

Abstract
The design of artificial metalloenzymes has achieved tremendous progress, although few designs can achieve catalytic performances comparable to that of native enzymes. Moreover, the structure and function of artificial metalloenzymes in non‐native states has rarely been explored. Herein, we found that a c‐type cytochrome b 5 (Cyt b 5), N57C/S71C Cyt b 5, with heme covalently attached to the protein matrix through two Cys–heme linkages, adopts a non‐native state with an open heme site after guanidine hydrochloride (Gdn⋅HCl)‐induced unfolding, which facilitates H2O2 activation and substrate binding. Stopped‐flow kinetic studies further revealed that c‐type Cyt b 5 in the non‐native state exhibited impressive peroxidase activity comparable to that of native peroxidases, such as the most efficient horseradish peroxidase. This study presents an alternative approach to the design of functional artificial metalloenzymes by exploring enzymatic functions in non‐native states.
Keywords: heme proteins, metalloenzymes, non-native state, peroxidase, protein design
The rational design of functional artificial metalloenzymes has received much attention in the last decade, not only to elucidate the structural and functional relationship of natural metalloenzymes, but also to provide the ability to create novel biocatalysts for potential applications.1 Although tremendous progress has been made in the design of metalloenzymes from single to multiple active sites, the catalytic performance of designed artificial metalloenzymes is limited and few designs can achieve a catalytic rate comparable to that of native enzymes. For example, Lu and co‐workers2 redesigned G65Y‐CuBMb, a biosynthetic model of heme–copper oxidases in myoglobin (Mb),3 by introducing three Lys residues at the surface to enhance the binding affinity for its redox partner, cytochrome b 5 (Cyt b 5), which reduced O2 to water at a rate of 52 s−1 and achieved the activity of native Cyt cbb 3 oxidase (50 s−1). Matsuo et al.4 constructed a H64D Mb mutant and reconstituted it with a heme derivative with a carboxylate‐based cluster attached to one propionate group, which exhibited an overall catalytic activity for guaiacol oxidation (k cat /K m=85 000 m −1 s−1) that exceeded that of native horseradish peroxidase (HRP) (k cat /K m=72 000 m −1 s−1).5 It is thus attractive to develop other methods to design functional artificial metalloenzymes with highly efficient catalytic performances comparable to native enzymes.
In addition to native states, metalloproteins possess inherent conformational variability, which may confer new functions for these proteins in the non‐native state.6 For example, an electron‐transfer protein, cytochrome c (Cyt c), was also found to carry out functions beyond respiration, such as peroxidase activity due to the dissociation of one heme axial ligand (Met80).7 The structure and function of heme proteins in non‐native states have attracted much attention in recent years because they may be as important as those in the native states and are essential for their biological functions.6, 8 Itoh and co‐workers have shown that an O2 carrier protein, hemocyanin, exhibited significantly enhanced monooxygenase activity upon addition of a denaturant.9 Therefore, exploration of the catalytic performance of artificial metalloenzymes in non‐native states may be a potential alternative approach to the design of functional artificial metalloenzymes.
To test our speculation, we focused herein on a well‐studied small electron‐transfer protein, Cyt b 5, which binds a heme group noncovalently with a bis‐His coordination (His39/His63; see Figure 1).10 In a previous study,11 we showed that the Cyt c‐like Cyt b 5 (N57C/S71C Cyt b 5) can be formed in vivo through the introduction of two cysteine residues (Cys57 and Cys71) close to the two heme vinyl groups. Due to the spatial position, Cys57 forms a typical thioether linkage with a heme 4‐vinyl group, whereas Cys71 forms an unusual [heme‐CO‐CH2‐S‐CH2‐Cα] linkage with a heme 2‐vinyl group (Figure 1). Molecular modeling indicated that covalent heme attachment to the polypeptide chain does not alter the overall structure of Cyt b 5.12 The c‐type Cyt b 5, which differs from wild‐type (WT) Cyt b 5 by only two covalent linkages, provides an ideal protein model to investigate the structure and function of heme proteins in non‐native states and also the consequences of the heme‐to‐protein attachment.
Figure 1.
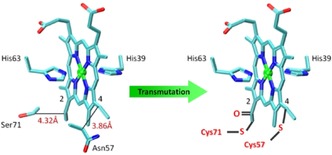
Conversion of Cyt b 5 into a Cyt c‐like protein by the introduction of two cysteine residues close to the heme vinyl groups to form covalent linkages.
The double‐mutant N57C/S71C Cyt b 5 was expressed and purified as previously described.11 With heme covalently linked to the protein matrix through two c‐type‐like linkages, N57C/S71C Cyt b 5 showed a Soret band (λ=409 nm) that was blueshifted relative to that of WT Cyt b 5 (λ=413 nm). To probe the stabilization role of the covalent linkages, we performed guanidine hydrochloride (Gdn⋅HCl)‐induced unfolding studies, which showed that as the concentration of Gdn⋅HCl was increased, the Soret band of N57C/S71C Cyt b 5 decreased and shifted from λ=409 to 413 nm (Figure 2A). Concurrently, the visible bands (λ=529 and 556 nm) characteristic of bis‐His coordination decreased, and a broad visible band appeared at λ≈620 nm. This observation suggests that N57C/S71C Cyt b 5 underwent conformational changes to generate a non‐native state with a different coordination state. A control experiment with WT Cyt b 5 showed that the Soret band decreased dramatically upon addition of Gdn⋅HCl and converted to a broad peak at λ≈370 nm, which indicated dissociation of the heme group from the heme‐binding pocket (Figure 2B). Analysis of the unfolding curves (Figure 2, insets) by plotting the absorbance of Soret band versus the concentration of Gdn⋅HCl showed that N57C/S71C Cyt b 5 has a half‐denaturation concentration (C m) of (3.03±0.07) m, which is slightly larger than that for WT Cyt b 5 ((2.90±0.05) m) under the same conditions. These results suggest that the heme‐to‐protein attachment prevents the dissociation of heme upon unfolding, whereas both proteins have similar stability for the ligation of the heme group in the initial stage of protein unfolding.
Figure 2.
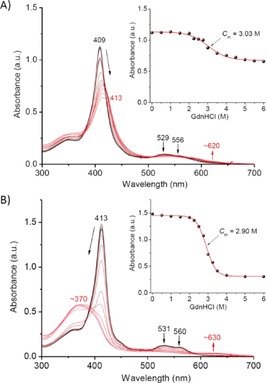
UV/Vis spectra of Gdn⋅HCl‐induced unfolding of A) N57C/S71C Cyt b 5 and B) WT Cyt b 5 in potassium phosphate buffer (100 mm, pH 7.0) at 25 °C. Insets: The changes in the Soret band versus Gdn⋅HCl concentration.
To provide structural information for N57C/S71C Cyt b 5 and WT Cyt b 5 in both native and Gdn⋅HCl‐induced non‐native states, we collected circular dichroism (CD) spectra in the absence and presence of 6 m Gdn⋅HCl. The spectrum of N57C/S71C Cyt b 5 in the far‐UV region (λ=190–250 nm) exhibits two negative peaks at λ=218 and 207 nm (Figure 3A), similar to that of WT Cyt b 5 (Figure 3C). On unfolding, the negative ellipticity decreased and became a single peak at λ=214 nm, which indicated a conversion from α‐helix (from 36.8 to 9.6 %) to β‐strand (from 12.3 to 39.6 %), as calculated by using the K2D2 server13 (Table S1). In the unfolded WT Cyt b 5, the α‐helix proportion deceased to approximately 2 %, with a majority of β‐strand (≈49 %). These observations suggest that the covalently linked heme contributes to the stabilization of local secondary structure of N57C/S71C Cyt b 5, although the overall protein structure was dramatically disrupted in the non‐native state.
Figure 3.
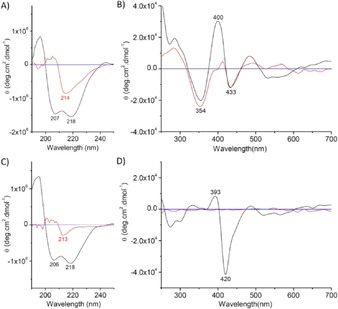
A, B) CD spectra of N57C/S71C Cyt b 5 and C, D) WT Cyt b 5 in the far‐UV (A, C) and near‐UV/Vis (B, D) regions in the absence (black line) and presence (red line) of 6 m Gdn⋅HCl in potassium phosphate buffer (100 mm, pH 7.0) at 25 °C. The horizontal line with a value of zero is shown in blue.
The spectrum of N57C/S71C Cyt b 5 in the native state in the near‐UV/Vis region (λ=250–700 nm; Figure 3B) showed a large positive peak at λ=400 nm and a negative peak at λ=433 nm in the Soret band region, which is distinct from that of WT Cyt b 5 (λ=393 and 400 nm; Figure 3D), which suggested that the formation of Cys–heme covalent linkages slightly altered the local conformation for the heme site. Moreover, large differences in the near‐UV region (λ=250 to 300 nm) suggested conformational rearrangements for hydrophobic core 2, in which aromatic amino acids (Tyr and Trp) are located (Figure S1 in the Supporting Information). Note that the negative peaks (λ=433 and 354 nm) of N57C/S71C Cyt b 5 were well retained in the Gdn⋅HCl‐induced non‐native state, albeit with a decreased positive peak (Figure 3B, red line). In contrast, almost no ellipticity was observed for unfolded WT Cyt b 5 in this region (Figure 3D, red line). These observations indicate that the heme group in unfolded N57C/S71C Cyt b 5, as covalently attached to the protein matrix, was chirally oriented by the surrounding amino acids to form a unique non‐native state. This is different for unfolded WT Cyt b 5, from which the heme dissociated in the absence of covalent heme–protein linkage.
Moreover, to provide information for the heme coordination in the Gdn⋅HCl‐induced non‐native state, we performed electron paramagnetic resonance (EPR) studies for both N57C/S71C Cyt b 5 and WT Cyt b 5. As shown in Figure 4, the EPR spectrum of N57C/S71C Cyt b 5 in the unfolded state (line a) exhibits signals at g ⊥=5.83 and g ∥=1.99, which are typical of a high‐spin ferric heme and similar to that of ferric Mb with His93/H2O coordination (g ⊥=5.94; g ∥=1.99).14 In comparison, unfolded WT Cyt b 5 exhibited a low intensity for the high‐spin signals (g ⊥=5.78; g ∥=1.99, line c), similar to that of free hemin in the presence of 6 m Gdn⋅HCl (g ⊥=5.80; g ∥=1.99, line e). Instead, low‐spin ferric heme signals (gx=3.02; gy≈2.16; gz was not detectable) was observed for WT Cyt b 5 (line d) in the native state, as well as for N57C/S71C Cyt b 5 (line b), which agrees with previous observations for Cyt b 5 (gx=3.1; gy=2.18),15 and L29E/F43H Mb in the non‐native state with a bis‐His coordination (His64/His93; gx=2.97; gy=2.20).16 Note that the signals in the double mutant are much broader, which indicates a mixture of different low‐spin species, compared with WT Cyt b 5 with a single species. These observations suggest that one axial ligand, presumably His39, in N57C/S71C Cyt b 5 is dissociated from the heme iron upon unfolding, whereas His63 might still act as an axial ligand with the help of two covalent linkages on both sides, which results in a high‐spin heme species. In WT Cyt b 5 without covalent linkages, the heme dissociated from the pocket, which might be in the free form or be coordinated by a single histidine residue at the surface, such as His26 and His80, or a residue exposed to the surface upon unfolding, such as His15 and His39/His63 (Figure S1).
Figure 4.
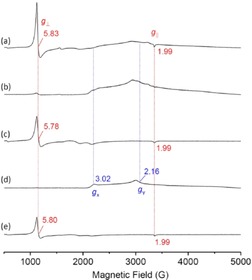
The EPR spectra of N57C/S71C Cyt b 5 and WT Cyt b 5 (0.3 mm) in a, c) the presence and b, d) absence of 6 m Gdn⋅HCl in potassium phosphate buffer (100 mm, pH 7.0) at 10 K. e) The EPR spectrum of hemin in the presence of 6 m Gdn⋅HCl is shown for comparison.
Inspired by the conversion of low spin to high spin for the heme active site upon Gdn⋅HCl‐induced unfolding, we were interested in exploring the catalytic performance of the c‐type Cyt b 5 in the non‐native state because alteration of the heme coordination state has been shown to be a prerequisite for the conversion of an electron‐transfer protein into a functional enzyme, such as an artificial peroxidase.7a–7d, 17 To test the peroxidase activity, we first chose guaiacol as a typical substrate and monitored its oxidation as catalyzed by WT Cyt b 5 and N57C/S71C Cyt b 5 in the absence and presence of 6 m Gdn⋅HCl with H2O2 as an oxidant. As shown in Figure S2, the oxidation product, with a characteristic absorption at λ=470 nm, was observed within a few seconds, particularly for unfolded N57C/S71C Cyt b 5, whereas no product was formed in the absence of Gdn⋅HCl, which indicated that an open heme site is critical for peroxidase activity. We then determined the kinetic parameters for steady‐state reactions at various guaiacol concentrations. Moreover, we chose 2,2′‐azino‐bis(3‐ethylbenzothiazoline‐6‐sulfonic acid) diammonium salt (ABTS) as another substrate and performed control experiments with WT Cyt b 5 and free hemin under the same conditions (Figure 5 and Table 1).
Figure 5.

Steady‐state rates of peroxidation as a function of A) guaiacol and B) ABTS concentration for N57C/S71C Cyt b 5 and WT Cyt b 5 (1 μm) with 6 m Gdn⋅HCl in potassium phosphate buffer (100 mm, pH 7.0) at 25 °C, with free hemin under the same conditions shown for comparison.
Table 1.
Kinetic parameters of guaiacol and ABTS oxidation with H2O2 catalyzed by WT Cyt b 5, c‐type Cyt b 5 and hemin in denaturing conditions in presence of 6 m Gdn⋅HCl. The parameters of some native peroxidases are also shown for comparison.
| Enzymes | Guaiacol | ABTS | ||||
|---|---|---|---|---|---|---|
| k cat [s−1] | K m [μm] | k cat/K m [m −1 s−1] | k cat [s−1] | K m [μm] | k cat/K m [m −1 s−1] | |
| N57C/S71C Cyt b5 | 1.41±0.01 | 38±1 | 37 100 | 122±1 | 86±2 | 1418 600 |
| WT Cyt b 5 | 0.46±0.01 | 40±2 | 11 500 | 10.6±0.1 | 122±2 | 86 890 |
| hemin | 0.43±0.01 | 134±3 | 3210 | 1.8±0.1 | 39±1 | 46 150 |
| CcP[a] | 4.1±0.3 | (53±6)×103 | 80 | – | – | – |
| lignin peroxidase[b] | 7.7 | 160 | 48 000 | – | – | – |
| HRP[c] | 420±40 | (5.8±0.7)×103 | 72 000 | 340±60 | 430±20 | 800 000 |
| HRP[d] | – | – | – | 332±18 | 233±21 | 1420 000 |
For guaiacol oxidation, the results showed that N57C/S71C Cyt b 5 exhibits a k cat value that is approximately threefold higher and a K m value that is similar when compared to the values for WT Cyt b 5. Although the overall catalytic efficiency (k cat/K m) of N57C/S71C Cyt b 5 is only approximately 3.2‐fold higher than that of WT Cyt b 5, it is much higher than that of native Cyt c peroxidase (CcP),18 close to that of lignin peroxidase,19 and almost half that of the efficient native peroxidase, HRP (Table 1).5 The enhanced activity is mainly attributable to the dramatically decreased K m value compared with those of native peroxidases, probably due to the exposure of the heme group that facilitates substrate binding, albeit with a comparatively lower k cat value. For ABTS oxidation, N57C/S71C Cyt b 5 exhibits a k cat value of 122 s−1, which is approximately 11.5‐fold and 67‐fold higher than that of WT Cyt b 5 and free hemin, respectively, which indicates the essential role of the covalently attached protein matrix in the activation of the oxidant H2O2. Although the k cat value is about threefold lower than that of native HRP, N57C/S71C Cyt b 5 has a K m value that is around three‐ to fivefold lower, which results in a catalytic efficiency of 1418 600 m −1 s−1, essentially equivalent to that of native HRP (1420 000 m −1 s−1).20 These observations demonstrate that c‐type Cyt b 5 is an efficient artificial peroxidase in the Gdn⋅HCl‐induced non‐native state.
The peroxidase reaction involves two steps, that is, formation of the reactive oxidation species (Compound I, oxoferryl heme π‐cation radical, and Compound II, oxoferryl heme) by reaction with H2O2, followed by a one‐electron oxidation of the substrates.4, 21 To provide more insights into the peroxidase activity of N57C/S71C Cyt b 5 in the non‐native state, we performed reactions with H2O2 and titration of the substrate separately. As presented in Figure 6A, stopped‐flow kinetic studies showed that N57C/S71C Cyt b 5 in 6 m Gdn⋅HCl readily reacts with H2O2 and results in a decrease in and redshift (4 nm) of the Soret bond from λ=413 to 417 nm. This observation is similar to that of Mb and its mutants when reacted with H2O2, which produces Compound II species.4, 14b,14c, 22 The time trace of the Soret band absorbance was found to be biphasic (Figure 6A, inset), with a rapid first phase (formation of Compound II) and a slower second phase, probably due to partial heme degradation, as observed previously for Mb mutants with a covalently linked heme that reacts with H2O2.14c The k obs for the first phase (0.24 s−1) was found to be about 3.8‐fold that of WT Cyt b 5 (0.064 s−1) under the same conditions (Figure S3A), which indicates that the covalently linked heme in N57C/S71C Cyt b 5 can efficiently produce oxidation species in the non‐native state.
Figure 6.
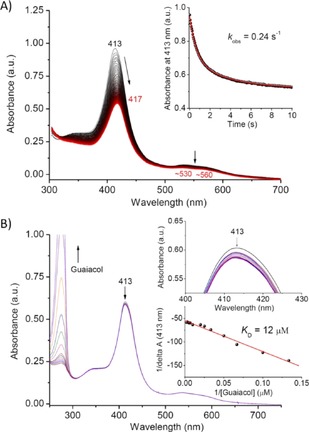
A) Stopped‐flow spectra of N57C/S71C Cyt b 5 (10 μm) in 6 m Gdn⋅HCl reacting with 100 mm H2O2 in potassium phosphate buffer (100 mm, pH 7.0) at 25 °C for 10 s. Inset: Time‐dependent change in the Soret band. B) UV/Vis spectra of titration of N57C/S71C Cyt b 5 (6 μm) in 6 m Gdn⋅HCl with guaiacol to a final concentration of 0.5 mm in the same buffer. Insets: The change in the Soret band and the double‐reciprocal plot of the change in Soret absorbance versus substrate concentration.
Conversely, upon titration of guaiacol in the absence of H2O2, the Soret band of N57C/S71C Cyt b 5 decreased slightly, which suggests the binding of guaiacol to the protein (Figure 6B). The double‐reciprocal plot of the Soret absorbance change versus substrate concentration showed that the binding has a K D value of 12 μm (Figure 6B, inset), which is about twofold lower that of WT Cyt b 5 under the same conditions (K D=22 μm; Figure S3B) and indicates a higher substrate binding affinity. This observation suggests that N57C/S71C Cyt b 5 in the non‐native state generates a heme pocket suitable for guaiacol binding. Moreover, a slight decrease in the Soret band of N57C/S71C Cyt b 5 was observed on titration with substrate ABTS (Figure S4). However, because ABTS has a broad strong absorption at around λ=345 nm, which overlaps that of free heme (λ≈370 nm), we did not perform titration of WT Cyt b 5 with ABTS.
Based on the above observations, the peroxidase activity of N57C/S71C Cyt b 5 in the non‐native state could be attributed to an uncovered heme distal site after Gdn⋅HCl‐induced unfolding, probably owing to dissociation of the axial His39 to a distal position and to the covalent heme attachment that involves the coordination of His63. The resultant conformation would facilitate the access and activation of H2O2 and also the binding of substrates. Moreover, the excess Gdn⋅HCl presumably plays a similar role to that of the conserved distal arginine in native peroxidases,5, 18, 19, 20 which works with a distal His as the well‐documented His–Arg pair to facilitate the formation of catalytic Compound II species for substrate oxidation.
In conclusion, we found that a c‐type Cyt b 5, N57C/S71C Cyt b 5, adopts a non‐native state with a covalently linked high‐spin heme after Gdn⋅HCl‐induced unfolding, presumably due to the uncovered distal site by dissociation of one axial ligand (His39). In this unique state, the protein exhibits an impressive peroxidase activity, which is essentially comparable to that of native peroxidases. This study demonstrates that an artificial metalloenzyme can be designed by exploring the catalytic performance of a metalloprotein in the non‐native state instead of the usual native state, which provides a novel alternative approach to traditional designs and might be generally applied to the design of artificial enzymes with advanced functions for potential applications.
Experimental Section
Experimental details, X‐ray crystal structure of WT Cyt b 5, and extra UV/Vis spectra of kinetic and titration studies are provided in the Supporting Information.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
It is a pleasure to acknowledge Prof. Zhong‐Xian Huang of Fudan University, China, for his kind gift of the Cyt b5 gene. EPR studies were performed at the Steady High Magnetic Field Facilities, High Magnetic Field Laboratory, CAS (Hefei, China). This work was supported by the National Science Foundation of China (31370812), the Hunan Provincial Natural Science Foundation (2015JJ1012), and the Scientific Research Fund of Hunan Provincial Education Department (15A158).
S. Hu, B. He, K.-J. Du, X.-J. Wang, S.-Q. Gao, Y.-W. Lin, ChemistryOpen 2017, 6, 325.
References
- 1.
- 1a. Lu Y., Yeung N., Sieracki N., Marshall N. M., Nature 2009, 460, 855–862; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1b. Nanda V., Koder R. L., Nat. Chem. 2010, 2, 15–24; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1c. Sontz P. A., Song W. J., Tezcan F. A., Curr. Opin. Chem. Biol. 2014, 19, 42–49; [DOI] [PubMed] [Google Scholar]
- 1d. Petrik I. D., Liu J., Lu Y., Curr. Opin. Chem. Biol. 2014, 19, 67–75; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1e. Dürrenberger M., Ward T. R., Curr. Opin. Chem. Biol. 2014, 19, 99–106; [DOI] [PubMed] [Google Scholar]
- 1f. Oohora K., Hayashi T., Curr. Opin. Chem. Biol. 2014, 19, 154–161; [DOI] [PubMed] [Google Scholar]
- 1g. Shoji O., Watanabe Y., J. Biol. Inorg. Chem. 2014, 19, 529–539; [DOI] [PubMed] [Google Scholar]
- 1h. Hu C., Chan S. I., Sawyer E. B., Yu Y., Wang J., Chem. Soc. Rev. 2014, 43, 6498–6510; [DOI] [PubMed] [Google Scholar]
- 1i. Matsuo T., Hirota S., Bioorg. Med. Chem. 2014, 22, 5638–5656; [DOI] [PubMed] [Google Scholar]
- 1j. Pordea A., Curr. Opin. Chem. Biol. 2015, 25, 124–132; [DOI] [PubMed] [Google Scholar]
- 1k. Peacock A. F., Curr. Opin. Chem. Biol. 2016, 31, 160–165; [DOI] [PubMed] [Google Scholar]
- 1l. Hyster T. K., Ward T. R., Angew. Chem. Int. Ed. 2016, 55, 7344–7357; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 7468–7482; [Google Scholar]
- 1m. Nastri F., Chino M., Maglio O., Bhagi-Damodaran A., Lu Y., Lombardi A., Chem. Soc. Rev. 2016, 45, 5020–5054; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1n. Hoarau M., Hureau C., Gras E., Faller P., Coord. Chem. Rev. 2016, 308, 445–459; [Google Scholar]
- 1o. Lin Y.-W., Coord. Chem. Rev. 2017, 336, 1–27. [Google Scholar]
- 2. Yu Y., Cui C., Liu X., Petrik I. D., Wang J., Lu Y., J. Am. Chem. Soc. 2015, 137, 11570–11573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Miner K. D., Mukherjee A., Gao Y. G., Null E. L., Petrik I. D., Zhao X., Yeung N., Robinson H., Lu Y., Angew. Chem. Int. Ed. 2012, 51, 5589–5592; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 5687–5690. [Google Scholar]
- 4. Matsuo T., Fukumoto K., Watanabe T., Hayashi T., Chem. Asian J. 2011, 6, 2491–2499. [DOI] [PubMed] [Google Scholar]
- 5. Savenkova M. I., Kuo J. M., Ortiz de Montellano P. R., Biochemistry 1998, 37, 10828–10836. [DOI] [PubMed] [Google Scholar]
- 6. Lin Y.-W., Wang J., J. Inorg. Biochem. 2013, 129, 162–171. [DOI] [PubMed] [Google Scholar]
- 7.
- 7a. Ying T., Wang Z.-H., Lin Y.-W., Xie J., Tan X., Huang Z.-X., Chem. Commun. 2009, 4512–4514; [DOI] [PubMed] [Google Scholar]
- 7b. Ying T., Wang Z.-H., Zhong F., Tan X., Huang Z.-X., Chem. Commun. 2010, 46, 3541–3543; [DOI] [PubMed] [Google Scholar]
- 7c. Wang Z., Matsuo T., Nagao S., Hirota S., Org. Biomol. Chem. 2011, 9, 4766–4769; [DOI] [PubMed] [Google Scholar]
- 7d. Wang Z., Ando Y., Nugraheni A. D., Ren C., Nagao S., Hirota S., Mol. Biosyst. 2014, 10, 3130–3137; [DOI] [PubMed] [Google Scholar]
- 7e. Kobayashi H., Nagao S., Hirota S., Angew. Chem. Int. Ed. 2016, 55, 14019–14022; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 14225–14228. [Google Scholar]
- 8.
- 8a. Du J.-F., Li W., Li L., Wen G.-B., Lin Y.-W., Tan X., ChemistryOpen 2015, 4, 97–101; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8b. Amacher J. F., Zhong F., Lisi G. P., Zhu M. Q., Alden S. L., Hoke K. R., Madden D. R., Pletneva E. V., J. Am. Chem. Soc. 2015, 137, 8435–8449; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8c. Pandiscia L. A., Schweitzer-Stenner R., J. Phys. Chem. B 2015, 119, 12846–12859; [DOI] [PubMed] [Google Scholar]
- 8d. Malyshka D., Schweitzer-Stenner R., Chemistry 2017, 23, 1151–1156. [DOI] [PubMed] [Google Scholar]
- 9.
- 9a. Morioka C., Tachi Y., Suzuki S., Itoh S., J. Am. Chem. Soc. 2006, 128, 6788–6789; [DOI] [PubMed] [Google Scholar]
- 9b. Suzuki K., Shimokawa C., Morioka C., Itoh S., Biochemistry 2008, 47, 7108–7115. [DOI] [PubMed] [Google Scholar]
- 10. Durley R. C., Mathews F. S., Acta Crystallogr. Sect. D 1996, 52, 65–76. [DOI] [PubMed] [Google Scholar]
- 11. Lin Y.-W., Wang W.-H., Zhang Q., Lu H.-J., Yang P.-Y., Xie Y., Huang Z.-X., Wu H.-M., ChemBioChem 2005, 6, 1356–1359. [DOI] [PubMed] [Google Scholar]
- 12. Lin Y.-W., Wu Y.-M., Liao L.-F., Nie C.-M., J. Mol. Model. 2012, 18, 1553–1560. [DOI] [PubMed] [Google Scholar]
- 13. Perez-Iratxeta C., Andrade-Navarro M. A., BMC Struct. Biol. 2008, 8, 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.
- 14a. Witting P. K., Mauk A. G., Lay P. A., Biochemistry 2002, 41, 11495–11503; [DOI] [PubMed] [Google Scholar]
- 14b. Yan D.-J., Li W., Xiang Y., Wen G.-B., Lin Y.-W., Tan X., ChemBioChem 2015, 16, 47–50; [DOI] [PubMed] [Google Scholar]
- 14c. Yan D.-J., Yuan H., Li W., Xiang Y., He B., Nie C.-M., Wen G.-B., Lin Y.-W., Tan X., Dalton Trans. 2015, 44, 18815–18822. [DOI] [PubMed] [Google Scholar]
- 15. Davydov R., Hoffman B. M., J. Biol. Inorg. Chem. 2008, 13, 357–369. [DOI] [PubMed] [Google Scholar]
- 16. Lin Y.-W., Yeung N., Gao Y.-G., Miner K. D., Lei L., Robinson H., Lu Y., J. Am. Chem. Soc. 2010, 132, 9970–9972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.
- 17a. Wang W.-H., Wang Y.-H., Lu J.-X., Wang J.-H., Xie Y., Huang Z.-X., Chem. Lett. 2002, 31, 674–675; [Google Scholar]
- 17b. Lin Y.-W., You X.-X., Chen L.-S., Wu Y.-M., Chem. Lett. 2012, 41, 1574–1575. [Google Scholar]
- 18. Murphy E. J., Metcalfe C. L., Nnamchi C., Moody P. C., Raven E. L., FEBS J. 2012, 279, 1632–1639. [DOI] [PubMed] [Google Scholar]
- 19. Koduri R. S., Tien M., J. Biol. Chem. 1995, 270, 22254–22258. [DOI] [PubMed] [Google Scholar]
- 20. Fruk L., Muller J., Niemeyer C. M., Chemistry 2006, 12, 7448–7457. [DOI] [PubMed] [Google Scholar]
- 21.
- 21a. Ozaki S.-i., Matsui T., Roach M. P., Watanabe Y., Coord. Chem. Rev. 2000, 198, 39–59; [Google Scholar]
- 21b. Hayashi T., Hisaeda Y., Acc. Chem. Res. 2002, 35, 35–43. [DOI] [PubMed] [Google Scholar]
- 22. Matsui T., Ozaki S., Liong E., G. N. Phillips, Jr. , Watanabe Y., J. Biol. Chem. 1999, 274, 2838–2844. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary