

Abstract
Schizophrenia is a devastating psychiatric disorder characterized by positive, negative and cognitive symptoms. While aberrant dopamine system function is typically associated with the positive symptoms of the disease, it is thought that this is secondary to pathology in afferent regions. Indeed, schizophrenia patients show dysregulated activity in the hippocampus and prefrontal cortex, two regions known to regulate dopamine neuron activity. These deficits in hippocampal and prefrontal cortical function are thought to result, in part, from reductions in inhibitory interneuron function in these brain regions. Therefore, it has been hypothesized that restoring interneuron function in the hippocampus and/or prefrontal cortex may be an effective treatment strategy for schizophrenia. In this article, we will discuss the evidence for interneuron pathology in schizophrenia and review recent advances in our understanding of interneuron development. Finally, we will explore how these advances have allowed us to test the therapeutic value of interneuron transplants in multiple preclinical models of schizophrenia.
Introduction
Schizophrenia is devastating psychiatric disorder that affects approximately 1% of the population1. Positive symptoms, such as paranoia, grandiosity, delusions, and hallucinations, are often the most striking features of the disorder; however, schizophrenia patients also display characteristic negative and cognitive symptoms, which can be severely debilitating. Negative symptoms, such as blunted affect, emotional withdrawal, and social avoidance and cognitive symptoms, including disruptions in working memory, attentional deficits, disorganized thought, and cognitive inflexibility, can negatively influence social and occupational functioning and diminish quality of life2–4. Currently prescribed antipsychotic medications, which act as antagonists at the dopamine D2 receptor5, have been somewhat effective in treating the positive symptoms of schizophrenia6. However, these drugs have been associated with side effects such as weight gain, sedation, and extrapyramidal symptoms, resulting in a high rate of discontinuation6. Further, even among patients that respond to antipsychotic treatment, disability rates remain high and functional outcomes poor, highlighting the need to treat the residual negative and cognitive symptoms2. Currently, there are no FDA-approved drugs specifically designed to target these symptom domains4, suggesting an urgent need for more effective treatment strategies that target all three symptom classes observed in schizophrenia patients.
The neuroanatomy of schizophrenia
The dopamine hypothesis is one of the longest-standing and most widely accepted hypotheses explaining the potential neurobiological mechanisms underlying schizophrenia. The original dopamine hypothesis suggests that schizophrenia symptoms are caused by excessive subcortical dopamine. Support for this hypothesis stems from the fact that currently prescribed antipsychotic medications, which can alleviate positive symptoms, act as antagonists at the D2 receptor5,7. Further, drugs that increase dopamine levels, such as amphetamine, can induce schizophrenia-like symptoms in otherwise healthy individuals8. When compared to controls, schizophrenia patients show potentiated amphetamine-induced dopamine release in the striatum9, which has been correlated with the severity of positive symptoms9. Despite this evidence, the dopamine hypothesis does not fully explain the complete array of symptoms present in schizophrenia. For example, antipsychotic medications have little effect on cognitive and negative symptoms2, suggesting that excessive subcortical dopamine may not underlie these symptom domains. More recently, the dopamine hypothesis has been revised to suggest that dopamine system hyperactivity and increased D2 receptor activation in the mesolimbic regions underlie positive symptoms, while hypoactive dopamine transmission and reduced D1 receptor activation in the prefrontal cortex are responsible for negative and cognitive symptoms10. While animal models demonstrate that D1 antagonism in the prefrontal cortex can impair working memory11, an effect that is reversed by co-administration of a D1 agonist12, human studies do not demonstrate consistent abnormalities in dopamine tissue concentrations or D1 receptor binding in the prefrontal cortex of schizophrenia patients13. The inability to identify significant pathology in the dopamine system of schizophrenia patients, has led some to suggest that the pathology actually lies in upstream brain regions that regulate the dopamine system14.
In animal models, the ventral hippocampus (vHipp) is one such brain region that has been shown to regulate dopamine signaling via a polysynaptic pathway from the nucleus accumbens (nAcc) to the ventral pallidum (VP), and ultimately to dopamine cells in the ventral tegmental area (VTA, Figure 1). Activation of the vHipp by the glutamate agonist NMDA, has been shown to activate dopamine neurons in the VTA, resulting in an increase in the number of spontaneously active cells15 and extracellular dopamine16. Administration of a glutamate antagonists into the nAcc blocks the increase in dopamine population activity caused by vHipp activation, suggesting that the vHipp sends glutamatergic projections to the nAcc15. The nAcc then sends inhibitory GABAergic projections to VP, and bicuculline (GABAA antagonist) injections into this brain region also block the increase in dopamine population activity induced by vHipp activation17. Further, inactivation of the ventral pallidum by muscimol/baclofen increased dopamine population activity, suggesting that under normal conditions, the VP maintains an inhibitory influence over the VTA17. Therefore, the vHipp can act through a multisynaptic pathway to regulate dopamine neuron population activity.
Figure 1.
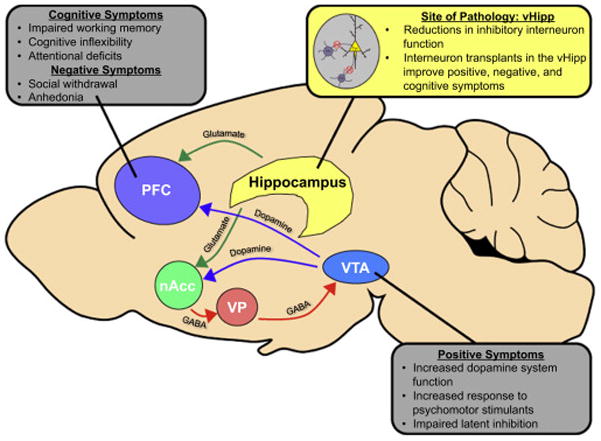
A change in dopamine population activity can influence both tonic dopamine levels in the nAcc and phasic dopamine release in response to motivationally relevant stimuli. 16,18,19. An increase in burst firing by dopamine neurons is thought to provide the post-synaptic signal that predicts and detects reward. Dopamine neurons that are not spontaneously active are held in a hyperpolarized state by GABAergic inputs from the VP, therefore only spontaneously active dopamine neurons can increase burst firing in response to motivationally relevant stimuli19. By regulating the number of spontaneously active dopamine neurons in the VTA, the vHipp can set the ‘gain’ of the dopamine system.
Work in multiple animal models of schizophrenia has demonstrated that hyperactivity in the vHipp can drive an increase in dopamine population activity. For example, the mitotoxin methylazoxymethanol acetate (MAM) has been administered to pregnant female rats at gestational day 17 to induce schizophrenia-like changes in anatomy, physiology, and behavior 20. MAM-treated rats show hyperactivity in the vHipp and an increase in the number of spontaneously active dopamine neurons in the VTA21. Inactivation of the vHipp was able to normalize dopamine cell population activity21, suggesting that the dysregulation of the vHipp can result in aberrant dopamine system function. The sub-chronic PCP model (5mg/kg, 2 times/day, 7d) has also been shown to induce behavioral symptoms that model the positive, negative and cognitive symptoms of schizophrenia22,23. We have demonstrated that sub-chronic PCP administration also produces an increase in the number of spontaneously active dopamine neurons in the VTA, an effect that can be reversed by inactivation of the vHipp24. Further, the cyclin D2 knock-out mouse model displays a relatively selective deficit in vHipp inhibition, accompanied by schizophrenia-like behavioral deficits25. Knock-out animals were shown to have an increase in dopamine population activity in the VTA25, suggesting that dysregulated activity in the vHipp can underlie aberrant dopamine signaling associated with schizophrenia-like behaviors. Thus, data from a diverse number of rodent models suggests a pathological enhancement in dopamine neuron population activity is directly attributable to an increased drive from the vHipp. The consequence of the increase in the number of spontaneously active dopamine neurons may be that normally benign stimuli are ascribed an abnormally high salience which may contribute to the paranoia and delusions associated with schizophrenia.
Consistent with our preclinical data, anatomical and physiological changes are consistently observed in the hippocampus of schizophrenia patients. The rodent hippocampus is organized along a dorsal to ventral axis, which corresponds to a posterior to anterior axis in humans26. Structurally, decreases in hippocampal volume are a consistent observation in both postmortem and imaging studies27,28. The decrease in hippocampal volume is seen in first episode patients but also appears to progress throughout the course of the illness29,30. Furthermore, decreases in hippocampal volume seem to be primarily limited to the anterior hippocampus31,32 and has been shown to predict deficits in executive function33.
In addition to the anatomical changes, schizophrenia patients also show differences in hippocampal activity. The hippocampus plays an important role in declarative memory and schizophrenia patients demonstrate impaired recruitment of the hippocampus during memory tasks34,35. Despite these seeming reductions in hippocampal function, unmedicated schizophrenia patients show increased hippocampal activity at rest34,36–38, an effect that can be attenuated by antipsychotic treatment38. This increase in hippocampal activity has been correlated with the severity of positive symptoms39, suggesting that hippocampal hyperactivity may drive increased activity in the dopamine system and underlie some symptoms of schizophrenia. Indeed, this is consistent with the preclinical data described above where inactivation of the vHipp can reduce dopamine population activity and normalize behavioral correlates of positive symptoms in a rodent model of schizophrenia21. Further, hippocampal connectivity is disrupted in schizophrenia patients40. Specifically, the anterior hippocampus shows decreased connectivity with regions such as the anterior cingulate and the medial prefrontal cortex40.
Finally, cellular and molecular changes have also been observed in the hippocampus of schizophrenia patients. For example, schizophrenia patients have a reduction in neural stem cell proliferation in the dentate gyrus, as determined by Ki-67, a marker of adult neurogenesis41. Further, schizophrenia patients show a reduced number of synapses between dentate gyrus mossy fibers and CA3 pyramidal neurons42,43, which is accompanied by changes in markers of synaptic plasticity44. Specifically, an increase in GluN2B-containing NMDA receptors, PSD95 protein, and spine density were observed in the CA3, but not CA1, region of the hippocampus of schizophrenia patients44. Together, these findings support the role of hippocampal pathology in schizophrenia.
The prefrontal cortex (PFC) has also been implicated in the pathology of schizophrenia45 (Figure 1). The dorsolateral prefrontal cortex (DLPFC) is one region of the PFC that is critically involved in working memory46, which is disrupted in schizophrenia47. Schizophrenia patients show reduced DLPFC activation during working memory tasks, with activation levels correlated to performance48. Even schizophrenia patients that perform normally on working memory tasks show higher levels of DLPFC activation compared to controls, suggesting a deficit in efficiency49. Further, anatomical changes, such as reduced dendritic spine density have been observed the DLPFC of schizophrenia patients50–52. Thus, it is likely that aberrant PFC activity contributes to the cognitive dysfunction observed in patients. The DLPFC has also been associated with negative symptoms of schizophrenia. For example, one study demonstrated a correlation between activity in the DLPFC and the severity of negative symptoms in schizophrenia patients53. Further, negative symptoms have also been associated with volumetric reductions in the prefrontal cortex54. Therefore, changes in PFC activity may also underlie the negative symptoms of schizophrenia.
Interestingly, the PFC and hippocampus are interconnected neuronal systems with the anterior hippocampus directly regulating activity in the PFC55. Moreover, deficits in connectivity between these regions have been observed in schizophrenia patients40,56,57. Indeed, we have previously demonstrated that hippocampal manipulations not only alter behavioral deficits consistent with positive symptoms, but can also regulate cognitive functions dependent on the PFC58. Together, these findings suggest that dysregulated prefrontal and hippocampal activity are present in schizophrenia patients and may underlie some of the symptoms of the disorder.
Interneuron dysfunction in schizophrenia
The cortex is composed of two primary cells types: excitatory pyramidal cells that use glutamate as their neurotransmitter and inhibitory interneurons that use GABA. A significant literature examining post mortem tissue from schizophrenia patients have demonstrated that the pathology of schizophrenia may involve a deficit in inhibitory signaling (Figure 1). As early as 1979, Bird and colleagues demonstrated that activity of glutamic acid decarboxylase (GAD), the enzyme required for the synthesis of GABA, was reduced across multiple brain regions in schizophrenia patients, including the prefrontal cortex and hippocampus. This study was confounded by the fact that most of the patients died of a protracted illness, which has also been shown to increase GAD activity59. However, there is now considerable evidence to suggest that the GABAergic system is disrupted in schizophrenia patients. For example, patients possess fewer GABA uptake sites60 and a reduction in non-pyramidal cells in the hippocampus61. In addition, multiple studies have demonstrated a decrease in GAD mRNA expression in the hippocampus62. However, it should be noted that other studies have not observed a decrease in GAD mRNA expression in the hippocampus62,63. Further, GAD67 protein levels have not been observed in the hippocampus64, leading some to suggest that the GABAergic deficits present in the hippocampus may be restricted to certain sub-classes of interneurons. Indeed, these changes seem to be restricted specifically to interneurons that express the Ca2+-binding protein, parvalbumin (PV), and those that express the neuropeptide, somatostatin (SST). For example, Zhang and Reynolds demonstrated a reduction in the number of PV-immunoreactive interneurons in the hippocampus of schizophrenia patients65 while the number of interneurons that express calretinin were not changed in any of the hippocampal subfields examined65. However, this result has not been replicated66. In addition, reductions in SST-positive interneurons have also been observed in the hippocampus of schizophrenia patients66. Reductions in particular classes of interneurons may be significant, as different interneuron sub-types have unique anatomical and physiological features, allowing these cells to differentially regulate pyramidal cell function67.
In animal models, deficits in PV-positive interneuron function have been shown to induce schizophrenia-like behaviors in otherwise normal animals. For example, GAD knock-out restricted to PV-positive cells causes deficits in pre-pulse inhibition, changes in social interaction and impaired fear conditioning68. Further, we have demonstrated that shRNA knock-down of PV specifically in the vHipp causes an increase in the number of spontaneously active dopamine neurons in the VTA and an increase in amphetamine-induced locomotor activity69. Using a pharmacogenetic approach, Nguyen and colleagues demonstrated that inhibition of PV-positive interneurons in the vHipp impairs prepulse inhibition and spontaneous alternation while inhibition of GAD65-positive cells increases amphetamine-induced locomotor activity and reduces spontaneous alternation70. These results suggest that the loss of interneurons observed in the vHipp of schizophrenic patients may underlie some symptoms of the disorder.
It is important to note that although hippocampal hyperactivity is consistently observed in schizophrenia patients, the mechanism by which this occurs is not universally accepted. As described above, interneuron deficits have not always been in the hippocampus of schizophrenia patients62–64,66. Instead, many believe that the increase in hippocampal hyperactivity is due to increases in glutamate signaling71. Regardless of the mechanism by which it occurs, the end result is an increase in hippocampal activity, which has been consistently observed in schizophrenia patients and across animal models of schizophrenia. Therefore, despite the mechanism of pathology, it is likely that enhancing interneuron function would reduce hippocampal hyperactivity and have a beneficial effect in schizophrenia.
Deficits in interneuron function have also been observed in the prefrontal cortex of schizophrenia patients. For example, schizophrenia patients have reductions in GAD67 mRNA in the DLPFC72–75. Interestingly, there does not appear to be a loss of cells, rather a subset of interneurons express less GAD72,75. This effect seems to be specific to psychosis, as patients with bipolar depression also show GAD reductions but those with unipolar depression do not73. In addition, the GAD reduction is independent of antipsychotic treatment75. More recent work has begun to explore changes in specific interneuron subtypes in the DLPFC. Hashimoto and colleagues found that schizophrenia patients show reduced mRNA expression of the neuropeptides SST and cholecystokinin76. Together, these results demonstrate a change in hippocampal and PFC interneuron function in schizophrenia and suggest that targeting the GABAergic system may be an effective treatment strategy for schizophrenia. Toward this end, multiple groups have begun to explore the use of cell-based therapies to replace dysfunctional interneurons.
Understanding interneuron development
Interneuron transplant therapy would not be possible without an understanding of the developmental origins of cortical interneurons and in recent years, fate-mapping experiments have provided insight into interneuron development. GABAergic interneurons are born in the telencephalic structures known as the ganglionic eminences (medial, lateral, and caudal). Interneuron precursors born in the ganglionic eminences have the remarkable ability to migrate tangentially across cortical regions77. The particular subclass of interneuron that develops is determined by a combination of spatial and temporal factors. For example, the medial ganglionic eminence (MGE) is the primary source of SST- and PV-positive interneurons77,78 while the caudal ganglionic eminence (CGE) gives rise to calretinin-positive interneurons79. Interneuron precursors in the MGE cease proliferation before migrating into the cortex80 and the fate of these interneuron precursors is determined by the signals that are present at the time the cells are born, rather than those that are encountered during migration79,81. Using BrdU to label MGE cells, it has been demonstrated that SST-positive cells are generally born before PV-positive interneurons82. Interneurons fill in the cortex in an inside-out pattern with deep layer interneurons born earlier than those in more superficial layers83, suggesting that PV- and SST-positive interneurons may demonstrate laminar targeting.
Spatial factors can also influence the particular interneuron sub-type that develops. The dorsal region of the MGE, for example, shows a bias toward SST-positive interneurons, while the more ventral region of the MGE shows a bias toward PV-positive cells, although both areas produce both cell types84. This biased interneuron development may result from a concentration gradient of the morphogen sonic hedgehog (SHH). SHH is expressed at higher levels in the more dorsal regions of the MGE, resulting in an increased percentage of SST-positive cells85. SHH is important for the activation and maintenance of NK2 transcription factor-related 2.1 (Nkx2.1)80, which is required for both the development of both PV- and SST-positive interneurons in the MGE86,87. Downstream, Nkx2.1 activates lim-homeodomain transcription factor (Lhx6). As PV- and SST-positive MGE cells exit the cell cycle and begin their migration, Nkx2.1 is down-regulated and Lhx6 expression increases and is maintained throughout the life of the cell88.
This enhanced understanding of interneuron development has allowed us to start evaluating the use of interneuron transplants in preclinical models of disease. In the first proof of concept experiment, Wichterle demonstrated that dissociated MGE cells from fetal brain tissue survived after being transplanted into the brain. Moreover, these cells migrated extensively into the striatum, thalamus, and neocortex77. Subsequent experiments have demonstrated that by 21 days after transplantation, MGE cells have migrated to their final destination and a majority (~70%) express GABA or GAD67. Excitingly, these cells display firing patterns similar to endogenous interneurons. Further, the cells integrate into the cortical circuitry, both increasing inhibitory input to and receiving excitatory input from surrounding pyramidal cells89. These experiments demonstrate that MGE tissue can be used to transplant GABAergic cells into preclinical models of disease.
The use of fetal tissue, however, is not a viable treatment strategy for humans. Therefore, many groups are working to develop interneurons from embryonic stem cells (ESCs). In 2005, Watanabe and colleagues developed a protocol for generating telencephalic precursors from ESCs, including those that express Nkx2.190. However, not all cells develop into GABAergic cells, and tumor formation is a significant limitation; therefore the development of transgenic ESC lines has allowed the exclusion of other cell types. For example, a mouse ESC line was modified to express GFP under the control of the Lhx6 promoter. The Lhx6::GFP cells were sorted by flow cytometry, then transplanted into the brain. These GFP+ cells survived in the brain and migrated throughout the cortex where they express markers of GABAergic cells, including PV and SST. Furthermore, these transplanted cells display firing properties reminiscent of endogenous PV- or SST- cells. Importantly, no tumor formation was observed in transplanted animals, even up to 240 days after the cells were transplanted91. The Lhx6::GFP ESC cell line was further modified to also express mCherry under the control of the Nkx2.1 promoter, allowing the differentiation of specific sub-classes of interneurons. Using these Lhx6::GFP::Nkx2.1::mCherry cells, a protocol employing differential expression of SHH and timing was developed to form enriched populations of PV- or SST-positive interneurons. SST-positive cells were exposed to high levels of SHH and cultured for a shorter period of time (12 days) while PV-positive cells were exposed to low levels of SHH and were cultured for 17 days, as this interneuron subtype is generally born earlier. Once transplanted into the brain, the PV- or SST-positive cells expressed interneuron markers and displayed firing properties similar to endogenous SST- and PV-positive interneurons92.
Using interneurons to treat schizophrenia
Multiple groups have begun to explore the use of interneuron transplants for the treatment of schizophrenia (Table 1). As detailed above, schizophrenia patients show reductions in GABAergic cells in the prefrontal cortex. Dysfunction in the prefrontal cortex is thought to be especially important for development of negative93 and cognitive symptoms94. Therefore, it is possible that replacing interneurons in the prefrontal cortex may have beneficial effects on negative and cognitive symptoms of schizophrenia. The first group to test this hypothesis used the phencyclidine (PCP) model of schizophrenia. PCP, an NMDA antagonist induces acute psychosis in humans95 and can produce schizophrenia-like deficits in rodents22,23. Using the sub-acute PCP model, Tanaka and colleagues demonstrated that deficits in novel object recognition and prepulse inhibition were prevented by transplanting MGE tissue into the medial prefrontal cortex (mPFC) of newborn animals96. These experiments suggested that interneuron transplants may be a useful strategy to prevent the development of negative and cognitive symptoms that model schizophrenia. While exciting, enthusiasm for these results was limited by two important considerations. First, there is no definitive biomarker to indicate that a person will develop schizophrenia, therefore preventative treatment using cell transplants is unlikely to become a viable treatment option for human patients. Further, the acute PCP model of schizophrenia does not recapitulate the developmental aspects of the disorder97.
Table 1.
Efficacy of interneuron transplants in preclinical models of schizophrenia
| Cell Source | Target Region | Rodent Model | Behavioral and Physiological Effects | Reference |
|---|---|---|---|---|
| MGE | mPFC | Acute PCP | Improved novel object recognition and normalized prepulse inhibition | Tanaka 2011 |
| MGE | mPFC | MAM | Improved reversal learning and increased social interaction time | Perez 2015 |
| MGE | vHipp | MAM | Reduced amphetamine-induced locomotor activity, normalized dopamine population activity, reduced hippocampal hyperactivity | Perez 2013 |
| MGE | vHipp | Cyclin D2 knock-out | Reduced amphetamine-induced locomotor activity, improved contextual fear conditioning normalized dopamine population activity, and reduced hippocampal hyperactivity | Giliani 2014 |
| ESC | vHipp | MAM | Normalized latent inhibition (PV, SST), increased social interaction time (PV), improved reversal learning (PV, SST) and extradimensional set shifting (PV), normalized dopamine population activity (PV, SST), reduced hippocampal hyperactivity (PV, SST) | Donegan 2015 |
Recently, a developmental model of schizophrenia was used to test the efficacy of MGE transplants. Injections of methylazoxymethanol (MAM) on gestational day 17 produces anatomical, behavioral, and physiological deficits that model schizophrenia20. Specifically, the MAM model produces deficits that model the negative and cognitive symptoms of schizophrenia, including deficits in cognitive flexibility58 and reductions in social interaction98. We have shown that MGE transplants into the mPFC of MAM rodents can restore cognitive flexibility and increase social interaction time99. Our results suggest that interneuron transplants may be an effective treatment strategy for negative and cognitive symptoms even after the pathology has developed.
The MAM model also produces deficits that model the positive symptoms of schizophrenia. MAM rodents show an increase in the firing rate of hippocampal pyramidal cells, an increase in dopamine population activity in the VTA, and enhanced amphetamine-induced locomotor activity, a model of the positive symptoms of schizophrenia. Interestingly, prenatal MAM treatment also produces a decrease in PV-positive interneurons in the ventral hippocampus100. Transplantation of fetal MGE tissue into the vHipp of MAM rats was able to reduce pyramidal cell activity in the hippocampus, decrease dopamine population activity in the VTA, and normalize amphetamine-induced locomotor activity, suggesting that interneuron transplants into the vHipp may be effective in reducing positive symptoms of schizophrenia101. However, the hippocampus projects directly to the prefrontal cortex55, therefore it is possible that transplants into the vHipp may influence negative and cognitive symptoms as well.
Recently, Giliani and colleagues demonstrated that hippocampal interneuron transplants may influence cognitive function in addition to alleviating positive symptoms. The cyclin D2 knock-out mouse has been used as a model of hippocampal hyperactivity. These animals display schizophrenia-like features, including an increase in cerebral blood volume in the ventral hippocampus, an increase in dopamine population activity in the VTA, and increased amphetamine-induced locomotor activity, all of which were reversed by MGE transplants into the vHipp. Interestingly, the MGE transplants also alleviated deficits in contextual fear conditioning, a hippocampal-dependent cognitive function25, suggesting that interneuron transplants into the vHipp may be able to influence the cognitive symptoms of schizophrenia, an area that is currently poorly treated.
The experiments described above are exciting and demonstrate that in multiple preclinical models, interneuron transplants effectively reduce schizophrenia-like symptoms. In order to move away from the use of fetal tissue to a more translational option, we have recently begun to explore the use of embryonic stem cells to grow interneurons. Specifically, we used the Lhx6::GFP::Nkx2.1::mCherry transgenic ESC line to grow enriched populations of SST- or PV-positive interneurons92. Interestingly, we demonstrated that the interneuron transplants into the vHipp integrated into the circuitry, effectively reducing spontaneous IPSCs in hippocampal pyramidal cells102. Further, both interneuron sub-types normalized the firing rate of putative hippocampal cells in MAM animals102. These hippocampal effects were accompanied by a reduction in dopamine population activity in the VTA102. Excitingly, the ESC-derived interneuron transplants also affected behavior, reducing not only positive but also cognitive and negative symptoms in the MAM model. However, the two interneuron subtypes had dramatically different effects on behavior. Latent inhibition is disrupted in schizophrenia patients and has been used as a model for the dopamine-dependent positive symptoms 103. We found that both SST- and PV-enriched transplants into the vHipp restored latent inhibition that was disrupted in the MAM animals102. Further, both interneuron subtypes alleviated MAM-induced deficits in reversal learning102, a form of cognitive flexibility mediated by the orbital frontal cortex104 and dependent on subcortical dopamine signaling105. PV-positive cells, however, were also able to increase social interaction time and restore extradimensional set-shifting performance in MAM rats102, two behaviors that critically depend on the mPFC93. These results suggest that interneuron, specifically PV-positive cell, transplants into the vHipp may be an effective treatment strategy to target positive, negative, and cognitive symptoms of schizophrenia (Figure 1).
Outstanding questions and future directions
These experiments provide evidence that interneuron transplants may be an effective treatment strategy for schizophrenia. However, before this strategy can move to the clinic, many questions must be answered. For example, the source of the cells will be a major issue. The use of MGE tissue from human fetuses has obvious moral implications; therefore, many labs have begun to develop protocols for generating interneuron precursors from human pluripotent stem cells106–108. Using patient-specific ESCs would have the additional advantage of eliminating the potential of rejection. It should be noted that human development is much longer than that of a rodent and therefore growing personalized interneurons from patient-derived stem cells would require a great deal of time and resources. Alternatively, some have proposed the development of stem cell banks, which would provide about 150 human ESC lines that would have sufficient homology for the use in more than 90% of the population109. Further, the ability to grow highly enriched populations of interneurons remains a challenge. In the experiments described above, flow cytometry was used to exclude cells that did not express interneuron-specific markers. Such an approach would likely be required for human cells as non-differentiated stem cell transplants can lead to the formation of tumors and other undesirable side effects110. In conclusion, the results presented here are promising and suggest that interneuron transplants may be an effective treatment strategy for schizophrenia. However, more work needs to be done at the preclinical level to understand the specific role of interneuron sub-types and the long-term effects of ESC transplants. Further, improved methods for generating large quantities of purified interneuron precursors rapidly will be required before this exciting new therapy can move to the clinic.
References
- 1.Stilo SA, Murray RM. The epidemiology of schizophrenia: replacing dogma with knowledge. Dialogues Clin Neurosci. 2010;12:305–315. doi: 10.31887/DCNS.2010.12.3/sstilo. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Strassnig MT, et al. Determinants of different aspects of everyday outcome in schizophrenia: The roles of negative symptoms, cognition, and functional capacity. Schizophrenia research. 2015 doi: 10.1016/j.schres.2015.03.033. doi: http://dx.doi.org/10.1016/j.schres.2015.03.033. [DOI] [PMC free article] [PubMed]
- 3.Green MF, Kern RS, Braff DL, Mintz J. Neurocognitive deficits and functional outcome in schizophrenia. Schizophrenia Bulletin. 2000;26:119–136. doi: 10.1093/oxfordjournals.schbul.a033430. [DOI] [PubMed] [Google Scholar]
- 4.Citrome L. Unmet needs in the treatment of schizophrenia: new targets to help different symptom domains. The Journal of clinical psychiatry. 2014;75(Suppl 1):21–26. doi: 10.4088/JCP.13049su1c.04. [DOI] [PubMed] [Google Scholar]
- 5.Seeman P, Chau-Wong M, Tedesco J, Wong K. Brain receptors for antipsychotic drugs and dopamine: direct binding assays. Proceedings of the National Academy of Sciences of the United States of America. 1975;72:4376–4380. doi: 10.1073/pnas.72.11.4376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Leucht S, et al. Comparative efficacy and tolerability of 15 antipsychotic drugs in schizophrenia: A multiple-treatments meta-analysis. The Lancet. 2013;382:951–962. doi: 10.1016/S0140-6736(13)60733-3. doi: http://dx.doi.org/10.1016/S0140-6736(13)60733-3. [DOI] [PubMed] [Google Scholar]
- 7.Meltzer HY, Creese I, Burt DR, Solomon HS. Dopamine Receptors and Average Clinical Doses. Science. 1976;194:545–546. doi: 10.2307/1742486. [DOI] [PubMed] [Google Scholar]
- 8.Janowsky DS, El-Yousef M, Davis JM, Sekerke H. Provocation of schizophrenic symptoms by intravenous administration of methylphenidate. Arch Gen Psychiatry. 1973;28:185–191. doi: 10.1001/archpsyc.1973.01750320023004. [DOI] [PubMed] [Google Scholar]
- 9.Abi-Dargham A, et al. Increased baseline occupancy of D2 receptors by dopamine in schizophrenia. Proceedings of the National Academy of Sciences of the United States of America. 2000;97:8104–8109. doi: 10.1073/pnas.97.14.8104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brisch R, et al. The role of dopamine in schizophrenia from a neurobiological and evolutionary perspective: old fashioned, but still in vogue. Frontiers in Psychiatry. 2014;5 doi: 10.3389/fpsyt.2014.00047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sawaguchi T, Goldman-Rakic P. D1 dopamine receptors in prefrontal cortex: involvement in working memory. Science. 1991;251:947–950. doi: 10.1126/science.1825731. [DOI] [PubMed] [Google Scholar]
- 12.Castner SA, Williams GV, Goldman-Rakic PS. Reversal of Antipsychotic-Induced Working Memory Deficits by Short-Term Dopamine D1 Receptor Stimulation. Science. 2000;287:2020–2022. doi: 10.1126/science.287.5460.2020. [DOI] [PubMed] [Google Scholar]
- 13.Goldman-Rakic PS, Castner SA, Svensson TH, Siever LJ, Williams GV. Targeting the dopamine D1 receptor in schizophrenia: insights for cognitive dysfunction. Psychopharmacology. 2004;174:3–16. doi: 10.1007/s00213-004-1793-y. [DOI] [PubMed] [Google Scholar]
- 14.Lodge DJ, Grace AA. Hippocampal dysregulation of dopamine system function and the pathophysiology of schizophrenia. Trends Pharmacol Sci. 2011;32:507–513. doi: 10.1016/j.tips.2011.05.001. doi: http://dx.doi.org/10.1016/j.tips.2011.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Floresco SB, Todd CL, Grace AA. Glutamatergic Afferents from the Hippocampus to the Nucleus Accumbens Regulate Activity of Ventral Tegmental Area Dopamine Neurons. The Journal of Neuroscience. 2001;21:4915–4922. doi: 10.1523/JNEUROSCI.21-13-04915.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Legault M, Wise RA. Injections of N-methyl-D-aspartate into the ventral hippocampus increase extracellular dopamine in the ventral tegmental area and nucleus accumbens. Synapse. 1999;31:241–249. doi: 10.1002/(SICI)1098-2396(19990315)31:4<241::AID-SYN1>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- 17.Floresco SB, West AR, Ash B, Moore H, Grace AA. Afferent modulation of dopamine neuron firing differentially regulates tonic and phasic dopamine transmission. Nat Neurosci. 2003;6:968–973. doi: 10.1038/nn1103. [DOI] [PubMed] [Google Scholar]
- 18.Blaha CD, Yang CR, Floresco SB, Barr AM, Phillips AG. Stimulation of the Ventral Subiculum of the Hippocampus Evokes Glutamate Receptor-mediated Changes in Dopamine Efflux in the Rat Nucleus Accumbens. Eur J Neurosci. 1997;9:902–911. doi: 10.1111/j.1460-9568.1997.tb01441.x. [DOI] [PubMed] [Google Scholar]
- 19.Lodge DJ, Grace AA. The Hippocampus Modulates Dopamine Neuron Responsivity by Regulating the Intensity of Phasic Neuron Activation. Neuropsychopharmacology : official publication of the American College of Neuropsychopharmacology. 2006;31:1356–1361. doi: 10.1038/sj.npp.1300963. [DOI] [PubMed] [Google Scholar]
- 20.Lodge DJ, Grace AA. Gestational methylazoxymethanol acetate administration: a developmental disruption model of schizophrenia. Behavioral Brain Research. 2009;204:306–312. doi: 10.1016/j.bbr.2009.01.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lodge DJ, Grace AA. Aberrant Hippocampal Activity Underlies the Dopamine Dysregulation in an Animal Model of Schizophrenia. The Journal of Neuroscience. 2007;27:11424–11430. doi: 10.1523/jneurosci.2847-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mouri A, Noda Y, Enomoto T, Nabeshima T. Phencyclidine animal models of schizophrenia: approaches from abnormality of glutamatergic neurotransmission and neurodevelopment. Neurochem Int. 2007;51:173–184. doi: 10.1016/j.neuint.2007.06.019. [DOI] [PubMed] [Google Scholar]
- 23.Neill JC, et al. Animal models of cognitive dysfunction and negative symptoms of schizophrenia: focus on NMDA receptor antagonism. Pharmacol Ther. 2010;128:419–432. doi: 10.1016/j.pharmthera.2010.07.004. [DOI] [PubMed] [Google Scholar]
- 24.Aguilar DD, Chen L, Lodge DJ. Increasing Endocannabinoid Levels in the Ventral Pallidum Restore Aberrant Dopamine Neuron Activity in the Subchronic PCP Rodent Model of Schizophrenia. International Journal of Neuropsychopharmacology. 2015;18 doi: 10.1093/ijnp/pyu035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gilani AI, et al. Interneuron precursor transplants in adult hippocampus reverse psychosis-relevant features in a mouse model of hippocampal disinhibition. Proceedings of the National Academy of Sciences. 2014;111:7450–7455. doi: 10.1073/pnas.1316488111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Strange BA, Witter MP, Lein ES, Moser EI. Functional organization of the hippocampal longitudinal axis. Nat Rev Neurosci. 2014;15:655–669. doi: 10.1038/nrn3785. http://www.nature.com/nrn/journal/v15/n10/abs/nrn3785.html - supplementary-information. [DOI] [PubMed] [Google Scholar]
- 27.Nelson MD, Saykin AJ, Flashman LA, Riordan HJ. Hippocampal volume reduction in schizophrenia as assessed by magnetic resonance imaging: A meta-analytic study. Arch Gen Psychiatry. 1998;55:433–440. doi: 10.1001/archpsyc.55.5.433. [DOI] [PubMed] [Google Scholar]
- 28.STEEN RG, MULL C, MCCLURE R, HAMER RM, LIEBERMAN JA. Brain volume in first-episode schizophrenia. 2006;188 doi: 10.1192/bjp.188.6.510. [DOI] [PubMed] [Google Scholar]
- 29.Velakoulis D, Wood SJ, Wong MH, et al. Hippocampal and amygdala volumes according to psychosis stage and diagnosis: A magnetic resonance imaging study of chronic schizophrenia, first-episode psychosis, and ultra–high-risk individuals. Arch Gen Psychiatry. 2006;63:139–149. doi: 10.1001/archpsyc.63.2.139. [DOI] [PubMed] [Google Scholar]
- 30.Chakos MH, et al. Duration of illness and treatment effects on hippocampal volume in male patients with schizophrenia. 2005;186 doi: 10.1192/bjp.186.1.26. [DOI] [PubMed] [Google Scholar]
- 31.Szeszko PR, et al. Smaller Anterior Hippocampal Formation Volume in Antipsychotic-Naive Patients With First-Episode Schizophrenia. Am J Psychiatry. 2003;160:2190–2197. doi: 10.1176/appi.ajp.160.12.2190. [DOI] [PubMed] [Google Scholar]
- 32.Schobel Scott A, et al. Imaging Patients with Psychosis and a Mouse Model Establishes a Spreading Pattern of Hippocampal Dysfunction and Implicates Glutamate as a Driver. Neuron. 2013;78:81–93. doi: 10.1016/j.neuron.2013.02.011. doi: http://dx.doi.org/10.1016/j.neuron.2013.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bilder RM, et al. Anterior hippocampal volume reductions predict frontal lobe dysfunction in first episode schizophrenia. Schizophrenia research. 1995;17:47–58. doi: 10.1016/0920-9964(95)00028-k. doi: http://dx.doi.org/10.1016/0920-9964(95)00028-K. [DOI] [PubMed] [Google Scholar]
- 34.Heckers S, et al. Impaired recruitment of the hippocampus during conscious recollection in schizophrenia. Nat Neurosci. 1998;1:318–323. doi: 10.1038/1137. [DOI] [PubMed] [Google Scholar]
- 35.Weiss AP, et al. Impaired hippocampal recruitment during normal modulation of memory performance in schizophrenia. Biol Psychiatry. 2003;53:48–55. doi: 10.1016/s0006-3223(02)01541-x. doi: http://dx.doi.org/10.1016/S0006-3223(02)01541-X. [DOI] [PubMed] [Google Scholar]
- 36.Schobel SA, et al. Anterior hippocampal and orbitofrontal cortical structural brain abnormalities in association with cognitive deficits in schizophrenia. Schizophrenia research. 2009;114:110–118. doi: 10.1016/j.schres.2009.07.016. doi: http://dx.doi.org/10.1016/j.schres.2009.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Malaspina D, et al. SPECT study of visual fixation in schizophrenia and comparison subjects. Biol Psychiatry. 1999;46:89–93. doi: 10.1016/s0006-3223(98)00306-0. doi: http://dx.doi.org/10.1016/S0006-3223(98)00306-0. [DOI] [PubMed] [Google Scholar]
- 38.Medoff DR, Holcomb HH, Lahti AC, Tamminga CA. Probing the human hippocampus using rCBF: Contrasts in schizophrenia. Hippocampus. 2001;11:543–550. doi: 10.1002/hipo.1070. [DOI] [PubMed] [Google Scholar]
- 39.Schobel SA, Lewandowski NM, Corcoran CM, et al. DIfferential targeting of the ca1 subfield of the hippocampal formation by schizophrenia and related psychotic disorders. Arch Gen Psychiatry. 2009;66:938–946. doi: 10.1001/archgenpsychiatry.2009.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Samudra N, et al. Alterations in hippocampal connectivity across the psychosis dimension. Psychiatry Research: Neuroimaging. 2015;233:148–157. doi: 10.1016/j.pscychresns.2015.06.004. doi: http://dx.doi.org/10.1016/j.pscychresns.2015.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Reif A, et al. Neural stem cell proliferation is decreased in schizophrenia, but not in depression. Mol Psychiatry. 2006;11:514–522. doi: 10.1038/sj.mp.4001791. [DOI] [PubMed] [Google Scholar]
- 42.Kolomeets NS, Orlovskaya DD, Rachmanova VI, Uranova NA. Ultrastructural alterations in hippocampal mossy fiber synapses in schizophrenia: A postmortem morphometric study. Synapse. 2005;57:47–55. doi: 10.1002/syn.20153. [DOI] [PubMed] [Google Scholar]
- 43.Kolomeets NS, Orlovskaya DD, Uranova NA. Decreased numerical density of CA3 hippocampal mossy fiber synapses in schizophrenia. Synapse. 2007;61:615–621. doi: 10.1002/syn.20405. [DOI] [PubMed] [Google Scholar]
- 44.Li W, et al. Synaptic Proteins In Schizophrenia Hippocampus Indicate Increased Neuronal Activity in CA3. The American journal of psychiatry. 2015;172:373–382. doi: 10.1176/appi.ajp.2014.14010123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bunney WE, Bunney BG. Evidence for a compromised dorsolateral prefrontal cortical parallel circuit in schizophrenia. Brain Res Rev. 2000;31:138–146. doi: 10.1016/s0165-0173(99)00031-4. doi: http://dx.doi.org/10.1016/S0165-0173(99)00031-4. [DOI] [PubMed] [Google Scholar]
- 46.Dudkin KN, Kruchinin VK, Chueva IV. Neurophysiological correlates of delayed visual differentiation tasks in monkeys: the effects of the site of intracortical blockade of NMDA receptors. Neurosci Behav Physiol. 2001;31:207–218. doi: 10.1023/a:1005224610354. [DOI] [PubMed] [Google Scholar]
- 47.Pantelis C, et al. Frontal-striatal cognitive deficits in patients with chronic schizophrenia. 1997;120 doi: 10.1093/brain/120.10.1823. [DOI] [PubMed] [Google Scholar]
- 48.Weinberger DR, Berman K, Zec RF. Physiologic dysfunction of dorsolateral prefrontal cortex in schizophrenia: I. regional cerebral blood flow evidence. Arch Gen Psychiatry. 1986;43:114–124. doi: 10.1001/archpsyc.1986.01800020020004. [DOI] [PubMed] [Google Scholar]
- 49.Callicott JH, et al. Complexity of prefrontal cortical dysfunction in schizophrenia: more than up or down. Am J Psychiatry. 2003;160:2209–2215. doi: 10.1176/appi.ajp.160.12.2209. [DOI] [PubMed] [Google Scholar]
- 50.Black JE, et al. Pathology of layer V pyramidal neurons in the prefrontal cortex of patients with schizophrenia. Am J Psychiatry. 2004;161:742–744. doi: 10.1176/appi.ajp.161.4.742. [DOI] [PubMed] [Google Scholar]
- 51.Garey LJ, et al. Reduced dendritic spine density on cerebral cortical pyramidal neurons in schizophrenia. Journal of Neurology, Neurosurgery & Psychiatry. 1998;65:446–453. doi: 10.1136/jnnp.65.4.446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Broadbelt K, Byne W, Jones LB. Evidence for a decrease in basilar dendrites of pyramidal cells in schizophrenic medial prefrontal cortex. Schizophrenia research. 2002;58:75–81. doi: 10.1016/s0920-9964(02)00201-3. doi: http://dx.doi.org/10.1016/S0920-9964(02)00201-3. [DOI] [PubMed] [Google Scholar]
- 53.Wolkin A, et al. Negative symptoms and hypofrontality in chronic schizophrenia. Arch Gen Psychiatry. 1992;49:959–965. doi: 10.1001/archpsyc.1992.01820120047007. [DOI] [PubMed] [Google Scholar]
- 54.Sanfilipo M, et al. Volumetric measure of the frontal and temporal lobe regions in schizophrenia: relationship to negative symptoms. Arch Gen Psychiatry. 2000;57:471–480. doi: 10.1001/archpsyc.57.5.471. [DOI] [PubMed] [Google Scholar]
- 55.Jay TM, Witter MP. Distribution of hippocampal CA1 and subicular efferents in the prefrontal cortex of the rat studied by means of anterograde transport of Phaseolus vulgaris-leucoagglutinin. J Comp Neurol. 1991;313:574–586. doi: 10.1002/cne.903130404. [DOI] [PubMed] [Google Scholar]
- 56.Fletcher P. The missing link: a failure of fronto-hippocampal integration in schizophrenia. Nat Neurosci. 1998;1:266–267. doi: 10.1038/1078. [DOI] [PubMed] [Google Scholar]
- 57.Meyer-Lindenberg AS, Olsen RK, Kohn PD, et al. REgionally specific disturbance of dorsolateral prefrontal–hippocampal functional connectivity in schizophrenia. Arch Gen Psychiatry. 2005;62:379–386. doi: 10.1001/archpsyc.62.4.379. [DOI] [PubMed] [Google Scholar]
- 58.Perez SM, Shah A, Asher A, Lodge DJ. Hippocampal deep brain stimulation reverses physiological and behavioural deficits in a rodent model of schizophrenia. Int J Neuropsychopharmacol. 2013;16:1331–1339. doi: 10.1017/S1461145712001344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bird ED, Spokes EGS, Iversen LL. INCREASED DOPAMINE CONCENTRATION IN LIMBIC AREAS OF BRAIN FROM PATIENTS DYING WITH SCHIZOPHRENIA. Brain. 1979;102:347–360. doi: 10.1093/brain/102.2.347. [DOI] [PubMed] [Google Scholar]
- 60.Simpson MD, Slater P, Deakin JF, Royston MC, Skan WJ. Reduced GABA uptake sites in the temporal lobe in schizophrenia. Neurosci Lett. 1989;107:211–215. doi: 10.1016/0304-3940(89)90819-7. [DOI] [PubMed] [Google Scholar]
- 61.Benes FM, Kwok EW, Vincent SL, Todtenkopf MS. A reduction of nonpyramidal cells in sector CA2 of schizophrenics and manic depressives. Biol Psychiatry. 1998;44:88–97. doi: 10.1016/s0006-3223(98)00138-3. doi: http://dx.doi.org/10.1016/S0006-3223(98)00138-3. [DOI] [PubMed] [Google Scholar]
- 62.Heckers S, et al. DIfferential hippocampal expression of glutamic acid decarboxylase 65 and 67 messenger rna in bipolar disorder and schizophrenia. Arch Gen Psychiatry. 2002;59:521–529. doi: 10.1001/archpsyc.59.6.521. [DOI] [PubMed] [Google Scholar]
- 63.Straub RE, et al. Allelic variation in GAD1 (GAD67) is associated with schizophrenia and influences cortical function and gene expression. Mol Psychiatry. 2007;12:854–869. doi: 10.1038/sj.mp.4001988. doi: http://www.nature.com/mp/journal/v12/n9/suppinfo/4001988s1.html. [DOI] [PubMed] [Google Scholar]
- 64.Stan AD, et al. Magnetic resonance spectroscopy and tissue protein concentrations together suggest lower glutamate signaling in dentate gyrus in schizophrenia. Mol Psychiatry. 2015;20:433–439. doi: 10.1038/mp.2014.54. [DOI] [PubMed] [Google Scholar]
- 65.Zhang ZJ, Reynolds GP. A selective decrease in the relative density of parvalbumin-immunoreactive neurons in the hippocampus in schizophrenia. Schizophrenia research. 2002;55:1–10. doi: 10.1016/s0920-9964(01)00188-8. doi: http://dx.doi.org/10.1016/S0920-9964(01)00188-8. [DOI] [PubMed] [Google Scholar]
- 66.Konradi C, et al. Hippocampal interneurons are abnormal in schizophrenia. Schizophrenia research. 2011;131:165–173. doi: 10.1016/j.schres.2011.06.007. doi: http://dx.doi.org/10.1016/j.schres.2011.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Markram H, et al. Interneurons of the neocortical inhibitory system. Nat Rev Neurosci. 2004;5:793–807. doi: 10.1038/nrn1519. doi: http://www.nature.com/nrn/journal/v5/n10/suppinfo/nrn1519_S1.html. [DOI] [PubMed] [Google Scholar]
- 68.Brown JA, et al. Inhibition of parvalbumin-expressing interneurons results in complex behavioral changes. Mol Psychiatry. 2015 doi: 10.1038/mp.2014.192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Boley AM, Perez SM, Lodge DJ. A fundamental role for hippocampal parvalbumin in the dopamine hyperfunction associated with schizophrenia. Schizophrenia research. 2014;157:238–243. doi: 10.1016/j.schres.2014.05.005. doi: http://dx.doi.org/10.1016/j.schres.2014.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Nguyen R, et al. Parvalbumin and GAD65 Interneuron Inhibition in the Ventral Hippocampus Induces Distinct Behavioral Deficits Relevant to Schizophrenia. The Journal of Neuroscience. 2014;34:14948–14960. doi: 10.1523/jneurosci.2204-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Tamminga CA, Southcott S, Sacco C, Wagner AD, Ghose S. Glutamate Dysfunction in Hippocampus: Relevance of Dentate Gyrus and CA3 Signaling. Schizophrenia Bulletin. 2012 doi: 10.1093/schbul/sbs062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Akbarian S, Kim JJ, Potkin SG, et al. GEne expression for glutamic acid decarboxylase is reduced without loss of neurons in prefrontal cortex of schizophrenics. Arch Gen Psychiatry. 1995;52:258–266. doi: 10.1001/archpsyc.1995.03950160008002. [DOI] [PubMed] [Google Scholar]
- 73.Guidotti A, Auta J, Davis JM, et al. Decrease in reelin and glutamic acid decarboxylase67 (gad67) expression in schizophrenia and bipolar disorder: A postmortem brain study. Arch Gen Psychiatry. 2000;57:1061–1069. doi: 10.1001/archpsyc.57.11.1061. [DOI] [PubMed] [Google Scholar]
- 74.Hashimoto T, et al. Gene Expression Deficits in a Subclass of GABA Neurons in the Prefrontal Cortex of Subjects with Schizophrenia. The Journal of Neuroscience. 2003;23:6315–6326. doi: 10.1523/JNEUROSCI.23-15-06315.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Volk DW, Austin MC, Pierri JN, Sampson AR, Lewis DA. DEcreased glutamic acid decarboxylase67 messenger rna expression in a subset of prefrontal cortical γ-aminobutyric acid neurons in subjects with schizophrenia. Arch Gen Psychiatry. 2000;57:237–245. doi: 10.1001/archpsyc.57.3.237. [DOI] [PubMed] [Google Scholar]
- 76.Hashimoto T, et al. Alterations in GABA-related transcriptome in the dorsolateral prefrontal cortex of subjects with schizophrenia. Mol Psychiatry. 2007;13:147–161. doi: 10.1038/sj.mp.4002011. doi: http://www.nature.com/mp/journal/v13/n2/suppinfo/4002011s1.html. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Wichterle H, Turnbull DH, Nery S, Fishell G, Alvarez-Buylla A. In utero fate mapping reveals distinct migratory pathways and fates of neurons born in the mammalian basal forebrain. Development. 2001;128:3759–3771. doi: 10.1242/dev.128.19.3759. [DOI] [PubMed] [Google Scholar]
- 78.Anderson SA, Marin O, Horn C, Jennings K, Rubenstein JL. Distinct cortical migrations from the medial and lateral ganglionic eminences. Development. 2001;128:353–363. doi: 10.1242/dev.128.3.353. [DOI] [PubMed] [Google Scholar]
- 79.Butt SJB, et al. The Temporal and Spatial Origins of Cortical Interneurons Predict Their Physiological Subtype. Neuron. 2005;48:591–604. doi: 10.1016/j.neuron.2005.09.034. doi: http://dx.doi.org/10.1016/j.neuron.2005.09.034. [DOI] [PubMed] [Google Scholar]
- 80.Xu Q, Wonders CP, Anderson SA. Sonic hedgehog maintains the identity of cortical interneuron progenitors in the ventral telencephalon. Development. 2005;132:4987–4998. doi: 10.1242/dev.02090. [DOI] [PubMed] [Google Scholar]
- 81.Valcanis H, Tan SS. Layer Specification of Transplanted Interneurons in Developing Mouse Neocortex. The Journal of Neuroscience. 2003;23:5113–5122. doi: 10.1523/JNEUROSCI.23-12-05113.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Inan M, Welagen J, Anderson SA. Spatial and Temporal Bias in the Mitotic Origins of Somatostatin- and Parvalbumin-Expressing Interneuron Subgroups and the Chandelier Subtype in the Medial Ganglionic Eminence. Cereb Cortex. 2012;22:820–827. doi: 10.1093/cercor/bhr148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Fairen A, Cobas A, Fonseca M. Times of generation of glutamic acid decarboxylase immunoreactive neurons in mouse somatosensory cortex. J Comp Neurol. 1986;251:67–83. doi: 10.1002/cne.902510105. [DOI] [PubMed] [Google Scholar]
- 84.Wonders CP, et al. A spatial bias for the origins of interneuron subgroups within the medial ganglionic eminence. Dev Biol. 2008;314:127–136. doi: 10.1016/j.ydbio.2007.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Xu Q, et al. Sonic Hedgehog Signaling Confers Ventral Telencephalic Progenitors with Distinct Cortical Interneuron Fates. Neuron. 2010;65:328–340. doi: 10.1016/j.neuron.2010.01.004. doi: http://dx.doi.org/10.1016/j.neuron.2010.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Xu Q, Cobos I, De La Cruz E, Rubenstein JL, Anderson SA. Origins of Cortical Interneuron Subtypes. The Journal of Neuroscience. 2004;24:2612–2622. doi: 10.1523/jneurosci.5667-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Butt SJB, et al. The Requirement of Nkx2-1 in the Temporal Specification of Cortical Interneuron Subtypes. Neuron. 2008;59:722–732. doi: 10.1016/j.neuron.2008.07.031. doi: http://dx.doi.org/10.1016/j.neuron.2008.07.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Wonders CP, Anderson SA. The origin and specification of cortical interneurons. Nat Rev Neurosci. 2006;7:687–696. doi: 10.1038/nrn1954. [DOI] [PubMed] [Google Scholar]
- 89.Alvarez-Dolado M, et al. Cortical Inhibition Modified by Embryonic Neural Precursors Grafted into the Postnatal Brain. The Journal of Neuroscience. 2006;26:7380–7389. doi: 10.1523/jneurosci.1540-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Watanabe K, et al. Directed differentiation of telencephalic precursors from embryonic stem cells. Nat Neurosci. 2005;8:288–296. doi: 10.1038/nn1402. [DOI] [PubMed] [Google Scholar]
- 91.Maroof AM, Brown K, Shi S-H, Studer L, Anderson SA. Prospective Isolation of Cortical Interneuron Precursors from Mouse Embryonic Stem Cells. The Journal of Neuroscience. 2010;30:4667–4675. doi: 10.1523/jneurosci.4255-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Tyson JA, et al. Duration of culture and sonic hedgehog signaling differentially specify PV versus SST cortical interneuron fates from embryonic stem cells. Development. 2015;142:1267–1278. doi: 10.1242/dev.111526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Yizhar O, et al. Neocortical excitation/inhibition balance in information processing and social dysfunction. Nature. 2011;477:171–178. doi: 10.1038/nature10360. doi: http://www.nature.com/nature/journal/v477/n7363/abs/nature10360.html - supplementary-information. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Birrell JM, Brown VJ. Medial frontal cortex mediates perceptual attentional set shifting in the rat. J Neurosci. 2000;20:4320–4324. doi: 10.1523/JNEUROSCI.20-11-04320.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Garey RE. PCP (phencyclidine): an update. J Psychedelic Drugs. 1979;11:265–275. doi: 10.1080/02791072.1979.10471408. [DOI] [PubMed] [Google Scholar]
- 96.Tanaka DH, Toriumi K, Kubo K-i, Nabeshima T, Nakajima K. GABAergic Precursor Transplantation into the Prefrontal Cortex Prevents Phencyclidine-Induced Cognitive Deficits. The Journal of Neuroscience. 2011;31:14116–14125. doi: 10.1523/jneurosci.2786-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Lodge DJ, Grace AA. Developmental pathology, dopamine, stress, and schizophrenia. Int J Dev Neurosci. 2011;29:207–213. doi: 10.1016/j.ijdevneu.2010.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Flagstad P, et al. Disruption of Neurogenesis on Gestational Day 17 in the Rat Causes Behavioral Changes Relevant to Positive and Negative Schizophrenia Symptoms and Alters Amphetamine-Induced Dopamine Release in Nucleus Accumbens. Neuropsychopharmacology : official publication of the American College of Neuropsychopharmacology. 2004;29:2052–2064. doi: 10.1038/sj.npp.1300516. [DOI] [PubMed] [Google Scholar]
- 99.Perez SM, Boley AM, Lodge DJ. Improved Cognitive Flexibility and Increased Social Interaction Observed in a Rodent Model of Schizophrenia Following mPFC Interneuron Transplants. Article in revision. 2015 [Google Scholar]
- 100.Lodge DJ, Behrens MM, Grace AA. A Loss of Parvalbumin-Containing Interneurons Is Associated with Diminished Oscillatory Activity in an Animal Model of Schizophrenia. J Neurosci. 2009;29:2344–2354. doi: 10.1523/jneurosci.5419-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Perez SM, Lodge DJ. Hippocampal interneuron transplants reverse aberrant dopamine system function and behavior in a rodent model of schizophrenia. Mol Psychiatry. 2013;18:1193–1198. doi: 10.1038/mp.2013.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Donegan JJ, et al. Stem cell derived interneuron transplants as a treatment for schizophrenia: preclinical validation in a rodent model. Mol Psychiatry. doi: 10.1038/mp.2016.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Weiner I, Arad M. Using the pharmacology of latent inhibition to model domains of pathology in schizophrenia and their treatment. Behav Brain Res. 2009;204:369–386. doi: 10.1016/j.bbr.2009.05.004. doi: http://dx.doi.org/10.1016/j.bbr.2009.05.004. [DOI] [PubMed] [Google Scholar]
- 104.Dias R, Robbins TW, Roberts AC. Dissociation in prefrontal cortex of affective and attentional shifts. Nature. 1996;380:69–72. doi: 10.1038/380069a0. [DOI] [PubMed] [Google Scholar]
- 105.Clarke HF, Hill GJ, Robbins TW, Roberts AC. Dopamine, but not serotonin, regulates reversal learning in the marmoset caudate nucleus. The Journal of Neuroscience. 2011;31:4290–4297. doi: 10.1523/jneurosci.5066-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Maroof Asif M, et al. Directed Differentiation and Functional Maturation of Cortical Interneurons from Human Embryonic Stem Cells. Cell Stem Cell. 2013;12:559–572. doi: 10.1016/j.stem.2013.04.008. doi: http://dx.doi.org/10.1016/j.stem.2013.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Germain ND, Banda EC, Becker S, Naegele JR, Grabel LB. Derivation and isolation of NKX2.1-positive basal forebrain progenitors from human embryonic stem cells. Stem Cells Dev. 2013;22:1477–1489. doi: 10.1089/scd.2012.0264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Goulburn AL, Stanley EG, Elefanty AG, Anderson SA. Generating GABAergic cerebral cortical interneurons from mouse and human embryonic stem cells. Stem Cell Research. 2012;8:416–426. doi: 10.1016/j.scr.2011.12.002. doi: http://dx.doi.org/10.1016/j.scr.2011.12.002. [DOI] [PubMed] [Google Scholar]
- 109.Steinbeck Julius A, Studer L. Moving Stem Cells to the Clinic: Potential and Limitations for Brain Repair. Neuron. 2015;86:187–206. doi: 10.1016/j.neuron.2015.03.002. doi: http://dx.doi.org/10.1016/j.neuron.2015.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Politis M, et al. Serotonergic neurons mediate dyskinesia side effects in Parkinson’s patients with neural transplants. Sci Transl Med. 2010;2:38ra46. doi: 10.1126/scitranslmed.3000976. [DOI] [PubMed] [Google Scholar]