

Abstract
CXCR4 is a cell membrane receptor that is overexpressed in triple-negative breast cancers and implicated in growth and metastasis of this disease. Using electrohydrodynamic cojetting, we prepared multicompartmental drug delivery carriers for CXCR4 targeting. The particles are comprised of a novel poly(lactide-co-glycolide) derivative that allows for straightforward immobilization of 1,1′-[1,4-phenylenebis-(methylene)]bis[1,4,8,11-tetraazacyclotetradecane] (Plerixafor), a small molecule with affinity for CXCR4. Targeted nanocarriers are selectively taken up by CXCR4-expressing cells and effectively block CXCR4 signaling. This study suggests that CXCR4 may be an effective target for nanocarrier-based therapies.
Graphical abstract
INTRODUCTION
While the most recent treatments for receptor-positive breast cancers have had a significant impact on patient care, triple-negative breast cancers (TNBC) generally do not respond to these therapeutic interventions.1 Triple-negative breast cancers (TNBCs) are notoriously aggressive and difficult to treat, often having poor prognosis. TNBCs have a worse short-term prognosis than other types of breast cancer and can have a more aggressive progression.1,2 Currently, the standard of care includes various chemotherapeutics delivered as neoadjuvants or adjuvants with variable outcomes in terms of survival and recurrence of disease.1,3 Recently, progress has been made to understand the biology and pathophysiology of TNBC. In particular, pathways involving CXCR4 have been implicated in both TNBC and metastatic breast cancer.3,4 CXCR4, a cell surface receptor overexpressed in TNBC, promotes cell proliferation and chemotaxis.5,6 In TNBC, among several other cancers, the CXCR4 pathway has been shown to be involved in tumor growth and metastasis.7 Additionally, the degree of CXCR4 overexpression in TNBC positively correlates with worse prognosis.8
Micro- and nanoscale carriers, loaded with therapeutics that actively target tumor sites, have great potential to address the challenges in the treatment of refractory diseases, especially aggressive or resistant cancers.9–11 Effective targeting may allow for a more favorable biodistribution of a drug, resulting in higher tolerable doses and decreased systemic toxicity. Given the overexpression of CXCR4 in TNBC and its role in growth and metastatic potential, this receptor may be an effective target for particle-based therapy. In addition to its targeting potential, blocking CXCR4 signaling has been shown to prevent invasion and metastasis of breast cancer cells,12,13 and CXCR4-targeted nanocarriers have been used for imaging breast cancer in lymph nodes.14 Several targeting ligands and CXCR4 antagonists have been developed, including antibodies, small peptides, and CXCL12 analogs.15,16 In particular, 1,1′-[1,4-phenylenebis-(methylene)]bis[1,4,8,11-tetraazacyclotetradecane], also known as Plerixafor, is a small molecule with affinity for CXCR4 that has been shown to block CXCR4 signaling in in vivo models and in clinical trials.4,17,18 However, to date, few studies have employed this molecule as a targeting ligand on nanocarriers.19,20 Given its high binding affinity to CXCR417 and relative nontoxicity,21 nanocarriers decorated with Plerixafor may become attractive therapeutic modalities.
Mulicompartmental particles, comprised of different polymers with different functionalities within a single carrier, may be used for targeted drug delivery. In particular, electro-hydrodynamic (EHD) cojetting is a versatile technique that allows for preparation of multicompartmental carriers.22–25 Briefly, in EHD cojetting, two or more polymeric solutions are extruded parallel to one another under laminar flow.22 A DC voltage is applied between the orifice and a grounded plate some distance away from the orifice. Upon application of a voltage, the solution is pumped out at of the orifice and leads to a distorted Taylor cone, from which a fine jet of small droplets is emitted toward the grounded plate.26 Because of high surface area to volume ratios, the solvent evaporates rapidly, resulting in solid particulates that can be collected on the counter-electrode. We have shown that the particle architecture is determined by the configuration and composition of fluids used during electrohydrodynamic cojetting.24
EHD cojetting allows for the fabrication of carriers that display multiple orthogonal functionalities and payloads in different compartments, such as siRNA or small cancer drugs.27,28 If responsive bulk materials are used, carriers can be designed to respond to environmental changes, such as light, oxidative stress, or local pH.25,28 Chemically reactive polymers may also be incorporated in selected compartments for selective surface conjugation of targeting moieties.29
Here we demonstrate the ability to selectively immobilize Plerixafor to the surface of nanoparticles by using a novel acrylate-functionalized poly(D,L-lactide-co-glycolic acid) (PLGA-acrylate) incorporated into EHD cojetted carriers, allowing for selective surface immobilization of Plerixafor via Michael addition taking advantage of the free acrylate groups. Furthermore, we show that such carriers can not only target CXCR4, but also inhibit CXCL12-induced signaling in an in vitro model of CXCR4-expressing breast cancer cells.30 Finally, we present data suggesting that Plerixafor-modified carriers may be able to more effectively deliver a drug and cause an effect, in this case, gene silencing, via delivery of siRNA.
EXPERIMENTAL SECTION
Materials
Poly(D,L-lactide-co-glycolic acid) (PLGA, 85:15 lactide/glycolide, 50–75 kg/mol), polyethylenimine (PEI, Mn = 60 kg/mol, Mw = 760 kg/mol), blue fluorescent poly[(m-phenylenevinylene)-alt-(2,5-dihexyloxy-p-phenylenevinylene)] (PMPDHPV), Tween 20, and diiospropylamine (DIA), and all solvents were purchased from Sigma. Plerixafor was obtained from Tocris Bioscience. Dithiobis-(succinimidyl propionate) (DSP) and paraformaldehyde were obtained from Thermo Scientific. Phosphate buffered saline (PBS), ProLong Gold, Dulbecco’s Modified Eagle Medium (DMEM), fetal bovine serum (FBS), nonessential amino acids (NEAA), and penicillin-streptomycin (pen-strep) were obtained from Invitrogen. A total of 20 base pair DNA conjugated with AlexaFluor 546 (20bpDNA-AF546) was obtained from Integrated DNA Technologies. MDA-MB-231 cells expressing a CXCR4-GFP fusion protein were prepared as described previously.30
Synthesis of Acrylate Functionalized PLGA
Deprotection of a copolymer of lactic acid and benzyl glycolide results in a hydroxylated polymer, hydroxyl-PLGA, the synthesis of which has been described previously.29 Acrylate-functionalized PLGA, PLGA-acrylate, was synthesized from hydroxyl-PLGA, as shown in Scheme 1. Hydroxyl-PLGA (5.0 g, 0.31 mmol, Mn,theo = 15800 g/mol) was dissolved in CH2Cl2 (100 mL). To this solution, triethylamine (3.28 mL, 0.023 mol) was added, and the reaction mixture was cooled to 0 °C. Acryloyl chloride (1.27 mL, 0.0157 mol) in CH2Cl2 (20 mL) was added dropwise to this solution over 30 min under argon. The mixture was stirred at 0 °C for 1 h and then overnight at room temperature under a nitrogen atmosphere. The solvent was evaporated and the remaining viscous polymer was dissolved in CHCl3 and precipitated into large amount of methanol. The dissolution–precipitation procedure was repeated two times. After decantation of methanol, the remaining sticky polymer was dissolved in CHCl3 and evaporated to dryness to give the polymer as a brown solid. Yield: 5 g (93%). Mn,GPC = 10050 g/mol; Mw/Mn = 1.57 (based on PS standards). 1H NMR (400 MHz, CDCl3, δ): 6.5–6.3 (m, CH2═CHC═O), 6.2–6.0 (m, CH2═CHC═O), 5.9–5.7 (m, CH2═CHC═O), 5.4–5.3 (m, CHCH2O), 5.2–5.0 (m, C═OCHCH3O), 4.8–4.4 (m, CHCH2OC═O and C═OCH2OC═O), 1.7–1.5 (bs, C═OCHCH3O).
Scheme 1.

Synthesis of a PLGA-Acrylate from Hydroxyl-PLGA
Plerixafor Immobilization Studies
For optimal imaging, large microcylinders with PLGA-acrylate in one compartment and a blue fluorescent dye in the other were made by a previously described procedure of the cryosectioning of EHD cojetted fibers.31–33 The fibers were manufactured by the cojetting of the following solutions at 0.05 mL/h and around 12.75 kV: (1) 30 w/v% PLGA with <0.05 mg/mL PMPDHV, and (2) 33 w/v% 10:1 w/w PLGA/PLGA-acrylate, both in 95:5 CHCl3/DMF. The microcylinders were then incubated in 2–3 mg/mL of Plerixafor in a total of 2 mL PBS (with 0.01 v/v% Tween 20); 14 μL of DIA was added, and the mixture was rotated, allowing it to react for 16 h at ambient conditions. After washing three times by centrifugation and resuspension with fresh 0.01 v/v% Tween 20 in PBS, the microcylinders were then incubated in 1 mL of PBS with 0.01 v/v% Tween 20 and 20 μL of 0.13 mg/mL 20bpDNA-AF546, and rotated at ambient conditions for 3 h. After incubation, the samples were washed three times. Fluorescent confocal imaging was performed on a FluoView 500 Laser Scanning Confocal Microscope (CLSM).
Fabrication of Plerixafor-Modified Nanocarriers
Nanoparticles were synthesized, as described previously,25 with the incorporation of PLGA-acrylate by the cojetting of the following solutions at 30 cm, 0.1 mL/h, and 9.75–10.05 kV: (1) 6.25 w/v% 4:1 w/w PLGA/PLGA-acrylate (for signaling study, 5.5 w/v% 10:1 w/w PLGA/PLGA-acrylate was used instead) with <0.05 mg/mL PMPDHV, and (2) 5.25 w/v% 1:1:0.1 w/w/w PLGA/PEI/DSP, both in 1:1 v/v CHCl3/DMF; after jetting the particles were allowed to cross-link for at least 72 h at room temperature. Due to the basic conditions required for the Plerixafor conjugation, the reaction time was shortened to around 3 h for immobilization on nanoparticles to avoid their degradation; additionally, 0.1 v/v% Tween 20 in PBS was used as the solvent for conjugation and washing. Control nanoparticles were made by placing in same conditions for conjugation, but with no Plerixafor. Nanoparticles were characterized with an AMRAY 1910 field emission scanning electron microscope (SEM) and by dynamic light scattering (DLS) with a Malvern Zetasizer Nano ZSP.
In Vitro Targeting Studies
MDA-MB-231 cells expressing CXCR4 were cultured in DMEM supplemented with 10% FBS, 1× NEAA, and 1× pen-strep. For uptake studies, cells were seeded on circular glass coverslips in 12-well plates. The seeded cells were cultured for 24 h, and then incubated with control and Plerixafor-modified nanocarriers for 1 h, both at a concentration of 10 μg/mL. They were then washed three times with PBS, incubated in 4% paraformaldehyde for 30 min, washed four more times, and mounted on glass slides with ProLong Gold. Imaging was performed using a FluoView 500 CLSM. CXCR4 signaling studies were performed, as described before34 by measuring bioluminescence from association of CXCR4 with β-arrestin 2 after incubation with varying permutations of CXCL12, nanocarriers, and free Plerixafor for 20 min.
Gene Silencing Studies
CXCR4 expressing MDA-MB-231 cells were cultured in DMEM with 10% FBS, 1× NEAA, and 1× pen-strep. For targeting study, cells were seeded at 5000 cells/well in a 96 well plate. The following day cells were incubated with different concentrations of Plerixafor immobilized and unmodified control carriers, both loaded with siRNA-GFP, for 1 h. 48 h after incubation, GFP fluorescence intensities of the cells were measured using a Biotek Synergy2 plate reader.
RESULTS AND DISCUSSION
We first synthesized a novel base polymer, PLGA-acrylate, for subsequent immobilization of targeting ligands to the surface of the carrier particles. As shown in Scheme 1, we synthesized a PLGA derivative that contained pendant acrylate groups. The latter were introduced by reacting the hydroxyl groups of hydroxyl-PLGA, a biodegradable polymer with pendant hydroxyl groups, with acryloyl chloride. Afterwards, the acrylate groups may react with amines from the Plerixafor via Michael addition. Synthesis of the desired product was confirmed by NMR spectroscopy (Figure S1). The molecular weight (Mn) was determined to be 10050 g/mol by gel permeation chromatography (GPC), with an Mw/Mn = 1.57 (based on polystyrene standards).
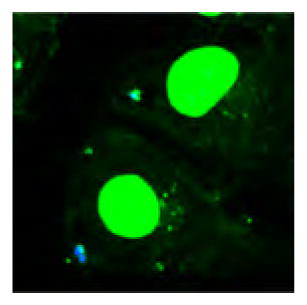
We then conducted a series of model studies to identify optimal conditions for the immobilization of Plerixafor to the acrylate groups on particles made of acrylate-PLGA. Once optimal conditions were established, we would then use those conditions for the decoration of nanocarriers with Perixafor. Microparticles that incorporate a functional polymer in one compartment allow for facile visualization of the selective immobilization using confocal microscopy, as we have shown previously.29 Following an approach established for other surface chemistries in the past,29 we decided to directly visualize the surface immobilization with confocal fluorescence microscopy. To ensure optimum imaging, we prepared a series of microcylinders using electrohydrodynamic cojetting that (i) had a size of 50 μm and (ii) were monodisperse and defect-free. To verify that Plerixafor can react with PLGA-acrylate via spatially controlled surface modification, we prepared the larger Janus microcylinders that displayed free acrylate groups in one hemisphere only. The anisotropic microcylinders were fabricated by first manufacturing PLGA fibers with PLGA-acrylate in one compartment via EHD cojetting. To clearly distinguish the two compartments, a blue fluorescent dye was incorporated into the second compartment. The fibers were subsequently cryosectioned to produce microcylinders. Michael addition of Plerixafor at the surface of the Janus particles followed the approach outlined in Figure 1a. We next incubated the Plerixafor-displaying microcylinders with a fluorescently labeled DNA, 20bpDNA-AF546 to demonstrate selective binding of the DNA between electrostatic interactions of the amines from Plerixafor and the negatively charged DNA. As seen from the confocal images in Figure 1b–d, the red fluorescence from the 20bpDNA-AF546 is compartmentalized on the surface of the microcylinders. The minimal overlap between the red and blue fluorescence indicates (i) preferential binding of the DNA to the surface-modified Plerixafor and (ii) only low levels of nonspecific DNA binding in areas without Plerixafor.
Figure 1.

(a) Reaction scheme for selective immobilization of Plerixafor and fluorescently labeled DNA. (b, c) Confocal images of a microcylinder (blue on one side) after Plerixafor immobilization and labeling with fluorescent DNA (red). (d) Overlay of (b) and (c). Scale bars are 20 μm.
After confirming that Plerixafor can be selectively immobilized onto anisotropic carriers, we then returned back to manufacturing endosome-sensing nanocarriers with PLGA-acrylate, and conjugated Plerixafor to their surfaces using the same procedure (Figure 2). These endosome-sensing carriers are bicompartmental nanoparticles comprised of a PLGA-based compartment and a PEI-based compartment. The PEI-based compartment allows for differential swelling based on the pH of the environment; the carriers swell under acidic conditions, such as those found in endosomes. When these carriers are endocytosed and enter an acidic environment, they subsequently swell, rupturing the endosomes by osmotic swelling or mechanical force. The carriers hence are able to reach the cytosol and deliver a particular payload. We have manufactured such carriers in the past and have shown them to be effective in the delivery of siRNA to the cytosol of cells.25 In this study, we now incorporate PLGA-acrylate into the PLGA compartment, allowing for surface immobilization of Plerixafor. Thus, such carriers may be used for selective targeting and gene silencing of CXCR4-overexpressing cells.
Figure 2.

(a) SEM image and (b) DLS data of cojetted carriers containing PLGA and PLGA-acrylate on one side, and PLGA, PEI, and DSP on the other side. Scale bar is 500 nm.
We prepared the nanocarriers comprised of a blend of PLGA and PLGA-acrylate in one compartment and PLGA and PEI in the other compartment. As shown in Figure 2, the cojetted carriers have a biomodal distribution, with the majority of particles falling in the range of 50–100 nm. We then assessed their in vitro behavior by incubating these particles with MDA-MB-231 cells, which express CXCR4 as a cell surface marker. To visualize the particles during the cell studies, a blue fluorescent dye was incorporated into the carriers. As seen in Figure S2, we observe a higher density of blue fluorescence, from Plerixafor-modified carriers, colocalized with the green fluorescence from the cells than compared to unmodified carriers. We may hence infer that Plerixafor-modified carriers are uptaken by the CXCR4 expressing cells at a higher rate than unmodified carriers.
In addition to the selective uptake of these carriers, we also explored a direct effect of the Plerixafor-modified nanoparticles on CXCR4 signaling. CXCL12 binding to CXCR4 results in recruitment of the cytosolic adapter protein β-arrestin 2 as part of signaling, which we quantified with a luciferase complementation reporter previously described by our group.34
For in vitro CXCR4 targeting and siRNA delivery, PLGA/PEI carriers were fabricated similar to a previously described procedure,25 by cojetting the following solutions at 0.1 mL/h, 30 cm, and ~11.25 kV: (1) 5.5 w/v% 10:1 w/w PLGA/PLGA-acrylate with <0.05 mg/mL PMPDHPV, and (2) 5.25 w/v% 10:10:1 w/w/w PLGA/PEI/DSP with 5 nmol/mL siRNA-GFP, both in 1:1 v/v CHCl3/DMF. The PEI-based carriers were allowed to cross-link for 72 h at room temperature before surface modification. Plerixafor-modified carriers were synthesized by incubating PEI-based carriers (2 mg) with Plerixafor (~2–3 mg) in 2 mL of solvent, followed by adding DIA (14 μL). The mixture was rotated for 3 h and then washed three times by centrifugation and resuspension. Control carriers were synthesized under the same conditions as Plerixafor-modified carriers, except that no Plerixafor was added.
As shown in Figure 3, incubation of cells with 300 ng/mL CXCL12 results in a ~100% increase in bioluminescence from association of CXCR4 with β-arrestin 2. Treatment with CXCL12 and Plerixafor-modified carriers limited the change in signal to only a ~25% increase, whereas carriers lacking Plerixafor had no such effect. The extent of inhibition with Plerixafor-modified carriers is similar to competitive inhibition by free Plerixafor. Although the relative concentration of Plerixafor represented by the modified carriers is considerably less than 1 μM, they nonetheless provide a similar inhibition of signaling; this may be attributed to the multivalency of the carriers, allowing for relatively higher affinity for CXCR4 receptors. Hence, the targeted nanocarriers not only show selective uptake in CXCR4 expressing cells, but also are able to inhibit CXCR4 signaling. These carriers may have dual benefits of selectively targeting TNBCs for the delivery of therapeutics and simultaneously directly blocking CXCR4 reduce further tumor growth and metastasis.
Figure 3.

Measurement of β-arrestin levels after incubation with CXCL12 and various carriers. Negative Control represents cells that were not incubated with CXCL12, Plerixafor, or carriers. Positive Control represents cells incubated only with CXCL12. Plerixafor-immobilized Carriers represents cells incubated with 1 μg/mL targeted carriers. Control Carriers represents cells incubated with 1 μg/mL unmodified carriers. Free Plerixafor represents cells incubated with 1 μM Plerixafor. *** denotes p < 0.001 compared to the positive control.
Finally, we prepared Plerixafor-modified (targeted) and control (untargeted) carriers, both loaded with siRNA against GFP, and incubated them at various concentrations with CXCR4-GFP expressing breast cancer cells for 1 h. As seen in Figure 4, after 48 h we observe that both carriers cause GFP knockdown at a concentration of 100 μg/mL. Moreover, cells incubated with Plerixafor-modified carriers expressed less GFP than those incubated with untargeted carriers, indicating that the targeted carriers may be taken up at a higher rate, resulting in higher delivery of siRNA. It is also may be noted that at 50 μg/mL, cells incubated with targeted carriers have significant GFP knockdown, while cells incubated with untargeted carriers do not. Therefore, targeting may allow for lower doses to achieve GFP silencing, hence, increasing the therapeutic index relative to untargeted carriers.
Figure 4.

GFP fluorescence intensities of cells measured by plate reader 48 h after 1 h incubation with Plerixafor-modified and untargeted carriers. P values based on a t test are shown with brackets indicating which samples are compared. Stand-alone asterisks represent comparisons to control (concentration = 0 μg/mL). *p < 0.05, **p < 0.01, ***p < 0.001.
CONCLUSIONS
The reported results show the potential for Plerixafor-modified carriers for targeting CXCR4 overexpressing cancers, such as TNBC. Effective surface immobilization of Plerixafor via Michael addition has been enabled by novel acarylate-functionalized PLGA. Furthermore, increased uptake of these particles by CXCR4-expressing breast cancer cells has been demonstrated, allowing for the potential of targeted delivery of therapeutics. Additionally, we establish that Plerixafor-modified carriers can suppress CXCR4 signaling. Taken together, this work suggests that this, or similar carrier formulations, may be used to help prevent growth and metastasis of TNBCs. Loading such targeted carriers with siRNA allows for effective gene silencing in vitro more than their corresponding untargeted carriers, and may be used effectively at lower doses than what is currently used in carrier-free approaches.25 Bicompartmental carriers may be further evaluated in vivo in future studies.
Acknowledgments
The authors acknowledge funding the Multidisciplinary University Research Initiative of the Department of Defense, the Army Research Office (W911NF-10-1-0518), the DOD Idea Award (W81XWH-11-1-0111), the EU-Project SAVVY (310445), and United States National Institutes of Health Grant R01CA170198.
Footnotes
Notes
The authors declare no competing financial interest.
Supporting Information
Characterization of polymer, particles, and additional in vitro studies are provided. The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.biomac.5b00653.
References
- 1.Foulkes WD, Smith IE, Reis JS. N Engl J Med. 2010;363:1938–1948. doi: 10.1056/NEJMra1001389. [DOI] [PubMed] [Google Scholar]
- 2.Dent R, Trudeau M, Pritchard KI, Hanna WM, Kahn HK, Sawka CA, Lickley LA, Rawlinson E, Sun P, Narod SA. Clin Cancer Res. 2007;13:4429–4434. doi: 10.1158/1078-0432.CCR-06-3045. [DOI] [PubMed] [Google Scholar]
- 3.Bosch A, Eroles P, Zaragoza R, Vina JR, Lluch A. Cancer Treat Rev. 2010;36:206–215. doi: 10.1016/j.ctrv.2009.12.002. [DOI] [PubMed] [Google Scholar]
- 4.Smith MCP, Luker KE, Garbow JR, Prior JL, Jackson E, Piwnica-Worms D, Luker GD. Cancer Res. 2004;64:8604–8612. doi: 10.1158/0008-5472.CAN-04-1844. [DOI] [PubMed] [Google Scholar]
- 5.Mizell J, Smith M, Li BDL, Ampil F, Chu QYD. Ann Surg Oncol. 2009;16:2711–2716. doi: 10.1245/s10434-009-0551-0. [DOI] [PubMed] [Google Scholar]
- 6.Luker KE, Luker GD. Cancer Lett. 2006;238:30–41. doi: 10.1016/j.canlet.2005.06.021. [DOI] [PubMed] [Google Scholar]
- 7.Luker KE, Lewin SA, Mihalko LA, Schmidt BT, Winkler JS, Coggins NL, Thomas DG, Luker GD. Oncogene. 2012;31:4750–4758. doi: 10.1038/onc.2011.633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chu QYD, Panu L, Holm NT, Li BDL, Johnson LW, Zhang SL. J Surg Res. 2010;159:689–695. doi: 10.1016/j.jss.2008.09.020. [DOI] [PubMed] [Google Scholar]
- 9.Brannon-Peppas L, Blanchette JO. Adv Drug Delivery Rev. 2012;64:206–212. doi: 10.1016/j.addr.2004.02.014. [DOI] [PubMed] [Google Scholar]
- 10.Peer D, Karp JM, Hong S, Farok Hzad OC, Margalit R, Langer R. Nat Nanotechnol. 2007;2:751–760. doi: 10.1038/nnano.2007.387. [DOI] [PubMed] [Google Scholar]
- 11.Petros RA, DeSimone JM. Nat Rev Drug Discovery. 2010;9:615–627. doi: 10.1038/nrd2591. [DOI] [PubMed] [Google Scholar]
- 12.Liang ZX, Wu H, Reddy S, Zhu AZ, Wang S, Blevins D, Yoons YH, Zhang YW, Shim HS. Biochem Biophys Res Commun. 2007;363:542–546. doi: 10.1016/j.bbrc.2007.09.007. [DOI] [PubMed] [Google Scholar]
- 13.Liang ZX, Yoon YH, Votaw J, Goodman MM, Williams L, Shim H. Cancer Res. 2005;65:967–971. [PMC free article] [PubMed] [Google Scholar]
- 14.Bunschoten A, Buckle T, Kuil J, Luker GD, Luker KE, Nieweg OE, van Leeuwen FWB. Biomaterials. 2012;33:867–875. doi: 10.1016/j.biomaterials.2011.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Burger JA, Peled A. Leukemia. 2009;23:43–52. doi: 10.1038/leu.2008.299. [DOI] [PubMed] [Google Scholar]
- 16.Tamamura H, Hori A, Kanzaki N, Hiramatsu K, Mizumoto M, Nakashima H, Yamamoto N, Otaka A, Fujii N. FEBS Lett. 2003;550:79–83. doi: 10.1016/s0014-5793(03)00824-x. [DOI] [PubMed] [Google Scholar]
- 17.De Clercq E. Nat Rev Drug Discovery. 2003;2:581–587. doi: 10.1038/nrd1134. [DOI] [PubMed] [Google Scholar]
- 18.Burger JA, Kipps TJ. Blood. 2006;107:1761–1767. doi: 10.1182/blood-2005-08-3182. [DOI] [PubMed] [Google Scholar]
- 19.Le Bon B, Van Craynest N, Daoudi JM, Di Giorgio C, Domb AJ, Vierling P. Bioconjugate Chem. 2004;15:413–423. doi: 10.1021/bc034220o. [DOI] [PubMed] [Google Scholar]
- 20.Li J, Zhu Y, Hazeldine ST, Li CY, Oupicky D. Angew Chem, Int Ed. 2012;51:8740–8743. doi: 10.1002/anie.201203463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cashen AF, Nervi B, DiPersio J. Future Oncol. 2007;3:19–27. doi: 10.2217/14796694.3.1.19. [DOI] [PubMed] [Google Scholar]
- 22.Roh KH, Martin DC, Lahann J. Nat Mater. 2005;4:759–763. doi: 10.1038/nmat1486. [DOI] [PubMed] [Google Scholar]
- 23.Roh KH, Martin DC, Lahann J. J Am Chem Soc. 2006;128:6796–6797. doi: 10.1021/ja060836n. [DOI] [PubMed] [Google Scholar]
- 24.Bhaskar S, Pollock KM, Yoshida M, Lahann J. Small. 2010;6:404–411. doi: 10.1002/smll.200901306. [DOI] [PubMed] [Google Scholar]
- 25.Misra AC, Bhaskar S, Clay N, Lahann J. Adv Mater. 2012;24:3850–3856. doi: 10.1002/adma.201200372. [DOI] [PubMed] [Google Scholar]
- 26.Ganan-Calvo AM. Phys Rev Lett. 1997;79:217–220. [Google Scholar]
- 27.Park TH, Eyster TW, Lumley JM, Hwang S, Lee KJ, Misra A, Rahmani S, Lahann J. Small. 2013;9:3051–3057. doi: 10.1002/smll.201201921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rahmani S, Park TH, Dishman AF, Lahann J. J Controlled Release. 2013;172:239–245. doi: 10.1016/j.jconrel.2013.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rahmani S, Saha S, Durmaz H, Donini A, Misra AC, Yoon J, Lahann J. Angew Chem, Int Ed. 2014;53:2332–2338. doi: 10.1002/anie.201310727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Song JW, Cavnar SP, Walker AC, Luker KE, Gupta M, Tung YC, Luker GD, Takayama S. PLoS One. 2009;4:e5756. doi: 10.1371/journal.pone.0005756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bhaskar S, Hitt J, Chang SWL, Lahann J. Angew Chem, Int Ed. 2009;48:4589–4593. doi: 10.1002/anie.200806241. [DOI] [PubMed] [Google Scholar]
- 32.Lee KJ, Yoon J, Rahmani S, Hwang S, Bhaskar S, Mitragotri S, Lahann J. Proc Natl Acad Sci U S A. 2012;109:16057–16062. doi: 10.1073/pnas.1213669109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sokolovskaya E, Yoon J, Misra AC, Brase S, Lahann J. Macromol Rapid Commun. 2013;34:1554–1559. doi: 10.1002/marc.201300427. [DOI] [PubMed] [Google Scholar]
- 34.Luker KE, Gupta M, Luker GD. Anal Chem. 2008;80:5565–5573. doi: 10.1021/ac8005457. [DOI] [PMC free article] [PubMed] [Google Scholar]