

Abstract
Mesenchymal stromal cells (MSCs) can modulate inflammation and contribute to tissue regeneration and, thus, have emerged as a promising option for cell-based therapy. However, the ability of MSCs to migrate to injured tissues still needs to be improved. In this study, we investigated whether genetically engineered MSCs could exhibit increased migratory properties and improved therapeutic efficacy. Using a mouse model of contact hypersensitivity (CHS), chemokine gene expression screening revealed that CXCL13 changed most significantly in injured tissue. Unfortunately, MSCs hardly express the corresponding receptor, CXCR5. Thus, CXCR5-overexpressing MSCs (MSCCXCR5) were generated that retained their abilities of proliferation, differentiation, and immunomodulation. Furthermore, MSCCXCR5 showed significantly increased migrating ability toward CXCL13. Importantly, systemic infusion of MSCCXCR5 dramatically suppressed CHS in mice, as evidenced by decreased levels of inflammatory cell infiltration and pro-inflammatory cytokine production. Numerous MSCCXCR5 migrated into inflamed ears, localized with T cells, inhibited T cell proliferation, promoted T cell apoptosis, and suppressed the production of T cell-derived pro-inflammatory factors. Collectively, these findings demonstrate that CXCR5 overexpression increases the ability of MSCs to respond to migratory stimuli and highly intensifies their immunomodulatory effects in vivo. This strategy for enhancing targeted stem/progenitor cell homing may improve the efficacy of MSC-based therapies.
Keywords: CXCR5, mesenchymal stromal cells, homing, immunomodulation, contact hypersensitivity
Graphical Abstract
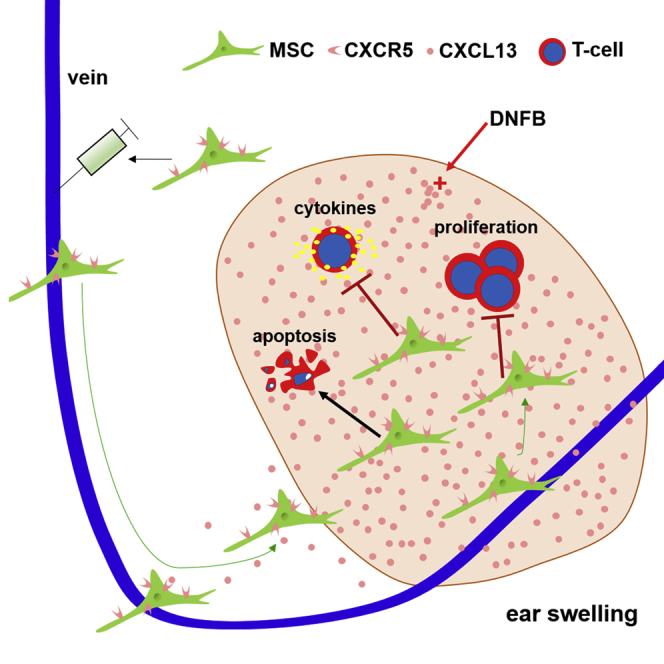
Xiang et al. describe that genetically engineered MSCs with CXCR5 significantly increase their migration to diseased organs/tissues and improve therapeutic efficacy in a mouse model of contact hypersensitivity. This strategy for enhancing targeted stem/progenitor cell homing may provide a potential strategy to improve the efficacy of MSC-based therapeutics.
Introduction
Mesenchymal stromal cells (MSCs) have attracted great interest for cell therapy because of their self-renewal capacity,1 multipotency,2 and potent immunomodulatory effects on both innate and adaptive immune cells.3, 4, 5, 6, 7 Although numerous preclinical and clinical studies have shown that MSCs can be therapeutically relevant for a variety of inflammatory and autoimmune diseases, certain obstacles still limit the translation of stem cell therapy into practice.8, 9, 10 For example, the therapeutic efficacy of MSCs largely depends on their ability to migrate to injured tissues.11, 12 After intravenous injection, MSCs typically distribute to the lungs and are detected at only low frequencies in injured tissues.13, 14 To overcome this limitation, we need to understand the molecular and cellular mechanisms that form the basis for MSC trafficking under physiological and pathological conditions.
For MSCs to home to a specific (i.e., injured) tissue, they require the right combination and interactions of tissue-secreted chemokines and the corresponding chemokine receptors on MSCs. MSCs show relatively good homing when freshly isolated, but they exhibit decreased homing capacities after ex vivo expansion.15 For instance, the CXCR4 chemokine receptor, which recognizes CXCL12 (also termed SDF-1α), is highly expressed on bone marrow MSCs but is lost upon culturing.15, 16 There are also other examples of mismatches between tissues and MSCs, such as when specific chemokines are upregulated in infarcted myocardium but the expression levels of the corresponding chemokine receptors (e.g., CCR1 and CXCR2) on MSCs remain very low.17 Therefore, several studies have sought to genetically modify MSCs with specific chemokine receptors needed for efficient homing in an effort to enhance their targeting ability. As an example, Bobis-Wozowicz et al.18 found that overexpression of CXCR4 significantly increased the motility, invasiveness, and homing of MSCs to the bone marrow of non-obese diabetic (NOD)/severe combined immunodeficiency (SCID) mice. Moreover, CCR7-modified MSCs showed increased migration to secondary lymphoid organs (SLOs) and remarkably alleviated murine graft versus host disease (GvHD).19 MSCs overexpressing CCR1 were associated with a significant reduction in infarct size, reduced cardiomyocyte apoptosis, and increased capillary density in injured myocardium.17 Importantly, different types of injured organs secrete specific inflammatory cytokines and chemokines.20, 21, 22 Therefore, elucidating the interactions between tissue-specific chemokines and the corresponding receptors on MSCs should provide new strategies for improving the homing and therapeutic efficacy of these cells.
Contact hypersensitivity (CHS), which is a T cell-mediated antigen-specific skin inflammation induced by skin exposure of sensitized mice to haptens, is an experimental model for human allergic contact dermatitis (ACD).23, 24, 25 Although previous studies demonstrated that MSCs could alleviate CHS,26, 27 their therapeutic efficacy still needs to be improved; for example, by enhancing their homing ability.
Here we hypothesized that genetically modifying MSCs to enhance the levels of specific chemokine receptors should improve the engraftment of such cells to damaged tissues, thereby improving their therapeutic effects in the mouse model of CHS.
Results
CXCL13 Is Highly Upregulated in Inflamed Ears of CHS mice
The murine CHS model consists of three stages: the sensitization stage, the challenge stage, and the resolution/regulation stage.25 We compared the mRNA expression levels of many common skin inflammation-related chemokines, such as CCL2, CCL8, CCL17, CCL27, CXCL2, CXCL10, and CXCL12, in the ears of unchallenged mice with the CHS-inflamed ears of challenged mice.24, 28 CCL19, CCL21, and CXCL13 were of interest to examine because recent studies demonstrated that they are strongly correlated with skin inflammation.28, 29, 30, 31
Figure 1A shows the fold changes of CCL2, CCL8, CCL17, CCL27, CXCL2, CXCL10, CXCL12, CCL19, CCL21, and CXCL13 in inflamed ears (2 days post-challenge, when the ear edema/spongiosis peaked) compared with unchallenged ears.24 The results indicated that the CXCL13 mRNA expression level was almost 100 times higher in inflamed ears and exhibited the most significant change among the tested chemokines (Figure 1A). Western blotting and in situ immunofluorescence staining confirmed that CXCL13 was intensely upregulated in the inflamed ears of CHS mice (Figures 1B and 1C). Furthermore, time course analysis during the challenge stage showed that CXCL13 expression peaked on day 2 post-challenge and then decreased gradually, and the changed trend was consistent with the clinical symptoms (Figure 1D).
Figure 1.

CXCL13 Expression in the Inflamed Ears of CHS Mice
(A) The expression levels of various chemokines involved in CHS were analyzed by qRT-PCR of mRNA samples extracted from the normal and inflamed ears of three independent mice per condition. Data are presented as the mean ± SEM for each group. Fold change represents the expression of each chemokine in inflamed ears on day 2 post-challenge compared with control ears. (B) Total tissue lysates of normal and inflamed ears were subjected to western blot analysis of mouse CXCL13 expression. The experiment was repeated three times with tissues isolated from independent mice; a representative blot is shown. (C) Representative confocal images of normal and inflamed ear sections stained for CXCL13 (red) on day 2 post-challenge. The experiment was repeated three times with tissues isolated from independent mice; a representative image is shown. Nuclei were visualized by DAPI staining (blue). Scale bars, 100 μm. (D) CXCL13 expression was analyzed by qRT-PCR, and mRNA was extracted from both normal and inflamed ears of three independent mice from day 0 to day 3 post-challenge (mean ± SEM). GAPDH was detected as an internal control for mRNA. In inflamed ears, CXCL13 expression peaks 48 hr post-challenge and then decreases.
Chemokine Receptors Exhibit Low-Level Expression on Human MSCs
Lesion-secreted chemokines and their MSC-expressed receptors might play important roles in regulating the homing of MSCs to inflamed tissues. Accordingly, we analyzed the mRNA expression of common CC chemokine receptors (CCR1–10) and CXC chemokine receptors (CXCR1–7) through genome-wide RNA sequencing (RNA-seq) of MSCs from three different donors (global transcriptional profiling data to be published elsewhere). As shown in Figure 2A, human MSCs at the fifth passage expressed extremely low chemokine receptors at the mRNA level. These results were confirmed by qRT-PCR analysis of additional human MSC samples from six donors (Figure 2B). Because we observed intense upregulation of CXCL13 in the inflamed ears of CHS mice, we examined the expression of its receptor, CXCR5, on MSCs specifically. The qRT-PCR results showed that the expression of CXCR5 was feeble in MSCs compared with human peripheral blood mononuclear cells (hPBMCs) (Figure 2C). Likewise, cell surface CXCR5 protein expression on MSCs was virtually undetectable when analyzed by flow cytometric analyses at passage 5 (Figure 2D). Collectively, these results indicate that chemokine receptors exhibit low-level expression on long-term culture human MSCs, further suggesting that MSCs might not efficiently home to the injured target tissue through the chemokine/chemokine-receptor axis.
Figure 2.

Expression of Chemokine Receptors on Human Bone Marrow-Derived MSCs
(A) Histograms represent the RNA-seq-derived expression profiles of CC and CXC chemokine receptors on fifth-passage MSCs of three independent donors (mean ± SEM). MSC surface markers (CD13, CD29, CD44, CD73, CD105, and CD90) were detected as a positive control. (B) The mRNA expression levels of CC and CXC chemokine receptors from fifth-passage MSCs of additional six independent donors were analyzed by qRT-PCR (mean ± SEM). GAPDH was detected as an internal control for mRNA. (C) qRT-PCR analysis was used to detect CXCR5 mRNA expression levels in MSCs and hPBMCs (a positive control) (mean ± SEM). GAPDH was detected as an internal control for mRNA. (D) Flow cytometric analysis was used to detect CXCR5 proteins on the cell surfaces of MSCs from six independent donors. hPBMCs served as a positive control. All experiments were repeated at least three times.
CXCR5-Overexpressing MSCs Retain the Characteristics of hMSCs
To determine whether overexpression of CXCR5 chemokine receptors in MSCs augmented their migration ability, MSCs were transduced with lentiviral vectors encoding CXCR5 (referred to as MSCCXCR5) or EGFP (referred to as MSCEGFP) (Figures S1A and S1B). We detected significantly higher expression of CXCR5 at the mRNA, protein, and cell surface levels of MSCCXCR5 compared with those of MSCEGFP (Figures 3A–3C). The MSCCXCR5 and MSCEGFP were all EGFP-positive and isolated by fluorescence-activated cell sorting (FACS) as described in Materials and Methods.
Figure 3.

Overexpression of CXCR5 in MSCs and the Effects of MSCEGFP and MSCCXCR5 on CD3+ T Cells In Vitro
(A) MSCs were transfected with the CXCR5-encoding or control gene, and qRT-PCR was used to analyze the mRNA expression of the CXCR5 transcript in MSCEGFP and MSCCXCR5. The experiment was repeated three times. Data are presented as the mean ± SEM for each group. (B) Total cell lysates of MSCEGFP and MSCCXCR5 were analyzed by western blotting of CXCR5 and EGFP proteins. The experiment was performed three times with tissues isolated from independent mice; a representative blot is shown. (C) Flow cytometry was used to analyze the levels of the CXCR5 protein on the surfaces of MSCEGFP and MSCCXCR5. The experiment was repeated three times. (D) T cells were cultured with or without MSCEGFP and MSCCXCR5 for 3 days, and their proliferation was analyzed by flow cytometry, whereas their production of TNF-α and IFN-γ was examined by flow cytometry. The results are representative of three independent experiments. Bar graphs show percentage inhibition of the frequency of CD3+ T cells and TNF-α- and IFN-γ-producing CD3+ T cells after coculture with MSCEGFP and MSCCXCR5. Cumulative results are expressed as mean ± SEM. Significant differences are indicated as follows: **p < 0.01 and ***p < 0.001.
To examine whether CXCR5 modification affected the characteristics of MSCs, we first used flow cytometry to analyze MSC surface markers. Compared with MSCEGFP, MSCCXCR5 expressed the same panel of surface markers, including CD29, CD44, CD73, CD90, CD105, and CD166, and did not express the hematopoietic stem cell markers CD34 or CD45, indicating that transduced cells maintained the phenotype of MSCs (Figures S1C and S1D). To demonstrate the multipotency of MSCCXCR5, we cultured cells under conditions that promote differentiation into osteogenic, adipogenic, or chondrogenic lineages. As confirmed by alizarin red S staining, oil red O staining, or Aggrecan staining, respectively, MSCCXCR5 showed no change in mesodermal differentiation capacity compared with MSCEGFP (Figures S1E and S1F). The transgenic MSCs do not undesirably alter the intrinsic characteristic of MSCs.
To investigate the immunoregulatory abilities of MSCs after transgenic manipulation, we detected the effects of MSCCXCR5 and MSCEGFP on the proliferation and pro-inflammatory cytokine production of CD3+ T cells. As shown in Figure 3D, both MSCCXCR5 and MSCEGFP significantly inhibited the proliferation of CD3+ T cells (83.09% ± 3% versus 25.05% ± 9.53%, 24.19% ± 5.53%; p < 0.001) and suppressed the percentages of tumor necrosis factor α (TNF-α)−producing (35.2% ± 8.18% versus 5.55% ± 3.5%, 5.01% ± 2.11%; p < 0.001) and interferon γ (IFN-γ)-producing (15.06% ± 5.12% versus 7.1% ± 2.08%, 7.13% ± 1.28%; p < 0.01) CD3+ T cells, respectively. Collectively, these results indicate that CXCR5 overexpression did not influence the intrinsic characteristics of MSCs.
MSCCXCR5 Exhibit Enhanced Migration toward Human and Murine CXCL13 In Vitro
We next investigated whether CXCR5-modified MSCs exhibited increased migration toward the ligand CXCL13. Indeed, a chemotaxis assay showed that MSCCXCR5, but not MSCEGFP, strongly responded to human CXCL13 (hCXCL13) at 5 ng/mL and murine CXCL13 (mCXCL13) at 50 ng/mL (Figure 4A). In the absence of CXCL13, MSCCXCR5 and MSCEGFP showed similar, relatively low levels of non-specific migration (Figure 4A). In addition, significant increases (nearly two times) in the chemotaxis response of MSCCXCR5 were observed over culture time from 5 hr to 10 hr (Figure 4B).
Figure 4.

MSCCXCR5 Exhibit Enhanced Migration toward hCXCL13 and mCXCL13 In Vitro and to Inflamed Ears In Vivo
(A) In vitro migration of MSCCXCR5 and MSCEGFP toward human CXCL13 (hCXCL13) or murine CXCL13 (mCXCL13). Transwell filters were stained with 0.1% crystal violet and observed under bright-field microscopy. Scale bars, 100 μm. (B) Quantification of migrated cells. Data are presented as the mean ± SEM. ***p < 0.001. (C) MSCEGFP and MSCCXCR5, both expressing green fluorescence, were intravenously injected into CHS mice. On days 1, 3, and 5 post-injection, the presence of EGFP-expressing MSCs was examined by cryosectioning. Signals: EGFP, green; DAPI, blue. Scale bars, 100 μm. (D) EGFP-positive cells were quantified per microscopic field of ear cryosections in triplicate mice. Data are presented as the mean ± SEM for each group. The data are representative of three independent experiments. ***p < 0.001 versus the CHS+MSCEGFP group on the corresponding day.
It has been reported that targeted cell movement corresponds to the formation of actin-filled projections.32 The Rho-like GTPase Rac1 reportedly contributes to the actin-related cytoskeletal alterations that may play important roles in cell activation, adhesion, migration, and invasion.33, 34, 35 In culture, MSCCXCR5 were spindle-shaped, and their actins were lined up in a parallel manner (Figure S2A, I). Cells formed lamellipodia protrusions, and more actins were rearranged along the cell membrane following the occurrence of migration (Figure S2A, II). As shown in Figure S2B, mCXCL13 increased the relative number of MSCCXCR5 carrying lamellipodia protrusions after migration. Finally, a PAK-1 pull-down assay, which was used to detect the activation of Rac1, revealed that the presence of mCXCL13 increased the activation of Rac1 in MSCCXCR5 (Figure S2C). To further confirm these observations, we also treated MSCCXCR5 with hCXCL13 and found that both the lamellipodia protrusions and the activation of Rac1 had significantly increased (Figures S2D–S2F).
MSCCXCR5 Exhibit an Enhanced Capacity for Targeted Migration to Inflamed Ears In Vivo
Because MSCCXCR5 showed increased migration toward CXCL13 in vitro, we then investigated whether they could migrate to inflamed ears following intravenous infusion in vivo. MSCCXCR5 and MSCEGFP (1 × 106 per mouse) were injected into CHS mice through the tail vein on day 2 after challenge. The inflamed ears were collected from each group on days 1, 3, and 5 post-injection and subjected to in situ immunofluorescence staining. Our results revealed that MSCEGFP were almost undetectable, whereas MSCCXCR5 were highly accumulated in the inflamed ears of CHS mice (Figures 4C and 4D). These results demonstrate that CXCR5 modification enhances the targeted migration of MSCs to inflamed sites in vivo, presumably by targeting CXCL13. To quantify the retention of the administered MSCCXCR5 and MSCEGFP to the inflamed ears of CHS mice, we detected human genomic DNA by amplifying the human-specific human beta-2 microglobulin (hB2M) sequence and calculated human cells in 1 μg genomic DNA of diseased ears (187.6 ± 9.9 versus 72.7 ± 7.8, p < 0.001). We also use the mRNA level of EGFP as an exogenous gene marker by qRT-PCR to calculate the cell number in 1 μg cDNA of diseased ears (132.6 ± 3.8 versus 40.7 ± 5.3, p < 0.001). Compared with MSCEGFP, two or three times as many MSCCXCR5 were distributed in the inflamed ears (Figures S3A–S3F).
MSCCXCR5 Infusion Dramatically Ameliorates CHS
To examine whether the enhanced migration of MSCCXCR5 to inflamed sites might improve the symptoms of CHS, we injected mice with MSCCXCR5 and MSCEGFP (1 × 106 per mouse) on day 2 post-challenge and measured swelling/inflammation (in terms of external ear thickness) every 24 hr for the next 5 days. As shown in Figure 5A, MSCCXCR5 displayed better treatment efficacy than MSCEGFP, as characterized by their ability to decrease ear thickness and leukocyte infiltration. MSCCXCR5 significantly attenuated CHS as early as 24 hr post-injection (CHS+MSCEGFP versus CHS, p < 0.05; CHS+MSCCXCR5 versus CHS, p < 0.05) and had even greater effects 48 hr post-injection (CHS+MSCCXCR5 versus CHS, p > 0.05; CHS+MSCCXCR5 versus CHS, p < 0.01) (Figure 5B).
Figure 5.

Injection of MSCCXCR5 Attenuates CHS In Vivo
(A) Representative photos and H&E-stained ear samples from mice of each group. 72 hr post-injection, ear tissues were collected, paraffin-embedded, sectioned, and stained with H&E. Photos were taken of at least six sections per tissue. Scale bars, 100 μm. (B) Ear swelling response (mean ± SEM). The degree of swelling was calculated as the thickness of the right ear (challenged ear) minus the baseline thickness of the left ear (unchallenged ear). Control mice were challenged with 0.2% DNFB without prior sensitization. Experimental mice were injected i.v. with 1 × 106 MSCCXCR5 or MSCEGFP on day 2 of CHS. n = 6 mice/group from three independent experiments. (C) MPO activity was examined by colorimetric assay of ear tissue homogenates obtained from each group during DNFB-induced CHS (72 hr post-injection). Data are presented as the mean ± SEM for individual mice. **p < 0.01. (D) The mRNA levels of TNF-α, IFN-γ, IL-17, IL-6, IL-4, and IL-10 were analyzed by qRT-PCR. mRNA was extracted from the inflamed ears of each group 72 hr post-injection. GAPDH was detected as an internal mRNA control. The results are representative of three experiments. Data are presented as the mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001. (E) The concentrations of TNF-α, IFN-γ, IL-17, IL-6, IL-4, and IL-10 in homogenates of inflamed ears obtained from each group 72 hr post-injection were determined by ELISA. Data are presented as the mean ± SEM for each group. n = 6 mice/group from three independent experiments. *p < 0.05, **p < 0.01, ***p < 0.001.
Because myeloperoxidase (MPO) activity can reflect the degree of neutrophil infiltration and thus may be used as an index for evaluating the severity of CHS,36 we examined the MPO activity of ear homogenates obtained from CHS mice 72 hr post-injection. Compared with the control group, MPO activity was notably decreased in the MSCCXCR5 group and differed slightly in the MSCEGFP group (Figure 5C). We also used qRT-PCR and ELISA to examine the levels of pro-inflammatory (IFN-γ, interleukin-17 [IL-17], TNF-α, and IL-6) and anti-inflammatory (IL-4 and IL-10) cytokines in the inflamed ears. We observed significant decreases in the expression levels of pro-inflammatory cytokines in the inflamed ears of the MSCCXCR5 treatment group, whereas only slight changes were seen in the MSCEGFP treatment group versus the control group. The expression of pro-inflammatory and anti-inflammatory cytokines in the MSCCXCR5 treatment group was close to the normal levels in the control group (Figures 5D and 5E). These results demonstrate that MSCCXCR5 are better able to improve the symptoms of CHS, mainly via their ability to suppress pro-inflammatory cytokines.
MSCCXCR5 Localize Near T Cells and Exert Potent Immunosuppressive Effects on These Cells in the Inflamed Ears of CHS Mice
MSCs can modulate inflammation and contribute to tissue regeneration through potent immunomodulatory effects on both innate and adaptive immune cells. Thus, we hypothesized that genetically modifying MSCs to enhance the levels of a specific chemokine receptor (CXCR5) should improve the engraftment of such cells to the inflamed ear, thereby improving their therapeutic effects in the mouse model of CHS. As shown in Figures 4 and 5, compared with MSCEGFP, MSCCXCR5 showed significantly increased migration ability toward CXCL13. More importantly, systemic infusion of MSCCXCR5 dramatically suppressed CHS in mice. Furthermore, many more MSCCXCR5 migrated into the inflamed ears and localized in close proximity with T cells (Figures 6A and 6B). Both of these results may identify the hypothesis that the transduced CXCR5 in MSCs improves their targeting ability to damaged tissues. In addition, frozen inflamed ear sections were fixed and stained for the β chain of the T cell receptor (TCR) complex (red) and costained for CD3 (white), EGFP (green), and nuclei (blue) with specific antibodies. The results demonstrated that more MSCCXCR5 migrated to the inflamed ear and located in close proximity with CD3+/TCR+ T cells. The corresponding data were added as Figure S4A. Moreover, we assessed the colocalization of CD4 or CD8 with CD3 in the sections and found that most of the CD4- or CD8-positive cells were CD3-positive cells. These results further confirmed that the frequencies of CD4+ or CD8+ T cell populations were much lower in the CHS+ MSCCXCR5 group compared with the CHS or CHS+ MSCEGFP groups of mice. The corresponding data are presented in Figure S4B.
Figure 6.

Possible Mechanisms through which MSCCXCR5 Could Attenuate CHS
(A) Sections were obtained from challenged ears 1 day after injections and stained with anti-CD4 (violet) or anti-CD8 (red). EGFP-expressing MSCEGFP and MSCCXCR5 were observed in challenged ears. Nuclei were visualized by DAPI staining (blue). n = 6 mice/group from three independent experiments, and pictures were taken of at least six sections per tissue. (B) Immunohistochemical staining of the challenged ears of CHS mice (left), CHS+MSCEGFP mice (center), and CHS+MSCCXCR5 mice (right). The mice were sacrificed 72 hr post-injection, and challenged ears were sampled, sectioned, and stained with anti-CD4 (violet) or anti-CD8 (red), EGFP (green), and DAPI (blue). n = 6 mice/group from three independent experiments, and pictures were taken of at least six sections per tissue. (C) The percentages of EdU-incorporated cells among the CD4+, CD8+ cell populations were assessed using flow cytometry. The data are representative of three independent experiments. (D) The percentages of Annexin-V-positive cells in the CD4+, CD8+ cell populations were measured by flow cytometry. The data are representative of three independent experiments. (E and F) The cells were stimulated with PMA (50 ng/mL) and ionomycin (500 ng/mL) for 6 hr. During this period, BFA (10 μg/mL) was used to inhibit the secretion of cytokines (all from Sigma-Aldrich), and the IL-17/IFN-γ-positive CD4+, CD8+ T cells were analyzed by flow cytometry. The numbers indicate the percentages of positive cells in the gated CD4+ or CD8+ T cell populations. The data are representative of three independent experiments.
We then tried to study the potential mechanisms of the underlying cellular mechanism. CHS is a T cell-mediated immune response that is induced by topical skin immunization with small molecules (i.e., haptens). Allergen-specific effector CD4+ and CD8+ T cells are activated and produce a plethora of inflammatory cytokines and mediators that contribute to the appearance of eczematous lesions. Previous studies have demonstrated that MSCs can inhibit T cell proliferation, induce T cell apoptosis, and decrease the release of pro-inflammatory cytokines by T cells.37, 38 Therefore, we asked whether more MSCCXCR5 migrated to damaged tissue could affect T cell numbers and functions and show more obvious therapeutic effects against CHS in vivo. Thus, we used ethynyl-2′-deoxyuridine (EdU) incorporation to detect T cell proliferation and found that the CHS+MSCCXCR5 group exhibited the most apparent decreased proliferation of CD4+/EdU+ and CD8+/EdU+ T cells compared with the CHS+MSCEGFP group and the CHS group (Figure 6C). Also, to quantitatively analyze apoptotic cell death, total cells isolated from the inflamed ears were stained with Annexin V, anti-CD4, and anti-CD8 and performed FACS analysis. Compared with the CHS and MSCEGFP groups, MSCCXCR5 significantly increased the apoptosis percentages of CD4+ and CD8+ T cells (Figure 6D).
Finally, consistent with reports that T cell-derived cytokines, especially IL-17 and IFN-γ, may play important roles in the progress of CHS,39, 40 we found that the proportions of IL-17- or IFN-γ-producing CD4+ and CD8+ T cells were reduced in the CHS+MSCCXCR5 group 24 hr post-infusion relative to the other groups (Figures 6E and 6F).
Taken together, compared with MSCEGFP, more MSCCXCR5 migrated into the inflamed ears, localized close to T cells, inhibited T cell proliferation, promoted T cell apoptosis, and suppressed the production of T cell-derived pro-inflammatory factors. These findings demonstrate that CXCR5 overexpression increases the ability of MSCs to respond to migratory stimuli and highly intensifies their immunomodulatory effects in vivo.
Discussion
In this study, we identified CXCL13 as being highly expressed in the inflamed ears of our CHS model and transduced CXCR5 into MSCs in an effort to improve their targeting ability. Indeed, we found that systemic infusion of MSCCXCR5 dramatically attenuated CHS in the mouse model.
Because of their multipotency, immunomodulatory properties, and ease of isolation and ex vivo expansion, MSCs have become promising candidates for cellular therapy and regenerative medicine.41, 42 However, inadequate engraftment of the cells to the tissue of injury limits the therapeutic efficacy of MSCs. It has been shown that, with higher passage number, the engraftment efficiency of MSCs decreases.15 In addition, it is believed that the therapeutic effects of MSCs depend mainly on cell-to-cell interaction and/or juxtacrine activity. Therefore, the therapeutic potential of injected MSCs may be closely bound up with their localization. It is anticipated that strategies that enhance local recruitment will improve the effectiveness of cellular therapy through acceleration of tissue recovery.43, 44 However, the predominant primary signaling pathways that orchestrate local engraftment of MSCs remain to be fully elucidated.
Chemokines and their receptors have been identified as major mediators for cell migration and homing to target tissues. Therefore, injection of genetically manipulated MSCs that overexpress chemokine receptors may provide a novel strategy for improvement of stem cell homing. Previous studies also demonstrated that genetic modifications of MSCs with chemokine receptors such as CXCR445, 46 and integrin-alpha447 significantly increased the mobilization of the cell to the site of tissue injury and enhanced the therapeutic efficacy of MSCs. Although the CXCR4/CXCL12 axis has emerged as an important regulator of cell migration, it seems that this might not be a unique axis to enhance MSC homing. For example, human MSCs overexpressing CXCR4 did not improve cell migration, indicating that there might be other mechanisms participating in MSC engraftment.48 For MSCs to home to a target tissue, they require the right combination of interactions of chemokines secreted by the injured tissue and the corresponding chemokine receptors on MSCs. The chemokine expression profiles of lesions may vary with different organs and diseases and be regulated temporally and spatially during the disease process. Hence, elucidating the chemokine expression profiles of specific disease microenvironments will provide exact clues for MSC homing. Here we reported that the CXCR5/CXCL13 axis is the disease-specific regulator for MSC homing in CHS. CXCL13 is a highly conserved chemokine expressed constitutively in secondary lymphoid tissues and is reported to be a homeostatic chemokine involved in lymphoid organogenesis.49 CXCL13 could be also observed in many inflammatory tissues, including lesions of the skin.31 Indeed, we found that CXCL13 acts as a prominent feature in the challenge phase of CHS in the inflamed ear that guides the migration of CXCR5-expressing MSCs to lesions in vivo. However, the expression of CXCR5 was low in MSCs. Thus, CXCR5-modified MSCs may serve as a disease-specific therapeutic strategy to enhance MSC homing for treating CHS.
CHS is a T cell-mediated immune response that is induced by topical skin immunization with small molecules (i.e., haptens). It typically peaks 24–48 hr post-challenge with the symptoms of cutaneous antigen-primed T cell infiltration and edema/spongiosis.24 In the elicitation phase, various types of innate immune cells, particularly allergen-specific effector CD8+ and CD4+ T cells, are activated and produce a plethora of inflammatory cytokines and mediators that contribute to the appearance of eczematous lesions. Recently, He et al.40 have claimed that IFN-γ and IL-17 mediate the elicitation of CHS by different mechanisms and that both cytokines are required for optimal responses. In this study, we found that more CXCR5-modified MSCs aggregated near T cells at the elicitation sites of CHS mice. Moreover, the results from in vitro and in vivo studies indicated that MSCs could suppress T cell responses; for example, inhibit T cell proliferation and suppress the production of the T cell-derived pro-inflammatory factors IFN-γ and IL-17. In addition, engrafted MSCs at the elicitation sites could promote apoptosis of invasive T cells in the inflamed ear. This may explain how the modified MSCs showed more obvious therapeutic effects against CHS in vivo.
In summary, here we performed ex vivo genetic manipulation of MSCs and found that CXCR5 overexpression strongly enhances the migration, engraftment into the injured tissue, and in vivo immunomodulatory effects of MSCs. Although the therapeutic potential of MSCs has not yet been fully translated into the clinic, this work advances our understanding of the chemokine/receptor axis and stem/progenitor cell biology and may offer a new strategy for enabling MSCs to be quickly and precisely delivered to disease sites, thereby maximizing their therapeutic benefits.
Materials and Methods
Mice
BALB/c male mice were purchased from the Animal Center of Sun Yat-sen University. All animal studies were carried out in accordance with the guidelines of the Sun Yat-sen University Institutional Animal Care and Use Committee.
RNA Library Preparation and Illumina Sequencing
MSC samples from three different donors were used for the genome-wide RNA-seq analysis. Then, RNA library preparation and sequencing were performed as recommended by the manufacturer (Genome Analyzer Iix, Illumina). Sequencing data were processed using Consensus Assessment of Sequence and Variation (CASAVA, version 1.8.2, Illumina) using the default settings. In brief, clusters were located using the raw images, cluster intensity and position parameters were obtained as output, and the noise for each cluster was estimated. The program determines the base sequences read from each cluster, the confidence level for each base, and whether the read passed filtering. The resulting bcl files were converted into fastq.gz files. Sequence reads were mapped to transcripts annotated in the NCBI database and used to calculate overall gene expression in terms of reads per kilobase of exon per million mapped reads (RPKM). The global transcriptional profiling data will be published elsewhere.
Isolation and Characterization of MSCs
Human bone marrow samples were obtained from healthy donors along with their informed consent. MSCs were isolated from the bone marrow and cultured as described previously.42 In brief, 20–30 mL of bone marrow was diluted 1:1 with human MSC growth medium consisting of low-glucose DMEM (L-DMEM, HyClone) and 10% fetal bovine serum (FBS, HyClone). Marrow mononuclear cells were separated by Ficoll-Paque (1.077 g/mL, Amersham Biosciences) density gradient centrifugation and seeded at a density of 1 × 105/cm2 into T75 cell culture flasks. At 80% confluence, the cells were detached by 0.25% trypsin-EDTA and designated as passage 1. The cells were further passaged at a ratio of 1:3. The culture-expanded MSCs exhibited surface expression of CD29, CD44, CD73, CD90, CD105, and CD166 (MSC markers) but not CD34 or CD45 (hematopoietic markers). After the fourth passage, the multiple differentiation capacity of the MSCs was confirmed by their forced differentiation to osteoblasts, chondrocytes, or adipocytes, which was performed as described previously.50, 51, 52
RNA Isolation, Reverse Transcription, and Real-Time qRT-PCR
Total RNA was extracted using an RNeasy mini kit (QIAGEN) according to the manufacturer’s protocol. Reverse transcription was performed using oligo-dT primers (Fermentas) and quantitative real-time qRT-PCR was performed using SYBR PCR Master Mix (Toyobo) according to the manufacturer’s instructions. qRT-PCR was conducted in duplicate for each sample, and three independent experiments were performed. Signals were detected using a Light Cycler 480 detection system (Roche). The primer sequences are listed in Table S1.
Western Blot Analysis
Cell and tissue lysates were prepared and centrifuged at 4°C. The proteins were separated by SDS-PAGE and transferred to a polyvinylidene fluoride (PVDF) membrane. The membrane was blocked, incubated with rabbit anti-CXCR5 antibody (Santa Cruz Biotechnology) or goat anti-CXCL13 antibody (R&D Systems), and then incubated at room temperature with secondary antibodies. Antigen-antibody complexes were detected by enhanced chemiluminescence (GE Healthcare).
Immunofluorescence Staining
At the indicated time points, tissues were harvested, fixed with 4% paraformaldehyde (Sigma), washed in PBS, and dehydrated overnight in 30% sucrose (Sigma). The samples were embedded in Tissue-Tek OCT compound (Bio-Optica) and frozen in an ethanol dry ice bath. Sections (7 mm thick) were placed on glass slides (Bio-Optica), blocked for 30 min with PBS-Tween 0.05% plus 0.5% FBS, and then incubated with the appropriate primary and secondary antibodies (Tables S2 and S3; diluted in blocking solution) overnight at 4°C and for 30 min at room temperature, respectively. Nuclei were visualized with DAPI (Fluka). Images were acquired under fluorescence or confocal microscopy. The primary antibodies and secondary antibodies are listed in Tables S2 and S3.
Lentiviral Vector Construction and Transduction
Entry vectors were generated by flanking the EF1α promoter, CXC motif chemokine receptor 5 (CXCR5), internal ribosomal entry site (IRES)-EGFP) gene with attB4/B1r, attB1/B2, and attB2r/B3 sites, respectively, by PCR (Figure S5A). The promoter PCR product was cloned into pDONR P4-P1r (Invitrogen) using the Gateway BP recombination method following the manufacturer’s instructions. The att-flanked CXCR5 fragment and IRES-EGFP fragment were cloned into pDONR 221 and pDONR P2r-P3 (Invitrogen) using the same method (Figure S5B). The resulting vectors, termed pUp-EF1a, pDown-CXCR5, and pTail-IRES-EGFP, were next recombined into the pDest-puro vector using a recognized LR recombination reaction protocol described in the Gateway LR kit and a clonase enzyme mix (Invitrogen) (Figure S5C). The final lentiviral expression vector was designated pLV/puro-EF1α-CXCR5-IRES-EGFP. The vector pLV/puro-EF1α-EGFP was constructed in a similar way (Figures S6A–S6C).
Lentiviruses were prepared by transient cotransfection of 293FT cells with the pLV/puro-EF1α-CXCR5-EGFP or pLV/puro-EF1α-EGFP construct together with Vira Power lentiviral packaging mix (Invitrogen) using Lipofectamine 2000 (Invitrogen). Three days after transfection, supernatants containing viral particles were harvested, filtered through polyether sulfone membranes (pore size, 0.45 μm), and titered.
For lentiviral transduction, MSCs were dissociated into single-cell suspensions using 0.125% TrypLE Select (Invitrogen) and then replated with lentiviral particles and 5 μg/mL Polybrene (Sigma-Aldrich, http://www.sigmaaldrich.com). Three days after transduction, 83.18% ± 5.83% of MSCCXCR5 cells expressed EGFP (in comparison with a transduction efficiency of 85.79% ± 6.28% of MSCEGFP) (Figure S7A). MSCCXCR5 and MSCEGFP were purified by FACS (Influx, Becton Dickinson). Stably transfected MSCs were cultured continuously after sorting.
Flow Cytometry
MSCs were transduced with vectors encoding human CXCR5 and EGFP incubated for 30 min with the appropriate antibody (Table S4) in the dark at 4°C and then analyzed by flow cytometry. Each analysis was performed on at least three separate cell preparations. Flow cytometric analyses were performed with Influx (BD) or Gallios (Beckman Coulter) flow cytometers, and the data were analyzed with the FlowJo7.5 (Tree Star) or Kaluza (Beckman Coulter) software packages.
Proliferation Assay
Isolated CD3+ T lymphocytes were stained using a CellTrace CFSE cell proliferation kit (stain concentration, 5 μM; Invitrogen) according to the manufacturer’s instructions. Labeled CD3+ T cells were cultured in replicate wells with or without MSCs and stimulated with an anti-CD3 monoclonal antibody (mAb) (0.2 μg/mL; BD Biosciences Pharmingen) and an anti-CD28 mAb (1 μg/mL; BD Biosciences Pharmingen) for 96 hr. T cell proliferation was evaluated by flow cytometric analysis of 5,6-carboxyfluorescein diacetate succinimidyl ester (CFSE) dilutions.
Survival Assay
Sorted CD3+ T lymphocytes were cultured in 24-well plates with or without MSCs (5:1 ratio). After 96 hr of culture, the extent of T cell survival was measured by flow cytometric analysis of Annexin V and propidium iodide (PI) staining (BD Biosciences Pharmingen).
Intracellular Cytokine Staining
CD3+ T cells were sorted from healthy donors, resuspended in RPMI 1640 medium (HyClone), and cultured in duplicate wells with or without MSCs for 72 hr. Brefeldin A (BFA, 10 μg/mL, Sigma), phorbol-12-myristate-13-acetate (PMA, 50 ng/mL, Sigma), and ionomycin (1 μg/mL, Sigma) were added, and the cells were cultured for an additional 6 hr. The cells were fixed, permeabilized, and stained for cell surface CD3, cytoplasmic TNF-α, and IFN-γ and analyzed by flow cytometry.
Migration Assays
Migration was assessed using a transwell chamber system of 8-μm-pore membrane filters (PIEP12R48, Millipore). Each upper chamber was loaded with serum-starved MSCs (2 × 105/well), whereas each lower chamber was loaded with 500 μL serum-free medium with or without hCXCL13 (5 ng/mL, PeproTech) or mCXCL13 (50 ng/mL, PeproTech). After 5–10 hr of incubation at 37°C in 5% CO2, the cells remaining on the upper surfaces were removed with a cotton wool swab, and the filters were fixed and stained with 0.1% crystal violet. The cells that had migrated to the lower surface were counted under microscopy.
CHS Model Induced by DNFB and Ear Thickness Measurements
The mouse ear swelling test was performed as described elsewhere.53, 54 Briefly, 2,4-dinitro-1-fluorobenzene (DNFB, D1529, Sigma) was used as a contact-sensitizing agent. DNFB (0.5% dissolved in 4:1 acetone/olive oil) was applied to the shaved back of each mouse (sensitization). 5 days later, the mice were challenged by epicutaneous application of 0.2% DNFB on the right ear. The control group consisted of age- and sex-matched syngeneic naive mice challenged with 0.2% DNFB on the right ear without prior sensitization.
Ear thickness was measured for both the left and right sides daily for 7 days post-challenge using a dial thickness gauge (Mitutoyo). The degree of swelling was calculated as the thickness of the right ear (challenged ear) minus the baseline thickness of the left ear (unchallenged ear). To compare the immunosuppressive effects of MSCEGFP and MSCCXCR5 on the CHS response, the mice were subjected to intravenous (i.v.) injection of one million MSCs on day 2 post-challenge.
Measurement of MPO Activity
72 hr after MSC injection, ear tissues were harvested and homogenized in 0.5% cetyltrimethylammonium chloride (Sigma). The homogenates were passed through a nylon mesh, centrifuged at 3000 rpm for 20 min at 4°C, and collected. MPO activity was assessed with the MPO kit (Jiancheng) according to the manufacturer’s instructions. MPO activity is shown as the absorbance per gram of total protein in ear tissue homogenates.
ELISA
Commercially available ELISA kits were used to measure the levels of TNF-α, IFN-γ, IL-17, IL-10, IL-4 (all from eBioscience), and IL-6 (RayBiotech) in the inflamed ears of each group according to the manufacturer’s recommended procedures.
In Vivo Distribution of Transplanted MSCs
To detect the distribution of transplanted MSCEGFP or MSCCXCR5 in vivo, samples were collected from the left and right ears of recipients on days 1, 3, and 5 post-injection. Cryosections were prepared and counterstained with 1.0 μg/ml DAPI in PBS for 20 min at room temperature in the dark.
Flow Cytometric Detection of Apoptosis
The DNFB-sensitized ears of each group were harvested 24 hr post-injection, and primed T cells were isolated and stained with anti-mouse CD3, anti-mouse CD4, or anti-mouse CD8 (BD Biosciences) in 100 μL staining buffer for 20 min in the dark at 4°C. The cells were then washed with cold FACS buffer (0.1% sodium azide and 0.5% BSA in PBS) and stained for 15 min with Annexin V and PI in binding buffer according to the manufacturer’s instructions.
Analysis of EdU Incorporation
MSC-injected mice were administered a single intraperitoneal injection of EdU (50 mg/kg body weight, Invitrogen). 24 hr after EdU injection, the mice were sacrificed, and challenged ears were collected and minced into small pieces. The cells were dissociated by digestion with 1 mg/ml collagenase type IV (Invitrogen) in RPMI 1640 medium (Invitrogen) at 37°C for 20 min. RPMI 1640 medium containing 5% FBS (HyClone) was added to stop the collagenase activity, and the samples were centrifuged at 1,500 rpm for 10 min at room temperature. The cells were stained with Alexa Fluor 647-conjugated anti-EdU antibody (Ab) using the EdU kit (Invitrogen) according to the manufacturer’s instructions. Finally, the cells were analyzed by flow cytometry.
Measurement of Cytokine Production by Hapten-Primed T Cells
Primed T cells were harvested from the DNFB-sensitized ears of each group 24 hr post-injection, and cytokines were measured by flow cytometry. For detection of intracellular cytokines, the primed T cells were stimulated with PMA (50 ng/mL), ionomycin (500 ng/mL), and BFA(10 μg/mL) for 6 hr and then collected, fixed, permeabilized, and stained for cell surface CD3, CD4, or CD8 or for cytoplasmic IL-17 or IFN-γ. The stained cells were analyzed by flow cytometry.
Statistics
All data are presented as the mean ± SEM obtained from at least three independent experiments. Comparisons between groups were performed using ANOVA with Newman-Keuls post hoc comparison. p < 0.05 was considered statistically significant. All statistical analyses were performed with the aid of SPSS version 14.0 (SPSS).
Author Contributions
X.Z., J.R., and A.P.X. designed the study, analyzed and interpreted the data, and wrote the paper. W.H. performed RNA library preparation and Illumina sequencing. X.C. performed the western blot and qRT-PCR experiments. Y.L. performed the animal experiments and migration assays. J.W. performed immunofluorescence staining and lentiviral vector construction and transduction. C.C. performed cell isolation and staining for the analysis of survival, proliferation, and cytokines. L.H. and T.W. performed ELISA and measurement of MPO activity.
Conflicts of Interest
The authors declare no conflict of interest.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (81425016 and 81371554); the Natural Science Foundation of Guangdong Province (S2013030013305); the Key Scientific and Technological Projects of Guangdong Province (2013B021800092, 2014A030310049, and 2015B020228001); the Natural Science Foundation of Guangdong Province (2014A030310049); the Key Scientific and Technological Program of Guangzhou City (201400000003-3, 201300000089, and 201604020132); and Guangdong Province Universities and Colleges Pearl River Scholar Funded Scheme (GDUPS, 2013).
Footnotes
Supplemental Information includes seven figures and four tables and can be found with this article online at http://dx.doi.org/10.1016/j.ymthe.2017.04.004.
Contributor Information
Jie Ren, Email: 13925155583@126.com.
Andy Peng Xiang, Email: xiangp@mail.sysu.edu.cn.
Supplemental Information
References
- 1.Salem H.K., Thiemermann C. Mesenchymal stromal cells: current understanding and clinical status. Stem Cells. 2010;28:585–596. doi: 10.1002/stem.269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Barzilay R., Melamed E., Offen D. Introducing transcription factors to multipotent mesenchymal stem cells: making transdifferentiation possible. Stem Cells. 2009;27:2509–2515. doi: 10.1002/stem.172. [DOI] [PubMed] [Google Scholar]
- 3.Comoli P., Ginevri F., Maccario R., Avanzini M.A., Marconi M., Groff A., Cometa A., Cioni M., Porretti L., Barberi W. Human mesenchymal stem cells inhibit antibody production induced in vitro by allostimulation. Nephrol. Dial. Transplant. 2008;23:1196–1202. doi: 10.1093/ndt/gfm740. [DOI] [PubMed] [Google Scholar]
- 4.Dazzi F., Lopes L., Weng L. Mesenchymal stromal cells: a key player in ‘innate tolerance’? Immunology. 2012;137:206–213. doi: 10.1111/j.1365-2567.2012.03621.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.English K., Ryan J.M., Tobin L., Murphy M.J., Barry F.P., Mahon B.P. Cell contact, prostaglandin E(2) and transforming growth factor beta 1 play non-redundant roles in human mesenchymal stem cell induction of CD4+CD25(High) forkhead box P3+ regulatory T cells. Clin. Exp. Immunol. 2009;156:149–160. doi: 10.1111/j.1365-2249.2009.03874.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kapoor S., Patel S.A., Kartan S., Axelrod D., Capitle E., Rameshwar P. Tolerance-like mediated suppression by mesenchymal stem cells in patients with dust mite allergy-induced asthma. J. Allergy Clin. Immunol. 2012;129:1094–1101. doi: 10.1016/j.jaci.2011.10.048. [DOI] [PubMed] [Google Scholar]
- 7.Spaggiari G.M., Capobianco A., Abdelrazik H., Becchetti F., Mingari M.C., Moretta L. Mesenchymal stem cells inhibit natural killer-cell proliferation, cytotoxicity, and cytokine production: role of indoleamine 2,3-dioxygenase and prostaglandin E2. Blood. 2008;111:1327–1333. doi: 10.1182/blood-2007-02-074997. [DOI] [PubMed] [Google Scholar]
- 8.Ben-Ami E., Berrih-Aknin S., Miller A. Mesenchymal stem cells as an immunomodulatory therapeutic strategy for autoimmune diseases. Autoimmun. Rev. 2011;10:410–415. doi: 10.1016/j.autrev.2011.01.005. [DOI] [PubMed] [Google Scholar]
- 9.François M., Galipeau J. New insights on translational development of mesenchymal stromal cells for suppressor therapy. J. Cell. Physiol. 2012;227:3535–3538. doi: 10.1002/jcp.24081. [DOI] [PubMed] [Google Scholar]
- 10.Ankrum J., Karp J.M. Mesenchymal stem cell therapy: Two steps forward, one step back. Trends Mol. Med. 2010;16:203–209. doi: 10.1016/j.molmed.2010.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Eggenhofer E., Luk F., Dahlke M.H., Hoogduijn M.J. The life and fate of mesenchymal stem cells. Front. Immunol. 2014;5:148. doi: 10.3389/fimmu.2014.00148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kang S.K., Shin I.S., Ko M.S., Jo J.Y., Ra J.C. Journey of mesenchymal stem cells for homing: strategies to enhance efficacy and safety of stem cell therapy. Stem Cells Int. 2012;2012:342968. doi: 10.1155/2012/342968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Barbash I.M., Chouraqui P., Baron J., Feinberg M.S., Etzion S., Tessone A., Miller L., Guetta E., Zipori D., Kedes L.H. Systemic delivery of bone marrow-derived mesenchymal stem cells to the infarcted myocardium: feasibility, cell migration, and body distribution. Circulation. 2003;108:863–868. doi: 10.1161/01.CIR.0000084828.50310.6A. [DOI] [PubMed] [Google Scholar]
- 14.Lin P., Correa D., Kean T.J., Awadallah A., Dennis J.E., Caplan A.I. Serial transplantation and long-term engraftment of intra-arterially delivered clonally derived mesenchymal stem cells to injured bone marrow. Mol. Ther. 2014;22:160–168. doi: 10.1038/mt.2013.221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rombouts W.J., Ploemacher R.E. Primary murine MSC show highly efficient homing to the bone marrow but lose homing ability following culture. Leukemia. 2003;17:160–170. doi: 10.1038/sj.leu.2402763. [DOI] [PubMed] [Google Scholar]
- 16.Honczarenko M., Le Y., Swierkowski M., Ghiran I., Glodek A.M., Silberstein L.E. Human bone marrow stromal cells express a distinct set of biologically functional chemokine receptors. Stem Cells. 2006;24:1030–1041. doi: 10.1634/stemcells.2005-0319. [DOI] [PubMed] [Google Scholar]
- 17.Huang J., Zhang Z., Guo J., Ni A., Deb A., Zhang L., Mirotsou M., Pratt R.E., Dzau V.J. Genetic modification of mesenchymal stem cells overexpressing CCR1 increases cell viability, migration, engraftment, and capillary density in the injured myocardium. Circ. Res. 2010;106:1753–1762. doi: 10.1161/CIRCRESAHA.109.196030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bobis-Wozowicz S., Miekus K., Wybieralska E., Jarocha D., Zawisz A., Madeja Z. Genetically modified adipose tissue-derived mesenchymal stem cells overexpressing CXCR4 display increased motility, invasiveness, and homing to bone marrow of NOD/SCID mice. Exp. Hematol. 2011;39:686–696.e4. doi: 10.1016/j.exphem.2011.03.004. [DOI] [PubMed] [Google Scholar]
- 19.Li H., Jiang Y., Jiang X., Guo X., Ning H., Li Y., Liao L., Yao H., Wang X., Liu Y. CCR7 guides migration of mesenchymal stem cell to secondary lymphoid organs: a novel approach to separate GvHD from GvL effect. Stem Cells. 2014;32:1890–1903. doi: 10.1002/stem.1656. [DOI] [PubMed] [Google Scholar]
- 20.Castor M.G., Pinho V., Teixeira M.M. The role of chemokines in mediating graft versus host disease: opportunities for novel therapeutics. Front. Pharmacol. 2012;3:23. doi: 10.3389/fphar.2012.00023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Guabiraba R., Besnard A.G., Menezes G.B., Secher T., Jabir M.S., Amaral S.S., Braun H., Lima-Junior R.C., Ribeiro R.A., Cunha F.Q. IL-33 targeting attenuates intestinal mucositis and enhances effective tumor chemotherapy in mice. Mucosal Immunol. 2014;7:1079–1093. doi: 10.1038/mi.2013.124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Roy I., Evans D.B., Dwinell M.B. Chemokines and chemokine receptors: update on utility and challenges for the clinician. Surgery. 2014;155:961–973. doi: 10.1016/j.surg.2014.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fonacier L.S., Dreskin S.C., Leung D.Y. Allergic skin diseases. J. Allergy Clin. Immunol. 2010;125(2, Suppl 2):S138–S149. doi: 10.1016/j.jaci.2009.05.039. [DOI] [PubMed] [Google Scholar]
- 24.Honda T., Egawa G., Grabbe S., Kabashima K. Update of immune events in the murine contact hypersensitivity model: toward the understanding of allergic contact dermatitis. J. Invest. Dermatol. 2013;133:303–315. doi: 10.1038/jid.2012.284. [DOI] [PubMed] [Google Scholar]
- 25.Vocanson M., Hennino A., Rozières A., Poyet G., Nicolas J.F. Effector and regulatory mechanisms in allergic contact dermatitis. Allergy. 2009;64:1699–1714. doi: 10.1111/j.1398-9995.2009.02082.x. [DOI] [PubMed] [Google Scholar]
- 26.Kim H.S., Yun J.W., Shin T.H., Lee S.H., Lee B.C., Yu K.R., Seo Y., Lee S., Kang T.W., Choi S.W. Human umbilical cord blood mesenchymal stem cell-derived PGE2 and TGF-β1 alleviate atopic dermatitis by reducing mast cell degranulation. Stem Cells. 2015;33:1254–1266. doi: 10.1002/stem.1913. [DOI] [PubMed] [Google Scholar]
- 27.Su W.R., Zhang Q.Z., Shi S.H., Nguyen A.L., Le A.D. Human gingiva-derived mesenchymal stromal cells attenuate contact hypersensitivity via prostaglandin E2-dependent mechanisms. Stem Cells. 2011;29:1849–1860. doi: 10.1002/stem.738. [DOI] [PubMed] [Google Scholar]
- 28.Mitsui G., Mitsui K., Hirano T., Ohara O., Kato M., Niwano Y. Kinetic profiles of sequential gene expressions for chemokines in mice with contact hypersensitivity. Immunol. Lett. 2003;86:191–197. doi: 10.1016/s0165-2478(03)00017-8. [DOI] [PubMed] [Google Scholar]
- 29.Bao L., Zhang H., Mohan G.C., Shen K., Chan L.S. Differential expression of inflammation-related genes in IL-4 transgenic mice before and after the onset of atopic dermatitis skin lesions. Mol. Cell. Probes. 2016;30:30–38. doi: 10.1016/j.mcp.2015.11.001. [DOI] [PubMed] [Google Scholar]
- 30.Han J.H., Suh C.H., Jung J.Y., Nam J.Y., Kwon J.E., Yim H., Kim H.A. Association of CXCL10 and CXCL13 levels with disease activity and cutaneous manifestation in active adult-onset Still’s disease. Arthritis Res. Ther. 2015;17:260. doi: 10.1186/s13075-015-0773-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li Z., Hodgkinson T., Gothard E.J., Boroumand S., Lamb R., Cummins I., Narang P., Sawtell A., Coles J., Leonov G. Epidermal Notch1 recruits RORγ(+) group 3 innate lymphoid cells to orchestrate normal skin repair. Nat. Commun. 2016;7:11394. doi: 10.1038/ncomms11394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Delorme V., Machacek M., DerMardirossian C., Anderson K.L., Wittmann T., Hanein D., Waterman-Storer C., Danuser G., Bokoch G.M. Cofilin activity downstream of Pak1 regulates cell protrusion efficiency by organizing lamellipodium and lamella actin networks. Dev. Cell. 2007;13:646–662. doi: 10.1016/j.devcel.2007.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jaffe A.B., Hall A. Rho GTPases: biochemistry and biology. Annu. Rev. Cell Dev. Biol. 2005;21:247–269. doi: 10.1146/annurev.cellbio.21.020604.150721. [DOI] [PubMed] [Google Scholar]
- 34.Williams M.J. Rho GTPases central regulators of cell migration. Small GTPases. 2012;3:1. doi: 10.4161/sgtp.20335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Murali A., Rajalingam K. Small Rho GTPases in the control of cell shape and mobility. Cell. Mol. Life Sci. 2014;71:1703–1721. doi: 10.1007/s00018-013-1519-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tewari P., Roy R., Mishra S., Mandal P., Yadav A., Chaudhari B.P., Chaturvedi R.K., Dwivedi P.D., Tripathi A., Das M. Benzanthrone induced immunotoxicity via oxidative stress and inflammatory mediators in Balb/c mice. Immunobiology. 2015;220:369–381. doi: 10.1016/j.imbio.2014.10.011. [DOI] [PubMed] [Google Scholar]
- 37.Gieseke F., Böhringer J., Bussolari R., Dominici M., Handgretinger R., Müller I. Human multipotent mesenchymal stromal cells use galectin-1 to inhibit immune effector cells. Blood. 2010;116:3770–3779. doi: 10.1182/blood-2010-02-270777. [DOI] [PubMed] [Google Scholar]
- 38.Li X., Xu Z., Bai J., Yang S., Zhao S., Zhang Y., Chen X., Wang Y. Umbilical Cord Tissue-Derived Mesenchymal Stem Cells Induce T Lymphocyte Apoptosis and Cell Cycle Arrest by Expression of Indoleamine 2, 3-Dioxygenase. Stem Cells Int. 2016;2016:7495135. doi: 10.1155/2016/7495135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pennino D., Eyerich K., Scarponi C., Carbone T., Eyerich S., Nasorri F., Garcovich S., Traidl-Hoffmann C., Albanesi C., Cavani A. IL-17 amplifies human contact hypersensitivity by licensing hapten nonspecific Th1 cells to kill autologous keratinocytes. J. Immunol. 2010;184:4880–4888. doi: 10.4049/jimmunol.0901767. [DOI] [PubMed] [Google Scholar]
- 40.He D., Wu L., Kim H.K., Li H., Elmets C.A., Xu H. IL-17 and IFN-gamma mediate the elicitation of contact hypersensitivity responses by different mechanisms and both are required for optimal responses. J. Immunol. 2009;183:1463–1470. doi: 10.4049/jimmunol.0804108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jiang Y., Cai Y., Zhang W., Yin Z., Hu C., Tong T., Lu P., Zhang S., Neculai D., Tuan R.S., Ouyang H.W. Human Cartilage-Derived Progenitor Cells From Committed Chondrocytes for Efficient Cartilage Repair and Regeneration. Stem Cells Transl. Med. 2016;5:733–744. doi: 10.5966/sctm.2015-0192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Peng Y., Chen X., Liu Q., Zhang X., Huang K., Liu L., Li H., Zhou M., Huang F., Fan Z. Mesenchymal stromal cells infusions improve refractory chronic graft versus host disease through an increase of CD5+ regulatory B cells producing interleukin 10. Leukemia. 2015;29:636–646. doi: 10.1038/leu.2014.225. [DOI] [PubMed] [Google Scholar]
- 43.Kavanagh D.P., Robinson J., Kalia N. Mesenchymal stem cell priming: fine-tuning adhesion and function. Stem Cell Rev. 2014;10:587–599. doi: 10.1007/s12015-014-9510-7. [DOI] [PubMed] [Google Scholar]
- 44.Kavanagh D.P., Suresh S., Newsome P.N., Frampton J., Kalia N. Pretreatment of Mesenchymal Stem Cells Manipulates Their Vasculoprotective Potential While Not Altering Their Homing Within the Injured Gut. Stem Cells. 2015;33:2785–2797. doi: 10.1002/stem.2061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yang J.X., Zhang N., Wang H.W., Gao P., Yang Q.P., Wen Q.P. CXCR4 receptor overexpression in mesenchymal stem cells facilitates treatment of acute lung injury in rats. J. Biol. Chem. 2015;290:1994–2006. doi: 10.1074/jbc.M114.605063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cheng Z., Ou L., Zhou X., Li F., Jia X., Zhang Y., Liu X., Li Y., Ward C.A., Melo L.G., Kong D. Targeted migration of mesenchymal stem cells modified with CXCR4 gene to infarcted myocardium improves cardiac performance. Mol. Ther. 2008;16:571–579. doi: 10.1038/sj.mt.6300374. [DOI] [PubMed] [Google Scholar]
- 47.Kumar S., Ponnazhagan S. Bone homing of mesenchymal stem cells by ectopic alpha 4 integrin expression. FASEB J. 2007;21:3917–3927. doi: 10.1096/fj.07-8275com. [DOI] [PubMed] [Google Scholar]
- 48.Wiehe J.M., Kaya Z., Homann J.M., Wöhrle J., Vogt K., Nguyen T., Rottbauer W., Torzewski J., Fekete N., Rojewski M. GMP-adapted overexpression of CXCR4 in human mesenchymal stem cells for cardiac repair. Int. J. Cardiol. 2013;167:2073–2081. doi: 10.1016/j.ijcard.2012.05.065. [DOI] [PubMed] [Google Scholar]
- 49.Koning J.J., Mebius R.E. Interdependence of stromal and immune cells for lymph node function. Trends Immunol. 2012;33:264–270. doi: 10.1016/j.it.2011.10.006. [DOI] [PubMed] [Google Scholar]
- 50.Salero E., Blenkinsop T.A., Corneo B., Harris A., Rabin D., Stern J.H., Temple S. Adult human RPE can be activated into a multipotent stem cell that produces mesenchymal derivatives. Cell Stem Cell. 2012;10:88–95. doi: 10.1016/j.stem.2011.11.018. [DOI] [PubMed] [Google Scholar]
- 51.Ke H., Wang P., Yu W., Liu X., Liu C., Yang F., Mao F.F., Zhang L., Zhang X., Lahn B.T., Xiang A.P. Derivation, characterization and gene modification of cynomolgus monkey mesenchymal stem cells. Differentiation. 2009;77:256–262. doi: 10.1016/j.diff.2008.09.021. [DOI] [PubMed] [Google Scholar]
- 52.Pinho S., Lacombe J., Hanoun M., Mizoguchi T., Bruns I., Kunisaki Y., Frenette P.S. PDGFRα and CD51 mark human nestin+ sphere-forming mesenchymal stem cells capable of hematopoietic progenitor cell expansion. J. Exp. Med. 2013;210:1351–1367. doi: 10.1084/jem.20122252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Varona R., Villares R., Carramolino L., Goya I., Zaballos A., Gutiérrez J., Torres M., Martínez-A C., Márquez G. CCR6-deficient mice have impaired leukocyte homeostasis and altered contact hypersensitivity and delayed-type hypersensitivity responses. J. Clin. Invest. 2001;107:R37–R45. doi: 10.1172/JCI11297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Garrigue J.L., Nicolas J.F., Fraginals R., Benezra C., Bour H., Schmitt D. Optimization of the mouse ear swelling test for in vivo and in vitro studies of weak contact sensitizers. Contact Dermat. 1994;30:231–237. doi: 10.1111/j.1600-0536.1994.tb00650.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.