

Abstract
Missense mutations which introduce or remove cysteine residues in receptor tyrosine kinases (RTKs) are believed to cause pathologies by stabilizing the active RTK dimers. However, the magnitude of this stabilizing effect has not been measured for full-length receptors. Here, we characterize the dimer stabilities of three full-length fibroblast growth factor (FGFR) mutants harboring pathogenic cysteine substitutions: the C178S FGFR1 mutant, the C342R FGFR2 mutant, and the C228R FGFR3 mutant. We find that the three mutations stabilize the FGFR dimers. We further see that the mutations alter the configuration of the FGFR transmembrane (TM) dimers. Thus, both aberrant dimerization and perturbed dimer structure likely contribute to the pathological phenotypes arising due to these mutations.
Keywords: FGFR, cysteine mutation, disulfide bond, dimer stability, structural change
Fibroblast growth factor receptors (FGFR1–4) comprise a family of transmembrane tyrosine kinases which bind 18 fgf ligands with high affinity, in the presence of heparan sulfate1. They are composed of extracellular (EC) portions built of three Ig-like domains (D1, D2, and D3), single transmembrane (TM) domains, and intracellular kinase domains2,3. These receptors play key roles in the regulation of cell differentiation, migration, proliferation and apoptosis1. They are required for embryonic development, lung morphogenesis, osteogenesis and limb bud development4,5.
The FGF receptors belong to the large family of receptor tyrosine kinases (RTKs)6. Like all RTKs, they are activated upon lateral dimerization in the cellular membrane, which brings the two kinases in close proximity1,6,7. In the dimer, the two kinases activate each other by cross-phosphorylating the tyrosines in the kinase activation loop. For many years, dimerization was believed to occur only in response to ligand binding1. Recent work has demonstrated, however, that dimerization can also occur in the absence of ligand because the FGF receptors have intrinsic sequence-specific propensities for lateral interactions8,9.
The FGF receptors are known to harbor many pathogenic mutations10,11. As germ-line mutations, they cause developmental abnormalities of the skeletal system1,4,10,12–15. As somatic mutations, that have been linked to various cancers16,17. Some of these mutations are believed to cause pathologies by stabilizing the active FGFR dimers18. In particular, mutations which introduce or remove cysteine residues are believed to belong to this category19. Indeed, the unpaired cysteines can form disulfide bonds that bridge two receptors, leading to constitutive dimerization.
Here we study the effect of three cysteine mutations in FGFR1, FGFR2, and FGFR3 on the stability of full-length FGF dimers. The first mutation that we investigate is the C178S mutation in the first Ig-like domain (D1) of FGFR1, associated with Kallman syndrome20. The phenotype includes severe ear anomalies (hypoplasia of the external ear), failure to start puberty, infertility, and complete lack of sense of smell. It occurs in 1 in 10,000 men and 1 in 50,000 women21. The second mutation that we study is the C342R mutation in the third Ig-like domain (D3) of FGFR2. This mutation is found in individuals with Crouzon syndrome, Jackson-Weiss syndrome (JWS), Pfeiffer syndrome, and Antley-Bixler-like syndrome (ABS2)11,14,22–31. Crouzon syndrome is characterized by premature fusion of skull sutures (craniosynostosis), and has an incident rate of 1 in 2500 individuals32. The JWS phenotype is characterized by craniosynostosis and foot abnormalities. The features of Pfeiffer syndrome are short fingers and soft-tissue syndactyly, while the ABS2 phenotype includes craniofacial and limb abnormalities13,14,24,33. The third mutation that we investigate is C228R, located in the second Ig-like domain (D2) of FGFR3 and linked to colorectal cancer carcinoma33, the most common type of intestinal cancer with 140,000 new cases each year in the United States alone.
These three pathogenic mutations substitute a cysteine in an Ig domain in the extracellular portions of the receptors. The Ig domains in the wild-type FGF receptors (D1, D2, D3) are each stabilized by two intra-molecular disulfide bonds. Thus, the loss of a cysteine in an Ig domain creates an unpaired cysteine. Specifically, the introduction of the studied mutations leaves C230 in FGFR1, C278 in FGFR2, and C176 in FGFR3 unpaired. These unpaired cysteines can then engage in inter-molecular disulfide bond formation, which can lead to dimer stabilization.
Here we investigate if the stabilities of the full-length FGFR dimers carrying the mutations are increased. In particular, we measure the stabilities of the mutant FGFR dimers using a quantitative FRET method termed Quantitative Imaging FRET (QI-FRET)34,35, and compare them to previously published stabilities of the wild-type FGF receptor dimers8. The QI-FRET method yields dimer stabilities and structural insights about the dimers, based on FRET detection35,35–37. While not a single molecule technique, this method allows us to acquire data over a broad receptor concentration range, and collect binding curves. The technique also allows us to monitor structural perturbations on the cytoplasmic side of the membrane as a result of pathogenic mutations in the extracellular domain of the receptors8. The technique has been described previously as a detailed step-by-step protocol34. Non-interacting monomeric RTK variants, namely truncated ErbB receptors lacking the intracellular domains, have been identified and serve as negative controls in the QI-FRET experiments38. Furthermore, sequence-specific interactions with FGFR homodimers have been demonstrated through extensive mutagenesis and deletions8,36,39.
The three mutations, C178S in FGFR1, C342R in FGFR2, and C228R in FGFR3 were introduced in the full length and truncated FGFR-YFP and FGFR-mCherry plasmids, which have been used in previous work8. YFP and mCherry comprise a FRET pair that allows FRET detection of FGFR dimerization. The experiments were performed in plasma membrane vesicles derived from CHO cells, which bud-off from cells when the cells are incubated with an osmotic buffer40. These vesicles are a simplified, yet highly relevant model of the plasma membrane41.
CHO cells were co-transfected with either FGFR1C178S-YFP and FGFR1C178S-mCherry, FGFR2C342R-YFP and FGFR2C342R-mCherry, or FGFR3C228R-YFP and FGFR3C228R-mCherry. Twenty four hours after transfection, the cells were treated with chloride salt buffer overnight at 37°C to induce shedding of plasma membrane vesicles bearing the receptors40. Vesicles were collected in 4 chambered slides and imaged in a confocal laser scanning microscope34. Each vesicle was imaged in 3 scans: a donor scan, a FRET scan, and an acceptor scan, as previously described34. The intensity of each vesicle image was quantified using a Matlab program, which yields the donor concentration, the acceptor concentration and the FRET efficiency for each vesicle34 (see Supplementary Information). Figure 1, top row, shows the FRET efficiency as a function of total receptor (donor label + acceptor label) concentration. Figure 1, middle row, shows the donor versus acceptor concentration in each vesicle. For each construct, more than 300 individual vesicles were imaged in at least 3 independent experiments. As discussed in detail previously, such large number of data points is required for robust analysis of the FRET data35. Because the technique uses a standard confocal microscope, we have white noise associated with image acquisition (discussed in34). Since the underlying factor is white noise, the uncertainties can be reduced by acquiring a large number of data points34.
Figure 1.
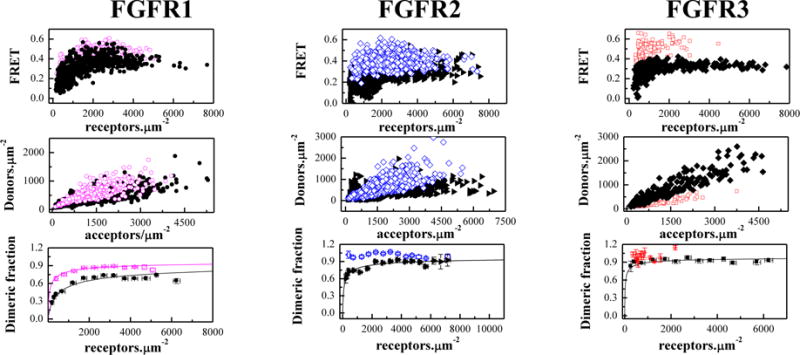
The cysteine mutations stabilize full-length FGFR dimers in the absence of ligands. Open magenta circles: C178R FGFR1; Open blue diamonds: C342R FGFR2; Open red squares: C228R FGFR3). Top panels: FRET efficiencies for the mutants (open color symbols) and the wild-types (solid black symbols). Each data point corresponds to a single vesicle. Middle panels: Donor concentration versus acceptor concentration in each vesicle. The DNA ratios in transfections varied from 1:2 to 1:5, and the acceptor concentration exceeds the donor concentration in all cases. This difference in expression is necessary in our experiments because of the different quantum yields of the donor (0.61) and the acceptor (0.22), and is fully accounted for in the data analysis. Bottom panels: Dimeric fraction as a function of total receptor concentration (donors+acceptors). The C178 mutation stabilizes FGFR1 dimers by −1.2 ± 0.2 kcal/mol). The C342R FGFR2 and C228R FGFR3 form constitutive dimers. Data for the wild-type receptors are from8. The QI-FRET method, used for data collection and analysis, has been described in detail in refs. 34,35,50
A monomer-dimer equilibrium model with two optimizable parameters, the dimerization constant K and the Intrinsic FRET efficiency (I-FRET) was fitted to the data in Figure 1 (see equation (8)), yielding the optimal K and I-FRET values for the receptors. As described in Methods, the dimerization constant reports on the propensities of the receptors to form dimers, while the Intrinsic FRET depends on the separation between the fluorescent proteins and thus reports on the dimer structure8,37.
Next, each data set in Figure 1 was divided by the optimal I-FRET to obtain the dimeric fraction in each vesicle as a function of the total receptor concentration in the vesicle. Then, the dimeric fractions for similar receptor concentrations were averaged within bins of width 500 receptors/μm2 in Figure 1, bottom row. The theoretical dimeric fraction is plotted with a solid line for each receptor, for the optimal value of K determined in the fit. The optimal values for the dimerization free energy and the Intrinsic FRET are shown in Table 1.
Table 1.
Dissociation constants, Kdiss, and Intrinsic FRET efficiencies, obtained from least-square parameter fits to the FRET data for mutant and wild-type full-length and truncated EC+TM FGF receptors. Also shown are the dimerization free energies (dimer stabilities), ΔG. The average distance between the fluorescent proteins in the dimers, d, is calculated under the assumption of free fluorescent protein rotation (an assumption that is not necessarily correct). The uncertainties shown are the 67% confidence intervals (standard errors), determined from the fits. The QI-FRET method, used for data collection and analysis, has been described in detail in refs.34,35,50, and is briefly discussed in Supplementary Information
| FGFR variant | Kdiss (rec.μm−2) | ΔG (kcal.mol−1) | ΔΔGMUT (kcal.mol−1) | I-FRET | d(Å) |
|---|---|---|---|---|---|
| Full FGFR1 C178S | 98 (78 to 117) | −5.5(−5.4 to −5.6) | −1.2 ± 0.2 | 0.69 (0.68 to 0.7) | 46.5 (46.1 to 46.8) |
| Full FGFR1 WT | 710 (630 to 826) | −4.3 (−4.2 to −4.4) | 0.66 (0.65 to 0.68) | 47.6 (46.8 to 47.9) | |
| Full FGFR2 C342R | 100% dimer | 100% dimer | >1.6 | 0.55 (0.54 to 0.56) | 51.3 (51 to 51.7) |
| Full FGFR2 WT | 111 (100 to 146) | −5.4 (−5.3 to −5.5) | 0.43 (0.42 to 0.44) | 55.7 (55.3 to 56.1) | |
| Full FGFR3 C228R | 100% dimer | 100% dimer | >0.7 | 0.63 (0.62 to 0.64) | 48.6 (48.3 to 49) |
| Full FGFR3 WT | 24 (14 to 34) | −6.3 (−6.1 to −6.5) | 0.55 (0.54 to 0.56) | 51.3 (51.0 to 51.7) | |
| ECTM FGFR1 C178S | 141 (122 to 160) | −5.3(−5.2 to −5.4) | −0.7(−0.6 to −0.8) | 0.68 (0.66 to 0.7) | 46.8 (46.1 to 47.6) |
| ECTM FGFR1 WT | 428 (370 to 540) | −4.6(−4.5 to −4.7) | 0.5 (0.47 to 0.51) | 53.1(52.8 to 54.2) | |
| ECTM FGFR2 C342R | 100% dimer | 100% dimer | > −3.6 | 0.72 (0.71, 0.73) | 45.4 (45 to 45.8) |
| ECTM FGFR2 WT | 3235 (2726 to 3809) | −3.4(−3.3 to −3.5) | 0.57 (0.55 to 0.61) | 50.7 (49.3to 51.4) | |
| ECTM FGFR3 C228R | 63 (50 to 100) | −5.7(−5.5 to −5.9) | −2.3± 0.2 | 0.7 (0.69 to 0.72) | 46.1 (45.4 to 46.5) |
| ECTM FGFR3 WT | 3235 (2670 to 3660) | −3.4(−3.3 to −3.5) | 0.52 (0.49 to 0.55) | 52.4(51.4 to 53.4) |
We see that the three mutations stabilize the FGFR dimers. The C178S FGFR1 mutation increases the dimerization propensity of FGFR1 by −1.2 kcal/mol (Table 1). The FRET data for the other two mutants do not depend on the concentration, indicative of constitutive association. This association here is dimerization.
FGFR receptors are known to form dimers, and we thus expect that FGFR2C342R and FGFR3C228R form constitutive dimers. In Figure S5 in Supplementary Information, the FRET efficiency for FGFR2C342R and FGFR3C228R is plotted as a function of the acceptor fraction. The data are averaged in acceptor fraction bins of width 0.05. For constitutive oligomers, the dependence of FRET efficiency on the acceptor fraction is known to be linear for a dimer and non-linear for higher order oligomers42–44. We see that the data in Figure S5 are well described by a linear function (p<0.01), a finding that is consistent with the expectation for dimer formation.
The fact that we observe constitutive dimers for FGFR2C342R and FGFR3C228R means that the monomer concentration is too low to be measured experimentally over the concentration range accessible in our experiments. The QI-FRET method, as applied to our microscope system, cannot resolve stabilities that exceed −7 kcal/mole and thus the best estimates for the effect of the mutations on dimerization are −1.2 kcal/mole, > −1.6 kcal/mole, and > −0.7 kcal/mole for full-length FGFR1, FGFR2, and FGFR3, respectively. Literature values for the energetic contributions of disulfide bonds to protein-protein interactions are in the range of 2–4 kcal/mol45,46. Therefore, the measured effects for the full-length FGF receptors are in the same general range. It is possible, however, that the effect are larger than what can be measured with the QI-FRET method, due to the −7 kcal/mole experimental cut-off.
As shown in Table 1, the Intrinsic FRET value for FGFR1 C178S is 0.69 ± 0.01 and is similar to the Intrinsic FRET value for wild-type FGFR1 (0.66 ± 0.02). The Intrinsic FRET value for the C342R FGFR2 mutant is 0.55 ± 0.01, and is different from the value for wild-type FGFR2 (0.43 ± 0.01). The Intrinsic FRET value measured for the FGFR3 C228R mutant is 0.63 ± 0.01, different from the Intrinsic FRET value for wild-type FGFR3, 0.55 ± 0.01. The increase in Intrinsic FRET for two of the mutants shows that the distance between the fluorescent proteins in the FGFR dimer decreases. Thus, both the C342R mutation in FGFR2 and the C228R mutation in FGFR3 induce conformational changes in the dimers which propagate from the EC domains all the way to the C-terminal tails of the receptors. While no such effect is observed for the FGFR1 C178S mutation, it cannot be excluded that the structural changes due to the C178S mutation are too subtle to be measured in the FRET assay. Furthermore, note that the fluorescent proteins are attached to the receptor C-terminal tails, which are likely highly unstructured.
Previous work has highlighted the importance of the TM domain configuration for the activation of the FGF receptors8. In particular, the QI-FRET technique, along with biochemical and NMR data, has revealed that the configurations of FGFR TM domains control the kinase activity8. With this knowledge in mind, here we assess whether the three cysteine mutations, described above, perturb the FGFR TM dimer configurations in the plasma membrane. We therefore performed experiments with truncated FGFR3 constructs lacking the IC domain. In this constructs, the fluorescent proteins were attached to the TM domains via flexible (GGS)5 linkers, allowing us to monitor directly the TM domain dimer configurations. These constructs contained the Ec domain, the TM domain, the flexible linker, and the fluorescent proteins. The FRET results for the three truncated constructs are shown in Figure 2 and Table 1. The estimates for the effect of the mutations on the truncated receptors are −0.7 kcal/mole, >3.6 kcal/mole, and > −2.3 kcal/mole, respectively (Table 1).
Figure 2.
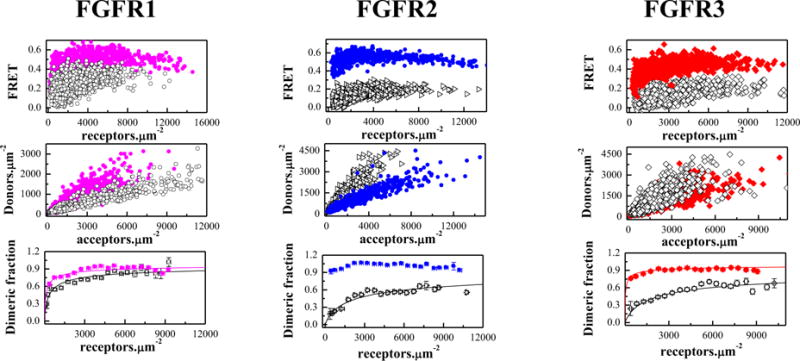
The cysteine mutations stabilize truncated EC+TM FGFR dimers in the absence of ligands. Solid magenta circles: C178R FGFR1; Solid blue diamonds: C342R FGFR2; Solid red squares: C228R FGFR3. Top panels: FRET efficiencies for the mutants (solid color symbols) and the wild-types (open black symbols,8). Middle panels: Donor concentration versus acceptor concentration in each vesicle. Bottom panels: Dimeric fraction as a function of total receptor concentration (donors+acceptors). The QI-FRET method, used for data collection and analysis, has been described in detail in refs. 34,35,50
While the Cys mutations stabilize both the full-length and the truncated FGF receptors (Table 1), the increase in dimer stability is not necessarily the same. For instance, the C178S FGFR1 mutation stabilizes the full length FGFR1 dimer by −1.2 ± 0.2 kcal/mol, while the stability of the truncated receptor is increased by −0.7 ± 0.2 kcal/mol only. The latter value is modest, and is reminiscent of previous measurements with truncated FGFR3 cysteine mutants linked to thanatophoric dysplasia36. These findings may suggest that the different RTK domains do not act independently during the dimerization process, but rather work in a synergistic manner as proposed previously47–49.
The Intrinsic FRET for the three truncated receptors was also increased, suggesting that the fluorescent proteins are closer in the mutant dimers as compared to the wild-type dimers. Indeed, the Intrinsic FRET for the wild-types varied between 0.50 and 0.57, while the values for the mutants were 0.68 to 0.72. This result suggests that these mutations bring the fluorescent proteins in the dimer closer together. Because the fluorescent proteins are attached directly to the C-termini of the TM domains via flexible linkers49, from these results we infer that the C-termini of the TM domains also move closer together due to the Cys mutations. Since in the full-length receptors the kinase domains are attached to the TM domains via the juxtamembrane domains, the results suggest that the Cys mutations in the extracellular domain bring the kinase domains in the dimer closer together. The increase in Intrinsic FRET, measured for two of the full-length FGFR, further supports the view that the Cys-induced structural changes which originate in the extracellular domains are propagated along the entire length of the FGFR dimer.
To test if disulfide bonds contribute to the observed dimer stabilization and structural perturbations, we performed SDS gel electrophoresis under reducing and non-reducing conditions, followed by Western blot staining for both full-length and truncated FGF receptors. While the native contacts that occur in the native plasma membrane between the two receptors in the dimer are eliminated in the SDS environment, this experiment can provide insight as to whether disulfide bonds play a role in dimer stabilization. All the mutants exhibited dimeric bands on the non-reducing gels, but not on the reducing gels (Figure 3), demonstrating the formation of inter-molecular disulfide bonds by the unpaired cysteines in all cases. Thus, disulfide bonds readily form due to the mutations, and the observed stabilization effects in the plasma membrane are likely a consequence of inter-molecular disulfide bond formation.
Figure 3.
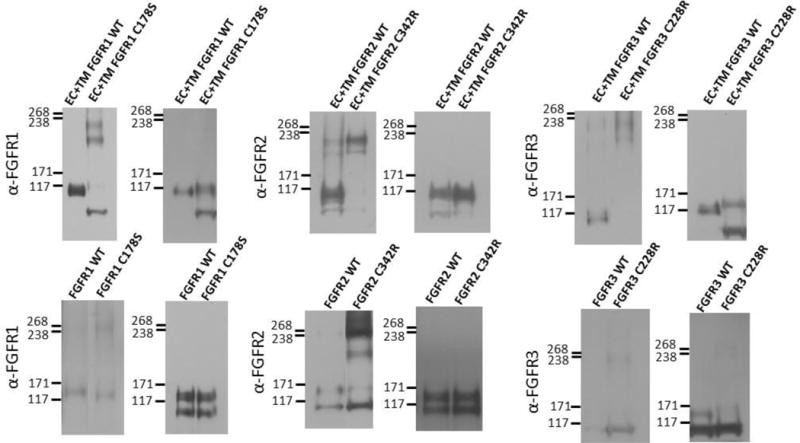
Anti-FGFR staining of (left) non-reducing and (right) reducing western blots for wild-type and mutant FGF receptors. Top row: truncated (EC+TM+YFP) receptors. Bottom row: full-length receptors. Typically, two monomeric and two dimeric bands can be observed, depending on the relative expression of the two forms, and the exposure. The lower bands correspond to immature, partially glycosylated receptors, while the higher molecular weight bands correspond to the mature, fully glycosylated forms of the receptors (~110 kDa for EC+TM+YFP and ~130 kDa for full length FGFR1–3)51–54. In these experiments, cells were starved in serum-free medium for 24 h following transfection with 1–3 μg of DNA encoding full length FGFRs. All experiments were performed with CHO cells, with the exception of the full-length FGFR3 blot, which was performed with HEK 293T cells due to poor expression of the C228 mutant in CHO cells. They were then treated with lysis buffer (25 mM Tris-HCl, 0.5% Triton X-100, 20mM NaCl, 2 mM EDTA, phosphatase inhibitor and protease inhibitor, Roche Applied Science). Lysates were collected following centrifugation at 15,000 g for 15 min at 4°C and loaded onto 3–8%NuPAGE®Novex®Tris-Acetatemini gels (Invitrogen, CA). The proteins were transferred onto a nitrocellulose membrane, and blocked using 5% milk in TBS. FGFR total protein levels were assessed using antibodies raised against the N-terminal epitope of FGFR3 (H-100; sc-9007), FGFR2 (H-80; sc-20735) and FGFR1 (H-76; sc-7945), all from Santa Cruz Biotechnology. This was followed by anti-rabbit HRP conjugated antibodies (W4011, Promega). The proteins were detected using the Amersham ECL detection system (GE Healthcare).
In summary, here we show directly and for the first time that three pathogenic Cys mutations stabilize full-length FGFR dimers, via a mechanism that involves intermolecular disulfide bonds. We also demonstrate that the pathogenic Cys mutations perturb the FGFR dimer structure in the plasma membrane, as shown previously for other FGFR mutations8,36. While future work should focus on the activity of these Cys mutants, the work presented here suggest that the three studied Cys pathogenic mutations affect FGFR signaling by perturbing both FGFR dimerization propensity and FGFR dimer structure.
Supplementary Material
Acknowledgments
This work was supported by NIH grant GM068619. We thank Dr. Daniel Donoghue for the full length FGFR3 plasmid, and Dr. Moosa Mohammadi for the full-length FGFR1 and FGFR2 plasmids.
Reference List
- 1.L’Horte CGM, Knowles MA. Cell responses to FGFR3 signaling: growth, differentiation and apoptosis. Experim Cell Res. 2005;304:417–431. doi: 10.1016/j.yexcr.2004.11.012. [DOI] [PubMed] [Google Scholar]
- 2.Mohammadi M, Olsen SK, Ibrahimi OA. Structural basis for fibroblast growth factor receptor activation. Cytokine & Growth Factor Reviews. 2005;16:107–137. doi: 10.1016/j.cytogfr.2005.01.008. [DOI] [PubMed] [Google Scholar]
- 3.Goetz R, Mohammadi M. Exploring mechanisms of FGF signalling through the lens of structural biology. Nat Rev Mol Cell Biol. 2013;14:166–180. doi: 10.1038/nrm3528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Foldynova-Trantirkova S, Wilcox WR, Krejci P. Sixteen Years and Counting: The Current Understanding of Fibroblast Growth Factor Receptor 3 (FGFR3) Signaling in Skeletal Dysplasias. Human Mutation. 2012;33:29–41. doi: 10.1002/humu.21636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Aviezer D, Golembo M, Yayon A. Fibroblast growth factor receptor-3 as a therapeutic target for achondroplasia - Genetic short limbed dwarfism. Current Drug Targets. 2003;4:353–365. doi: 10.2174/1389450033490993. [DOI] [PubMed] [Google Scholar]
- 6.Fantl WJ, Johnson DE, Williams LT. Signaling by Receptor Tyrosine Kinases. Annu Rev Biochem. 1993;62:453–481. doi: 10.1146/annurev.bi.62.070193.002321. [DOI] [PubMed] [Google Scholar]
- 7.Lemmon MA, Schlessinger J. Cell Signaling by Receptor Tyrosine Kinases. Cell. 2010;141:1117–1134. doi: 10.1016/j.cell.2010.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sarabipour S, Hristova K. Mechanism of FGF receptor dimerization and activation. Nat Commun. 2016;7:10262. doi: 10.1038/ncomms10262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ahmed Z, Lin CC, Suen KM, Melo FA, Levitt JA, Suhling K, Ladbury JE. Grb2 controls phosphorylation of FGFR2 by inhibiting receptor kinase and Shp2 phosphatase activity. J Cell Biol. 2013;200:493–504. doi: 10.1083/jcb.201204106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vajo Z, Francomano CA, Wilkin DJ. The molecular and genetic basis of fibroblast growth factor receptor 3 disorders: The achondroplasia family of skeletal dysplasias, Muenke craniosynostosis, and Crouzon syndrome with acanthosis nigricans. Endocrine Reviews. 2000;21:23–39. doi: 10.1210/edrv.21.1.0387. [DOI] [PubMed] [Google Scholar]
- 11.Passos-Bueno MR, Wilcox WR, Jabs EW, Sertié AL, Alonso LG, Kitoh H. Clinical spectrum of fibroblast growth factor receptor mutations. Human Mutation. 1999;14:115–125. doi: 10.1002/(SICI)1098-1004(1999)14:2<115::AID-HUMU3>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 12.Horton WA, Hall JG, Hecht JT. Achondroplasia. Lancet. 2007;370:162–172. doi: 10.1016/S0140-6736(07)61090-3. [DOI] [PubMed] [Google Scholar]
- 13.Meyers GA, Orlow SJ, Munro IR, Przylepa KA, Jabs EW. Fibroblast-Growth-Factor-Receptor-3 (Fgfr3) Transmembrane Mutation in Crouzon-Syndrome with Acanthosis Nigricans. Nat Genet. 1995;11:462–464. doi: 10.1038/ng1295-462. [DOI] [PubMed] [Google Scholar]
- 14.Park WJ, Bellus GA, Jabs EW. Mutations in Fibroblast Growth-Factor Receptors - Phenotypic Consequences During Eukaryotic Development. American Journal of Human Genetics. 1995;57:748–754. [PMC free article] [PubMed] [Google Scholar]
- 15.Mcintosh I, Bellus GA, Jabs EW. The pleiotropic effects of fibroblast growth factor receptors in mammalian development. Cell Structure and Function. 2000;25:85–96. doi: 10.1247/csf.25.85. [DOI] [PubMed] [Google Scholar]
- 16.van Rhijn BWG, van Tilborg AAG, Lurkin I, Bonaventure J, De Vries A, Thiery JP, van der Kwast TH, Zwarthoff EC, Radvanyi F. Novel fibroblast growth factor receptor 3 (FGFR3) mutations in bladder cancer previously identified in non-lethal skeletal disorders. European Journal of Human Genetics. 2002;10:819–824. doi: 10.1038/sj.ejhg.5200883. [DOI] [PubMed] [Google Scholar]
- 17.van Rhijn BWG, Montironi R, Zwarthoff EC, Jobsis AC, van der Kwast TH. Frequent FGFR3 mutations in urothelial papilloma. Journal of Pathology. 2002;198:245–251. doi: 10.1002/path.1202. [DOI] [PubMed] [Google Scholar]
- 18.Webster MK, Donoghue DJ. FGFR activation in skeletal disorders: Too much of a good thing. Trends Genet. 1997;13:178–182. doi: 10.1016/s0168-9525(97)01131-1. [DOI] [PubMed] [Google Scholar]
- 19.Adar R, Monsonego-Ornan E, David P, Yayon A. Differential activation of cysteine-substitution mutants of fibroblast growth factor receptor 3 is determined by cysteine localization. Journal of Bone and Mineral Research. 2002;17:860–868. doi: 10.1359/jbmr.2002.17.5.860. [DOI] [PubMed] [Google Scholar]
- 20.Dode C, Levilliers J, Dupont JM, De Paepe A, Le Du N, Soussi-Yanicostas N, Coimbra RS, Delmaghani S, Compain-Nouaille S, Baverel F, Pecheux C, Le Tessier D, Cruaud C, Delpech M, Speleman F, Vermeulen S, Amalfitano A, Bachelot Y, Bouchard P, Cabrol S, Carel JC, Delemarre-van de Waal H, Goulet-Salmon B, Kottler ML, Richard O, Sanchez-Franco F, Saura R, Young J, Petit C, Hardelin JP. Loss-of-function mutations in FGFR1 cause autosomal dominant Kallmann syndrome. Nat Genet. 2003;33:463–465. doi: 10.1038/ng1122. [DOI] [PubMed] [Google Scholar]
- 21.Zenaty D, Bretones P, Lambe C, Guemas I, David M, Leger J, de Roux N. Paediatric phenotype of Kallmann syndrome due to mutations of fibroblast growth factor receptor 1 (FGFR1) Mol Cell Endocrinol. 2006:254–255. doi: 10.1016/j.mce.2006.04.006. [DOI] [PubMed] [Google Scholar]
- 22.Reardon W, Winter RM, Rutland P, Pulleyn LJ, Jones BM, Malcolm S. Mutations in the Fibroblast Growth-Factor Receptor-2 Gene Cause Crouzon-Syndrome. Nat Genet. 1994;8:98–103. doi: 10.1038/ng0994-98. [DOI] [PubMed] [Google Scholar]
- 23.Park WJ, Meyers GA, Li X, Theda C, Day D, Orlow SJ, Jones MC, Jabs EW. Novel Fgfr2 Mutations in Crouzon and Jackson-Weiss Syndromes Show Allelic Heterogeneity and Phenotypic Variability. Human Molecular Genetics. 1995;4:1229–1233. doi: 10.1093/hmg/4.7.1229. [DOI] [PubMed] [Google Scholar]
- 24.Rutland P, Pulleyn LJ, Reardon W, Baraitser M, Hayward R, Jones B, Malcolm S, Winter RM, Oldridge M, Slaney SF, Poole MD, Wilkie AOM. Identical Mutations in the Fgfr2 Gene Cause Both Pfeiffer and Crouzon Syndrome Phenotypes. Nat Genet. 1995;9:173–176. doi: 10.1038/ng0295-173. [DOI] [PubMed] [Google Scholar]
- 25.Meyers GA, Day D, Goldberg R, Daentl DL, Przylepa KA, Abrams LJ, Graham JM, Feingold M, Rawnsley E, Scott AF, Jabs EW. FCFR2 exon IIIa and IIIe mutations in Crouzon, Jackson-Weiss, and Pfeiffer syndromes: Evidence for missense changes, insertions, and a deletion due to alternative RNA splicing. American Journal of Human Genetics. 1996;58:491–498. [PMC free article] [PubMed] [Google Scholar]
- 26.Passos-Bueno MR, Sertie AL, Richieri-Costa A, Alonso LG, Zatz M, Alonso N, Brunoni D, Ribeiro SFM. Description of a new mutation and characterization of FGFR1, FGFR2, and FGFR3 mutations among Brazilian patients with syndromic craniosynostoses. American Journal of Medical Genetics. 1998;78:237–241. [PubMed] [Google Scholar]
- 27.Kress W, Collmann H, Busse M, Halliger-Keller B, Mueller CR. Clustering of FGFR2 gene mutations inpatients with Pfeiffer and Crouzon syndromes (FGFR2-associated craniosynostoses) Cytogenet Cell Genet. 2000;91:134–137. doi: 10.1159/000056833. [DOI] [PubMed] [Google Scholar]
- 28.Reardon W, Smith A, Honour JW, Hindmarsh P, Das D, Rumsby G, Nelson I, Malcolm S, Ades L, Sillence D, Kumar D, DeLozier-Blanchet C, Mckee S, Kelly T, McKeehan WL, Baraitser M, Winter RM. Evidence for digenic inheritance in some cases of Antley-Bixler syndrome? Journal of Medical Genetics. 2000;37:26–32. doi: 10.1136/jmg.37.1.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tsai FJ, Yang CF, Wu JY, Tsai CH, Lee CC. Mutation analysis of Crouzon syndrome and identification of one novel mutation in Taiwanese patients. Pediatrics International. 2001;43:263–266. doi: 10.1046/j.1442-200x.2001.01392.x. [DOI] [PubMed] [Google Scholar]
- 30.Lajeunie E, Heuertz S, El Ghouzzi V, Martinovic J, Renier D, Le Merrer M, Bonaventure J. Mutation screening in patients with syndromic craniosynostoses indicates that a limited number of recurrent FGFR2 mutations accounts for severe forms of Pfeiffer syndrome. European Journal of Human Genetics. 2006;14:289–298. doi: 10.1038/sj.ejhg.5201558. [DOI] [PubMed] [Google Scholar]
- 31.Ke R, Yang X, Tianyi C, Ge M, Lei J, Mu X. The C342R Mutation in FGFR2 Causes Crouzon Syndrome With Elbow Deformity. J Craniofac Surg. 2015;26:584–586. doi: 10.1097/SCS.0000000000001472. [DOI] [PubMed] [Google Scholar]
- 32.Hirsch JF, Renier D, Sainterose C. Intracranial-Pressure in Craniostenosis. Monographs in Paediatrics. 1982;15:114–118. [Google Scholar]
- 33.Greenman C, Stephens P, Smith R, Dalgliesh GL, Hunter C, Bignell G, Davies H, Teague J, Butler A, Edkins S, O’Meara S, Vastrik I, Schmidt EE, Avis T, Barthorpe S, Bhamra G, Buck G, Choudhury B, Clements J, Cole J, Dicks E, Forbes S, Gray K, Halliday K, Harrison R, Hills K, Hinton J, Jenkinson A, Jones D, Menzies A, Mironenko T, Perry J, Raine K, Richardson D, Shepherd R, Small A, Tofts C, Varian J, Webb T, West S, Widaa S, Yates A, Cahill DP, Louis DN, Goldstraw P, Nicholson AG, Brasseur F, Looijenga L, Weber BL, Chiew YE, Defazio A, Greaves MF, Green AR, Campbell P, Birney E, Easton DF, Chenevix-Trench G, Tan MH, Khoo SK, Teh BT, Yuen ST, Leung SY, Wooster R, Futreal PA, Stratton MR. Patterns of somatic mutation in human cancer genomes. Nature. 2007;446:153–158. doi: 10.1038/nature05610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chen LR, Novicky L, Merzlyakov M, Hristov T, Hristova K. Measuring the Energetics of Membrane Protein Dimerization in Mammalian Membranes. J Am Chem Soc. 2010;132:3628–3635. doi: 10.1021/ja910692u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sarabipour S, Del Piccolo N, Hristova K. Characterization of Membrane Protein Interactions in Plasma Membrane Derived Vesicles with Quantitative Imaging Forster Resonance Energy Transfer. Acc Chem Res. 2015;48:2262–2269. doi: 10.1021/acs.accounts.5b00238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Del Piccolo N, Placone J, Hristova K. Effect of Thanatophoric Dysplasia Type I Mutations on FGFR3 Dimerization. Biophys J. 2015;108:272–278. doi: 10.1016/j.bpj.2014.11.3460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sarabipour S, Ballmer-Hofer K, Hristova K. VEGFR-2 conformational switch in response to ligand binding. Elife. 2016;5 doi: 10.7554/eLife.13876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.King C, Sarabipour S, Byrne P, Leahy DJ, Hristova K. The FRET signatures of non-interacting proteins in membranes: simulations and experiments. Biophys J. 2014;106:1309–1317. doi: 10.1016/j.bpj.2014.01.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chen L, Placone J, Novicky L, Hristova K. The extracellular domain of fibroblast growth factor receptor 3 inhibits ligand-independent dimerization. Science Signaling. 2010;3:ra86. doi: 10.1126/scisignal.2001195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Del Piccolo N, Placone J, He L, Agudelo SC, Hristova K. Production of plasma membrane vesicles with chloride salts and their utility as a cell membrane mimetic for biophysical characterization of membrane protein interactions. Anal Chem. 2012;84:8650–8655. doi: 10.1021/ac301776j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sarabipour S, Chan RB, Zhou B, Di Paolo G, Hristova K. Analytical characterization of plasma membrane-derived vesicles produced via osmotic and chemical vesiculation. Biochim Biophys Acta. 2015;1848:1591–1598. doi: 10.1016/j.bbamem.2015.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Adair BD, Engelman DM. Glycophorin a helical transmembrane domains dimerize in phospholipid bilayers - a resonance energy transfer study. Biochemistry. 1994;33:5539–5544. doi: 10.1021/bi00184a024. [DOI] [PubMed] [Google Scholar]
- 43.Li M, Reddy LG, Bennett R, Silva ND, Jr, Jones LR, Thomas DD. A fluorescence energy transfer method for analyzing protein oligomeric structure: Application to phospholamban. Biophys J. 1999;76:2587–2599. doi: 10.1016/S0006-3495(99)77411-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schick S, Chen LR, Li E, Lin J, Koper I, Hristova K. Assembly of the M2 Tetramer Is Strongly Modulated by Lipid Chain Length. Biophys J. 2010;99:1810–1817. doi: 10.1016/j.bpj.2010.07.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Betz SF. Disulfide bonds and the stability of globular proteins. Protein Sci. 1993;2:1551–1558. doi: 10.1002/pro.5560021002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.MaAuley A, Jacob J, Kolvenbach CG, Westland K, Lee HJ, Brych SR, Rehder D, Kleemann GR, Brems DN, Matsumura M. Contributions of a disulfide bond to the structure, stability, and dimerization of human IgG1 antibody C(H)3 domain. Protein Sci. 2008;17:95–106. doi: 10.1110/ps.073134408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Macdonald JL, Pike LJ. Heterogeneity in EGF-binding affinities arises from negative cooperativity in an aggregating system. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:112–117. doi: 10.1073/pnas.0707080105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Macdonald-Obermann JL, Pike LJ. The Intracellular Juxtamembrane Domain of the Epidermal Growth Factor (EGF) Receptor Is Responsible for the Allosteric Regulation of EGF Binding. J Biol Chem. 2009;284:13570–13576. doi: 10.1074/jbc.M109.001487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sarabipour S, Hristova K. FGFR3 Unliganded Dimer Stabilization by the Juxtamembrane Domain. J Mol Biol. 2015;427:1705–1714. doi: 10.1016/j.jmb.2015.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Li E, Placone J, Merzlyakov M, Hristova K. Quantitative measurements of protein interactions in a crowded cellular environment. Anal Chem. 2008;80:5976–5985. doi: 10.1021/ac800616u. [DOI] [PubMed] [Google Scholar]
- 51.Chen F, Degnin C, Laederich MB, Horton AW, Hristova K. The A391E mutation enhances FGFR3 activation in the absence of ligand. Biochimica et Biophysica Acta-Biomembranes. 2011;1808:2045–2050. doi: 10.1016/j.bbamem.2011.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chen F, Sarabipour S, Hristova K. Multiple Consequences of a Single Amino Acid Pathogenic RTK Mutation: The A391E Mutation in FGFR3. PLoS ONE. 2013;8:e56521. doi: 10.1371/journal.pone.0056521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.He L, Horton WA, Hristova K. The physical basis behind achondroplasia, the most common form of human dwarfism. J Biol Chem. 2010;285:30103–30114. doi: 10.1074/jbc.M109.094086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.He L, Wimley WC, Hristova K. FGFR3 heterodimerization in achondroplasia, the most common form of human dwarfism. J Biol Chem. 2011;286:13272–13281. doi: 10.1074/jbc.M110.205583. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.