

Abstract
Small-molecule inhibitors of Bromodomain and Extra Terminal proteins (BET), including BRD2, BRD3 and BRD4 proteins have therapeutic potential for the treatment of human cancers and other diseases and conditions. In this paper, we report the design, synthesis and evaluation of γ-carboline-containing compounds as a new class of small molecule BET inhibitors. The most potent inhibitor (compound 18, RX-37) obtained from this study binds to BET bromodomain proteins (BRD2, BRD3 and BRD4) with Ki values of 3.2–24.7 nM and demonstrates high selectivity over other non-BET bromodomain-containing proteins. Compound 18 potently and selectively inhibits cell growth in human acute leukemia cell lines harboring the rearranged mixed lineage leukemia 1 gene. We have determined a co-crystal structure of 18 in complex with BRD4 BD2 at 1.4 Å resolution, which provides a solid structural basis for the compound’s high binding affinity and for its further structure-based optimization. Compound 18 represents a promising lead compound for the development of a new class of therapeutics for the treatment of human cancer and other conditions.
Introduction
There are 46 bromodomain-containing proteins encoded in the human genome, and these are divided approximately into 8 subfamilies1. By binding to acetylated lysine residues (Ac-K) on histone tails, bromodomain proteins function as epigenetic “readers” and play a key role in epigenetic regulation of gene transcription2. One bromodomain subfamily, the Bromodomain and Extra Terminal (BET) family proteins (BRD2, BRD3, BRD4 and BRDT), has emerged as a class of new therapeutic targets for human diseases and conditions, including cancers, diabetes, HIV infection, inflammation, acute heart failure and male contraception3–7.
Several classes of potent and specific small-molecular BET bromodomain inhibitors (BET inhibitors) have recently been reported8–17, and include JQ-1 (1)3, I-BET 762 (2)4, I-BET 151 (3)18 and OTX015 (4)19 (Figure 1). These potent and selective BET inhibitors have provided a set of powerful pharmacological tools which support further investigation of the roles of BET proteins in different cellular processes and the therapeutic potential of BET inhibition in human diseases and conditions3, 4, 18, 19. For example, JQ-1 and other BET inhibitors are particularly effective in vitro and in vivo against NUT-midline carcinoma (NMC), a rare cancer characterized by the NUT-BRD4 chromosomal rearrangement20. BET inhibitors are also very effective in vitro and in vivo against acute leukemia harboring the mixed lineage leukemia 1 (MLL1) gene rearrangement, in which BRD4 functions as a co-activator for MLL1 gene transcription18. BET inhibitors may have a promising therapeutic potential for the treatment of multiple myeloma by down-regulation of the oncogene c-Myc21. By interacting directly with the androgen receptor (AR), BET BRD2- BRD4 proteins function as co-activators of AR, and inhibition of these BET proteins is effective in vitro and in vivo against castration-resistant prostate cancer22. Several BET inhibitors, including I-BET-762 (2), OTX015 (4), TEN-010 (structure not disclosed), CPI- 0610 (structure not disclosed), and BAY1238097 (structure not disclosed) have recently been advanced into human clinical trials for the treatment of different forms of human cancer23–27.
Figure 1.
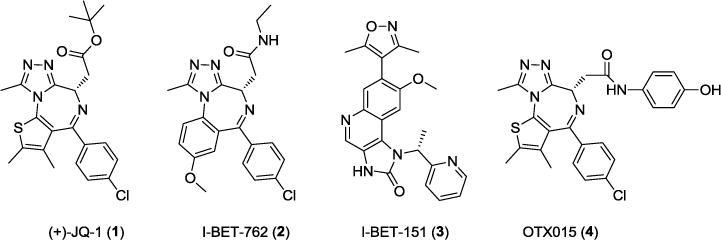
Structures of known BET bromodomain inhibitors.
Despite the discovery of these BET inhibitors, new, potent and specific BET inhibitors with different chemotypes are needed in order fully to exploit the therapeutic potential of BET inhibition in different human diseases. It is well known that drugs with the same therapeutic targets can demonstrate different toxicity and pharmacological profiles, due for example to their on-target and off-target toxicities and different pharmacokinetics, resulting in different clinical applications. In the present study, we report the design, synthesis and evaluation of a new class of potent and specific small molecule BET inhibitors, the [6,5,6] tricyclic 5H-pyrido[4,3-b]indoles.
Results and Discussion
Design of novel BET inhibitors containing a [6,5,6] tricyclic 5H-pyrido[4,3-b]indole system
The design of a new class of BET inhibitors began with an analysis of the co-crystal structure of 3 complexed with the BRD4 BD1 protein (Figure 2A)18. The interactions of 3 with the BRD4 BD1 protein involve three distinct regions: (1) the “head”, a 3,5-dimethylisoxazole fragment, which participates in a direct or water-bridged hydrogen bonding network with N140 and Y97 of BRD4 BD1 in the histone acetyl lysine (Ac-K) binding pocket; (2) the “body”, a [6,6,5] tricyclic ring system which interacts with the channel in which the four-carbon hydrophobic portion of the lysine residue on the histone tails also binds; and (3) the “tail”, a pyridyl group which interacts with a surface hydrophobic pocket, formed by TRP81, PRO82, MET149, and ILE146 of BRD4 BD128–35. Compound 3 also binds to the BRD4 BD2 protein with high affinity29, 36 but the co-crystal structure of 3 complexed with the BRD4 BD2 protein is not available and accordingly, we modeled the complex of 3 with the BRD4 BD2 protein (Figure 2B), to gain a structural understanding of their interactions. Comparison of the modeled structure (Figure 2B) with the co-crystal structure (Figure 2A) shows that the interactions of 3 with the BD1 and BD2 domains of BRD4 are similar but not identical.
Figure 2.

(A). Co-crystal structure of compound 3 (I-BET 151) in a complex with BRD4 BD1 (PDBID: 3ZYU). (B). Modeled structure of I-BET151 in a complex with BRD4 BD2. I-BET-151 is shown in stick with carbon atoms colored in orange in (A) or in yellow in (B), oxygen atoms in red and nitrogen atoms in blue. Water molecules are shown as red spheres and the hydrogen bonds are denoted by red dash lines. Three regions of BRD4 BD1 interacting with I-BET 151 are labeled as Head, Body and Tail. Figures were prepared using PyMOL.
For our design of new classes of BET inhibitors, we decided first to keep the 3,5-dimethylisoxazole “head” group unchanged but to replace the [6,6,5] tricyclic group in 3 with other chemical scaffolds. As discussed above, the [6,6,5] tricyclic group in 3 binds to a hydrophobic channel in the BRD4 BD1 and BD2 proteins and the nitrogen atom in the central ring in 3 forms water-bridged hydrogen bonds with the proteins by functioning as a hydrogen bond acceptor. We explored the replacement of the [6,6,5] tricyclic system in 3 with a [6,5,6] tricyclic 5H-pyrido[4,3-b]indole, also called γ-carboline system in our design. Our modeling (Figure 3) suggested that the phenyl group in our [6,5,6] tricyclic system can closely mimic the phenyl ring in the [6,6,5] tricyclic system in 3. The NH of the indole ring, functions as a hydrogen donor, mimicking the pyridine nitrogen in the [6,6,5] system in the water-bridged hydrogen bonding network. Additionally, the pyridine ring in the new [6,5,6] tricyclic system enjoys close interactions with the conserved TRP residue in BD1 (TRP81) and BD2 (TRP374) domains of BRD4. It is notable that this new [6,5,6] tricyclic system has balanced physiochemical properties.
Figure 3.
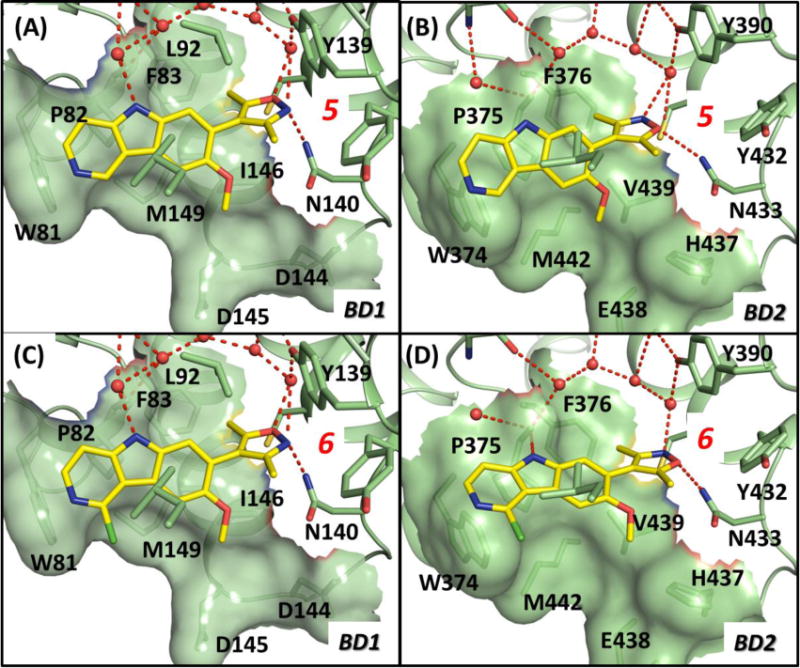
Predicted binding models of compounds 5 and 6 complexed with BRD4 BD1 and BD2 domains. (A). Binding model for compound 5 in a complex with BRD4 BD1. (B). Binding model for compound 5 in a complex with BRD4 BD2. (C). Binding model for compound 6 in a complex with BRD4 BD1. (D). Binding model for compound 6 in a complex with BRD4 BD2. Crystal structures of BRD4 BD1 (PDB ID: 3ZYU) and BRD4 BD2 (PDB ID: 2YEM) were used for model construction. Compounds are shown in stick with yellow color for carbon atoms, blue color for nitrogen atom and red color for oxygen atoms. Side chains of key interacting residues are shown in stick. Figures were prepared using PyMOL.
To test experimentally whether this tricyclic system is suitable for the design of a new class of BET inhibitors, we first synthesized compounds 5 and 6 (Figure 4), which contain only the “head” group and the new [6,5,6] tricyclic system as the “body”. We evaluated compounds 5 and 6 for their binding affinities to the BD1 and BD2 domain proteins of BRD4 using our optimized competitive fluorescence-polarization (FP) assays. Since both 5 and 6 lack an appropriate hydrophobic group to occupy the hydrophobic pocket in both BD1 and BD2 domains of BRD4, we expected that they may bind to BRD4 BD1 and BD2 proteins with only moderate affinities.
Figure 4.

Chemical structure of new BET inhibitors.
Our binding data confirm this, showing that 5 binds to BRD4 BD1 and BD2 domain proteins with Ki values of 1644 nM and 824 nM, respectively, while 6 has Ki values of 305 nM and 194 nM, respectively, to the BD1 and BD2 domains in BRD4 (Table 1). While both compounds 5 and 6 are much less potent than those optimized BET inhibitors 1–4 in binding to BRD4, we expected that their binding affinities could be improved significantly upon installation of an appropriate group to interact with the hydrophobic pocket in the BRD4 BD1 and BD2 proteins.
Table 1.
Structure-activity relationship studies of the “head group”.
| ID | BRD4 BD1 | BRD4 BD2 | ||
|---|---|---|---|---|
| IC50 (nM) | Ki (nM) | IC50 (nM) | Ki (nM) | |
| 1 | 28.7±1.7 | 7.6±0.4 | 35.7±3.6 | 10.7±1.1 |
| 2 | 156±15 | 38.8±5.0 | 98.4±3.3 | 32.3±3.8 |
| 3 | 31.7±7.7 | 9.0±2.9 | 226±44 | 74.8±8.6 |
| 4 | 25.5±1.0 | 10.9±0.6 | 16.6±1.1 | 6.0±0.3 |
| 5 | 4592±72 | 1644±71 | 2691±460 | 824±25 |
| 6 | 862±100 | 305±26 | 579±71 | 194±24 |
| 7 | 3868±1972 | 1243±549 | 1817±136 | 478±69 |
| 8 | 9443±3797 | 2814±782 | 13826±3706 | 2182±132 |
| 9 | >10000 | > 10000 | 73970±7844 | 8322±1272 |
| 10 | 4874±320 | 1726±17 | 2580±80 | 867±107 |
| 11 | 14022±781 | 4842±29 | 6175±549 | 1948±175 |
Structure-activity relationship studies of the “head group”
In our modeled structure of compound 6 complexed with BD1 of BRD4 (Figure 3C), the 3,5-dimethylisoxazole moiety in 6, the “head group”, forms hydrogen bonds with N140 and Y97 via a bridging water molecule in the Ac-K binding pocket of the protein and one of the two methyl groups has hydrophobic contacts with the protein. Similar interactions were observed in the predicted model of compound 6 in a complex with the BRD4 BD2 protein (Figure 3D).
To probe the structure-activity relationships at this site, we synthesized a number of new compounds (Figure 4) with different head groups and determined their binding affinities to BRD4 BD1 and BD2 proteins. The results are summarized in Table 1.
Replacing the 3-methyl or the 5-methyl group in 6 with an ethyl group yielded 7 and 8, respectively. Compound 7 binds to BRD4 BD1 and BD2 with Ki values of 1243 nM and 478 nM respectively, and is 2–4 times weaker than compound 6. Compound 8 binds to BRD4 BD1 and BD2 with Ki values of 2814 nM and 2182 nM, and is >5 times weaker than 6. Replacing both the 3-methyl and 5-methyl groups with ethyl resulted in 9, which has a very weak binding affinity to both BRD4 BD1 and BD2 domains. Changing the 3,5-dimethylisoxazole group in 6 to 3,5-dimethyl-1H-pyrazole led to compound 10 which, with Ki values of 1726 nM and 867 nM to BRD4 BD1 and BD2 is 4-times less potent than 6. Replacing the 3,5-dimethyl-1H-pyrazole group in 10 with 1,3,5-trimethyl-1H-pyrazole yielded 11, which is 2-times less potent than 10 in binding to both BRD4 BD1 and BD2 domains.
Hence, among these head groups we explored, the 3,5-dimethylisoxazole group remains the best for binding to both BRD4 BD1 and BD2 proteins.
Structure-activity relationship studies of the “tail” group
Comparison of the modeled structure of 6 in a complex with the BRD4 BD1 protein (Figure 3C) with the co-crystal structure of 3 complexed with the BRD4 BD1 protein (Figure 2) clearly showed that 6 lacks a group that can interact with the hydrophobic pocket formed by the hydrophobic residues TRP81, PRO82, MET149, and ILE146. Hence, introduction of an appropriate group in 6 targeting this hydrophobic pocket should enhance its binding affinity to BRD4.
Analysis of our modeled structures for compounds 5 and 6 in a complex with the BD1 and BD2 domains of BRD4 showed that the Cl substitution in 6 is directly above the hydrophobic pocket, making this an attractive site at which to append an appropriate hydrophobic group for interactions with those hydrophobic residues in the pocket. Interestingly, modeling suggested that replacement of the Cl atom in compound 6 with a 3,5-dimethylisoxazole group which can occupy the hydrophobic pocket, resulted in 12 (Figure 5A and 5B) which was found to bind to BRD4 BD1 and BD2 proteins with Ki values of 47.8 nM and 70.1 nM respectively, and is thus 3-times more potent than compound 6.
Figure 5.

Predicted binding models of 12 and 13 in a complex with BRD4 BD1 and BD2 domains. (A). Binding model for compound 12 complexed with BRD4 BD1. (B). Binding model for compound 12 in a complex with BRD4 BD2. (C). Binding model for compound 13 in a complex with BRD4 BD1. (D). Binding model for compound 13 in a complex with BRD4 BD2. Crystal structures of BRD4 BD1 (PDB ID: 3ZYU) and BRD4 BD2 (PDB ID: 2YEM) were used for model construction. Compounds are shown in stick with yellow color for carbon atoms, blue color for nitrogen atom and red color for oxygen atoms. Side chains of key interacting residues are shown in stick. Figures were prepared using PyMOL.
We next synthesized compound 13 (Figure 4), in which the 3,5-dimethylisoxazole group was replaced by 3,5-dimethyl-1H-pyrazole. The resulting compound (13) has Ki values of 98.8 nM and 100 nM to BRD4 BD1 and BD2 domains, respectively, and is slightly less potent than 12.
Since analogs of 1H-pyrazoles are structurally more versatile than those of isoxazoles, 1H-pyrazole ring systems were used for further modifications at this site. Modeling suggested that one of the two methyl groups in the 3,5-dimethyl-1H-pyrazole moiety in compound 13 could interact with hydrophobic residues in BD1 and BD2 of BRD4, while the other methyl group is exposed to the solvent environment (Figure 5C and 5D). To investigate the roles of these methyl groups, we synthesized compound 14, in which a 1H-pyrazole group replaces the 3,5-dimethyl-1H-pyrazole group in 13. Compound 14 has Ki values of 247 nM and 201 nM to BRD4 BD1 and BD2 domains, respectively, and is thus 2-times less potent than 13. Changing the 3,5-dimethyl-1H-pyrazole group in 13 to 1,3,5-trimethyl-1H-pyrazole led to 15, which has affinities to both BRD4 BD1 and BD2 domains similar to those of 13. Replacing the 3,5-dimethyl-1H-pyrazole group in 13 with 3,5-dimethyl-1-phenyl-1H-pyrazole resulted in compound 16, which has Ki values of 103 nM and 98.1 nM to BRD4 BD1 and BD2 domains, respectively, and is equipotent with 13 (Table 2).
Table 2.
Structure-activity relationship of compounds with modifications of the “tail” group
| ID | BRD4BD1 | BRD4 BD2 | ||
|---|---|---|---|---|
| IC50 (nM) | Ki (nM) | IC50 (nM) | Ki (nM) | |
| 3 | 31.7±7.7 | 9.0±2.9 | 226±44 | 74.8±8.6 |
| 12 | 134±7 | 47.8±1.0 | 221±38 | 70.1±2.0 |
| 13 | 276±44 | 98.8±11.6 | 324±8 | 100±16 |
| 14 | 702±53 | 247±29 | 658±67 | 201±5 |
| 15 | 327±11 | 116±5 | 481±86 | 134±42 |
| 16 | 301±37 | 103±3 | 316±24 | 98.1±6.1 |
| 17 | 131±11 | 44.1±6.4 | 61.9±13.9 | 16.1±2.8 |
| 18 | 75.5±6.2 | 24.7±1.0 | 36.5±8.9 | 12.2±1.6 |
| 19 | 80.6±1 | 26.9±1.0 | 129±45 | 38.0±2.2 |
Analysis of the binding models for compound 13 in complexes with BRD4 BD1 and BD2 domains suggested that replacement of one of the methyl groups in 13 with a larger group such as ethyl or cyclopropyl may further enhance the hydrophobic interactions with BD1 and BD2 domains of BRD4. Indeed compound 17, in which the 3,5-dimethyl-1H-pyrazole group in 13 is replaced by a 3,5-diethyl-1H-pyrazole group has Ki values of 44.1 nM and 16.1 nM to BRD4 BD1 and BD2 domains, respectively. Compound 17 is therefore 2- and 6-times more potent than 13 in binding to BRD4 BD1 and BD2 domains, respectively. Changing the 3,5-dimethyl-1H-pyrazole group in 13 to a 3-cyclopropyl-5-methyl-1H-pyrazole group yielded compound 18, which has Ki values of 24.7 nM and 12.2 nM to BRD4 BD1 and BD2 domains, respectively. Replacement of the 3,5-dimethyl-1H-pyrazole group in 13 with a 3-phenyl-5-methyl-1H-pyrazole group led to compound 19, which has Ki values of 26.9 nM and 38.0 nM to BRD4 BD1 and BD2 domains, respectively. Hence, our modifications at this site have yielded compounds 17, 18 and 19 with much improved binding affinities over those of our initial compounds 5 and 6.
Assessment of the binding affinities to BRD2 and BRD3 proteins
In addition to BRD4, the BRD2 and BRD3 proteins have also been implicated as important therapeutic targets in a number of human diseases37–39. We determined the binding affinities of 17, 18 and 19, our three most potent BET inhibitors to both the BD1 and BD2 domain proteins of BRD2 and BRD3, with compounds 1–4 included as the positive controls. The results are summarized in Table 3.
Table 3.
Binding affinities of reference BET inhibitors and compounds 17–19 to BRD2, BRD3 and BRD4 BD1 and BD2 proteins.
| ID | Ki (nM) | |||||
|---|---|---|---|---|---|---|
| BRD2 | BRD3 | BRD4 | ||||
| BD1 | BD2 | BD1 | BD2 | BD1 | BD2 | |
| 1 | 13.2±4.5 | 12.5±2.7 | 6.6±1.2 | 8.9±1.6 | 7.6±1.6 | 10.71.67 |
| 2 | 56.8±10.0 | 49.2±6.5 | 53.9±6.0 | 30.3±4.5 | 38.8±5.0 | 32.3±3.8 |
| 3 | 9.0±4.3 | 49.6±10.8 | 7.2±3.0 | 22.3±3.5 | 9.0±2.9 | 74.8±8.6 |
| 4 | 16.6±6.6 | 5.4±.46 | 10.7±0.7 | 4.0±.07 | 10.9±0.9 | 6.0±.09 |
| 17 | 21.0±3.3 | 15.4±3.2 | 12.9±2.9 | 4.2±0.4 | 44.1±6.4 | 16.1±2.8 |
| 18 | 11.1±1.0 | 11.7±3.0 | 7.3±0.1 | 3.2±0.5 | 24.7±1.0 | 12.2±1.6 |
| 19 | 12.2±1.7 | 22.2±2.8 | 10.4±1.0 | 9.4±1.0 | 26.9±1.0 | 38.0±2.2 |
Compounds 17, 18 and 19 have high affinities to both the BD1 and BD2 domain proteins in BRD2 and BRD3. Compound 18 is the most potent inhibitor with Ki values of 11.1 nM and 11.7 nM to BRD2 BD1 and BD2 domains, and 7.3 nM and 3.2 nM to BRD3 BD1 and BD2 domains, respectively.
Further assessment of the binding affinities of compound 18 to BRD2-4 proteins and the binding specificity over other non-BET bromodomain containing proteins
We evaluated the binding affinities of compound 18 to BRD2-4 BD1 and BD2 domain proteins and 9 representative non-BET bromodomain-containing proteins from 7 other subfamilies using Bio-Layer Interferometry, a label-free technique for determination of the binding affinities (Kd) between molecules39, 40. Bio-Layer Interferometry allows further assessment of the binding affinities of our most potent compound 18 to BRD2-4 BD1 and BD2 domains and importantly, its selectivity over other subfamilies of bromodomain-containing proteins. Compounds 1–3 were included in these experiments as control compounds and the results are shown in Table 4.
Table 4.
Binding affinities of reference compounds 1–3 and new compounds 18 to BET proteins and representative bromodomain proteins in other 7 sub-families using Bio-layer Interferometry method.
| KD (nM) | ||||
|---|---|---|---|---|
| 1 | 2 | 3 | 18 | |
| BRD2(BD1) | 14.7±1.9 | 159±11 | 54.8±7.8 | 48.1±4.0 |
| BRD2(BD2) | 6.2±1.8 | 45.4±1.0 | 70.3±7.3 | 29.6±6.9 |
| BRD3 (BD1) | 13.6±1.0 | 60.7±10.3 | 29.8±7.3 | 17.7±3.6 |
| BRD3 (BD2) | 11.7±1.5 | 40.7±8.5 | 40.5±7.2 | 16.3±4.1 |
| BRD4 (BD1) | 12.8±2.9 | 99.4±5.8 | 52.8±9.2 | 47.0±14.8 |
| BRD4 (BD2) | 6.7±0.7 | 47.6±2.7 | 215±29 | 44.6±22.1 |
| CREBBP | > 10000 | 3084 | 670 | |
| ATAD2A | > 10000 | > 10000 | > 10000 | |
| ATAD2B | > 10000 | |||
| TRIM24 | ~ 10000 | |||
| BAZ2B | > 10000 | |||
| MLL1 | 9500 | |||
| TAF1B2 | > 10000 | |||
| BRG1 | > 10000 | |||
| PB1BR5 | > 10000 | |||
Compound 18 has Kd values of 16.3–48.1 nM with BRD2-BRD4 BD1 and BD2 domain proteins, which are consistent with the data obtained from the FP-based, competitive binding assays (Table 3). Nine non-BET bromodomain-containing proteins were evaluated and compound 18 was found to display a moderate affinity to CREBBP protein (Kd = 670 nM) but has very low affinities (Kd values ≥ 10,000 nM) for the 8 other bromodomain proteins. The CREBBP and BRD2-4 proteins, it should be noted, share a high sequence homology and have similar binding pockets1. Hence, compound 18 displays high binding affinities to BRD2-4 BD1 and BD2 domains and shows an excellent selectivity over other non-BET bromodomain-containing proteins with the exception of CREBBP.
Determination of co-crystal structure of compound 18 in a complex with the BRD4 BD2 protein
To understand the structural basis for the high binding affinity of compound 18 to BRD4 BD1 and BD2 proteins, we determined the co-crystal structure of 18 in complexes with both BD1 and BD2 domains of BRD4. We successfully obtained a co-crystal structure of 18 in a complex with BRD4 BD2 at 1.4 Å resolution (Figure 6).
Figure 6.

Crystal structure of BRD4 BD2 bound to 18. Orthogonal views of 18 (green sticks) in the binding site of BRD4 BD2 are shown in panels A and B. The dashed black lines in panel A depict the hydrogen bonds formed between three bridging water molecules (red spheres) and the protein. One water molecule bridges the backbone carbonyl of K378 with the indole NH group of the tricyclic system; a second bridges a pyrrole nitrogen with E438, and a third mediates the interaction between the oxygen and nitrogen of the 3,5-dimethylisoxazole group and Y390. The dashed line in panel B depicts the hydrogen bond that exists between the oxygen of the 3,5-dimethylisoxazole group and N433. Panel C shows an omit map of the protein refined without compound 18 present and is shown as a green grid contoured at 3σ. The coordinates have been deposited into the Protein Data Bank with code ID: 4Z93.
The co-crystal structure shows that both the nitrogen and oxygen atoms in the 3,5-dimethylisoxazole group of 18 form hydrogen bonds with the hydroxyl group of Try390 of BRD4 BD2, through a “conserved” water molecule, which has been found in all the co-crystal structures with different classes of BET inhibitors. The oxygen atom in the 3,5-dimethylisoxazole group also forms a weak hydrogen bond with the NH2 group of Asn343. One of the methyl groups has hydrophobic interactions with Pro375 and Phe376 and the other methyl group is involved in hydrophobic contacts with Leu387, Tyr390 and Tyr432 residues. The [6,5,6] tricyclic system binds to the channel occupied by the hydrophobic portion of the Lys side chain in the histone peptide and is sandwiched between Leu385 on one side and Pro375 and Trp374 on the other. The indole NH group in the center of the tricyclic ring system forms a hydrogen bond with the backbone carbonyl group of Lys378 via a bridge water molecule. The cyclopropyl group in the “tail” inserts into a well-defined pocket, formed by Val439 and Met442 at the bottom and Pro375, Trp374, Glu438 and His437 at the sides.
Recent studies have suggested that the BD1 and BD2 domains of BRD4 have different biological functions and the design of small-molecule inhibitors that can selectively target either of these two domains is of interest. In an effort to gain an understanding of the similarities and differences in their interactions with these two different domains of BRD4, we compared the co-crystal structure of 18 complexed with BRD4 BD2 (Figure 6) with the co-crystal structure of 3 complexed with BRD4 BD1 (Figure 1A). Superposition of these two co-crystal structures showed that compounds 3 and 18 assume binding models that are very similar but have some key differences. The methoxyl group and the cyclopropyl group in 18 have interactions with His437 in BD2, which is replaced by Asp144 in BD1. The Glu438 in BD2, together with His437, forms the outside wall of a well-defined binding pocket for interactions with the cyclopropyl group in 18. Because Glu438 in BD2 is replaced by Asp145 in BD1 and His437 in BD2 is replaced by Asp144 in BD1, the BD1 binding pocket where the pyridine group in 3 binds is much shallower than the corresponding pocket in BD2. These differences may be explored for the design of small-molecule inhibitors selective for either the BD1 or the BD2 domain.
Determination of cell growth inhibition activity and specificity of BET inhibitors in acute leukemia cell lines
Potent and specific BET inhibitors such as 1, 2 and 3 effectively inhibit cell growth in human acute leukemias such as the MV4;11 and MOLM-13 cell lines,18 which contain the MLL1 chromosomal rearrangement. Importantly, specific BET inhibitors should demonstrate cellular selectivity over human acute leukemia cell lines harboring different gene rearrangements such as the K562 cell line, which harbors the BCR-ABL fusion protein but no MLL1 fusion protein40. We tested our three most potent BET inhibitors (17–19) for their cellular activity and specificity in the MV4;11, MOLM-13 and K562 cell lines and compared them directly to compounds 1–3 in these experiments. The data are summarized in Table 5.
Table 5.
Cell growth inhibitory activity of reference BET inhibitors 1–3 and new compounds 17–19 in acute leukemia cell lines.
| Compound | IC50 (nM) | ||
|---|---|---|---|
| MV4; 11 | MOLM-13 | K562 | |
| 1 | 24±19 | 56±24 | >2000 |
| 2 | 93±45 | 241±58 | >2000 |
| 3 | 162±112 | 228±52 | >2000 |
| 17 | 23±10 | 78±12 | >2000 |
| 18 | 20±9 | 66±14 | >2000 |
| 19 | 34±15 | 144±31 | >2000 |
Compounds 17–19 potently inhibit the viability of MV4;11 and MOLM-13 cells. For example, 18 has IC50 values of 20 nM and 66 nM in inhibition of cell viability in the MV4;11 and MOLM-13 cell lines, respectively. Compound 17 has similar potencies in the same cell lines when compared to 18 but 19 is slightly less potent than 17 and 18 in both cell lines. Significantly, compounds 17–19 have IC50 values of >2,000 nM in inhibition of cell growth in the K562 cell line harboring BCR-ABL fusion protein, thus displaying excellent cellular specificity.
Chemistry
The general synthetic route to compounds 6–11 is shown in Scheme 1. Compound 20 was prepared in four steps following a published method41. Selective bromination of 20 gave the 7-bromo compound 21. Suzuki coupling of 21 with diverse pinacol boronates (22–27) furnished 6–11. Removal of the Cl atom of 6 using Pd/C in a hydrogen atmosphere gave 5 (Scheme 2). Compounds 12–19 were obtained after a second Suzuki coupling reaction of 6 with pinacol boronates (Scheme 2). The syntheses of pinacol boronates 23–25, 29–32 employed similar methods42, 43 and are described in Supporting Information (Scheme S1).
Scheme 1.

Synthesis of compounds 6–11.
Reaction conditions: (a) Br2, NaOAc, AcOH, overnight; (b) pd(PPh3)4, K2CO3, Dimethoxyethane, H2O, reflux.
Scheme 2.

Synthesis of compounds 5, 12–19.
Reaction conditions: (a) H2, Pd/C in MeOH for 12 hours. (b) pd(PPh3)4, K2CO3, Dimethoxyethane, H2O, reflux, overnight.
Summary
In this study, we report our structure-based design, synthesis and evaluation of a new class of BET bromodomain inhibitors. Based upon the co-crystal structure of compound 3 (I-BET-151) in a complex with the BRD4 BD1 protein, we designed a new class of compounds with a [6,5,6] γ-carboline tricyclic scaffold. The most potent compound (18) binds to the BD1 and BD2 domains of the BRD2-BRD4 proteins with Ki values of 3.2–24.7 nM and shows high specificity over other non-BET bromodomain-containing proteins with the exception of CREBBP, for which Kd = 670 nM. Determination of a high-resolution co-crystal structure of 18 in a complex with BRD4 BD2 provided a structural basis for the high binding affinity and for further structure-based optimization. Compound 18 potently inhibits cell growth in MV4;11 and MOLM-13 acute leukemia cell lines harboring the MLL1 fusion gene with IC50 values of 20 and 66 nM, respectively, and demonstrates excellent specificity over the K562 acute leukemia cell line containing the BCR-ABL fusion gene. Furthermore, compound 18, with a low molecular weight of 413.5 has excellent physiochemical properties with a calculated cLogP of 2.68. Taken together, these data indicate that compound 18 represents a promising, new class of BET inhibitors. Further optimization of 18 may ultimately yield a new class of therapeutics for the treatment of human cancers and other diseases.
Experimental Section
1. Chemistry
General Methods
Proton nuclear magnetic resonance (1H NMR) spectroscopy and carbon nuclear magnetic resonance (13C NMR) spectroscopy were performed in Bruker Avance 300 NMR spectrometers. 1H chemical shifts are reported with CHD2OD (3.31 ppm) or HDO (4.70 ppm) as internal standards. The final products were purified by C18 reverse phase semi-preparative HPLC column with solvent A (0.1% TFA in H2O) and solvent B (0.1% TFA in CH3CN) as eluents. The final compounds were isolated as their TFA salts, and the purities were confirmed by analytical HPLC to be >95% in all cases.
Synthesis of target compound 5
4-(8-Methoxy-5H-pyrido[4,3-b]indol-7-yl)-3,5-dimethylisoxazole (5)
10% Pd-C (5 mg) was suspended in a MeOH solution of 6 (15 mg, 0.046 mmol). The reaction was stirred for 26 h under a H2 balloon at room temperature. The Pd-C was removed by filtration and the filtrate was purified by semi-preparative HPLC to give 4 mg (30%) of 5 as a colorless powder after being lyophilized for 24 h. 1H NMR (300 MHz, MeOD-d4) δ 9.61 (s, 1H), 8.54 (d, 1H, J=6.9 Hz), 8.11 (s, 1H), 7.97 (s, 1H), 7.61 (s, 1H), 4.00 (s, 3H), 2.36 (s, 3H), 2.19 (s, 3H). ESIMS m/z [M+H]+ calcd. = 294.33; found = 294.75.
Synthesis of target compounds 6–19
4-(1-Chloro-8-methoxy-5H-pyrido[4,3-b]indol-7-yl)-3,5-dimethylisoxazole (6)
Compounds 21 (157 mg, 0.5 mmol), 22 (655 mg, 2.0 mmol), and K2CO3 (345 mg, 2.5 mmol) were dissolved individually in a dimethoxyethane/H2O (50 mL: 25 mL) mixture. The reaction system was vacuumed, followed by addition of tetrakin (triphenylphosphine) palladium(0), then vacuumed again and filled with N2. After heating at reflux overnight, the mixture was extracted with EtOAc, and the organic fraction was concentrated before purification by preparative HPLC. 57 mg (35%) of compound 6 was obtained as pale yellow powder after being lyophilized for 24 h. 1H NMR (300 MHz, MeOD-d4) δ 8.26 (d, 1H, J= 6.0 Hz), 8.09 (s, 1H), 7.60 (d, 1H, J= 6.3 Hz), 7.49 (s, 1H), 3.98 (s, 3H), 2.63 (s, 3H), 2.20 (s, 3H). 13C NMR (75 MHz, MeOD-d4), δ168.07, 161.30, 155.05, 148.78, 143.09, 141.11, 136.57, 122.59, 122.28, 118.84, 116.16, 115.01, 108.08, 105.09, 56.69, 11.69, 10.84. ESIMS m/z [M+H]+ calcd. = 328.77; found = 328.83.
4-(1-Chloro-8-methoxy-5H-pyrido[4,3-b]indol-7-yl)-3-ethyl-5-methylisoxazole (7)
This compound was prepared by coupling 21 and 23 under conditions similar to those used for the preparation of 6. 1H NMR (300 MHz, MeOD-d4) δ 8.27 (d, 1H, J= 6.3 Hz), 8.02 (s, 1H), 7.64 (d, 1H, J= 6.3 Hz), 7.48 (s, 1H), 3.93 (s, 3H), 2.70 (q, 2H, J= 7.5 Hz), 2.13 (s, 3H), 1.20 (t, 3H, J=7.5 Hz). ESIMS m/z [M+H]+ calcd. = 342.80; found = 342.42.
4-(1-Chloro-8-methoxy-5H-pyrido[4,3-b]indol-7-yl)-5-ethyl-3-methylisoxazole (8)
This was prepared by coupling 21 and 24 under conditions similar to those used for the preparation of 6. 1H NMR (300 MHz, MeOD-d4) δ 8.19 (d, 1H, J= 5.7 Hz), 8.08 (s, 1H), 7.49 (d, 1H, J= 5.7 Hz), 7.43 (s, 1H), 3.96(s, 3H), 2.64 (q, 2H, J= 7.5 Hz), 2.34 (s, 3H), 1.12 (t, 3H, J= 7.5 Hz). ESIMS m/z [M+H]+ calcd. = 342.80; found = 342.67.
4-(1-Chloro-8-methoxy-5H-pyrido[4,3-b]indol-7-yl)-3,5-diethylisoxazole (9)
This molecule was prepared by coupling 21 and 25 under conditions similar to those used for the preparation of 6. 1H NMR (300 MHz, DMSO-d6) δ 12.06 (s, 1H), 8.22 (d, 1H, J= 5.7 Hz), 7.96 (s, 1H), 7.52 (d, 1H, J= 5.7 Hz), 7.49 (s, 1H), 3.89 (s, 3H), 2.65 (q, 2H, J= 7.5 Hz), 2.53 (q, 2H, J= 7.5 Hz), 1.15 (t, 3H, J= 7.5 Hz), 1.04 (t, 3H, J= 7.5 Hz). 13C NMR (75 MHz, DMSO-d6), δ 169.85, 163.67, 152.08, 145.84, 143.62, 143.55, 133.92, 119.93, 119.20, 116.06, 114.30, 111.86, 106.71, 103.19, 55.71, 18.94, 18.49, 11.81. ESIMS m/z [M+H]+ calcd. = 356.67; found = 356.83.
1-Chloro-7-(3,5-dimethyl-1H-pyrazol-4-yl)-8-methoxy-5H-pyrido[4,3-b]indole (10)
This was prepared by coupling 21 with 26 under conditions similar to those used for the preparation of 6. 1H NMR (300 MHz, MeOD-d4) δ 8.24 (d, 1H, J= 6.0 Hz), 8.10 (s, 1H), 7.56 (d, 1H, J= 6.0 Hz), 7.49 (s, 1H), 3.97 (s, 3H), 2.31 (s, 6H). 13C NMR (75 MHz, MeOD-d4), δ 154.63, 148.56, 145.51, 142.58, 136.32, 122.52, 121.86, 118.63, 118.34, 116.15, 107.94, 105.19, 101.44, 56.69, 10.75. ESIMS m/z [M+H]+ calcd. = 327.79; found = 327.92.
1-Chloro-8-methoxy-7-(1,3,5-trimethyl-1H-pyrazol-4-yl)-5H-pyrido[4,3-b]indole (11)
This molecule was prepared by coupling 21 and 27 under conditions similar to those used for the preparation of 6. 1H NMR (300 MHz, MeOD-d4) δ 8.26 (d, 1H, J= 6.0 Hz), 8.07 (s, 1H), 7.61 (d, 1H, J= 6.3 Hz), 7.45 (s, 1H), 3.95 (s, 3H), 3.88 (s, 3H), 2.24 (s, 3H), 2.20 (s, 3H). ESIMS m/z [M+H]+ calcd. = 341.81; found = 342.33.
4,4′-(8-Methoxy-5H-pyrido[4,3-b]indole-1,7-diyl)bis(3,5-dimethylisoxazole) (12)
This molecule was prepared by coupling 6 and 22 under conditions similar to those used for the preparation of 6. 1H NMR (300 MHz, MeOD-d4) δ 8.63 (d, 1H, J=6.6 Hz), 8.04 (d, 1H, J=6.6 Hz), 7.67 (s, 1H), 7.02 (s, 1H), 3.78 (s, 3H), 2.52 (s, 3H), 2.34 (s, 3H), 2.28 (s, 3H), 2.17 (s, 3H). 13C NMR (75 MHz, MeOD-d4), δ 172.28, 168.28, 161.12, 160.42, 155.77, 149.23, 138.54, 137.66, 136.81, 124.16, 122.24, 121.46, 117.29, 114.52, 109.87, 109.42, 103.77, 56.42, 12.05, 11.68, 10.79, 10.60. ESIMS m/z [M+H]+ calcd. = 389.43; found = 389.50.
4-(1-(3,5-Dimethyl-1H-pyrazol-4-yl)-8-methoxy-5H-pyrido[4,3-b]indol-7-yl)-3,5-dimethylis-oxazole (13)
This molecule was prepared in 75% yield by coupling 6 and 26 under conditions similar to those used for the preparation of 6. 1HNMR (300 MHz, MeOD-d4) δ 8.54 (d, 1H, J=6.9 Hz), 7.95 (d, 1H, J= 6.9 Hz), 7.63 (s, 1H), 7.02 (s, 1H), 3.74 (s, 3H), 2.34 (s, 3H), 2.29 (s, 6H), 2.16 (s, 3H). 13C NMR (75 MHz, MeOD-d4), δ 168.23, 161.18, 155.58, 148.99, 146.03, 143.05, 137.39, 136.23, 123.63, 122.87, 121.08, 116.98, 114.63, 108.64, 103.98, 56.29, 11.68, 11.41, 10.79. ESIMS m/z [M+H]+ calcd. = 388.44; found = 388.42.
4-(8-Methoxy-1-(1H-pyrazol-4-yl)-5H-pyrido[4,3-b]indol-7-yl)-3,5-dimethylisoxazole (14)
This molecule was prepared by coupling 6 and 28 under conditions similar to those used for the preparation of 6. 1H NMR (300 MHz, MeOD-d4) δ 8.48 (s, 2H), 8.45 (d, 1H, J=6.9 Hz), 7.89 (d, 1H, J=6.9 Hz), 7.63 (s, 1H), 7.61 (s, 1H), 3.82 (s, 3H), 2.35 (s, 3H), 2.17 (s, 3H). 13C NMR (75 MHz, MeOD-d4), δ 168.18, 161.18, 155.28, 149.11, 143.30, 137.25, 135.69, 123.48, 123.01, 119.19, 116.80, 114.66, 113.69, 108.22, 104.50, 56.37, 11.67, 10.80. ESIMS m/z [M+H]+ calcd. =360.39; found =361.17.
4-(8-Methoxy-1-(1,3,5-trimethyl-1H-pyrazol-4-yl)-5H-pyrido[4,3-b]indol-7-yl)-3,5-dimethylisoxazole (15)
This molecule was prepared by coupling 6 and 27 under conditions similar to those used for the preparation of 6. 1HNMR (300 MHz, MeOD-d4) δ 8.54 (d, 1H, J=6.9 Hz), 7.95 (d, 1H, J= 6.9 Hz), 7.63 (s, 1H), 7.04 (s, 1H), 3.97 (s, 3H), 3.75 (s, 3H), 2.34 (s, 3H), 2.32 (s, 3H), 2.21 (s, 3H), 2.16 (s, 3H). ESIMS m/z [M+H]+ calcd. = 402.47; found = 402.75.
4-(1-(3,5-Dimethyl-1-phenyl-1H-pyrazol-4-yl)-8-methoxy-5H-pyrido[4,3-b]indol-7-yl)-3,5-dimethylisoxazole (16)
This molecule was prepared by coupling 6 and 29 under conditions similar to those used for the preparation of 6. 1H NMR (300 MHz, MeOD-d4) δ 8.60 (d, 1H, J=6.6 Hz), 8.00 (d, 1H, J= 6.9 Hz), 7.64–7.66 (m, 6H), 7.11 (s, 1H), 3.78 (s, 3H), 2.35 (s, 3H), 2.33 (s, 3H), 2.31 (s, 3H), 2.17 (s, 3H). 13C NMR (75 MHz, MeOD-d4), δ 168.26, 161.16, 155.67, 149.62, 149.10, 142.49, 142.22, 140.20, 137.48, 136.42, 131.02, 130.49, 126.75, 123.83, 122.75, 121.30, 117.06, 114.60, 112.68, 108.87, 103.95, 56.38, 12.56, 11.74, 11.69, 10.81. ESIMS m/z [M+H]+ calcd. = 464.54; found = 464.42.
4-(1-(3,5-Diethyl-1H-pyrazol-4-yl)-8-methoxy-5H-pyrido[4,3-b]indol-7-yl)-3,5-dimethyl-isoxazole (17)
This molecule was prepared in 35% yield by coupling 6 and 30 under conditions similar to those used for the preparation of 6. 1H NMR (300 MHz, MeOD-d4) δ 8.55 (d, 1H, J=6.9 Hz), 7.97 (d, 1H, J= 6.9 Hz), 7.64 (s, 1H), 6.92 (s, 1H), 3.70 (s, 3H), 2.65 (m, 4H). 2.34 (s, 3H), 2.16 (s, 3H), 1.10 (t, 6H, J= 7.5 Hz). 13C NMR (75 MHz, MeOD-d4), δ 168.25, 155.57, 148.83, 143.32, 137.42, 136.27, 123.78, 122.91, 121.45, 117.08, 114.59, 108.86, 103.85, 56.30, 30.88, 24.41, 20.51, 13.97, 11.69, 10.81. ESIMS m/z [M+H]+ calcd. = 416.50; found = 416.42.
4-(1-(3-Cyclopropyl-5-methyl-1H-pyrazol-4-yl)-8-methoxy-5H-pyrido[4,3-b]indol-7-yl)-3,5-dimethylisoxazole (18)
This molecule was prepared in 19% yield by coupling 6 and 31 under conditions similar to those used for the preparation of 6. 1HNMR (300 MHz, MeOD-d4) δ 8.55 (d, 1H, J= 6.9 Hz), 7.96 (d, 1H, J= 6.6 Hz), 7.63 (s, 1H), 7.12 (s, 1H), 3.76 (s, 3H), 2.35 (s, 3H), 2.28 (s, 3H), 2.17 (s, 3H), 1.73 (m,1H), 0.87 (m, 4H). 13C NMR (75 MHz, MeOD-d4), δ 168.18, 161.15, 155.46, 152.39, 148.94, 145.13, 143.14, 137.35, 136.19, 123.61, 122.93, 121.20, 116.85, 114.63, 110.46, 108.60, 104.41, 56.32, 11.66, 11.01, 10.77, 9.22, 8.45, 8.25. ESIMS m/z [M+H]+ calcd. = 414.48; found = 414.50.
4-(8-Methoxy-1-(5-methyl-3-phenyl-1H-pyrazol-4-yl)-5H-pyrido[4,3-b]indol-7-yl)-3,5-dimethyl-isoxazole (19)
This molecule was prepared by coupling 6 and 32 under conditions similar to those used for the preparation of 6. 1H NMR (300 MHz, MeOD-d4) δ 8.54 (d, 1H, J= 6.9 Hz), 7.98 (d, 1H, J= 6.9 Hz), 7.57 (s, 1H), 7.34 (m, 2H), 7.25 (m, 3H), 6.98 (s, 1H), 3.70 (s, 3H), 2.33 (s, 3H), 2.30 (s, 3H), 2.12 (s, 3H). 13C NMR (75 MHz, MeOD-d4), δ 168.18, 161.13, 155.42, 148.93, 143.24, 137.32, 136.38, 130.28, 128.12, 123.68, 122.65, 121.39, 116.84, 114.58, 108.90, 104.08, 56.33, 11.65, 10.88, 10.76. ESIMS m/z [M+H]+ calcd. = 450.51; found = 450.75.
7-Bromo-1-chloro-8-methoxy-5H-pyrido[4,3-b]indole (21)
Compound 20 (377 mg, 1.6 mmol) and NaOAc (197 mg, 2.4 mmol) were dissolved in AcOH (40 mL). Bromine (389 mg, 2.4 mmol) was added dropwise to the reaction system. After stirring at room temperature overnight, the reaction was quenched with Na2SO3 solution. AcOH was then removed under reduced pressure and H2O was added followed by extraction with EtOAc. The combined organic fractions were concentrated and purified by preparative HPLC to give 157 mg (31%) of 21 as a colorless powder. 1H NMR (300 MHz, MeOD-d4) δ 8.21 (d, 1H, J= 5.7 Hz), 8.00 (s, 1H), 7.84 (s, 1H), 7.50 (d, J=6.0 Hz), 4.03 (s, 3H).
Synthesis of intermediates 23–25 and 29–32
t-Butyl 3-cyclopropyl-5-methyl-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazole-1-carboxylate (31)
Compound S20 (1.312 g, 3.77 mmol) was dissolved in THF (30 mL) and cooled to −78 °C under N2. n-Butyl lithium (0.362 g, 5.65 mmol) was added slowly to the solution which was then stirred for 30 min at −78 °C. S23 (0.77 g, 4.14 mmol) was added to the reaction system, and the mixture was stirred at −78 °C for an additional 2 hours. Upon completion, the reaction was quenched by adding NH4Cl saturated solution. The mixture was then extracted with EtOAc and dried over anhydrous Na2SO4. Purification by flash column chromatography (EtOAc: hexane = 1: 10) gave 31 as a colorless oil (0.459 mg, 35% yield). 1H NMR (300 MHz, CDCl3), δ 2.65 (s, 3H), 2.28 (m, 1H), 1.62 (s, 9H), 1.33 (s, 12H), 0.99 (m, 2H), 0.88 (m, 2H).
3-Ethyl-5-methyl-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)isoxazole (23)
This molecule was prepared by treatment of S13 and S23 under conditions similar to those used for the preparation of 31. Compound 23 was obtained as a mixture with 24. 1H NMR (300 MHz, CDCl3), δ 2.93 (q, J=7.5 Hz, 2H), 2.35 (s, 3H), 1.31 (s, 12H), 1.26 (m, 3H).
5-Ethyl-3-methyl-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)isoxazole (24)
This molecule was prepared by treatment of S14 and S23 under conditions similar to those used for the preparation of 31. 24 was prepared as a mixture with 23. 1H NMR (300 MHz, CDCl3), δ 2.77 (q, J=7.5 Hz, 2H), 2.53 (s, 3H), 1.31(s, 12H), 1.26(m, 3H).
t-Butyl 5-cyclopropyl-3-methyl-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazole-1-carboxylate (25)
This molecule was prepared by treatment of S12 and S23 under conditions similar to those used for the preparation of 31. 1H NMR (300 MHz, CDCl3), δ 2.93 (q, J=7.5 Hz, 2H), 2.77 (q, J=7.5 Hz, 2H), 1.31 (s, 12H), 1.26 (m, 6H).
3,5-Dimethyl-1-phenyl-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazole (29)
This molecule was prepared by treatment of S22 and S23 under conditions similar to those used for the preparation of 31. 1H NMR (300 MHz, CDCl3), δ 7.35–7.50 (m, 5H), 2.45 (s, 3H), 2.44 (s, 3H), 1.34 (s, 12H).
t-Butyl 3,5-diethyl-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazole-1-carboxylate (30)
This was prepared by treatment of S19 and S23 under conditions similar to those used for the preparation of 31. 1H NMR (300 MHz, CDCl3), δ3.15 (q, J=7.5 Hz, 2H), 2.74 (q, J=7.5 Hz, 2H), 1.61 (s, 9H), 1.27 (s, 12H), 1.17 (m, 6H).
t-Butyl 5-methyl-3-phenyl-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazole-1-carboxylate (32)
This molecule was prepared by in 39% yield treatment of S21 and S23 under conditions similar to those used for the preparation of 31. 1H NMR (300 MHz, CDCl3), δ 7.81 (m, 2H), 7.37 (m, 3H), 2.76 (s, 3H), 1.67 (s, 9H), 1.32 (s, 12H).
2. Molecular modeling
The co-crystal structures of BRD4 BD1 in a complex with compound 3 (PDB entry: 3ZYU), BRD4 BD2 in a complex with GW841819X (PDB entry: 2YEM) and BRD4 BD2 in a complex with compound 6 were used to model the binding poses of designed compounds with BRD4 BD1 and BRD4 BD2. Chain A of the BRD4 BD1 crystal structure was first extracted and protons were added using the “protonate 3D” module in MOE44 where protonation states of His residues at pH 7.0 were determined by considering neighboring residues in the structure. All water molecules from the crystal structure were saved. The same procedures were used for BRD4 BD2 protein. The designed compound structures were drawn and optimized using the MOE program. All the binding poses of designed compounds with BRD4 BD1 were modeled using the GOLD program (version 4.0.1)45, 46. The center of the binding site for BRD4 BD1 was set at C136 and for BRD4 BD2 at C429. The radius of the binding site in both cases was defined as 11 Å, large enough to cover the entire binding pocket. At the binding site, water molecules A2129, A2137 and A2178 of BRD4 BD1 (PDB entry: 3ZYU) were included during the docking simulations where flags of on, toggle, and toggle were set individually and allowed to spin for optimal hydrogen bond interaction. In each genetic algorithm (GA) run, a maximum number of 200,000 operations were performed on a population of 5 islands of 100 individuals each. Operator weights for crossover, mutation and migration were set to 95, 95 and 10 respectively. The docking simulations were terminated after 20 runs for each ligand. ChemScore implemented in Gold 4.0.1 was used as the fitness function to evaluate the docked poses. The 20 conformations ranked highest by each fitness function were saved for analysis of the predicted docking modes.
3. Biochemical binding assays
3a. Expression and purification of bromodomain-containing proteins
BRD2 BD1 (residues 72–205), BRD2 BD2 (residues 349–460), BRD3 BD1 (residues 24–144), BRD3 BD2 (residues 306–417), BRD4 BD1 (residues 44–168), BRD4 BD2 (residues 333–460) and CREBBP (residues 1043–1159) were cloned into N-terminal His6-TEV vectors and transformed into Rosetta2 DE3 cells. Cultures were grown to an OD600 of 1.0 – 2.5 at 37 °C in Terrific Broth, induced with 0.4 mM IPTG and expressed for 4 h at 37 °C. All proteins were purified from the soluble fraction using a standardized protocol implementing Ni-NTA resin (Qiagen) followed by gel filtration on a Superdex75 column (GE Healthcare). Cells were sonicated in 25mM Tris pH 7.5, 200mM NaCl, 0.1% βME with 40 μL of Leupeptin/Aprotinin mixture and pelleted at 17,000 rpm for 45 min. The soluble fraction was applied to a Ni-NTA for 1 h, then washed with 25 mM Tris pH 7.5, 200 mM NaCl, 10 mM imidazole and protein eluted with 25 mM Tris pH 7.5, 200 mM NaCl, 300 mM imidazole. The protein was applied to a size exclusion column equilibrated with 25 mM Tris pH 7.5, 150 mM NaCl and 0.1 % 2-mercaptoethanol (βME) or 2 mM tris(2-carboxyethyl)phosphine (TCEP).
Trim24 (residues 790–972), BAZ2B (residues 2054–2168), TAF1B2 (residues 155–276), BRG1 (residues 59–180) and PB1BR5 (residues 645–766) were cloned, transformed, grown and induced as described above with expression overnight at 20 °C. Cells were resuspended and sonicated in Buffer A (50 mM Hepes pH 7.5 and 500 mM NaCl) containing 10 mM imidazole, 5% glycerol and protease inhibitors, then pelleted by centrifugation. The soluble fractions were applied to Ni-NTA. The matrix was washed with Buffer A plus 30 mM imidazole and the proteins eluted with Buffer A plus 350 mM imidazole. The proteins were concentrated and each was applied to a Superdex 75 column equilibrated with 10 mM Hepes pH 7.5, 200 mM NaCl and 5% glycerol. Aliquots of the purified proteins were stored at −80 °C.
ATAD2A (residues 981–1108) and ATAD2B (residues 953–1086) were obtained from the Structural Genomics Consortium (Toronto, Canada), and precloned into a His6-TEV expression vector. The clones were transformed, grown and induced as described above with expression overnight at 18 °C. The cells were resuspended and sonicated in Buffer A plus 5 mM imidazole, 5% glycerol and protease inhibitors. The soluble fractions were applied to Ni-NTA, washed with Buffer A containing 30 mM imidazole and 5% glycerol and eluted in Buffer A plus 250 mM imidazole and 5% glycerol. The proteins were further purified on a Superdex 200 (GE Healthcare) column in Buffer A with 5% glycerol. Aliquots of the purified proteins were stored at −80 °C.
MLL1 (residues 1566–1784) was cloned into a His6-TEV expression vector and transformed into Rosetta2 cells. The cells were grown at 37 °C in 2xYT to an OD600 of 0.9, then induced with 0.4 mM IPTG and expressed overnight at 20 °C. 100 μM Zn(OAc)2 was added at the time of induction to ensure proper protein folding. Cells were resuspended in Buffer B (50 mM HEPES pH 7.5 and 500 mM NaCl) with 50 μM Zn(OAc)2 and protease inhibitors, then sonicated and pelleted by centrifugation. The soluble fraction was applied to an Ni-NTA column, washed with Buffer B plus 20 mM imidazole and 50 μM Zn(OAc)2, then the protein was eluted with Buffer B plus 500 mM imidazole and 50 μM Zn(OAc)2. The protein was then dialyzed overnight at 4 °C against 20 mM HEPES pH 7.5, 200 mM NaCl and 1 mM DTT in the presence of TEV protease. The tag was removed by implementing a second Ni-NTA column. The flow through containing MLL1 protein was run over Superdex 75 equilibrated with 20 mM HEPES pH 7.5, 150 mM NaCl and 1 mM TCEP. Aliquots of the purified protein were stored at −80 °C.
3b. Competitive Fluorescence Polarization (FP) binding assays
Competitive fluorescence polarization (FP) binding assays were performed to quantitate the binding affinities of compounds to BRD2-BRD4 BD1 and BD2 domains. For the development of competitive FP binding assays, we designed and synthesized a FAM-labeled fluorescent probe based upon ZBA248, a potent BET inhibitor we have developed whose chemical structure is provided in the Supplemental Information. Equilibrium dissociation constant (Kd) values of ZBA248 to these six proteins were determined from protein saturation experiments by monitoring the total fluorescence polarization of mixtures composed with the fluorescent probe at a fixed concentration and proteins with increasing concentrations up to full saturation. Serial dilutions of each tested protein were mixed with ZBA248 to a final volume of 200 μL in the assay buffer (100 mM phosphate buffer, pH = 6.5, with 0.01% Triton X-100). The final probe concentration in all assays was 1.5 nM. Plates were incubated at room temperature for 30 min with gentle shaking to assure equilibrium. FP values in millipolarization units (mP) were measured using the Infinite M-1000 plate reader (Tecan U.S., Research Triangle Park, NC) in Microfluor 1 96-well, black, round-bottom plates (Thermo Scientific, Waltham, MA) at an excitation wavelength of 485 nm and an emission wavelength of 530 nm. The Kd values for the interaction with ZBA248, calculated by fitting the sigmoidal dose-dependent FP increases as a function of protein concentrations using Graphpad Prism 6.0 software (Graphpad Software, San Diego, CA), are 2.0, 2.2, 6.5, 0.6, 5.5, and 3.0 nM to BRD2 BD1 and 2, BRD3 BD1 and 2, and BRD4 BD1 and 2, respectively.
The IC50 values of compounds were determined in competitive binding experiments. Mixtures of 10 μL of the compound in assay buffer with 40% ethylene glycol and 190 μL of preincubated protein/probe complex solution in the assay buffer were added into assay plates which were then incubated at room temperature for 30 min with gentle shaking. Final protein concentrations for the BRD2 BD1, BRD2 BD2, BRD3 BD1, BRD3 BD2, BRD4 BD1 and BRD4 BD2 assays were 3, 6, 15, 2, 10, and 6 nM, respectively. The final probe concentration in all competitive experiments was 1.5 nM. Negative controls containing protein/probe complex only (equivalent to 0% inhibition), and positive controls containing only free probes (equivalent to 100% inhibition), were included on each assay plate. FP values were measured as described above. IC50 values were determined by nonlinear regression fitting of the competition curves. Ki values of competitive inhibitors were obtained directly by nonlinear regression fitting, based upon the Kd values of the probe to different proteins, and concentrations of the proteins and probes in the competitive assays47, 48.
3c. Bio-layer Interferometry (BLI)
BLI experiments were performed using an OctetRED96 instrument from PALL/ForteBio. All experiments were performed at 25 °C using PBS (pH 7.4) as the assay buffer, to which 0.1% BSA and 0.01% Tween-20 were added to reduce nonspecific interactions. 0.5% DMSO was also introduced to increase compound solubility. Assays were conducted in Greiner 96 well black flat-bottom microplates containing the protein solutions, pure assay buffer for dissociation, and serial dilutions of compounds to be tested. During the experiment, sample plates were continuously shaken at 1000 RPM to eliminate the mass transport effect.
Biotinylated proteins prepared using the Thermo EZ-Link long-chain biotinylation reagent were immobilized on Super Streptavidin (SSA) biosensors by dipping sensors into plate wells containing protein solutions whose concentrations were predetermined from control experiments to achieve the best signal-to-noise ratio. Sensor saturation typically could be achieved in 10–15 min. Biotinylated blocked Streptavidin (SAV-B4) sensors were prepared as the inactive reference controls by following the protocol provided by the manufacturer. Sensors loaded with proteins were moved and dipped into wells with pure assay buffer and washed in the buffer for 10 min to eliminate loose nonspecific bound protein and establish a stable base line. Association-dissociation cycles of compounds were started by moving and dipping sensors to compound and pure buffer wells alternatively starting from the lowest concentration of compound. Association and dissociation times were carefully determined to ensure full association and dissociation.
Buffer-only reference was included in all assays. Raw kinetic data collected were processed with the Data Analysis software provided by the manufacturer using double reference subtraction in which both buffer-only reference and inactive protein reference were subtracted.
The resulting data were analyzed based on a 1:1 binding model from which kon and koff values were obtained and then Kd values were calculated.
4. Crystallization and structure determination of BRD4 BD2 with compound 18
A solution of BRD4 BD2 was concentrated to ~8 mg/mL and incubated with a 2-fold excess of compound 18 prior to crystallization. Crystals were grown in 50% polyethylene glycol 400, 0.01 M imidazole pH 8.0, and 0.01 M ATP. Data were collected at the Advance Photon Source at Argonne National Lab on the LS-CAT beamlines 21-ID-D. The complex crystallized in space group P21212 and contained 1 molecule per asymmetric unit. Data were processed with HKL200049 and the structure was solved by molecular replacement with Phaser50 using PDB code 2OUO as a model. The structures were refined with Buster51 and electron density maps fit with COOT52. Coordinates and restraints for the compounds were developed using Grade with the mogul+qm option51. Structures were validated using Molprobity53, Parvati54, and Whatcheck55. Ligand statistics were obtained from the Uppsala Electron-Density Server. Data refinement and statistics are given in Table S1. The coordinates were deposited in the PDB with the following ID: 4Z93 for compound 18.
5. Cell viability assay
Cell lines obtained from the American Type Culture Collection (ATCC) were used within three months of thawing fresh vials. Cells were maintained in the recommended culture medium with 10% FBS at 37 °C and an atmosphere of 5% CO2. The effect of BET inhibitors on cell viability was determined in a 4-day proliferation assay as described previously56. Cells were seeded in 96-well white opaque cell culture plates at a density of 3,000–10,000 cells/well in 75 μL of culture medium. Each compound tested was serially diluted in the appropriate medium, and 75 μL of a diluted solution containing the compound was added to the appropriate wells of the cell plate. After the addition of the tested compound, the cells were incubated at 37 °C in an atmosphere of 5% CO2 for 4 days. Cell viability was determined using the CellTiter-Glo® Luminescent Cell Viability Assay Kit (Promega, Madison, WI) according to the manufacturer’s instructions. The luminescent signal was measured using a Tecan Infinite M1000 multimode microplate reader (Tecan, Morrisville, NC). The readings were normalized to the DMSO-treated cells and the IC50 was calculated by nonlinear regression analysis using GraphPad Prism 5 software.
Supplementary Material
Acknowledgments
The financial support from the National Institutes of Health (P50 CA186786 and P30 CA046592) and the Prostate Cancer Foundation is greatly appreciated. Use of the Advanced Photon Source, an Office of Science User Facility operated for the U.S. Department of Energy (DOE) Office of Science by Argonne National Laboratory, was supported by the U.S. DOE under Contract No. DE-AC02-06CH11357. The Life Sciences Collaborative Access Team (LS-CAT) at Sector 21 of the Advanced Photon Source at Argonne National Laboratory was supported by the Michigan Economic Development Corporation and the Michigan Technology Tri-Corridor (Grant 085P1000817). We would like to thank Dr. David Smith of LS-CAT for his assistance with crystal screening and remote data collection.
List of non-standard abbreviations
- AR
Androgen Receptor
- BCR-ABL
Breakpoint Cluster Region protein- Abelson murine leukemia viral oncogene homolog 1
- BD1/BD2
Bromodomain 1/Bromodomain 2
- BET
Bromodomain and Extra Terminal protein
- BRD2/3/4
Bromodomain-containing protein 2, 3 or 4
- BSA
Bovine Serum Albumin
- COOT
Crystallographic Object-Oriented Toolkit
- FP
Fluorescence-Polarization
- GA
Genetic Algorithm
- MLL1
Mixed Lineage Leukemia 1
- mP
millipolarization unit
- TEV
Tobacco Etch Virus
Footnotes
Conflicts of Interests: The authors declare the following competing financial interest(s): Xu Ran, Yujun Zhao, Liu Liu, Longchuan Bai, Chao-Yie Yang, Bing Zhou, Jennifer L. Meagher, Jeanne A. Stuckey and Shaomeng Wang are inventors in a patent filed by the University of Michigan, which covers the BET inhibitors disclosed in the current study. The patent has been licensed by OncoFusion Therapeutics from the University of Michigan. The inventors receive royalties from the University of Michigan on the patent. Shaomeng Wang also owns stock in OncoFusion Therapeutics and serves as a consultant for OncoFusion Therapeutics.
References
- 1.Filippakopoulos P, Picaud S, Mangos M, Keates T, Lambert JP, Barsyte-Lovejoy D, Felletar I, Volkmer R, Muller S, Pawson T, Gingras AC, Arrowsmith CH, Knapp S. Histone recognition and large-scale structural analysis of the human bromodomain family. Cell. 2012;149:214–231. doi: 10.1016/j.cell.2012.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sanchez R, Meslamani J, Zhou MM. The bromodomain: From epigenome reader to druggable target. Biochimica et biophysica acta. 2014 doi: 10.1016/j.bbagrm.2014.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Filippakopoulos P, Qi J, Picaud S, Shen Y, Smith WB, Fedorov O, Morse EM, Keates T, Hickman TT, Felletar I, Philpott M, Munro S, McKeown MR, Wang Y, Christie AL, West N, Cameron MJ, Schwartz B, Heightman TD, La Thangue N, French CA, Wiest O, Kung AL, Knapp S, Bradner JE. Selective inhibition of BET bromodomains. Nature. 2010;468:1067–1073. doi: 10.1038/nature09504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nicodeme E, Jeffrey KL, Schaefer U, Beinke S, Dewell S, Chung CW, Chandwani R, Marazzi I, Wilson P, Coste H, White J, Kirilovsky J, Rice CM, Lora JM, Prinjha RK, Lee K, Tarakhovsky A. Suppression of inflammation by a synthetic histone mimic. Nature. 2010;468:1119–1123. doi: 10.1038/nature09589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhu J, Gaiha GD, John SP, Pertel T, Chin CR, Gao G, Qu H, Walker BD, Elledge SJ, Brass AL. Reactivation of latent HIV-1 by inhibition of BRD4. Cell reports. 2012;2:807–816. doi: 10.1016/j.celrep.2012.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Denis GV. Bromodomain coactivators in cancer, obesity, type 2 diabetes, and inflammation. Discovery medicine. 2010;10:489–499. [PMC free article] [PubMed] [Google Scholar]
- 7.Matzuk MM, McKeown MR, Filippakopoulos P, Li Q, Ma L, Agno JE, Lemieux ME, Picaud S, Yu RN, Qi J, Knapp S, Bradner JE. Small-molecule inhibition of BRDT for male contraception. Cell. 2012;150:673–684. doi: 10.1016/j.cell.2012.06.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhao L, Cao D, Chen T, Wang Y, Miao Z, Xu Y, Chen W, Wang X, Li Y, Du Z, Xiong B, Li J, Xu C, Zhang N, He J, Shen J. Fragment-based drug discovery of 2-thiazolidinones as inhibitors of the histone reader BRD4 bromodomain. Journal of Medicinal Chemistry. 2013;56:3833–3851. doi: 10.1021/jm301793a. [DOI] [PubMed] [Google Scholar]
- 9.Zhao L, Wang Y, Cao D, Chen T, Wang Q, Li Y, Xu Y, Zhang N, Wang X, Chen D, Chen L, Chen YL, Xia G, Shi Z, Liu YC, Lin Y, Miao Z, Shen J, Xiong B. Fragment-based drug discovery of 2-thiazolidinones as BRD4 inhibitors: 2 Structure-based optimization. J Med Chem. 2015;58:1281–1297. doi: 10.1021/jm501504k. [DOI] [PubMed] [Google Scholar]
- 10.Hewitt MC, Leblanc Y, Gehling VS, Vaswani RG, Cote A, Nasveschuk CG, Taylor AM, Harmange JC, Audia JE, Pardo E, Cummings R, Joshi S, Sandy P, Mertz JA, Sims RJ, 3rd, Bergeron L, Bryant BM, Bellon S, Poy F, Jayaran H, Tang Y, Albrecht BK. Development of methyl isoxazoleazepines as inhibitors of BET. Bioorg Med Chem Lett. 2015 doi: 10.1016/j.bmcl.2015.03.045. [DOI] [PubMed] [Google Scholar]
- 11.Wong C, Laddha SV, Tang L, Vosburgh E, Levine AJ, Normant E, Sandy P, Harris CR, Chan CS, Xu EY. The bromodomain and extra-terminal inhibitor CPI203 enhances the antiproliferative effects of rapamycin on human neuroendocrine tumors. Cell Death Dis. 2014;5:e1450. doi: 10.1038/cddis.2014.396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.McKeown MR, Shaw DL, Fu H, Liu S, Xu X, Marineau JJ, Huang Y, Zhang X, Buckley DL, Kadam A, Zhang Z, Blacklow SC, Qi J, Zhang W, Bradner JE. Biased multicomponent reactions to develop novel bromodomain inhibitors. J Med Chem. 2014;57:9019–9027. doi: 10.1021/jm501120z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gosmini R, Nguyen VL, Toum J, Simon C, Brusq JM, Krysa G, Mirguet O, Riou-Eymard AM, Boursier EV, Trottet L, Bamborough P, Clark H, Chung CW, Cutler L, Demont EH, Kaur R, Lewis AJ, Schilling MB, Soden PE, Taylor S, Walker AL, Walker MD, Prinjha RK, Nicodeme E. The discovery of I-BET726 (GSK1324726A), a potent tetrahydroquinoline ApoA1 up-regulator and selective BET bromodomain inhibitor. J Med Chem. 2014;57:8111–8131. doi: 10.1021/jm5010539. [DOI] [PubMed] [Google Scholar]
- 14.Jahagirdar R, Zhang H, Azhar S, Tobin J, Attwell S, Yu R, Wu J, McLure KG, Hansen HC, Wagner GS, Young PR, Srivastava RA, Wong NC, Johansson J. A novel BET bromodomain inhibitor, RVX-208, shows reduction of atherosclerosis in hyperlipidemic ApoE deficient mice. Atherosclerosis. 2014;236:91–100. doi: 10.1016/j.atherosclerosis.2014.06.008. [DOI] [PubMed] [Google Scholar]
- 15.Gehling VS, Hewitt MC, Vaswani RG, Leblanc Y, Cote A, Nasveschuk CG, Taylor AM, Harmange JC, Audia JE, Pardo E, Joshi S, Sandy P, Mertz JA, Sims RJ, 3rd, Bergeron L, Bryant BM, Bellon S, Poy F, Jayaram H, Sankaranarayanan R, Yellapantula S, Bangalore Srinivasamurthy N, Birudukota S, Albrecht BK. Discovery, Design, and Optimization of Isoxazole Azepine BET Inhibitors. ACS Med Chem Lett. 2013;4:835–840. doi: 10.1021/ml4001485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brand M, Measures AM, Wilson BG, Cortopassi WA, Alexander R, Hoss M, Hewings DS, Rooney TP, Paton RS, Conway SJ. Small molecule inhibitors of bromodomain-acetyl-lysine interactions. ACS Chem Biol. 2015;10:22–39. doi: 10.1021/cb500996u. [DOI] [PubMed] [Google Scholar]
- 17.Hay D, Fedorov O, Filippakopoulos P, Martin S, Philpott M, Picaud S, Hewings DS, Uttakar S, Heightman TD, Conway SJ, Knapp S, Brennan PE. The design and synthesis of 5-and 6-isoxazolylbenzimidazoles as selective inhibitors of the BET bromodomains. Medchemcomm. 2013;4:140–144. doi: 10.1039/C2MD20189E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dawson MA, Prinjha RK, Dittmann A, Giotopoulos G, Bantscheff M, Chan WI, Robson SC, Chung CW, Hopf C, Savitski MM, Huthmacher C, Gudgin E, Lugo D, Beinke S, Chapman TD, Roberts EJ, Soden PE, Auger KR, Mirguet O, Doehner K, Delwel R, Burnett AK, Jeffrey P, Drewes G, Lee K, Huntly BJ, Kouzarides T. Inhibition of BET recruitment to chromatin as an effective treatment for MLL-fusion leukaemia. Nature. 2011;478:529–533. doi: 10.1038/nature10509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bonetti P, Ponzoni M, Tibiletti MG, Stathis A, Heirat P, Zucca E, Bertoni F. The BRD-inhibitor OTX015 Shows Pre-clinical Activity in Diffuse Large B-cell Lymphoma (DLBCL) European Journal of Cancer. 2012;48:163–163. [Google Scholar]
- 20.Hewings DS, Rooney TP, Jennings LE, Hay DA, Schofield CJ, Brennan PE, Knapp S, Conway SJ. Progress in the development and application of small molecule inhibitors of bromodomain-acetyl-lysine interactions. Journal of Medicinal Chemistry. 2012;55:9393–9413. doi: 10.1021/jm300915b. [DOI] [PubMed] [Google Scholar]
- 21.Delmore JE, Issa GC, Lemieux ME, Rahl PB, Shi J, Jacobs HM, Kastritis E, Gilpatrick T, Paranal RM, Qi J, Chesi M, Schinzel AC, McKeown MR, Heffernan TP, Vakoc CR, Bergsagel PL, Ghobrial IM, Richardson PG, Young RA, Hahn WC, Anderson KC, Kung AL, Bradner JE, Mitsiades CS. BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell. 2011;146:904–917. doi: 10.1016/j.cell.2011.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Asangani IA, Dommeti VL, Wang X, Malik R, Cieslik M, Yang R, Escara-Wilke J, Wilder-Romans K, Dhanireddy S, Engelke C, Iyer MK, Jing X, Wu YM, Cao X, Qin ZS, Wang S, Feng FY, Chinnaiyan AM. Therapeutic targeting of BET bromodomain proteins in castration-resistant prostate cancer. Nature. 2014 doi: 10.1038/nature13229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.clinicaltrials.gov. NCT01587703.
- 24.Clinicaltrials.gov. NCT01713582.
- 25.Clinicaltrials.gov. NCT01987362.
- 26.Clinicaltrials.gov. NCT01949883.
- 27.Clinicaltrials.gov. NCT02369029.
- 28.Chung CW, Witherington J. Progress in the discovery of small-molecule inhibitors of bromodomain–histone interactions. Journal of biomolecular screening. 2011;16:1170–1185. doi: 10.1177/1087057111421372. [DOI] [PubMed] [Google Scholar]
- 29.Chung CW, Coste H, White JH, Mirguet O, Wilde J, Gosmini RL, Delves C, Magny SM, Woodward R, Hughes SA, Boursier EV, Flynn H, Bouillot AM, Bamborough P, Brusq JM, Gellibert FJ, Jones EJ, Riou AM, Homes P, Martin SL, Uings IJ, Toum J, Clement CA, Boullay AB, Grimley RL, Blandel FM, Prinjha RK, Lee K, Kirilovsky J, Nicodeme E. Discovery and characterization of small molecule inhibitors of the BET family bromodomains. Journal of Medicinal Chemistry. 2011;54:3827–3838. doi: 10.1021/jm200108t. [DOI] [PubMed] [Google Scholar]
- 30.Hewings DS, Wang M, Philpott M, Fedorov O, Uttarkar S, Filippakopoulos P, Picaud S, Vuppusetty C, Marsden B, Knapp S, Conway SJ, Heightman TD. 3,5-dimethylisoxazoles act as acetyl-lysine-mimetic bromodomain ligands. Journal of Medicinal Chemistry. 2011;54:6761–6770. doi: 10.1021/jm200640v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hewings DS, Fedorov O, Filippakopoulos P, Martin S, Picaud S, Tumber A, Wells C, Olcina MM, Freeman K, Gill A, Ritchie AJ, Sheppard DW, Russell AJ, Hammond EM, Knapp S, Brennan PE, Conway SJ. Optimization of 3,5-dimethylisoxazole derivatives as potent bromodomain ligands. Journal of Medicinal Chemistry. 2013;56:3217–3227. doi: 10.1021/jm301588r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fish PV, Filippakopoulos P, Bish G, Brennan PE, Bunnage ME, Cook AS, Federov O, Gerstenberger BS, Jones H, Knapp S, Marsden B, Nocka K, Owen DR, Philpott M, Picaud S, Primiano MJ, Ralph MJ, Sciammetta N, Trzupek JD. Identification of a chemical probe for bromo and extra C-terminal bromodomain inhibition through optimization of a fragment-derived hit. Journal of Medicinal Chemistry. 2012;55:9831–9837. doi: 10.1021/jm3010515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Seal J, Lamotte Y, Donche F, Bouillot A, Mirguet O, Gellibert F, Nicodeme E, Krysa G, Kirilovsky J, Beinke S, McCleary S, Rioja I, Bamborough P, Chung CW, Gordon L, Lewis T, Walker AL, Cutler L, Lugo D, Wilson DM, Witherington J, Lee K, Prinjha RK. Identification of a novel series of BET family bromodomain inhibitors: binding mode and profile of I-BET151 (GSK1210151A) Bioorganic & Medicinal Chemistry Letters. 2012;22:2968–2972. doi: 10.1016/j.bmcl.2012.02.041. [DOI] [PubMed] [Google Scholar]
- 34.Bamborough P, Diallo H, Goodacre JD, Gordon L, Lewis A, Seal JT, Wilson DM, Woodrow MD, Chung CW. Fragment-based discovery of bromodomain inhibitors part 2: optimization of phenylisoxazole sulfonamides. Journal of Medicinal Chemistry. 2012;55:587–596. doi: 10.1021/jm201283q. [DOI] [PubMed] [Google Scholar]
- 35.Chung CW, Dean AW, Woolven JM, Bamborough P. Fragment-based discovery of bromodomain inhibitors part 1: inhibitor binding modes and implications for lead discovery. Journal of Medicinal Chemistry. 2012;55:576–586. doi: 10.1021/jm201320w. [DOI] [PubMed] [Google Scholar]
- 36.Vollmuth F, Blankenfeldt W, Geyer M. Structures of the dual bromodomains of the P-TEFb-activating protein Brd4 at atomic resolution. The Journal of biological chemistry. 2009;284:36547–36556. doi: 10.1074/jbc.M109.033712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Feng Q, Zhang Z, Shea MJ, Creighton CJ, Coarfa C, Hilsenbeck SG, Lanz R, He B, Wang L, Fu X, Nardone A, Song Y, Bradner J, Mitsiades N, Mitsiades CS, Osborne CK, Schiff R, O’Malley BW. An epigenomic approach to therapy for tamoxifen-resistant breast cancer. Cell research. 2014 doi: 10.1038/cr.2014.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pastori C, Daniel M, Penas C, Volmar CH, Johnstone AL, Brothers SP, Graham RM, Allen B, Sarkaria JN, Komotar RJ, Wahlestedt C, Ayad NG. BET bromodomain proteins are required for glioblastoma cell proliferation. Epigenetics: official journal of the DNA Methylation Society. 2014;9:611–620. doi: 10.4161/epi.27906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Picaud S, Wells C, Felletar I, Brotherton D, Martin S, Savitsky P, Diez-Dacal B, Philpott M, Bountra C, Lingard H, Fedorov O, Muller S, Brennan PE, Knapp S, Filippakopoulos P. RVX-208, an inhibitor of BET transcriptional regulators with selectivity for the second bromodomain. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:19754–19759. doi: 10.1073/pnas.1310658110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zuber J, Shi J, Wang E, Rappaport AR, Herrmann H, Sison EA, Magoon D, Qi J, Blatt K, Wunderlich M, Taylor MJ, Johns C, Chicas A, Mulloy JC, Kogan SC, Brown P, Valent P, Bradner JE, Lowe SW, Vakoc CR. RNAi screen identifies Brd4 as a therapeutic target in acute myeloid leukaemia. Nature. 2011;478:524–528. doi: 10.1038/nature10334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bisagni E, Nguyen CH, Pierre A, Pepin O, Decointet P, Gros P. 1-Amino-Substituted 4-Methyl-5h-Pyrido[4,3-B]Indoles (Gamma-Carbolines) as Tricyclic Analogs of Ellipticines - a New Class of Antineoplastic Agents. Journal of Medicinal Chemistry. 1988;31:398–405. doi: 10.1021/jm00397a023. [DOI] [PubMed] [Google Scholar]
- 42.Griesbeck AG, Franke M, Neudorfl J, Kotaka H. Photocycloaddition of aromatic and aliphatic aldehydes to isoxazoles: Cycloaddition reactivity and stability studies. Beilstein Journal of Organic Chemistry. 2011;7:127–134. doi: 10.3762/bjoc.7.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Liverton N. Tricyclic compounds as mGluR4 modulators and their preparation and use for treatment and prevention of mGluR4-mediated diseases. PCT Int Appl. 2012 2012006760. [Google Scholar]
- 44.Muller PA, Vousden KH. Mutant p53 in cancer: new functions and therapeutic opportunities. Cancer Cell. 2014;25:304–317. doi: 10.1016/j.ccr.2014.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jones G, Willett P, Glen RC, Leach AR, Taylor R. Development and validation of a genetic algorithm for flexible docking. Journal of molecular biology. 1997;267:727–748. doi: 10.1006/jmbi.1996.0897. [DOI] [PubMed] [Google Scholar]
- 46.Verdonk ML, Cole JC, Hartshorn MJ, Murray CW, Taylor RD. Improved protein-ligand docking using GOLD. Proteins. 2003;52:609–623. doi: 10.1002/prot.10465. [DOI] [PubMed] [Google Scholar]
- 47.Wang ZX. An exact mathematical expression for describing competitive binding of two different ligands to a protein molecule. FEBS Lett. 1995;360:111–114. doi: 10.1016/0014-5793(95)00062-e. [DOI] [PubMed] [Google Scholar]
- 48.Zhang RM, Mayhood T, Lipari P, Wang YL, Durkin J, Syto R, Gesell J, McNemar C, Windsor W. Fluorescence polarization assay and inhibitor design for MDM2/p53 interaction. Analytical Biochemistry. 2004;331:138–146. doi: 10.1016/j.ab.2004.03.009. [DOI] [PubMed] [Google Scholar]
- 49.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Macromolecular Crystallography, Pt A. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 50.Mccoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. Journal of Applied Crystallography. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bricogne GBE, Brandl M, Flensburg C, Keller P, Paciorek W, Roversi P, Sharff A, Smart OS, Vonrhein C, Womack TO. Cambridge, United Kingdom: Global Phasing Ltd; BUSTER version X.Y.Z. 2011 [Google Scholar]
- 52.Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallographica Section D-Biological Crystallography. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chen VB, Arendall WB, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, Richardson DC. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallographica Section D-Biological Crystallography. 2010;66:12–21. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zucker F, Champ PC, Merritt EA. Validation of crystallographic models containing TLS or other descriptions of anisotropy. Acta Crystallographica Section D-Biological Crystallography. 2010;66:889–900. doi: 10.1107/S0907444910020421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hooft RWW, Vriend G, Sander C, Abola EE. Errors in protein structures. Nature. 1996;381:272–272. doi: 10.1038/381272a0. [DOI] [PubMed] [Google Scholar]
- 56.Bai L, Chen J, McEachern D, Liu L, Zhou H, Aguilar A, Wang S. BM-1197: a novel and specific Bcl-2/Bcl-xL inhibitor inducing complete and long-lasting tumor regression in vivo. PloS one. 2014;9:e99404. doi: 10.1371/journal.pone.0099404. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.