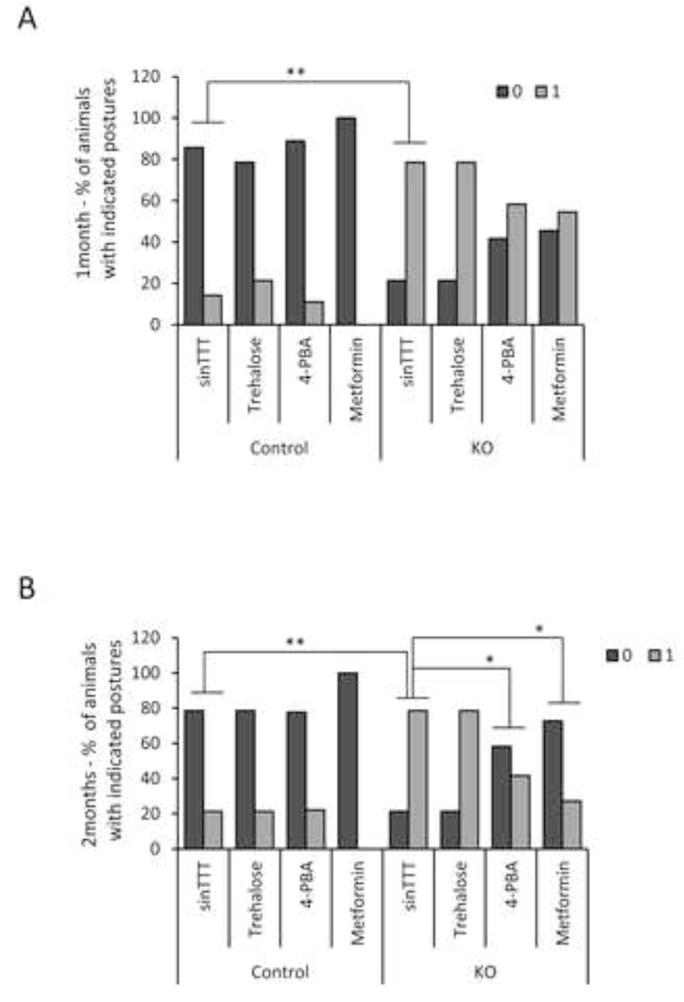
Tail suspension test of Epm2b+/− (control) and Epm2b−/− (KO) mice untreated (w/o TTT) or treated with trehalose, 4-PBA or metformin, during one (A) or two months (B). Percentage of Epm2b+/− (control) or Epm2b−/− (KO) mice showing normal posture (0) or altered (1) stereotypical clasping of the hind-limb upon tail suspension (n: 8–11 in each group). Significant differences between the groups were indicated (*p< 0.05, **p< 0.01) according to the Fisher exact test followed by Wilcoxon-Mann-Whitney test.
The vertical pole test was originally designed to quantify mouse movement disorders in animal models of Parkinson disease [29]. As observed in Fig. 7, untreated Epm2b−/− mice presented a significant worse performance than untreated control mice in the time to turn and locomotion activity parameters of the test (p=0.018 and p=0.028 after one month of treatment and p<0.001 for both parameters after two months of treatment). Treatment with trehalose decreased both parameters of this test (time to turn and locomotion activity) after one month of treatment (p<0.001 and p=0.002 respectively) and the amelioration was even higher after two months of treatment (p<0.001 for both parameters) (Fig. 7). 4-PBA also improved both parameters after one month of treatment (p<0.001 and p=0.03 respectively) and we observed a great amelioration of both the time to turn and locomotion activity parameters after the second month of treatment (p<0.001 for both) (Fig. 7). After one month, metformin treated animals showed a significant improvement in the time to turn parameter (p=0.049) and a clear tendency to improve the locomotion activity, although it was not statistically significant. However, after two month of treatment the improvement became clearly observed for both parameters (p=0.0038 and p=0.0053, for time to turn and locomotion activity, respectively) (Fig. 7).
Figure 7.
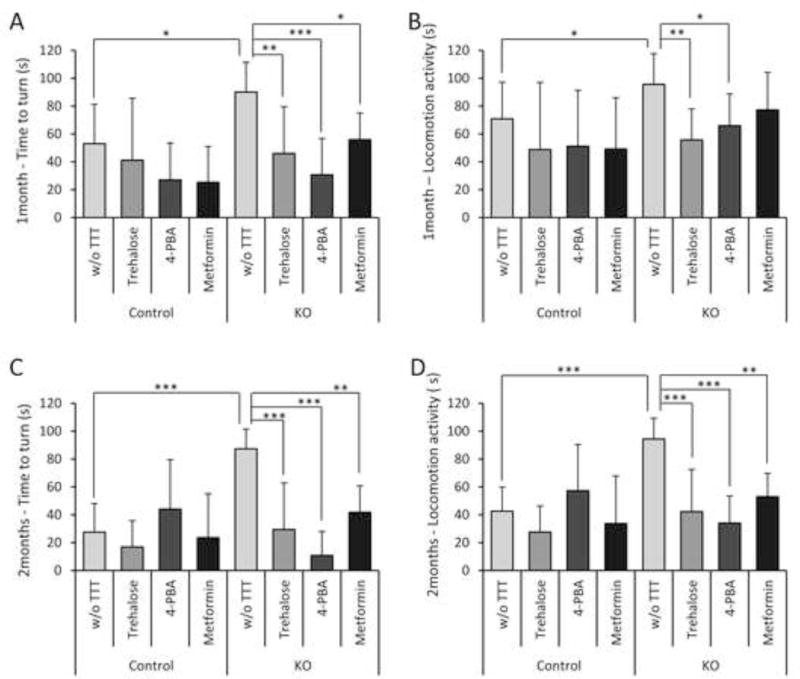
Vertical pole test for evaluation of motor coordination of Epm2b+/− (control) and Epm2b−/− (KO) mice untreated (w/o TTT) or treated with trehalose, 4-PBA or metformin, during one (A–B) or two months (C–D). After one month of treatment, the time taken by the mice to turn completely downward (time to turn) (A) and the total time taken to reach the floor (locomotion activity) (B) were recorded. After two months of treatment, the time to turn (C) and the locomotion activity (D) were also recorded. Bars indicate mean of values corresponding to eight to twelve mice ± SEM. Asterisks denote significant differences between the groups (*p< 0.05, **p< 0.01, ***p< 0.001) according to Kruskal-Wallis non-parametric test followed by Conover-Inman post-hoc test).
Animals were also subjected to additional behavioral tests (open field, used to assay general locomotion activity levels and anxiety, Fig. 8; beam balance, used to assay fine motor coordination and balance, Fig. 9), but we did not find any significant difference in the parameters of these tests between treated or untreated animals, possibly because the neurological deterioration present in the Epm2b−/− mice at five months of age could have affected some neuropsychological abilities but not others.
Figure 8.
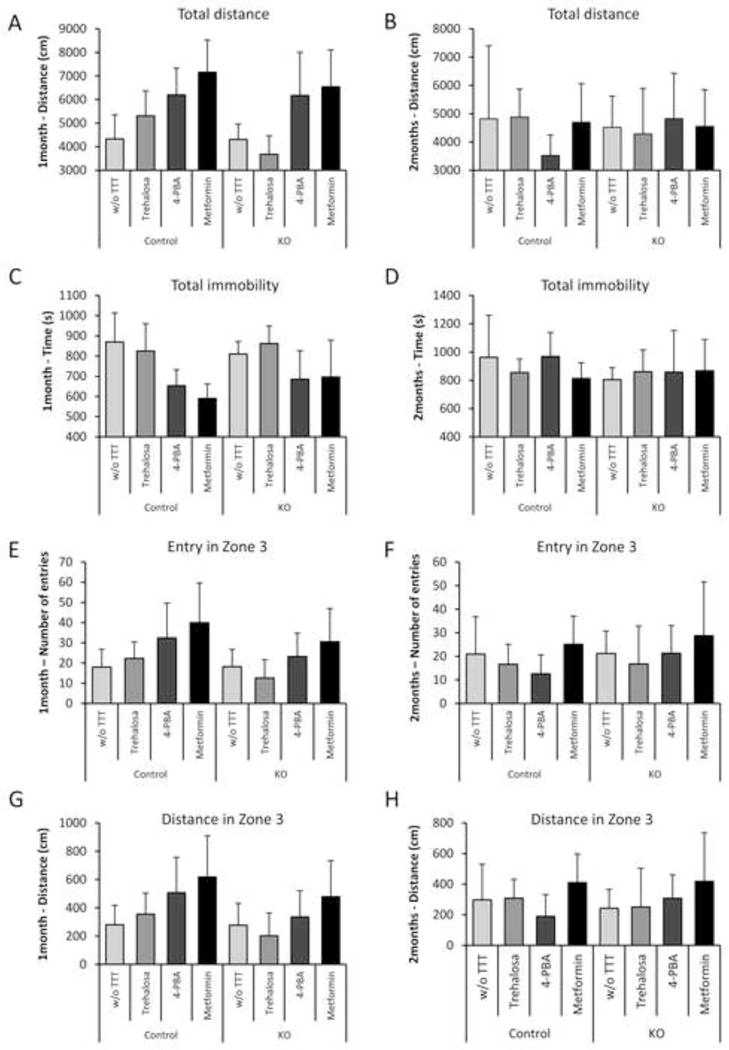
Open-field analyses of Epm2b+/− (control) and Epm2b−/− (KO) mice untreated (w/o TTT) or treated with the described drugs during one month (A, C, E, G) or two months (B, D, F, H). No significant differences between groups (n: 8–12 mice in each group) were observed in all recorded parameters: total distance roamed (A–B), total immobility time (C–D), number of entries in the more central zone (zone 3) (E–F), or the distance roamed in zone 3 (G–H), according to Kruskal-Wallis non-parametric test followed by Conover-Inman post-hoc test.
Figure 9.
Beam-balance analyses of Epm2b+/− (control) and Epm2b−/− (KO) mice untreated (w/o TTT) or treated with the described drugs during one month (A) or two months (B). No significant differences between groups (n: 8–12 mice in each group) were observed in the time to cross the beam in any of the beam sizes analyzed (5, 12 and 26 mm), according to Kruskal-Wallis non-parametric test followed by Conover-Inman post-hoc test.
DISCUSSION
More than a century after its description, the progressive myoclonus epilepsy of Lafora type is still a fatal disease leading to the death of patients usually within the decade after the apparition of the first symptoms. Unfortunately, up to now, available treatments are only palliatives and no curative drugs are available yet. Recent reports indicate that the elimination of the capacity of the cells to synthesize glycogen, either by depleting glycogen synthase or the protein targeting to glycogen (PTG) in animal models of LD ([35], [36], [37], [38]), not only results in the elimination of the accumulation of polyglucosans, but also in the amelioration of the neuropathological symptoms. These results have led those authors to suggest the use of glycogen synthase inhibitors as a putative strategy to treat LD. While the field awaits for the beneficial effects that these drugs could have in the future, in this work we have used an alternative strategy to find a beneficial treatment for LD based on the restoration of proteostasis, which is defective in this disease ([9], [10], [11]). We describe the use trehalose and 4-phenylbutyric acid (4-PBA), two chemical chaperones with recognized abilities to ameliorate misfolding of proteins ([17], [18], [19], [20], [21]). In addition, we have checked the effect of metformin, an activator of AMP-activated protein kinase (AMPK), as activation of this kinase plays a neuroprotective role in different neurodegenerative diseases ([24], [25], [26], [27]).
We first observed that the designed treatments affected the corresponding molecular targets in the brain of a mouse model of LD (Epm2b−/−, lacking malin): trehalose and 4-PBA increased the levels of the chaperone BIP/Grp78, involved in the amelioration of proteostasis dysfuction, and metformin induced the activation of the AMPK complex. Then, we analyzed the possible beneficial effect of these drugs on LD symptoms. We found that the administration of the chemical chaperone 4-PBA reduced the levels of polyglucosan (PAS+) and polyubiquitinated protein aggregates in different areas of the brain of Epm2b−/− treated mice. This beneficial effect could be due to its action as a chemical chaperone or to its ability to improve the expression of the endogenous chaperone BiP/GRP78. The improved chaperone content of the cell could diminish the formation of intracellular aggregates. The use of trehalose, another small molecule that acts as chemical chaperone also improved the expression of BiP/GRP78 but did not have a beneficial effect in reducing intracellular aggregates, perhaps because its role as chemical chaperone could not be as potent as 4-PBA. Trehalose and 4-PBA were effective in reducing gliosis in the different parts of the hippocampus, but only 4-PBA had in addition a beneficial effect in preventing neuronal loss. The different histological effect of these two drugs had a reflection in the performance of the treated animals when subjected to different neuropsychological tests: whereas 4-PBA treated animals improved the tail suspension and the pole tests, trehalose had only a beneficial effect on the pole test.
We also describe the beneficial effect of metformin, an activator of AMPK, in the treatment of LD. We observed a clear reduction in the number of polyglucosan (PAS+) and polyubiquitinated protein aggregates, a reduction of neuronal loss and gliosis in different hippocampal regions and an amelioration of the neuropsychological alterations of Epm2b−/− treated mice. One possible explanation for the positive effect of metformin on LD could be that it maintained a sustained activation of AMPK, and this resulted in neuroprotection. As AMPK is involved in the activation of the autophagy pathway, we checked whether treatment with metformin could have an effect on autophagy, but as shown in Supplementary Fig. S1, we observed no changes in the levels of two recognized autophagy markers, namely LC3B-II and p62, upon treatment (trehalose and 4-PBA treatment did not affect these parameters either). So, an enhancement of autophagy is not responsible for the beneficial effects of these compounds. On the other hand, activation of AMPK may inhibit glycogen synthase [39]. However, upon metformin treatment, we did not observe changes in the state of different enzymes related to glycogen homeostasis (protein levels and phosphorylated forms of glycogen synthase and glycogen synthase kinase 3β were similar in all the cases; Supplementary Fig. S2), (trehalose and 4-PBA treatment did not affect these parameters either). These results are in agreement with previous reports which indicate that the specific activity of glycogen synthase does not change in the brain of LD mouse models in comparison to controls, especially at young ages as the ones used in our studies (5 month old animals at the end of the treatments) ([40], [41], [42]). Thus, we conclude that the beneficial effect of metformin in LD could be due to alternative mechanisms.
In conclusion, we describe for the first time the beneficial effect of the administration of small molecule compounds on the neuropathological symptoms of LD. It is encouraging to see that the beneficial effect of these drugs is already present after one month of treatment and much better results are obtained if the treatment is prolonged for two months. As these compounds have good safety records and are already approved for clinical use on different neurological pathologies, we think their use in clinical practice based on drug repositioning could be straightforward.
Supplementary Material
Acknowledgments
We want to thank Carla Rubio-Villena and Maria Adelaida Garcia-Gimeno for their help in the analyses of glycogen related enzymes. This work was supported by grants from the Spanish Ministry of Education and Science SAF2011-27442, Fundació La Marato de TV3 (ref. 100130) and an ACCI2012 action from CIBERER. A.B. holds a postdoctoral fellowship from the Program “Junta para la Ampliatión de Estudios” (JAE-Doc) co-funded by the European Social Fund (ESF).
ABBREVIATIONS
- AMPK
AMP-activated protein kinase
- LB
Lafora bodies
- LD
Lafora disease
- 4-PBA
4-phenylbutyric acid
- SDS-PAGE
sodium dodecyl sulfate polyacrylamide gel electrophoresis
References
- 1.Delgado-Escueta AV. Advances in lafora progressive myoclonus epilepsy. Curr Neurol Neurosci Rep. 2007;7:428–433. doi: 10.1007/s11910-007-0066-7. [DOI] [PubMed] [Google Scholar]
- 2.Monaghan TS, Delanty N. Lafora disease: epidemiology, pathophysiology and management. CNS Drugs. 2010;24:549–561. doi: 10.2165/11319250-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 3.Minassian BA, Lee JR, Herbrick JA, Huizenga J, Soder S, et al. Mutations in a gene encoding a novel protein tyrosine phosphatase cause progressive myoclonus epilepsy. Nat Genet. 1998;20:171–174. doi: 10.1038/2470. [DOI] [PubMed] [Google Scholar]
- 4.Serratosa JM, Gomez-Garre P, Gallardo ME, Anta B, de Bernabe DB, et al. A novel protein tyrosine phosphatase gene is mutated in progressive myoclonus epilepsy of the Lafora type (EPM2) Hum Mol Genet. 1999;8:345–352. doi: 10.1093/hmg/8.2.345. [DOI] [PubMed] [Google Scholar]
- 5.Chan EM, Young EJ, Ianzano L, Munteanu I, Zhao X, et al. Mutations in NHLRC1 cause progressive myoclonus epilepsy. Nat Genet. 2003;35:125–127. doi: 10.1038/ng1238. [DOI] [PubMed] [Google Scholar]
- 6.Lohi H, Ianzano L, Zhao XC, Chan EM, Turnbull J, et al. Novel glycogen synthase kinase 3 and ubiquitination pathways in progressive myoclonus epilepsy. Hum Mol Genet. 2005;14:2727–2736. doi: 10.1093/hmg/ddi306. [DOI] [PubMed] [Google Scholar]
- 7.Vilchez D, Ros S, Cifuentes D, Pujadas L, Valles J, et al. Mechanism suppressing glycogen synthesis in neurons and its demise in progressive myoclonus epilepsy. Nat Neurosci. 2007;10:1407–1413. doi: 10.1038/nn1998. [DOI] [PubMed] [Google Scholar]
- 8.Solaz-Fuster MC, Gimeno-Alcaniz JV, Ros S, Fernandez-Sanchez ME, Garcia-Fojeda B, et al. Regulation of glycogen synthesis by the laforin-malin complex is modulated by the AMP-activated protein kinase pathway. Hum Mol Genet. 2008;17:667–678. doi: 10.1093/hmg/ddm339. [DOI] [PubMed] [Google Scholar]
- 9.Liu Y, Wang Y, Wu C, Liu Y, Zheng P. Deletions and missense mutations of EPM2A exacerbate unfolded protein response and apoptosis of neuronal cells induced by endoplasm reticulum stress. Hum Mol Genet. 2009;18:2622–2631. doi: 10.1093/hmg/ddp196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vernia S, Rubio T, Heredia M, Rodriguez de Cordoba S, Sanz P. Increased endoplasmic reticulum stress and decreased proteasomal function in lafora disease models lacking the phosphatase laforin. PLoS One. 2009;4:e5907. doi: 10.1371/journal.pone.0005907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zeng L, Wang Y, Baba O, Zheng P, Liu Y. Laforin is required for the functional activation of malin in endoplasmic reticulum stress resistance in neuronal cells. FEBS J. 2012;279:2467–2478. doi: 10.1111/j.1742-4658.2012.08627.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rao SN, Maity R, Sharma J, Dey P, Shankar SK, et al. Sequestration of chaperones and proteasome into Lafora bodies and proteasomal dysfunction induced by Lafora disease-associated mutations of malin. Hum Mol Genet. 2010;19:4726–4734. doi: 10.1093/hmg/ddq407. [DOI] [PubMed] [Google Scholar]
- 13.Aguado C, Sarkar S, Korolchuk VI, Criado O, Vernia S, et al. Laforin, the most common protein mutated in Lafora disease, regulates autophagy. Hum Mol Genet. 2010;19:2867–2876. doi: 10.1093/hmg/ddq190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Criado O, Aguado C, Gayarre J, Duran-Trio L, Garcia-Cabrero AM, et al. Lafora bodies and neurological defects in malin-deficient mice correlate with impaired autophagy. Hum Mol Genet. 2012;21:1521–1533. doi: 10.1093/hmg/ddr590. [DOI] [PubMed] [Google Scholar]
- 15.Hetz C, Mollereau B. Disturbance of endoplasmic reticulum proteostasis in neurodegenerative diseases. Nat Rev Neurosci. 2014;15:233–249. doi: 10.1038/nrn3689. [DOI] [PubMed] [Google Scholar]
- 16.Sinadinos C, Valles-Ortega J, Boulan L, Solsona E, Tevy MF, et al. Neuronal glycogen synthesis contributes to physiological aging. Aging Cell. 2014;13:935–945. doi: 10.1111/acel.12254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ricobaraza A, Cuadrado-Tejedor M, Perez-Mediavilla A, Frechilla D, Del Rio J, et al. Phenylbutyrate ameliorates cognitive deficit and reduces tau pathology in an Alzheimer’s disease mouse model. Neuropsychopharmacology. 2009;34:1721–1732. doi: 10.1038/npp.2008.229. [DOI] [PubMed] [Google Scholar]
- 18.Wiley JC, Meabon JS, Frankowski H, Smith EA, Schecterson LC, et al. Phenylbutyric acid rescues endoplasmic reticulum stress-induced suppression of APP proteolysis and prevents apoptosis in neuronal cells. PLoS One. 2010;5:e9135. doi: 10.1371/journal.pone.0009135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhou W, Bercury K, Cummiskey J, Luong N, Lebin J, et al. Phenylbutyrate up-regulates the DJ-1 protein and protects neurons in cell culture and in animal models of Parkinson disease. J Biol Chem. 2011;286:14941–14951. doi: 10.1074/jbc.M110.211029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tanaka M, Machida Y, Niu S, Ikeda T, Jana NR, et al. Trehalose alleviates polyglutamine-mediated pathology in a mouse model of Huntington disease. Nat Med. 2004;10:148–154. doi: 10.1038/nm985. [DOI] [PubMed] [Google Scholar]
- 21.Chen Q, Haddad GG. Role of trehalose phosphate synthase and trehalose during hypoxia: from flies to mammals. J Exp Biol. 2004;207:3125–3129. doi: 10.1242/jeb.01133. [DOI] [PubMed] [Google Scholar]
- 22.Hardie DG, Ross FA, Hawley SA. AMPK: a nutrient and energy sensor that maintains energy homeostasis. Nat Rev Mol Cell Biol. 2012;13:251–262. doi: 10.1038/nrm3311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Carling D, Thornton C, Woods A, Sanders MJ. AMP-activated protein kinase: new regulation, new roles? Biochem J. 2012;445:11–27. doi: 10.1042/BJ20120546. [DOI] [PubMed] [Google Scholar]
- 24.Poels J, Spasic MR, Callaerts P, Norga KK. Expanding roles for AMP-activated protein kinase in neuronal survival and autophagy. Bioessays. 2009;31:944–952. doi: 10.1002/bies.200900003. [DOI] [PubMed] [Google Scholar]
- 25.Han Y, Xie N, Cao L, Zhao X, Liu X, et al. Adenosine monophosphate-activated protein kinase and peroxisome proliferator-activated receptor gamma coactivator 1alpha signaling provides neuroprotection in status epilepticus in rats. Neurosci Lett. 2011;500:133–138. doi: 10.1016/j.neulet.2011.06.020. [DOI] [PubMed] [Google Scholar]
- 26.Dulovic M, Jovanovic M, Xilouri M, Stefanis L, Harhaji-Trajkovic L, et al. The protective role of AMP-activated protein kinase in alpha-synuclein neurotoxicity in vitro. Neurobiol Dis. 2014;63:1–11. doi: 10.1016/j.nbd.2013.11.002. [DOI] [PubMed] [Google Scholar]
- 27.Ashabi G, Khodagholi F, Khalaj L, Goudarzvand M, Nasiri M. Activation of AMP-activated protein kinase by metformin protects against global cerebral ischemia in male rats: Interference of AMPK/PGC-1alpha pathway. Metab Brain Dis. 2014;29:47–58. doi: 10.1007/s11011-013-9475-2. [DOI] [PubMed] [Google Scholar]
- 28.Steru L, Chermat R, Thierry B, Simon P. The tail suspension test: a new method for screening antidepressants in mice. Psychopharmacology (Berl) 1985;85:367–370. doi: 10.1007/BF00428203. [DOI] [PubMed] [Google Scholar]
- 29.Ogawa N, Hirose Y, Ohara S, Ono T, Watanabe Y. A simple quantitative bradykinesia test in MPTP-treated mice. Res Commun Chem Pathol Pharmacol. 1985;50:435–441. [PubMed] [Google Scholar]
- 30.Stanford SC. The Open Field Test: reinventing the wheel. J Psychopharmacol. 2007;21:134–135. doi: 10.1177/0269881107073199. [DOI] [PubMed] [Google Scholar]
- 31.Luong TN, Carlisle HJ, Southwell A, Patterson PH. Assessment of motor balance and coordination in mice using the balance beam. J Vis Exp. 2011;49:2376. doi: 10.3791/2376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.R-Core-Team. A language and environment for statistical computing. Foundation for Statistical Computing; 2014. http://wwwR-projectorg/ [Google Scholar]
- 33.Valles-Ortega J, Duran J, Garcia-Rocha M, Bosch C, Saez I, et al. Neurodegeneration and functional impairments associated with glycogen synthase accumulation in a mouse model of Lafora disease. EMBO Mol Med. 2011;3:667–681. doi: 10.1002/emmm.201100174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ortolano S, Vieitez I, Agis-Balboa RC, Spuch C. Loss of GABAergic cortical neurons underlies the neuropathology of Lafora disease. Mol Brain. 2014;7:7. doi: 10.1186/1756-6606-7-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Turnbull J, DePaoli-Roach AA, Zhao X, Cortez MA, Pencea N, et al. PTG depletion removes Lafora bodies and rescues the fatal epilepsy of Lafora disease. PLoS Genet. 2011;7:e1002037. doi: 10.1371/journal.pgen.1002037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pederson BA, Turnbull J, Epp JR, Weaver SA, Zhao X, et al. Inhibiting glycogen synthesis prevents lafora disease in a mouse model. Ann Neurol. 2013;74:297–300. doi: 10.1002/ana.23899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Turnbull J, Epp JR, Goldsmith D, Zhao X, Pencea N, et al. PTG protein depletion rescues malin-deficient Lafora disease in mouse. Ann Neurol. 2014;75:442–446. doi: 10.1002/ana.24104. [DOI] [PubMed] [Google Scholar]
- 38.Duran J, Gruart A, Garcia-Rocha M, Delgado-Garcia JM, Guinovart JJ. Glycogen accumulation underlies neurodegeneration and autophagy impairment in Lafora disease. Hum Mol Genet. 2014;23:3147–3156. doi: 10.1093/hmg/ddu024. [DOI] [PubMed] [Google Scholar]
- 39.Carling D, Hardie DG. The substrate and sequence specificity of the AMP-activated protein kinase. Phosphorylation of glycogen synthase and phosphorylase kinase. Biochim Biophys Acta. 1989;1012:81–86. doi: 10.1016/0167-4889(89)90014-1. [DOI] [PubMed] [Google Scholar]
- 40.Tagliabracci VS, Girard JM, Segvich D, Meyer C, Turnbull J, et al. Abnormal metabolism of glycogen phosphate as a cause for Lafora disease. J Biol Chem. 2008;283:33816–33825. doi: 10.1074/jbc.M807428200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.DePaoli-Roach AA, Tagliabracci VS, Segvich DM, Meyer CM, Irimia JM, et al. Genetic depletion of the malin E3 ubiquitin ligase in mice leads to lafora bodies and the accumulation of insoluble laforin. J Biol Chem. 2010;285:25372–25381. doi: 10.1074/jbc.M110.148668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Turnbull J, Wang P, Girard JM, Ruggieri A, Wang TJ, et al. Glycogen hyperphosphorylation underlies lafora body formation. Ann Neurol. 2010;68:925–933. doi: 10.1002/ana.22156. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.