

SUMMARY
Oxidative phosphorylation (OXPHOS) is the major pathway for ATP production in humans. Deficiencies in OXPHOS can arise from mutations in either mitochondrial or nuclear genomes and comprise the largest collection of inborn errors of metabolism. At present we lack a complete catalog of human genes and pathways essential for OXPHOS. Here we introduce a genome-wide CRISPR “death screen” that actively selects dying cells to reveal human genes required for OXPHOS, inspired by the classic observation that human cells deficient in OXPHOS survive in glucose but die in galactose. We report 191 high-confidence hits essential for OXPHOS, including 72 underlying known OXPHOS diseases. Our screen reveals a functional module consisting of NGRN, WBSCR16, RPUSD3, RPUSD4, TRUB2, and FASTKD2 that regulates the mitochondrial 16S rRNA and intra-mitochondrial translation. Our work yields a rich catalog of genes required for OXPHOS and, more generally, demonstrates the power of death screening for functional genomic analysis.
Graphical abstract
INTRODUCTION
In humans, the oxidative phosphorylation (OXPHOS) machinery consists of five macromolecular complexes comprising 90 proteins in total, 77 of which are nuclearly encoded and 13 of which are encoded by the mitochondrial genome (mtDNA). These structural components of the OXPHOS system rely heavily on accessory factors for their proper expression and function. Accordingly, mutations in structural subunits or in assembly factors and mtDNA maintenance, transcription, and translation pathways have been reported to cause mitochondrial OXPHOS disease (Diaz et al., 2011; Nunnari and Suomalainen, 2012; Wallace, 1997). These disorders are challenging to diagnose and manage in part because the age of onset, pattern of organ system involvement, and severity are so highly variable (Pfeffer et al., 2013; Vafai and Mootha, 2012). In humans, the exact number of OXPHOS accessory and regulatory factors remains unknown, but they have been comprehensively identified in S. cerevisiae through systematic genetic studies (Steinmetz et al., 2002; VanderSluis et al., 2014). However, the OXPHOS system has diverged in this model organism, with yeast, for example, lacking complex I and having an mtDNA organization distinct from that of humans. Therefore, although yeast data have significantly advanced our understanding of OXPHOS, additional pathways remain to be identified in humans.
It has long been known that human cells deficient in OXPHOS are viable in glucose-rich medium, but they cannot survive when glucose is replaced with galactose (Robinson et al., 1992). The striking difference in fitness between glucose and galactose has been attributed to the fact that cells growing in glucose utilize both glycolysis and OXPHOS for ATP production, whereas cells growing in galactose are almost entirely dependent on OXPHOS (Reitzer et al., 1979). Hence, although loss of OXPHOS leads to a cell growth disadvantage under glucose-rich conditions (Wheaton et al., 2014), these cells are not viable in galactose. This “metabolic state-dependent lethality” has previously been successfully exploited in arrayed chemical screens to reveal novel small molecule inhibitors of OXPHOS (Gohil et al., 2010). A prior pooled RNAi screen also employed negative growth selection in glucose and galactose media to search for OXPHOS-related genes but had limited success (Bayona-Bafaluy et al., 2011), probably for two reasons. First, RNAi often achieves only partial knockdown, and complete knockdown may be required to have a phenotypic consequence. Second, although such negative selection growth screens can be successful for identifying essential genes with a strong effect on growth (Hart et al., 2015; Luo et al., 2008), detecting the depletion of a gene in a highly complex library is particularly challenging when the fitness effect is modest between the two conditions being compared.
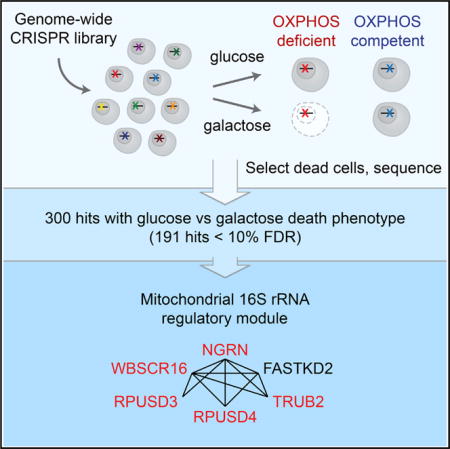
To overcome this challenge, we introduce a positive selection “death screen” that actively selects dying cells and subjects them to next-generation deep sequencing. We combine this approach with a genome-wide clustered regularly interspaced short palindromic repeats (CRISPR)/Cas9 library to systematically catalog OXPHOS factors in human cells. We report that K562 cells with a deficiency in OXPHOS undergo cell death when grown in galactose but not glucose. We exploit this property to purify dead cells from a genome-wide knockout population after culture in glucose or galactose to identify genes required for OXPHOS. In addition to recovering many known OXPHOS components and disease genes, our robust approach also reveals brand-new OXPHOS genes that are uncharacterized or non-mitochondrially localized. Five of the uncharacterized genes (NGRN, WBSCR16, RPUSD3, RPUSD4, and TRUB2), along with FASTKD2, encode components of a functional module that, as we now show, regulates mitochondrial 16S rRNA abundance and is required for intra-mitochondrial translation.
RESULTS
A Genome-wide CRISPR/Cas9 Screen for Death in Galactose versus Glucose
We sought to overcome the challenges inherent in negative selection growth screens by developing a genome-wide CRISPR/Cas9 screen employing positive selection and deep sequencing of dying cells. Annexin V is a well-established marker of cell death that binds phosphatidylserine, an intracellular molecule that becomes accessible during both apoptosis and necrosis (Martin et al., 1995). To confirm that cells with OXPHOS defects undergo cell death in galactose medium, we treated human myelogenous leukemia K562 cells cultured in either glucose or galactose with inhibitors to respiratory chain (RC) complex I (piericidin), complex III (antimycin), or complex V (oligomycin). Annexin V staining clearly shows that treatment with these drugs leads to 85%–95% cell death within 24 hr in galactose but not glucose (Figure 1A). We confirmed that the galactose-specific cell death phenotype also applied to genetic disruption of OXPHOS by testing cells in which we ablated the OXPHOS disease gene NDUFA9 using CRISPR/Cas9 (van den Bosch et al., 2012; Figure S1A). Thus, disruption of an OXPHOS disease gene is sufficient to induce cell death selectively in galactose versus glucose.
Figure 1. A Genome-wide CRISPR Death Screen to Identify Genes Necessary for OXPHOS.
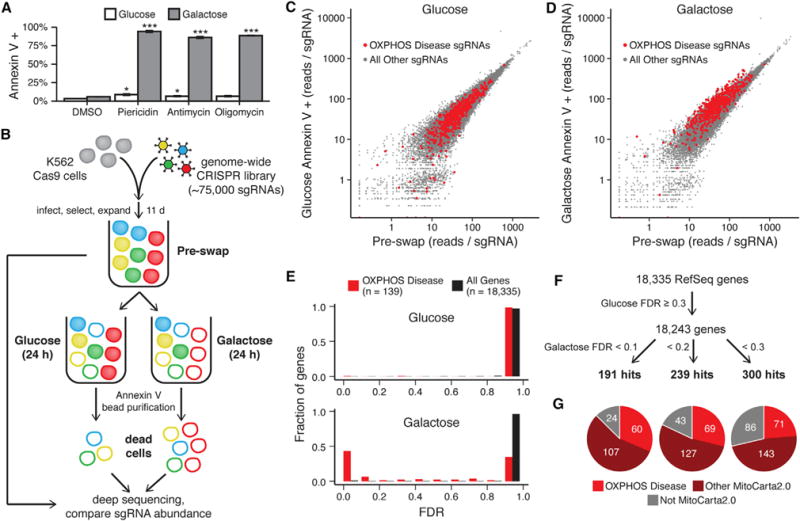
(A) Cell death was measured by Annexin V staining and flow cytometry in K562 cells cultured in glucose- or galactose-containing medium with the RC inhibitors piericidin (10 nM), antimycin (100 nM), or oligomycin (10 nM) or DMSO vehicle control. Data are expressed as mean ± SEM (n = 3). *p < 0.05, ***p < 0.001, t test relative to DMSO treatment in the corresponding medium.
(B) Schematic overview of the genome-wide CRISPR screen. K562 cells expressing Cas9 were infected with a lentiviral CRISPR sgRNA library, split into glucose-containing or galactose-containing medium, and cultured for 24 hr. Dead cells under each condition were purified using Annexin V, and the abundance of each sgRNA was compared with a reference sample of all cells from before the medium exchange (pre-swap). Red represents cells expressing an sgRNA for an OXPHOS disease gene.
(C and D) The median-normalized read count of each sgRNA averaged across two infection replicates is compared between the pre-swap cells (x axis) and the Annexin V+ dead cells after growth in glucose (C) or galactose (D) (y axis).
(E) Gene enrichment in Annexin V+ cells from glucose (top) and galactose medium (bottom) was scored using MAGeCK, and the fraction of known OXPHOS disease genes and all genes with a given FDR is shown.
(F) Schematic representing the approach used to identify hits in the screen and the number of hits below each galactose FDR threshold.
(G) The distribution of hits for each galactose FDR threshold between known OXPHOS disease genes (red), other mitochondrial genes as defined by MitoCarta2.0 (maroon), and non-mitochondrial genes (gray). See also Figure S1 and S2.
We developed our positive selection screen by isolating dead cells using Annexin V purification (Figure 1B). We employed the Avana lentiviral genome-wide CRISPR/Cas9 library with 74,687 single-guide RNAs (sgRNAs) to systematically target 18,335 distinct human genes in K562 cells (Doench et al., 2016). Infected cells were cultured in glucose-rich medium supplemented with uridine and pyruvate, which are required for the growth of some cells with RC defects (King and Attardi, 1989). After 11 days of growth to allow for CRISPR-mediated gene knockout, cells from two independent infection replicates were swapped into medium containing either glucose or galactose as the sole sugar source for 24 hr. Dead cells from each condition were isolated with Annexin V-conjugated microbeads and are termed “Annexin V+ cells.” The dead cells from the glucose and galactose conditions, along with the common pre-swap reference samples, were subjected to next-generation deep sequencing to determine the abundance of each sgRNA (Table S1).
We benchmarked the performance of the screen using a curated list of OXPHOS disease genes as positive controls (Table S2). The vast majority of sgRNAs under both conditions showed similar abundance levels in Annexin V+ cells compared with pre-swap cells (Figures 1C and 1D). However, sgRNAs targeting known OXPHOS disease genes were more abundant in Annexin V+ cells from galactose culture compared with their pre-swap levels. In contrast, those sgRNAs were indistinguishable from the background distribution when cells were grown in glucose. We then used the model-based analysis of genome-wide CRISPR-Cas9 knockout (MAGeCK) algorithm (Li et al., 2014) to collapse the sgRNA-level data and score the 18,335 human genes in our library for enrichment in Annexin V+ cells from glucose or galactose relative to the common pre-swap control (Table S3). We also observed this expected enrichment of positive controls at the gene level, and the known OXPHOS disease genes scored significantly better in galactose compared with glucose, as measured by the false discovery rate (FDR) (Figure 1E). Moreover, the fraction of OXPHOS disease genes below 10% FDR in galactose was enriched 39-fold compared with the background of all genes.
We used the MAGeCK scores to define a set of genes that were specifically necessary for survival in galactose relative to glucose. We filtered out 92 genes with an FDR below 30% in glucose medium because these likely represent broadly lethal genes that cause non-specific cell death. We then identified hit genes enriched for lethality in galactose at three FDR thresholds: 191 “high confidence” hits at 10%, an additional 48 hits between 10% and 20%, and 61 hits between 20% and 30% (Figures 1F and 2). Notably, a third of the hits lack identifiable homologs in S. cerevisiae, underscoring the importance of mammalian screening. We previously reported the MitoCarta2.0 inventory of human mitochondrial proteins (Calvo et al., 2016). To date, more than 95% of all known OXPHOS disease genes encode proteins resident within mitochondria and are found in the MitoCarta2.0 inventory. Accordingly, we observed in our screen that the vast majority of hits encode mitochondrial proteins annotated in MitoCarta2.0, and nearly two-thirds of those mitochondrial hits were not previously reported to be OXPHOS disease genes (Figure 1G). The non-mitochondrial hits are more likely to be false positives, and indeed their fraction correlates strongly with the overall FDR level, suggesting that the FDR statistic is well calibrated (Figure S1B). In total, our screen recovered 71 of 139 positive controls (OXPHOS disease genes) below 30% FDR, yielding a 51% sensitivity. Moreover, our identification of 229 hits not previously linked to OXPHOS disease more than doubled the number of human genes potentially implicated in OXPHOS.
Figure 2. Known and Candidate OXPHOS Disease Genes Identified by the Glucose/Galactose Death Screen.
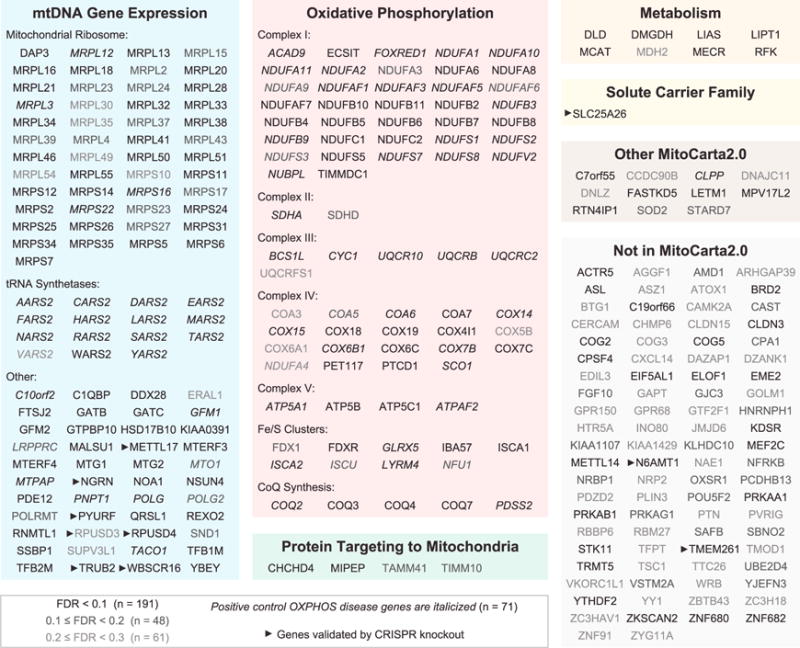
The 300 hits with a galactose FDR < 30% and a glucose FDR ≥ 30% are shown in manually curated functional categories.
We were curious to know whether our screening strategy could detect selective lethality in galactose versus glucose in cells that also exhibit impaired proliferation in glucose media because disruption of OXPHOS can reduce cell growth (Wheaton et al., 2014). We therefore compared the pre-swap samples to cells harvested 6 days earlier, immediately after drug selection for sgRNA-infected cells, and we identified 1,218 genes below 30% FDR whose disruption leads to a growth defect (Figure S2A; Tables S1 and S3). Growth defect hits were enriched for mitochondrial genes, with MitoCarta2.0 genes representing 19% of the hits versus 6% of all genes in the library (p < 10−56, Fisher’s test). Among the 300 genes that scored below 30% FDR for galactose lethality in the glucose/galactose death screen, 141 (47%) also led to a growth defect in rich media (Figure S2B). This fraction was higher among mitochondrial genes, where 126 of the 214 MitoCarta2.0 hits (59%) caused a growth defect as well. We also observed that ten of our positive control OXPHOS disease genes that did not score for death in galactose were detected as growth defect genes. Thus, although knockout of a small subset of OXPHOS genes likely did not score because they are essential under all conditions, our death screening approach is robust and detects selective lethality in galactose even in the context of cells with impaired growth in full glucose-containing media.
Pathway Analysis of Mitochondrial Genes Required for OXPHOS
We next asked which mitochondrial pathways are enriched or depleted in the hits from the glucose/galactose death screen. We partitioned all 1,135 MitoCarta2.0 genes present in our CRISPR library into eight non-overlapping categories (Figure 3A). Compared with the 19% of all MitoCarta2.0 genes that were hits below 30% FDR, four categories were enriched: Mitochondrial tRNA Synthetases, Mitochondrial Ribosome (MitoRibosome), OXPHOS, and Other mtDNA Gene Expression (p < 10−3, Fisher’s test). Compared with the MitoCarta2.0 background, three categories were significantly less likely to cause selective lethality in galactose: Solute Carrier Family, Metabolism, and uncategorized MitoCarta2.0 genes (p < 10−3, Fisher’s test). As stated earlier, although lesions to OXPHOS can lead to complete non-viability in galactose, they also lead to quantitative growth impairment in glucose. Indeed, we observed a similar pattern of enrichment or depletion for growth defect hits (Figure 3A). The one category that was notably an outlier was Protein Targeting to Mitochondria. This category was significantly enriched for genes required for growth (p = 0.013) but tended to be depleted (although not reaching significance, p = 0.24) for genes required for survival in galactose. It is notable that very few human OXPHOS disease genes encode members of the mitochondrial protein import machinery, consistent with the screening results here that these genes are likely generally essential.
Figure 3. Mitochondrial Pathways Underlying the Glucose/Galactose Death Phenotype.
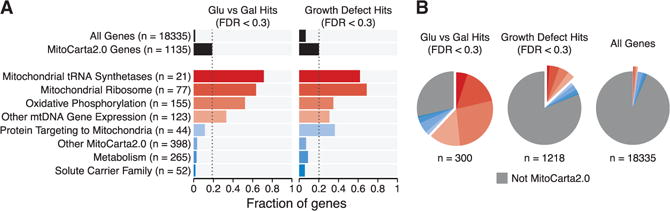
(A) The MitoCarta2.0 genes targeted by the CRISPR library were partitioned into eight distinct functional categories, and the fraction of genes in each category that were hits below 30% FDR is shown for the glucose/galactose death phenotype (left) or growth defect phenotype (right). The fractions of all genes and all MitoCarta2.0 genes that were hits are shown at top (black bars). Dashed lines indicate the fraction of MitoCarta2.0 hits.
(B) The distribution of hits below 30% FDR into the same eight MitoCarta2.0 categories and the non-MitoCarta2.0 category is shown. The distribution of all genes in the CRISPR library into those categories is shown for comparison.
Together, the four enriched categories (OXPHOS, MitoRibosome, Mitochondrial tRNA Synthetases, and Other mtDNA Gene Expression) show striking over-representation among the hits in the glucose/galactose death screen. Together, they comprised 62% of the genes with a glucose/galactose death phenotype (Figure 3B). In contrast, these same categories represent only 13% of all genes showing a growth defect and only 2% of all genes targeted by the genome-wide CRISPR library. These findings underscore the ability of the glucose/galactose death phenotype to recover OXPHOS-related genes.
Validation of the Glucose/Galactose Death Screen
We next sought to experimentally validate the results from the glucose/galactose death screen using individually targeted genetic knockouts. We used CRISPR/Cas9 and the sgRNA sequences from the screening library to disrupt the expression of nine genes (below 10% FDR) that were not previously associated with an OXPHOS disease or linked to the respiratory chain, including genes encoding mitochondrial proteins (NGRN, PYURF, SLC25A26, METTL17, and WBSCR16) and proteins not in MitoCarta2.0 (COG2, TMEM261, N6AMT1, and YTHDF2). Consistent with the 10% FDR threshold, we could confirm that depletion of eight of the nine genes led to significant and selective cell death in galactose, and only YTHDF2 failed to validate and was not studied further (Figure 4A; Figure S3A). COG2 and WBSCR16 knockouts showed a moderate but significant increase in cell death in glucose but to a lesser extent than in galactose. To determine whether OXPHOS was disrupted in the knockouts corresponding to the eight remaining genes, we quantified whole-cell oxygen consumption, which is primarily driven by OXPHOS activity. Of the eight genes tested, depletion of all but COG2 produced a significant defect in basal oxygen consumption, consistent with OXPHOS deficiency in those cells (Figure 4B). Loss of COG2 did not present any oxygen consumption deficiency, indicating that COG2 may be required for the survival of cells in galactose independent of OXPHOS. To provide biochemical evidence of OXPHOS disruption, we measured the relative abundance of RC subunits by immunoblotting (Figure 4C). All gene-disrupted cell lines had reduced levels of at least one RC complex subunit compared with control cells, again indicative of an OXPHOS deficiency. To further characterize the mitochondrial defects within these cells, we measured RC assembly and mitochondrial translation relative to control cells (Figures S3B and S3C). We observed defects for each disrupted gene, with phenotypes ranging from isolated complex I deficiency (e.g., PYURF) to combined respiratory chain deficiency (e.g., METTL17) (Figure 4C; Figures S3B and S3C). Importantly, during the validation of this screen, another study reported the association of SLC25A26 with OXPHOS disease, further credentialing our results (Kishita et al., 2015). All together, we validated eight of nine hits for the galactose-specific death phenotype and showed that disruption of seven of those eight genes reduced oxygen consumption.
Figure 4. Functional Validation of the Screening Results.
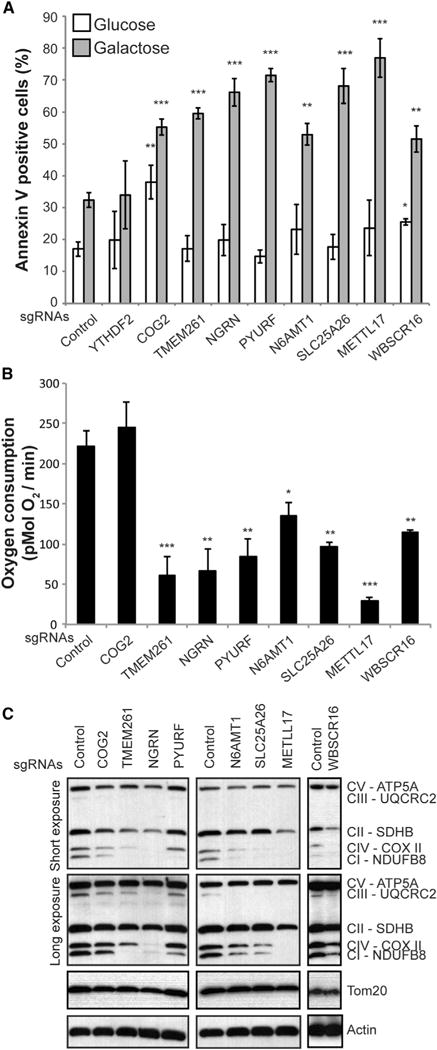
(A) Annexin V staining and flow cytometry analysis of K562 cell lines expressing Cas9 and sgRNAs directed against the genes indicated and grown for 24 hr in glucose or galactose-containing medium. Data are shown as mean ± SEM (n = 3, n = 6 for control). *p < 0.05, **p < 0.01, ***p < 0.001, t test relative to control sgRNA-treated cells in the corresponding medium.
(B) Basal whole-cell oxygen consumption was measured in the indicated K562 knockout cell lines. Data are shown as mean ± SEM (n = 4, n = 9 for control). *p < 0.05, **p < 0.01, ***p < 0.001, t test relative to control sgRNA-treated cells. (C) Protein immunoblot analysis of the indicated K562 cell lines using a multiplex antibody cocktail targeting a subunit of each respiratory complex (CI–CV).
See also Figure S3.
Analysis of Non-mitochondrial Pathways
As expected, the majority of our glucose/galactose death screen hits encode mitochondrial proteins, but 13% of our hits below 10% FDR and 29% of our hits below 30% FDR encode non-mitochondrial proteins. Although we anticipated that most of these non-mitochondrial hits were false discoveries, upon manual inspection, it was clear that multiple genes involved in the AMP kinase (AMPK) pathway, INO80/YY1 transcriptional control and Golgi apparatus were represented. Gene ontology (GO) analysis of the non-mitochondrial hits below 30% FDR confirmed that these three major pathways were strongly enriched: the INO80/YY1 transcriptional pathway (q < 10−5 with NFRKB, ACTR5, INO80, YY1 and TFPT), the AMPK pathway (q < 10−2 with PRKAA1, PRKAB1, and PRKAG1), and the COG machinery of the Golgi apparatus (q < 10−2 with COG2, COG3, and COG5) (Figure 5A). It is notable that the YY1 pathway was previously reported to regulate mitochondrial biogenesis (Cunningham et al., 2007). We experimentally confirmed the galactose-specific death phenotype in COG2-depleted cells above, although those cells did not exhibit decreased oxygen consumption. Using a small molecule AMPK inhibitor, Compound C (Zhou et al., 2001), we confirmed the synthetic lethality between growth in galactose and AMPK inhibition (Figures 5B and 5C). Although we could use a chemical inhibitor to demonstrate selective death in galactose versus glucose, further work is needed to determine whether AMPK directly regulates OXPHOS.
Figure 5. Non-mitochondrial Pathways Identified in the Screen.
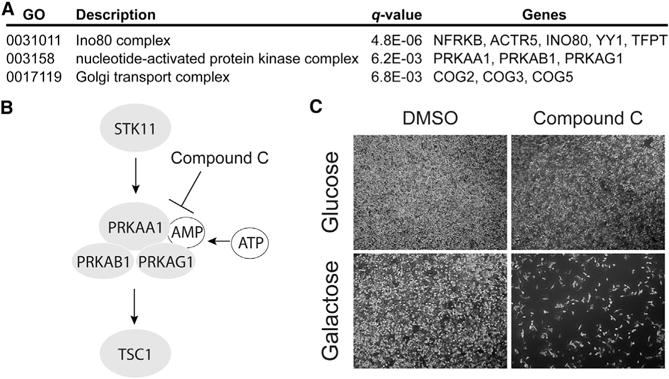
(A) Gene ontology enrichment analysis of the non-mitochondrial hits.
(B) Schematic representation of five proteins within the AMPK pathway that were hits in the screen, with pathway inhibition by Compound C indicated.
(C) Phase-contrast microscopy of HeLa cells grown in glucose- or galactose-containing medium and treated for 16 hr with 2 μM Compound C.
Identification of a Functional Module Regulating the 16S rRNA and Intra-mitochondrial Translation
Among the genes validated here, we further investigated NGRN because it encodes a protein with a highly conserved but unstudied Pfam domain suggestive of RNA binding (Castello et al., 2012; Ishigaki et al., 2000), hence providing a clue to its molecular function. We confirmed that NGRN localizes to mitochondria (Figure 6A; Figure S4A). When we disrupted NGRN using CRISPR, we observed a depletion of mitoribosome proteins (Figure 6B) and a loss of mitochondrial translation (Figure 6C). Expression of an sgRNA-resistant version of NGRN-3xFLAG could entirely restore these defects, confirming that they were not an off-target effect. Given its potential RNA binding, we hypothesized that NGRN might regulate mtDNA-encoded RNAs. In the absence of NGRN, we observed a significant depletion of the 16S rRNA, the mtDNA-encoded large mitochondrial rRNA (mt-RNR2), which could be fully restored by re-expressing NGRN-3xFLAG (Figure 6D). In contrast, the 12S small mitochondrial rRNA (mt-RNR1) and the mtDNA-encoded mRNAs were not significantly altered (Figure 6D; Figure S4B). When we co-immunoprecipitated all RNAs using NGRN-3xFLAG, we found that NGRN interacts with the 16S rRNA as well as its precursor 16S-tRNALeu but not with the 12S rRNA (Figures 6E and 6F). Collectively, these findings show that NGRN encodes a mitochondrial protein that interacts with the 16S rRNA and its precursor and that it is required for the maintenance of the mitochondrial ribosome and translation.
Figure 6. A Protein Module Required for 16S rRNA Stability and Intra-mitochondrial Translation.
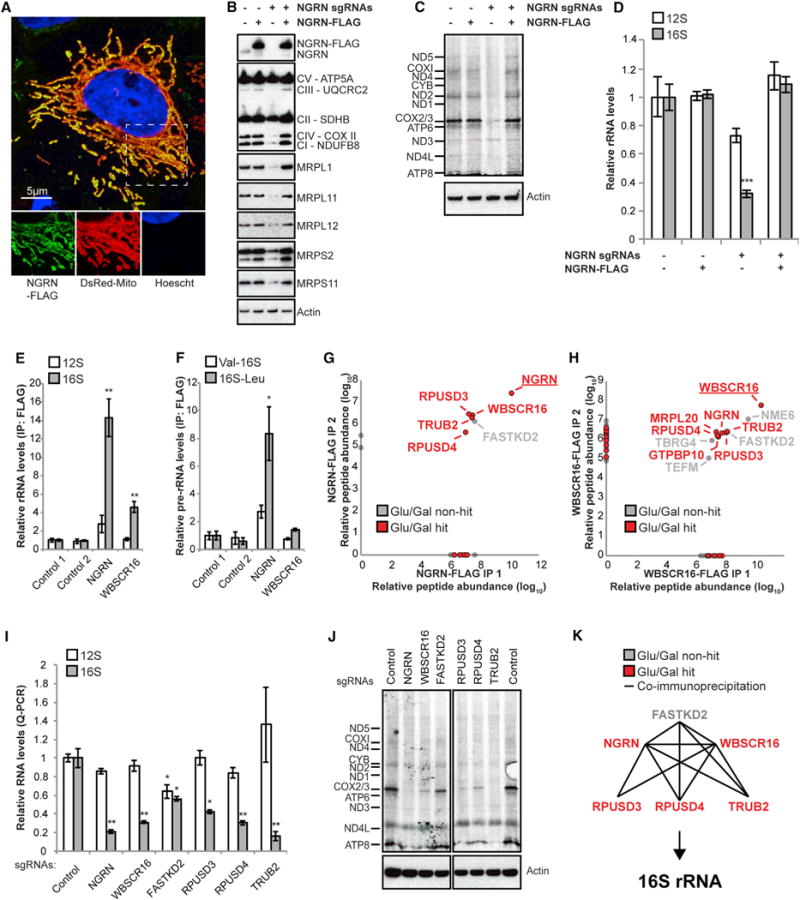
(A) Confocal microscopy analysis of HeLa cells expressing pDsRed2-Mito (a mitochondrial marker) and NGRN-3xFLAG and immunolabeled with antibodies to FLAG.
(B) Protein immunoblot analysis of a mitochondrion-rich fraction from K562 cells expressing sgRNAs targeting NGRN and/or an sgRNA-resistant version of human NGRN-3xFLAG cDNA.
(C) Autoradiography after 35S-methionine/cysteine labeling of mitochondrial translation on K562 cells expressing sgRNAs targeting NGRN and/or an sgRNA-resistant version of human NGRN-3xFLAG cDNA. All cells were treated with 200 μg/mL emetine.
(D) qPCR analysis of the small (12S, mt-RNR1) or large (16S, mt-RNR2) mitochondrial rRNAs from K562 cells expressing sgRNAs targeting NGRN and/or an sgRNA-resistant version of human NGRN-3xFLAG.
(E) qPCR analysis of the small (12S, mt-RNR1) or large (16S, mt-RNR2) mitochondrial rRNAs from NGRN-3xFLAG or WBSCR16-3xFLAG immunoprecipitations.
(F) qPCR analysis of the 16S 5′ (Val-16S) and 3′ (16S-Leu) rRNA precursors from NGRN-3xFLAG or WBSCR16-3xFLAG immunoprecipitations. (G and H) Relative peptide abundance of two independent NGRN-3xFLAG or WBSCR16-3xFLAG immunoprecipitations, respectively, and analysis by mass spectrometry. Baits are underlined. Genes identified in our screen (Figure 2) are shown in red.
(I) qPCR analysis of the small (12S, mt-RNR1) or large (16S, mt-RNR2) mitochondrial rRNAs from K562 cells expressing sgRNAs targeting the indicated genes.
(J) Autoradiography after 35S-methionine/cysteine labeling of mitochondrial translation on K562 cells expressing sgRNAs targeting the indicated genes. All cells were treated with 200 μg/mL emetine.
(K) A functional module that regulates the 16S rRNA and mitochondrial translation, with lines indicating protein-protein interactions detected by co-immunoprecipitation.
All bar plots (D–F and I) display mean ± SEM (n = 3) with asterisks indicating t test significance (*p < 0.05, **p < 0.01, ***p < 0.001). See also Figure S4.
To further characterize NGRN, we performed mass spectrometry analysis of the NGRN-3xFLAG immunoprecipitate. We found that NGRN is part of a previously unstudied protein module that surprisingly includes four other poorly characterized hits from our screen (WBSCR16, RPUSD3, RPUSD4, and TRUB2) as well as FASTKD2, a mitochondrial RNA-binding protein whose loss leads to infantile mitochondrial encephalomyopathy (Ghezzi et al., 2008; Figure 6G; Figures S4C and S4D; Table S4). Depletion of WBSCR16 phenocopies the mitochondrial translational defects we observed with NGRN loss (Figure S3C). Moreover, immunoprecipitation of WBSCR16 recovers the NGRN-interacting proteins (Figure 6H) and similarly shows interaction with the 16S rRNA (Figure 6E). Protein-protein interactions within the module were not sensitive to RNase treatment, indicating that these interactions are not mediated by the 16S rRNA (Figure S4C). Finally, we used CRISPR to disrupt all members of the module and found that they were all required for 16S rRNA abundance and mitochondrial translation (Figures 6I and 6J; Figures S4E and S4F). Thus, five of the previously unstudied hits from our CRISPR screen, combined with FASTKD2, appear to form a protein-RNA functional module (Figure 6K) that regulates the mitochondrial 16S rRNA and intra-mitochondrial translation.
DISCUSSION
We have introduced death screening, a technique that actively selects dying cells and subjects them to next-generation sequencing, and we employed this approach to systematically identify human genes required for OXPHOS. Using this positive selection screen, we report here the identification of 191 high-confidence genes required for OXPHOS. Our validation provides the first biochemical evidence for the role of previously uncharacterized mitochondrial (NGRN, RPUSD3, RPUSD4, TRUB2, WBSCR16, PYURF, and METTL17) and non-mitochondrial (TMEM261 and N6AMT1) genes in OXPHOS homeostasis. In particular, we identified a previously unrecognized protein-RNA functional module consisting of NGRN, WBSCR16, RPUSD3, RPUSD4, TRUB2, and FASTKD2 required for 16S rRNA abundance and intra-mitochondrial translation. Moreover, while this work was in progress, 17 additional hits from our screen, including SLC25A26, which we validated, were reported as bona fide OXPHOS disease genes in patients, providing further human genetic validation of our screening results (Table S5).
Our high-quality catalog of genes required for OXPHOS (Figure 2) will be an important resource for mitochondrial biologists, geneticists, and clinicians. In addition to expanding our understanding of this critical metabolic process and opening new avenues for exploration, it will immediately inform patient diagnosis. Whole-exome sequencing is emerging as a first-line diagnostic for OXPHOS disorders, but interpreting patient sequencing data remains an ongoing challenge. Although the MitoCarta inventory had a significant effect on OXPHOS disease diagnostics (Calvo et al., 2016; Pagliarini et al., 2008), about half of mitochondrial disease patients with whole-exome sequencing still do not receive a molecular diagnosis (Taylor et al., 2014). The inventory of candidate OXPHOS disease genes that we present here will help further prioritize variants discovered in patient sequencing data and improve diagnosis in two critical ways. First, current best practice is that all MitoCarta genes are examined for mutations, but our screen identified 24 high-confidence (< 10% FDR) and 62 lower-confidence (10%–30% FDR) non-MitoCarta genes that should now also be analyzed in all patients and that would otherwise have been missed. Even among MitoCarta genes, our list will help discover disease variants by prioritizing additional mitochondrial protein-encoding genes that merit higher scrutiny. Second, one of the biggest bottlenecks in diagnosis is establishing the pathogenicity of patient variants, and our data provide both preliminary functional evidence of a cellular OXPHOS phenotype as well as validated CRISPR reagents to rapidly confirm mutation pathogenicity (Table S3). Similar to our CRISPR-mediated depletion and rescue of NGRN described in Figure 6, we envision that patient variants could be reintroduced, and their pathogenicity could be tested for a simple and quantitative galactose-specific death phenotype. This approach presents an alternative to the need for invasive biopsies to obtain patient-derived fibroblasts and perform biochemical assays.
Although the majority of our hits encode mitochondrially localized proteins, a small number encode proteins localized to other compartments. Although many of these non-mitochondrial proteins likely represent false discoveries, several pathways localized outside mitochondria are high-scoring. The YY1/INO80 pathway was previously reported to regulate mitochondrial biogenesis at a transcriptional level (Cunningham et al., 2007). Five members of the AMPK signaling pathway were also identified in our screen. AMPK is traditionally viewed as a sensor of cellular energy charge and responsive to lesions to mitochondrial ATP production. Here we find that loss of the AMPK pathway itself leads to selective death in galactose versus glucose. Future studies will be required to determine why loss of AMPK leads to selective death in galactose versus glucose and whether this phenotype is related to OXPHOS.
Our inventory of newly identified genes essential for OXPHOS includes five previously uncharacterized proteins (NGRN, WBSCR16, RPUSD3, RPUSD4, and TRUB2) that, together with FASTKD2, we now show form a mitochondrial module required for intra-mitochondrial protein translation. In particular, we present here the first characterization of NGRN, an essential mitochondrial RNA-binding protein. We could confirm the mitochondrial localization of NGRN and its role in specifically regulating the mitochondrial 16S rRNA and intra-mitochondrial translation. Using protein immunoprecipitation and mass spectrometry, we identified and confirmed the interaction of NGRN with WBSCR16, RPUSD3, RPUSD4, TRUB2, and FASTKD2. Depletion of any of these genes disrupts mitochondrial translation by decreasing the level of the mitochondrial 16S rRNA. The role of FASTKD2 in regulating mitochondrial RNA metabolism has been corroborated by others (Antonicka and Shoubridge, 2015; Jourdain et al., 2015; Popow et al., 2015), but the absence of FASTKD2 was not sufficient to trigger a glucose/galactose death phenotype in K562 cells. Although at first this result appears to be inconsistent with reports that nonsense mutations in FASTKD2 are responsible for a familial case of mitochondrial encephalomyopathy, it should be noted that patients with FASTKD2 mutations present symptoms that are restricted to the muscle and CNS, and fibroblasts from the same patients are asymptomatic, thus illustrating the tissue specificity of certain mitochondrial diseases (Ghezzi et al., 2008). Among the other proteins interacting with NGRN, it is intriguing that RPUSD3, RPUSD4, and TRUB2 are putative pseudouridine synthases. Whether the functional module (Figure 6K) containing NGRN and the pseudouridine synthases is formed by a single or by multiple overlapping protein complexes has not been addressed here, but it is striking that depletion in any of these pseudouridine synthases leads to similar defects in 16S rRNA abundance. It has long been known that the mitochondrial 16S rRNA is itself pseudouridylated (Ofengand and Bakin, 1997), although the enzyme responsible for this modification has been elusive. Moreover, the purpose of pseudouridylation has not been clear, although it has been speculated to help provide RNA structure. Because NGRN binds to at least one of the precursors of the 16S rRNA in addition to the putative pseudouridine synthases, we speculate that the functional module we identified here could mediate 16S pseudouridylation and take part in an early step of mitochondrial ribosome biogenesis. Further work is required to ascertain this hypothesis and demonstrate the extent of pseudouridylation in mitochondria.
More generally, our conversion of a negative selection growth screen into a positive selection death screen should have broad utility. Traditional negative selection growth screens have limited power. Accurate quantification of depleted hits is challenging because of low signal, and large numbers of cells must be maintained to avoid losing genes at random because of genetic drift. Moreover, genes essential for robust cell growth cannot be assayed for growth phenotypes under other conditions because they are always strongly depleted. This limitation was a particular concern in our screen for OXPHOS genes because, although human cells can survive without OXPHOS in glucose, they would still be at a growth disadvantage compared with OXPHOS-competent cells. Here we have introduced a method by which we actively select dying cells and subject them to deep sequencing, and our death screening approach is widely applicable beyond metabolic studies. For example, in cancer biology, there is significant interest in identifying mechanisms of drug resistance and sensitivity, determining synthetic lethal pathways, and converting cytostatic drugs into cytotoxic agents. Death screening using either loss-of-function or gain-of-function genetic libraries is well suited for such studies.
EXPERIMENTAL PROCEDURES
Cell Lines
K562, HeLa, and HEK293T cells were obtained from the ATCC and maintained in DMEM (Invitrogen) with 25 mM glucose, 10% fetal bovine serum (FBS, Invitrogen), 1 mM sodium pyruvate, 50 μg/mL uridine, and 100 U/mL penicillin/streptomycin under 5% CO2 at 37°C.
Glucose and Galactose Cell Death Screening
K562 cells expressing Cas9 and EGFP were infected with the genome-wide Avana-4 CRISPR lentiviral library (Doench et al., 2016). Two infection replicates were performed. Infected cells were selected for 4 days with 2 μg/mL puromycin (Life Technologies), after which a sample of cells was collected for determining the initial sgRNA abundance. The cells were expanded for an additional 6 days, washed with PBS, and cultured in DMEM with 10 mM glucose or galactose, 10% dialyzed FBS (dFBS, Life Technologies), 1 mM sodium pyruvate, 50 μg/mL uridine, and 100 U/mL penicillin/streptomycin. Pre-swap cells were harvested prior to the medium swap. After 24-hr culture in glucose or galactose medium, Annexin V+ dead cells were purified using magnetic Annexin V microbeads and mass spectrometry (MS)-positive selection columns (Miltenyi Biotec). Genomic DNA was isolated from all samples, and the sgRNA sequences were amplified by PCR and sequenced on a HiSeq 2500 (Illumina). MAGeCK (v0.4.4) was used to score genes under each condition relative to the pre-swap reference samples (Li et al., 2014). See the Supplemental Experimental Procedures for details.
Statistical Analysis
Sample means between control and treatment conditions were compared by t test after confirming that data were normally distributed and samples had similar variance as estimated by SEM. The error bars in the figures represent SEM. All statistical tests were reported with two-sided p values.
Pathway Analysis
Mito Carta2.0 genes were divided into non-overlapping categories based on GO, Kyoto Encyclopedia of Genes and Genomes (KEGG), and manual annotation. Enrichment of mitochondrial hits in gene categories was assessed by Fisher’s test. To identify GO terms enriched among the non-mitochondrial hits from the glucose/galactose death screen, the GOrilla tool (http://cbl-gorilla.cs.technion.ac.il/) was used (Eden et al., 2009). Non-mitochondrial hits with a galactose FDR below 30% were used as the target genes, whereas all other non-mitochondrial genes in the CRISPR library were used as background genes. GO terms with q < 0.01 that were also the most specific of overlapping terms in the GO hierarchy were considered significant and are reported here.
Cell Death Analysis
Cell death in cells treated with RC inhibitors or gene-specific sgRNAs was measured by staining cells with Annexin V Alexa Fluor 647 conjugate (Invitrogen) according to the manufacturer’s protocol. Briefly, cells were washed in ice-cold PBS, incubated with stain for 15 min at room temperature, and then analyzed by flow cytometry on a BD Accuri C6 (BD Biosciences).
Oxygen Consumption
1.25 × 105 K562 cells were plated on a Seahorse plate coated with Cell-Tak™ Cell and Tissue Adhesive (Corning Life Sciences) in DMEM containing 1 g/L glucose and 50 μg/mL uridine, and oxygen consumption was recorded using a Seahorse XF96 Analyzer (Seahorse Biosciences).
AMPK Inhibition
HeLa cells were trypsinized, washed in PBS, and seeded in glucose or galactose-containing media in the presence of DMSO or 2 μM Compound C (Dorsomorphin, Abcam). Cells were imaged after 16 hr.
Mitochondrial Translation
106 K562 cells expressing the corresponding sgRNAs were washed in PBS and incubated for 30 min in 1 mL of labeling medium (10% dFBS, 1 mM sodium pyruvate, and 50 μg/mL uridine in DMEM without methionine/cysteine; Sigma). Emetine (Sigma) was added to a final concentration of 200 μg/mL, and cells were incubated for 5 min before addition of 50 μCi 35S-labeled methionine/cysteine mixture (PerkinElmer) and incubation for 1 hr. Cells were recovered and washed twice in PBS before lysis and quantification of protein abundance (DC protein assay, Bio-Rad). 50 μg of total proteins were loaded on a 10%–20% SDS-PAGE (Life Technologies) and transferred to a polyvinylidene fluoride (PVDF) membrane before autoradiography. The transfer step was included to allow sensitive immunoblotting with anti-actin antibodies to ensure equal loading. In all experiments, a replicate of the control lane was treated with chloramphenicol (50 μg/mL) to ensure the mitochondrial origin of the 35S signal. The name associated with each band is proposed based on their relative abundance and molecular weight and is informed by prior studies (Fernández-Silva et al., 2007).
qPCR
qPCR was performed using the TaqMan technology (Life Technologies). RNA was extracted from total cells with an RNeasy kit (QIAGEN) and DNase-I digested before murine leukemia virus (MLV) reverse transcription using random primers (Promega) and qPCR. All data were normalized to TBP.
Immunoprecipitation, Co-immunoprecipitation, and Mass Spectrometry
For immunoprecipitation, a 3xFLAG-tagged version of NGRN or WBSCR16 was cloned into pWPI-Neo (Addgene), and viruses were produced. HEK293T cells stably expressing GFP or FMC1-FLAG (an unrelated mitochondrial protein) were used as negative controls (control 1 and control 2, respectively). 24 hr post-infection, HEK293T cells were selected with 0.5 mg/mL Geneticin (Life Technologies) for 48 hr and expanded. A crude preparation of mitochondria was isolated by differential centrifugation and lysed in lysis buffer containing 50 mM Tris/HCl (pH 7.5), 150 mM NaCl, 1 mM MgCl2, 1% NP-40, 3 mM vanadylate RNase complex, 1× protease and phosphatase inhibitor (Cell Signaling Technology), and 40 U/mL RNAsin (Promega). RNAsin was omitted when RNase A (20 μg/mL, Thermo Scientific) was used. Washed FLAG M2 magnetic beads (Sigma) were added to the lysate and incubated overnight at 4°C. Beads were recovered after extensive washing, and the protein/RNA complexes were eluted with 100 μg/mL 3xFLAG peptide (Sigma). For RNA isolation, the eluate was DNase I-treated (Promega) for 1 hr at 37°C before purification of the RNA (RNeasy kit, QIAGEN). For protein isolation, the eluate was precipitated using trichloroacetic acid (TCA) and analyzed by mass spectrometry at the Whitehead proteomics facility or by protein immunoblotting. Peptides were identified and quantified using the Top 3 total ion current (TIC) method (Scaffold4). Interacting proteins were then selected based on the following criteria: presence in MitoCarta2.0 and >2-fold enrichment over either control. Replicates represent independent experiments done 4 months apart. For co-immunoprecipitation, RPUSD3-V5, RPUSD4-V5, and TRUB2-V5 cDNAs were obtained from the Broad ORFeome Collection and co-expressed with NGRN-3xFLAG in HEK293T cells. Three days after infection, whole cells were resuspended in lysis buffer and clarified by centrifugation (5 min, 2,000 × g). Immunoprecipitation was performed at room temperature with anti-FLAG (Sigma) or anti-V5 (Abcam) antibodies using a Dynabeads protein G immunoprecipitation kit (Thermo Fisher).
Supplementary Material
Highlights.
Death screening is a new method using active selection and sequencing of dead cells
A high-quality catalog of genes essential for human OXPHOS is reported
NGRN, WBSCR16, RPUSD3, RPUSD4, TRUB2, and FASTKD2 form a 16S rRNA regulatory module
Acknowledgments
We thank R. Gopal, X.R. Bao, and members of the V.K.M. laboratory for advice and feedback on the manuscript; E. Spooner for help with mass spectrometry; Y. Li for computational assistance; S. Schwartz and A. Regev for fruitful discussions; and L. Solomon for graphical design assistance. This work was supported by NIH grant R01-GM077465 (to V.K.M.), EMBO long-term fellowship ALTF 554-2015 (to A.A.J.), and the Next Generation Fund at the Broad Institute of MIT and Harvard (to J.G.D.). V.K.M. is an Investigator of the Howard Hughes Medical Institute.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information includes Supplemental Experimental Procedures, four figures, and five tables and can be found with this article online at http://dx.doi.org/10.1016/j.cmet.2016.08.017.
AUTHOR CONTRIBUTIONS
J.D.A., A.A.J., and V.K.M. conceived the project and designed the experiments. J.D.A., A.A.J., and C.A.B. performed the experiments. J.D.A., A.A.J., S.E.C., and J.G.D. performed data analysis. D.E.R. and V.K.M. supervised the work. J.D.A., A.A.J., and V.K.M. wrote the manuscript with input from all authors.
References
- Antonicka H, Shoubridge EA. Mitochondrial RNA Granules Are Centers for Posttranscriptional RNA Processing and Ribosome Biogenesis. Cell Rep. 2015;10:920–932. doi: 10.1016/j.celrep.2015.01.030. [DOI] [PubMed] [Google Scholar]
- Bayona-Bafaluy MP, Sánchez-Cabo F, Fernández-Silva P, Pérez-Martos A, Enríquez JA. A genome-wide shRNA screen for new OxPhos related genes. Mitochondrion. 2011;11:467–475. doi: 10.1016/j.mito.2011.01.007. [DOI] [PubMed] [Google Scholar]
- Calvo SE, Clauser KR, Mootha VK. MitoCarta2.0: an updated inventory of mammalian mitochondrial proteins. Nucleic Acids Res. 2016;44(D1):D1251–D1257. doi: 10.1093/nar/gkv1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castello A, Fischer B, Eichelbaum K, Horos R, Beckmann BM, Strein C, Davey NE, Humphreys DT, Preiss T, Steinmetz LM, et al. Insights into RNA biology from an atlas of mammalian mRNA-binding proteins. Cell. 2012;149:1393–1406. doi: 10.1016/j.cell.2012.04.031. [DOI] [PubMed] [Google Scholar]
- Cunningham JT, Rodgers JT, Arlow DH, Vazquez F, Mootha VK, Puigserver P. mTOR controls mitochondrial oxidative function through a YY1-PGC-1alpha transcriptional complex. Nature. 2007;450:736–740. doi: 10.1038/nature06322. [DOI] [PubMed] [Google Scholar]
- Diaz F, Kotarsky H, Fellman V, Moraes CT. Mitochondrial disorders caused by mutations in respiratory chain assembly factors. Semin Fetal Neonatal Med. 2011;16:197–204. doi: 10.1016/j.siny.2011.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doench JG, Fusi N, Sullender M, Hegde M, Vaimberg EW, Donovan KF, Smith I, Tothova Z, Wilen C, Orchard R, et al. Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nat Biotechnol. 2016;34:184–191. doi: 10.1038/nbt.3437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eden E, Navon R, Steinfeld I, Lipson D, Yakhini Z. GOrilla: a tool for discovery and visualization of enriched GO terms in ranked gene lists. BMC Bioinformatics. 2009;10:48. doi: 10.1186/1471-2105-10-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernández-Silva P, Acín-Pérez R, Fernández-Vizarra E, Pérez-Martos A, Enriquez JA. In vivo and in organello analyses of mitochondrial translation. Methods Cell Biol. 2007;80:571–588. doi: 10.1016/S0091-679X(06)80028-2. [DOI] [PubMed] [Google Scholar]
- Ghezzi D, Saada A, D’Adamo P, Fernandez-Vizarra E, Gasparini P, Tiranti V, Elpeleg O, Zeviani M. FASTKD2 nonsense mutation in an infantile mitochondrial encephalomyopathy associated with cytochrome c oxidase deficiency. Am J Hum Genet. 2008;83:415–423. doi: 10.1016/j.ajhg.2008.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gohil VM, Sheth SA, Nilsson R, Wojtovich AP, Lee JH, Perocchi F, Chen W, Clish CB, Ayata C, Brookes PS, Mootha VK. Nutrient-sensitized screening for drugs that shift energy metabolism from mitochondrial respiration to glycolysis. Nat Biotechnol. 2010;28:249–255. doi: 10.1038/nbt.1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hart T, Chandrashekhar M, Aregger M, Steinhart Z, Brown KR, MacLeod G, Mis M, Zimmermann M, Fradet-Turcotte A, Sun S, et al. High-Resolution CRISPR Screens Reveal Fitness Genes and Genotype-Specific Cancer Liabilities. Cell. 2015;163:1515–1526. doi: 10.1016/j.cell.2015.11.015. [DOI] [PubMed] [Google Scholar]
- Ishigaki S, Niwa J, Yoshihara T, Mitsuma N, Doyu M, Sobue G. Two novel genes, human neugrin and mouse m-neugrin, are upregulated with neuronal differentiation in neuroblastoma cells. Biochem Biophys Res Commun. 2000;279:526–533. doi: 10.1006/bbrc.2000.3971. [DOI] [PubMed] [Google Scholar]
- Jourdain AA, Koppen M, Rodley CD, Maundrell K, Gueguen N, Reynier P, Guaras AM, Enriquez JA, Anderson P, Simarro M, Martinou JC. A mitochondria-specific isoform of FASTK is present in mitochondrial RNA granules and regulates gene expression and function. Cell Rep. 2015;10:1110–1121. doi: 10.1016/j.celrep.2015.01.063. [DOI] [PubMed] [Google Scholar]
- King MP, Attardi G. Human cells lacking mtDNA: repopulation with exogenous mitochondria by complementation. Science. 1989;246:500–503. doi: 10.1126/science.2814477. [DOI] [PubMed] [Google Scholar]
- Kishita Y, Pajak A, Bolar NA, Marobbio CM, Maffezzini C, Miniero DV, Monné M, Kohda M, Stranneheim H, Murayama K, et al. Intra-mitochondrial Methylation Deficiency Due to Mutations in SLC25A26. Am J Hum Genet. 2015;97:761–768. doi: 10.1016/j.ajhg.2015.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Xu H, Xiao T, Cong L, Love MI, Zhang F, Irizarry RA, Liu JS, Brown M, Liu XS. MAGeCK enables robust identification of essential genes from genome-scale CRISPR/Cas9 knockout screens. Genome Biol. 2014;15:554. doi: 10.1186/s13059-014-0554-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo B, Cheung HW, Subramanian A, Sharifnia T, Okamoto M, Yang X, Hinkle G, Boehm JS, Beroukhim R, Weir BA, et al. Highly parallel identification of essential genes in cancer cells. Proc Natl Acad Sci USA. 2008;105:20380–20385. doi: 10.1073/pnas.0810485105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin SJ, Reutelingsperger CP, McGahon AJ, Rader JA, van Schie RC, LaFace DM, Green DR. Early redistribution of plasma membrane phosphatidylserine is a general feature of apoptosis regardless of the initiating stimulus: inhibition by overexpression of Bcl-2 and Abl. J Exp Med. 1995;182:1545–1556. doi: 10.1084/jem.182.5.1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nunnari J, Suomalainen A. Mitochondria: in sickness and in health. Cell. 2012;148:1145–1159. doi: 10.1016/j.cell.2012.02.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ofengand J, Bakin A. Mapping to nucleotide resolution of pseudouridine residues in large subunit ribosomal RNAs from representative eukaryotes, prokaryotes, archaebacteria, mitochondria and chloroplasts. J Mol Biol. 1997;266:246–268. doi: 10.1006/jmbi.1996.0737. [DOI] [PubMed] [Google Scholar]
- Pagliarini DJ, Calvo SE, Chang B, Sheth SA, Vafai SB, Ong SE, Walford GA, Sugiana C, Boneh A, Chen WK, et al. A mitochondrial protein compendium elucidates complex I disease biology. Cell. 2008;134:112–123. doi: 10.1016/j.cell.2008.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeffer G, Horvath R, Klopstock T, Mootha VK, Suomalainen A, Koene S, Hirano M, Zeviani M, Bindoff LA, Yu-Wai-Man P, et al. New treatments for mitochondrial disease-no time to drop our standards. Nat Rev Neurol. 2013;9:474–481. doi: 10.1038/nrneurol.2013.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popow J, Alleaume AM, Curk T, Schwarzl T, Sauer S, Hentze MW. FASTKD2 is an RNA-binding protein required for mitochondrial RNA processing and translation. RNA. 2015;21:1873–1884. doi: 10.1261/rna.052365.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reitzer LJ, Wice BM, Kennell D. Evidence that glutamine, not sugar, is the major energy source for cultured HeLa cells. J Biol Chem. 1979;254:2669–2676. [PubMed] [Google Scholar]
- Robinson BH, Petrova-Benedict R, Buncic JR, Wallace DC. Nonviability of cells with oxidative defects in galactose medium: a screening test for affected patient fibroblasts. Biochem Med Metab Biol. 1992;48:122–126. doi: 10.1016/0885-4505(92)90056-5. [DOI] [PubMed] [Google Scholar]
- Steinmetz LM, Scharfe C, Deutschbauer AM, Mokranjac D, Herman ZS, Jones T, Chu AM, Giaever G, Prokisch H, Oefner PJ, Davis RW. Systematic screen for human disease genes in yeast. Nat Genet. 2002;31:400–404. doi: 10.1038/ng929. [DOI] [PubMed] [Google Scholar]
- Taylor RW, Pyle A, Griffin H, Blakely EL, Duff J, He L, Smertenko T, Alston CL, Neeve VC, Best A, et al. Use of whole-exome sequencing to determine the genetic basis of multiple mitochondrial respiratory chain complex deficiencies. JAMA. 2014;312:68–77. doi: 10.1001/jama.2014.7184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vafai SB, Mootha VK. Mitochondrial disorders as windows into an ancient organelle. Nature. 2012;491:374–383. doi: 10.1038/nature11707. [DOI] [PubMed] [Google Scholar]
- van den Bosch BJ, Gerards M, Sluiter W, Stegmann AP, Jongen EL, Hellebrekers DM, Oegema R, Lambrichs EH, Prokisch H, Danhauser K, et al. Defective NDUFA9 as a novel cause of neonatally fatal complex I disease. J Med Genet. 2012;49:10–15. doi: 10.1136/jmedgenet-2011-100466. [DOI] [PubMed] [Google Scholar]
- VanderSluis B, Hess DC, Pesyna C, Krumholz EW, Syed T, Szappanos B, Nislow C, Papp B, Troyanskaya OG, Myers CL, Caudy AA. Broad metabolic sensitivity profiling of a prototrophic yeast deletion collection. Genome Biol. 2014;15:R64. doi: 10.1186/gb-2014-15-4-r64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace DC. Mitochondrial DNA in aging and disease. Sci Am. 1997;277:40–47. doi: 10.1038/scientificamerican0897-40. [DOI] [PubMed] [Google Scholar]
- Wheaton WW, Weinberg SE, Hamanaka RB, Soberanes S, Sullivan LB, Anso E, Glasauer A, Dufour E, Mutlu GM, Budigner GS, Chandel NS. Metformin inhibits mitochondrial complex I of cancer cells to reduce tumorigenesis. eLife. 2014;3:e02242. doi: 10.7554/eLife.02242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou G, Myers R, Li Y, Chen Y, Shen X, Fenyk-Melody J, Wu M, Ventre J, Doebber T, Fujii N, et al. Role of AMP-activated protein kinase in mechanism of metformin action. J Clin Invest. 2001;108:1167–1174. doi: 10.1172/JCI13505. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.