

Abstract
Accumulating evidence suggests that the experience of early life adversity is a risk factor for a range of poor outcomes across development, including poor physical health in adulthood. The biological embedding model of early adversity (Miller, Chen, & Parker, 2011) suggests that early adversity might become embedded within immune cells known as monocytes/macrophages, programming them to be overly aggressive to environmental stimuli and insensitive to inhibitory signals, creating a “proinflammatory phenotype” that increases vulnerability to chronic diseases across the life span. We tested this hypothesis in the present study. Adolescent girls (n = 147) had blood drawn every 6 months across a 2.5-year period. To assess inflammatory responses to challenge, their monocytes were stimulated in vitro with a bacterial product, and production of the cytokine interleukin-6 was quantified. Hydrocortisone was added to cultures to assess the cells’ sensitivity to glucocorticoids’ anti-inflammatory signal. Using cluster analyses, we found that early life adversity was associated with greater odds of displaying a proinflammatory phenotype characterized by relatively larger interleukin-6 responses and relatively less sensitivity to glucocorticoids. In contrast, ongoing social stress was not associated with increasing odds of being categorized in the proinflammatory cluster. These findings suggest that early life adversity increases the probability of developing a proinflammatory phenotype, which, if sustained, could forecast risk for health problems later in life.
The experience of early adversity, including exposure to maltreatment, poverty, and parental mental illness, is a risk factor for a diverse set of poor outcomes across development, including internalizing and externalizing symptoms, substance abuse, and adult psychopathology (e.g., Appleyard, Egeland, van Dulmen, & Sroufe, 2005; Kessler et al., 2010; Poulton et al., 2002; Shonkoff et al., 2012). In the last three decades, a growing number of studies have shed light on the possibility that adversity in childhood has a lasting influence on adult physical health, particularly chronic diseases associated with aging, such as cardiovascular disease, diabetes, arthritis, and some cancers (Gluckman & Hanson, 2006; Miller, Chen, & Parker, 2011; Repetti, Taylor, & Seeman, 2002; see Ehrlich, Miller, & Chen, 2016, for a review). Across a wide range of samples and indices of adversity, the findings consistently suggest that these early stressful experiences somehow become “biologically embedded” and contribute to physical health problems in adulthood.
The Biological Embedding Model of Early Adversity
In recent years, Miller, Chen, and colleagues have developed a biological embedding model of early adversity (Miller & Chen, 2013; Miller et al., 2011). This framework grows out of earlier theories of environmental programming, such as the fetal-origins literature (Barker, 1992), life-course models (Hertzmann, 1999; Lynch & Smith, 2005; Shonkoff, Boyce, & McEwen, 2009), epigenetics (Meaney, 2001), and behavioral immunology (Coe & Lubach, 2007; Raison & Miller, 2003). These earlier models set the foundation for exploring how social and contextual experiences can calibrate physiological processes in ways that have lasting implications for health.
The importance of exposure to early life adversity
The biological embedding model proposes that the experience of early adversity becomes embedded within certain types of immune cells called monocytes (and in their mature form, macrophages). In response to harsh social and environmental conditions, these cells are thought to be programmed to have a proinflammatory phenotype, which manifests in relatively aggressive inflammatory responses to stimuli and lower insensitivity to signals that dampen this response. Over time, this phenotype is thought to contribute to low-grade chronic inflammation. This kind of “nonresolving” inflammation has been implicated in the development and progression of age-related illnesses, including heart disease, stroke, and autoimmune disorders (Nathan & Ding, 2010). However, although the biological embedding model focuses on risk for physical illness as a consequence of the proinflammatory phenotype, emerging evidence suggests that inflamemation also plays a role in many psychiatric disorders, including depression, schizophrenia, and autism (Markham & Koenig, 2011; Meyer, Feldon, & Dammann, 2011).
The role of ongoing stressors
The biological embedding model also proposes that, in addition to the programming effects of early adversity, individuals from disadvantaged backgrounds are at risk for the proinflammatory phenotype through accentuating effects of interpersonal stressors and unhealthy behaviors across the life span. Children living in adverse conditions are often exposed to harsh parenting, uncontrollable and unpredictable stressors, and a lack of social support, all of which modulates brain development (Blair & Raver, 2012), particularly corticolimbic and corticostriatal regions that are involved in self-regulation skills, social information processing, and detection of threat and reward. As a result of early adversity, individuals may develop self-regulatory deficits, hostile attribution biases, and heightened vigilance to threat (Chen, Langer, Raphaelson, & Matthews, 2004; Dodge, Pettit, & Bates, 1994; Weiss, Dodge, Bates, & Pettit, 1992), ultimately leading to unhealthy behaviors like cigarette smoking, conflictual interactions with others, and poor-quality social relationships across development (Miller et al., 2011; Repetti, Robles, & Reynolds, 2011).
In addition to the social challenges that adolescents may face as a result of early adversity, adolescence is a period of major changes to close relationships that could accentuate the experience of stress during this period of development. During adolescence, several notable changes to adolescents’ social experiences are evident: there is a shift in time spent with family members to a larger proportion of time spent with peers (Csikszentmihalyi & Larson, 1984; Furman & Buhrmester, 1992), an increased focus on social relationships (Smetana, Campione-Barr, & Metzger, 2006), and the onset of early romantic relationships (Furman & Shaffer, 2003). These changes bring the potential for stress and strain as adolescents navigate their increasingly complex social lives, and these social stressors are thought to shape inflammatory activity.
The combination of early adversity and ongoing stress creates a feedback loop in which individuals from disadvantaged backgrounds are both exposed to more social stress and demonstrate greater reactivity to those stressors. Thus, ongoing social stressors may operate as a mediator of links between early adversity and proinflammatory processes as well as a moderator of these links, amplifying the effects of exposure to disadvantage early in life (e.g., John-Henderson, Marsland, Kamarck, Muldoon, & Manuck, 2016).
Inflammation and glucocorticoids
Inflammation plays an important role in protecting the body from infections and injuries. Monocytes and macrophages are cells that initiate and sustain inflammatory responses (Marques, Silverman, & Sternberg, 2009; Miller et al., 2008; Raison & Miller, 2003). When macrophages encounter microorganisms that contribute to infectious diseases or evidence of tissue damage, one of their initial responses is to secrete proteins, called proinflammatory cytokines (e.g., interleukin-6 [IL-6], IL-1β, and tumor necrosis factor-α). These proteins in turn attract cells to the site of the infection or injury, initiate processes to decapacitate the invading microorganisms, and stimulate the process of healing (Zhang & Mosser, 2008).
Glucocorticoids serve an important complementary role to the proinflammatory functions of monocytes and macrophages. The proinflammatory cytokine IL-1 signals the brain to activate the hypothalamus–pituitary–adrenal axis, leading to the release of cortisol (Rivest, 2010). Cortisol works first to coordinate and reinforce the inflammatory response, assisting in the mobilization and readying of the immune system in order to be responsive to microbial products and cellular damage (Busillo & Cidlowski, 2013). However, cortisol ultimately functions to downregulate the inflammatory response, thus playing a critical role in resolving acute inflammatory responses and helping to restore homeostasis to the system.
This inflammatory response is critical for surviving acute infections and injuries, but it must be carefully regulated so that inflammation does not lead to tissue damage or exacerbate disease (e.g., Hotamisligil, 2006; Libby & Theroux, 2005). When the system is out of balance (as a result of sustained activity or incomplete resolution of responses) chronic low-grade inflammation can ensue. This nonresolving inflammation (Nathan & Ding, 2010) has been related to many diseases of aging, such as diabetes, obesity-related problems, cardiovascular disease, autoimmune disease, and cancer, as well as early mortality and psychiatric illnesses (Danesh, Collins, Appleby, & Peto, 2000; Kaptoge et al., 2010; Libby, Ridker, & Hansson, 2009; Ridker, 2007; Yeh & Willerson, 2003; Zunszain, Hepgul, & Pariante, 2013).
Evidence for the Biological Embedding Model
Early adversity and inflammatory activity
Mounting evidence supports the idea that early life adversity is linked to measures of inflammatory processes. Some of the earliest evidence came from a study that sampled adults on the basis of early and current life socioeconomic status (Miller, Chen, et al., 2009). Participants’ peripheral blood mononuclear cells were cultured in vitro with a series of microbial products, and the magnitude of the cell responses to these stimuli was indexed by subsequent production of IL-6. Participants who experienced socioeconomic disadvantage as children produced more IL-6 in response to viral and bacterial stimuli relative to participants from families of high status, an effect that was independent of adult socioeconomic status. Using gene expression profiling, this study also found indications of lower sensitivity to the anti-inflammatory actions of cortisol in the cells of participants who experienced socioeconomic disadvantage as children.
Similar evidence for a link between socioeconomic status and production of cytokines after exposure to bacterial products comes from a sample of adolescents whose parents reported on their current family income (Schreier, Roy, Frimer, & Chen, 2014). Family income was marginally negatively associated with production of three cytokines after exposure to bacterial challenge. In addition to single indicators of socioeconomic status, one study found that trajectories of socioeconomic status were predictive of IL-6 production following exposure to bacterial products (Azad et al., 2012). This study found that children with persistently low status had the greatest IL-6 responses to bacterial stimuli, and children whose families improved in status over the course of the children’s lives had similar cytokine responses to children who were persistently high in status, suggesting that there could be some flexibility in the timing with which these biological programming effects take place.
However, other studies have not found direct links between childhood socioeconomic status and stimulated immune responses to bacterial challenge (e.g., John-Henderson et al., 2016). In this study, John-Henderson et al. did not find a connection between childhood socioeconomic status and IL-6 responses to bacterial product in their adult sample; but they did find a marginally significant interaction between childhood status and recent negative life events, such that childhood socioeconomic status was associated with greater IL-6 production when individuals are also experiencing a higher number of negative life events. These findings are consistent with a vulnerability framework of early adversity, suggesting that individuals who experienced early life adversity might be particularly susceptible to ongoing stressors.
Ongoing social stressors and inflammatory activity
Evidence suggests that social stress is an especially potent source of strain for adolescents, both for psychological well-being and for immune functioning. Abrasive social relationships, characterized by high levels of unresolved conflict, low support, and mistrust, have been associated with several markers of inflammatory activity, including IL-6 responses to bacterial challenge and glucocorticoid insensitivity (Ehrlich, Miller, Rohleder, & Adam, 2016; Miller, Chen, et al., 2009), biomarkers of low-grade inflammation (Dixon, Meng, Goldberg, Schneiderman, & Delamater, 2009; Fuligni et al., 2009), and pro-and anti-inflammatory gene expression (Murphy, Slavich, Chen, & Miller; 2015; Murphy, Slavich, Rohleder, & Miller, 2013).
Limitations of Existing Evidence
To date, however, research on the links between early adversity and inflammatory processes has been focused primarily on predicting inflammatory measures in isolation (e.g., biomarkers of low-grade inflammation and IL-6 response to bacterial stimuli) using traditional variable-centered analytical approaches (e.g., wherein the degree of early adversity is predicted to correlate with inflammatory measures in a roughly dose-response manner). There is certainly merit in this approach, and these models have shed light on the extent to which increases in early adversity map on to higher levels of inflammation, sometimes decades later in life. Nevertheless, this variable-centered approach falls short of modeling what is at the heart of the biological embedding model. As described earlier, the biological embedding model specifies that early adversity endows monocytes and macrophages with a tendency to (a) mount relatively large cytokine responses to threats and (b) be relatively insensitive to signals from cortisol to dampen this inflammatory response. When analytic models predict inflammatory outcomes in isolation, they are only examining a select portion of the proinflammatory phenotype, and these models are unable to examine the coupling of the features of cellular response that is thought to result from exposure to early adversity. This tradition of predicting variables in isolation becomes especially problematic when indices of inflammatory processes are only weakly correlated or are sometimes not correlated at all.
Based on the predictions of the biological embedding model, two features of cellular activity should be evident. First, indicators of the proinflammatory phenotype should aggregate to some degree, thus distinguishing individuals on the basis of their cellular reactivity (i.e., the phenotype should identify individuals who have both larger cytokine responses and are less sensitive to signals from cortisol). Second, if it is the case that the phenotype develops as a result of exposure to adverse conditions in early life, and results in the programming of cells to respond in an aggressive manner, then these phenotypes should be relatively stable over time. Given the model’s consideration of the role of ongoing stressors in continuing to shape inflammatory activity, however, it may be the case that individuals’ phenotype evolves as a result of changes in ongoing exposure to stressful experiences.
Unfortunately, most of the existing tests of the biological embedding model focus solely on either (a) the role of early exposures to adversity or (b) current stressors and fail to consider the possibility that both early and current exposures each play a role in shaping inflammatory activity (for an exception, see John-Henderson et al., 2016). On the one hand, this emphasis on the critical role of early experience is consistent with the large body of research on sensitive periods of development, during which certain experiences are thought to exert permanent changes to developing physiological systems (e.g., Black, Jones, Nelson, & Greenough, 1998). On the other hand, this relative lack of consideration of ongoing experiences fails to take into account the considerable evolutionary advantage that would result from having immunologic defenses that continue to be sensitive and flexible to ongoing environmental circumstances. From a survival perspective, the most advantageous immune defenses would be those capable of learning how to manage new threats in the environment over the course of the life span.
Finally, an additional limitation with the majority of research on early adversity and inflammatory processes is that much of this research has studied inflammatory activity in adulthood, rather than in childhood or adolescence. This approach makes intuitive sense, as a key element of the biological embedding model is that the changes to the endocrine and immune systems take time to unfold. Adulthood is also the time period in which one would expect to see variability in inflammation and health outcomes (e.g., diabetes and heart disease) that might have ties to early adversity. However, it is equally important to consider how exposure to adversity in childhood might be evident in changes to the immune system as early as adolescence, which could shed light on the processes that unfold along the progression to chronic disease.
The Present Study
In the present study, we had three research aims. First, we sought to evaluate whether we could categorize individuals using cluster analysis into groups that distinguished between more and less inflammatory phenotypes. This approach represents a departure from the field’s tradition of examining measures in isolation, and it allows us to identify individuals who may be particularly at risk for chronic health problems in the future due to the coupling of larger inflammatory cytokine responses and lower sensitivity to glucocorticoids. We created six sets of inflammatory clusters in a sample of adolescents who participated in a longitudinal study over a 2.5-year period, with laboratory visits every 6 months. This design allows us to examine the stability of inflammatory cluster membership across the six waves. We hypothesized that we could identify more and less inflammatory clusters at each time point, and these groups would differ significantly in the extent to which they showed elevations in inflammatory activity. Given the biological embedding model’s consideration of the impact of both early exposure to adversity and ongoing stressful life experiences, we expected moderate but not complete stability in cluster membership across the 2.5-year period.
Second, another aim was to examine whether exposure to early adversity was associated with cluster membership across the 2.5-year period. We hypothesized that early life adversity, characterized by low socioeconomic status and disruptions in the family, would be associated with a greater likelihood of being in the more inflammatory phenotype cluster, relative to the less inflammatory cluster, and we did not expect this association to change over time.
Third, in light of evidence that ongoing stressors, particularly ones that are social in nature, influence inflammatory activity, we examined whether ongoing social stressors influenced cluster membership at baseline and over time. First, we tested the role of social stress as a potential mediator of the link between early life adversity and inflammatory cluster membership. This analysis grows out of the notion that early adversity shapes personality development in ways that result in social difficulties across the life span (e.g., conflict, rejection, and isolation; see Miller et al., 2011; Repetti et al., 2002; Tolan, 2016). Second, drawing on the findings of John-Henderson et al. (2016), and more general notions that early adversity accentuates stress reactivity (Cameron et al., 2005; Lupien, McEwen, Gunnar, & Heim, 2009; Repetti et al., 2002), we tested whether social stressors interacted with early life adversity to predict cluster membership. We hypothesized that ongoing social stressors would be associated with a greater likelihood of membership in the proinflammatory cluster. We also hypothesized that a sensitizing effect would emerge, such that individuals who experienced early adversity and high levels of ongoing social stress would be especially likely to be categorized in the more inflammatory cluster compared to individuals who experienced early adversity but not high levels of chronic stress.
Method
Participants
Participants from the Vancouver, British Columbia, community were recruited for a larger study of depression and atherosclerosis among women at risk for affective disorders. We placed advertisements for the study in schools, newspapers, and local magazines. Interested adolescents were directed to a website, where they completed applications to determine eligibility for the study. Of those, 147 participants met criteria and were successfully enrolled in the study. Participants were between 15 and 19 years old at the start of the study (Mage=17.0, SD=1.3) and were primarily from European (48.3%) or Asian (42.9%) backgrounds. All participants were free of acute illness; reported no chronic medical conditions or standing medications, other than oral contraceptives; and were at high risk for developing a first episode of major depression. Girls were considered to be at high risk for depression if they reported that they had a first-degree relative with a history of affective disorder, scored in the top quartile on one of two indices of cognitive vulnerability to depression, or had both a family history of depression and cognitive vulnerability (for more study details, see Miller & Cole, 2012).
Measures
Early life adversity
We formed an early life adversity index using data from interviews at the first laboratory visit. One point was assigned for each of the following risks: (a) birth to a teenage mother, who was younger than 20 years old at delivery; (b) familial disruption before age 15, caused by the death of a parent, divorce, or separation from a parent that lasted more than 1 year; (c) a history of affective illness in parents/guardians; (d) low household education (guardians with a high school diploma or less); and (e) limited economic resources, as reflected by leasing (rather than owning) the family’s primary residence from birth through school entry. Scores on the childhood adversity index could range from 0 to 5.
Social stressors
At each laboratory visit, we assessed several indicators of social stress using the UCLA Life Stress Inter view—Adolescent Version (Hammen, 1991). The Life Stress Interview has been used to index ongoing stressors in an objective, contextually sensitive manner (Rudolph & Hammen, 1999; Shih, Eberhart, Hammen, & Brennan, 2006). This semistructured interview documents the experience of stressors across different domains, including interpersonal relationships. Trained interviewers inquire about the degree of trust, intimacy, support, and conflict in each of the adolescent’s major relationships (i.e., family, peer, and romantic relationships as well as the broader social network). Questions are framed to elicit concrete behavioral examples to provide evidence for the degree of ongoing stress within each context. Interviewers provided ratings of ongoing stress ranging from 1 to 5, with higher scores indicating evidence of conflict, mistrust, instability, or loneliness. Interviewers were reliable across the interpersonal domains, with intraclass correlations ranging from 0.65 (family stress) to 0.80 (peer stress). We averaged the scores from the four social stress domains into a single measure of social stress at each time point (Cronbach αs=0.53–0.71). This approach is consistent with previous work (e.g., Miller, Rohleder, & Cole, 2009; Shih et al., 2006), allows us to reduce the number of statistical analyses, and provides a comprehensive index of adolescents’ abrasive social experiences across relationship domains.
Inflammatory parameters
At each visit, we collected peripheral blood at the morning laboratory visits following an overnight fast to measure various aspects of inflammation. We assessed the extent to which participants’ monocytes reacted to microbial challenge by culturing whole blood with lipopolysaccharide (LPS), a bacterial stimulus that selectively engages these cells. Whole blood was drawn into lithium-heparin Vacutainers (Becton-Dickinson, Oakville, Ontario, Canada), diluted in a 10:1 ratio with saline, and incubated with 50 ng/ml of LPS (Sigma, St. Louis, MO) for 6 hr at 37°C in 5% carbon dioxide. The supernatants were collected and frozen at −80°C until analysis. We measured IL-6 production in duplicate with DuoSet ELISA Development Systems kits (R&D Systems, Minneapolis, MN), which have a minimum detection threshold of 0.7 ng/ml. Across runs, the average intra-assay coefficient of variation for duplicate IL-6 measurements was 1.85%.
Next, we measured the extent to which participants’ monocytes were sensitive to anti-inflammatory signals from cortisol. To do this, we quantified IL-6 production in cells that had been coincubated with LPS and cortisol. As noted, cortisol conveys anti-inflammatory signals to immune cells, and this assay measured the monocytes’ ability to respond to those signals by dampening IL-6 production. Blood was diluted in a 10:1 ratio with saline and dispensed into culture plates (Sigma Chemicals, St. Louis, MO) with LPS (50 ng/ml). Doses of hydrocortisone were added to four of the wells in varying concentrations (2.76×10−5 M, 2.76×10−6 M, 2.76×10−7 M, and 2.76×10−8 M). The fifth well contained only LPS. After 6 hr of incubation at 37 °C in 5% CO2, the supernatants were collected and frozen until analysis. IL-6 levels were measured in duplicate using the DuoSet ELISA Development Systems kits described above (R&D Systems). We created dose-response curves for each participant and used these curves to calculate the area under the curve. This value is inversely proportional to glucocorticoid sensitivity, and as such, larger values indicate that the immune cells are less sensitive to cortisol’s anti-inflammatory signals.
Results
Construction of clusters
Our first research aim was to evaluate whether we could create inflammatory phenotypes at each of the six laboratory visits using the inflammatory measures described earlier. For each laboratory visit, we created a set of clusters using the two inflammatory measures, sensitivity to glucocorticoids and production of IL-6 following LPS stimulation, using k-means clustering with n = 2 clusters. (We restricted the number of clusters to two given our sample size so that we could create groups that were of a meaningful size.) Prior to the construction of clusters, we examined patterns of missing data across the six laboratory visits. Little’s missing completely at random test revealed that the data were missing completely at random, χ2 (562) = 565.6, p .45. As such, we used maximum likelihood estimation and all available data to impute missing data. This approach is recommended for handling missing data in longitudinal studies because it improves power and results in less biased parameter estimates than other techniques (Graham, 2009; Jelicic, Phelps, & Lerner, 2009). Assessments of stimulated IL-6 production and sensitivity to glucocorticoids were not correlated within a time point (rs <.10, ps >.23), with the exception of measures at Visit 4 (r = .23, p = .005).
Table 1 describes the characteristics of the inflammatory phenotypes that emerged. At each visit, two clusters reliably emerged, and for ease of discussion, we will refer to these clusters as more inflammatory and less inflammatory to differentiate the two groups. Across the assessments, 23.1% were categorized in the same cluster at each laboratory visit, and 49.0% were categorized in the same cluster in at least five out of six visits. Across laboratory visits, clusters generally differed from each other in mean levels of stimulated IL-6 production and resistance to glucocorticoids, suggesting that the clustering procedure was successful in separating participants into two distinct inflammatory clusters.
Table 1.
Tests of mean differences in inflammatory measures across clusters
| Visit | Less Inflammatory Cluster
|
More Proinflammatory Cluster
|
t Tests
|
|||||
|---|---|---|---|---|---|---|---|---|
| n | Prod. of IL-6 Follow. LPS Stim | Sensitivity to Glucocort | n | Prod. of IL-6 Follow. LPS Stim | Sensitivity to Glucocort | Prod. of IL-6 Follow. LPS Stim | Sensitivity to Glucocort | |
| 1 | 78 | 32370.3 | 0.135 | 69 | 57304.6 | 0.166 | 16.8*** | 2.52* |
| 2 | 87 | 38408.3 | 0.118 | 60 | 62058.7 | 0.184 | 11.7*** | 5.36*** |
| 3 | 73 | 43973.3 | 0.096 | 74 | 48987.5 | 0.198 | 1.84† | 13.2*** |
| 4 | 98 | 40478.3 | 0.123 | 49 | 62521.6 | 0.220 | 10.7*** | 8.41*** |
| 5 | 69 | 37303.0 | 0.116 | 78 | 61208.8 | 0.169 | 12.3*** | 4.80*** |
| 6 | 44 | 37066.5 | 0.106 | 103 | 57499.8 | 0.168 | 8.45*** | 8.02*** |
Note: IL-6, Interleukin-6; LPS, lipopolysaccharide.
p <.10.
p <.05.
p <.01.
p <.001.
Early life adversity and cluster membership
Our second and third aims were to determine whether early life adversity was associated with membership in the more inflammatory cluster, and whether social stress over the follow-up mediated or moderated this association. We estimated a series of multilevel logistic models with HLM 6.08. Logit-linear structural models were used to predict the probability of being in the more inflammatory cluster given early life adversity and current social stress (Raudenbush, 2001; West, Ryu, Kwok, & Cham, 2011). The logit function also returns the odds ratio (log-odds) of being in the more inflammatory cluster as a function of each predictor. Odds ratios greater than one indicate greater odds of being in the more inflammatory cluster, and odds ratios less than one indicate reduced odds of being in the more inflammatory cluster. All models were estimated with random slopes, in which Level 2 error terms were allowed to vary freely, and models were estimated with robust standard errors.
Model 1 assessed whether early life adversity predicted inflammatory cluster membership at study entry and over the follow-up. The within-person (Level 1) models included a variable reflecting months since study entry (TIMEi,j; uncentered). The between-person (Level 2) models included early life adversity (ELAj; centered). We then tested whether the results from Model 1 persisted when between-person demographic variables, age at study entry and race, were also included at Level 2.
In Model 2, we assessed whether any associations between early life adversity and inflammatory cluster membership were mediated by social stress experienced over the follow-up. Model 2 again consisted of time at Level 1 and early life adversity at Level 2. To determine whether fluctuations in social stress across assessments mediated links between early life adversity and cluster membership, social stress at each assessment (SStressi;j) was entered as a within-person (group centered; Level 1) variable. To determine whether fluctuations in social stress across assessments moderated links between early life adversityand cluster membership, we inspected the interaction between social stress (Level 1) and early life adversity (Level 2). We then tested whether results for Model 2 persisted when demographics variables were also entered at Level 2.
Model 2 at Level 1,
| (1) |
At Level 2,
Does early life adversity predict inflammatory cluster membership?
A two-level model was run to determine whether early life adversity exposure predicted inflammatory cluster membership over the study follow-up. The results are presented in Table 2. At baseline, participants were more likely to be in the less versus more proinflammatory cluster, β10 = −0.461, SE=0.167, p = .007, OR = 0.631, 95% CI (0.454, 0.877), but over the follow-up there was an increasing probability of participants switching to the more inflammatory cluster, β10 = 0.023, SE = 0.007, p = .002, OR = 1.02, 95% CI (1.01, 1.04).
Table 2.
Prediction of inflammatory cluster membership at study entry and over time from ELA
| Variable | Coefficient | b | SE | p | Odds Ratio | 95% CI |
|---|---|---|---|---|---|---|
| For intercept | ||||||
| Intercept | B00 | −0.461 | 0.167 | .007 | 0.631 | 0.454, 0.877 |
| ELA | B01 | 0.507 | 0.197 | .012 | 1.66 | 1.24, 2.45 |
| For time slope | ||||||
| Time | B10 | 0.023 | 0.007 | .002 | 1.02 | 1.01, 1.04 |
| Time×ELA | B11 | −0.008 | 0.009 | .340 | 0.992 | 0.975, 1.01 |
Note: The results of the multilevel model predicting inflammatory cluster membership at study entry and over time from early life adversity (ELA) exposure are presented. Time (months since study entry) was entered uncentered at Level 1; ELA was entered grand mean centered at Level 2.
Early life adversity was associated with increased probability of being in the more inflammatory cluster at baseline, β01 = 0.507, SE = 0.197, p = .012, OR = 1.66, 95% CI (1.24, 2.45), but did not predict changes in cluster membership over the follow-up (β11 = −0.008, SE = 0.009, p =.340; see Figure 1). This finding suggests that early life adversity may be associated with a phenotypic “set point” that is consistent over time.
Figure 1.
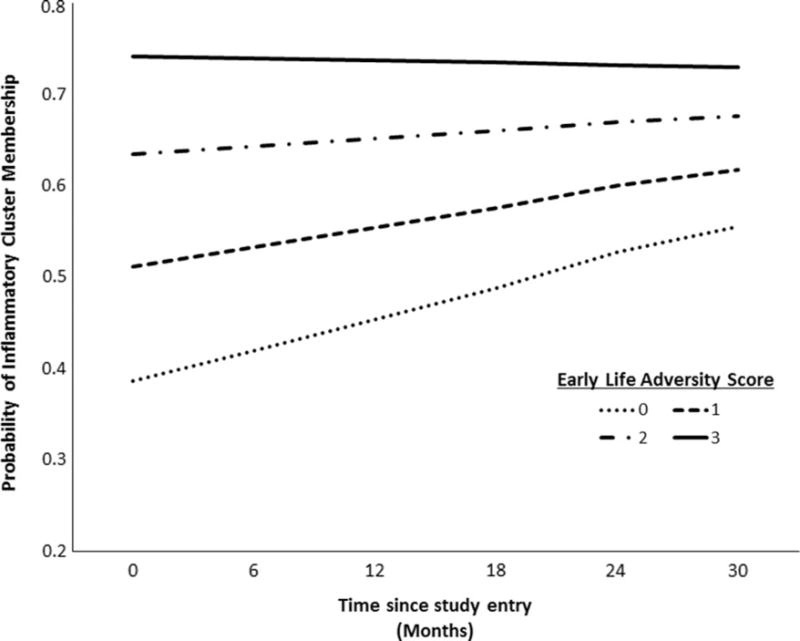
Early life adversity predicting inflammatory cluster membership across visits. The x-axis represents months from baseline, and y-axis represents the log-odds of being in the inflammatory cluster at a given point in time over the follow-up. Early life adversity predicts the probability of being in the inflammatory cluster at study entry, such that greater exposure to early life adversity is associated with increased probability of inflammatory cluster membership. Early life adversity did not predict probability of inflammatory cluster membership over the follow-up, however, suggesting a constant effect of early life adversity exposure over the follow-up.
This model was followed by entering participant demographics (age and race) at Level 2. Neither participant age nor race predicted inflammatory cluster membership, either at study entry or over the follow-up. Early life adversity continued to predict cluster membership at study entry with demographics included in the model.
Does social stress mediate or moderate the association between early life adversity and inflammatory cluster membership?
Next, a two-level model was used to determine whether social stress mediated or moderated the link between early life adversity and probability of membership in the more inflammatory cluster, as described above. The results are presented in Table 3.
Table 3.
Prediction of inflammatory cluster membership at study entry and over time from ELA and social stress
| Variable | Coefficient | b | SE | p | Odds Ratio | 95% CI |
|---|---|---|---|---|---|---|
| Intercept | ||||||
| Model intercept | B00 | −0.449 | 0.167 | .008 | 0.638 | 0.459, 0.887 |
| ELA | B01 | 0.514 | 0.198 | .011 | 1.67 | 1.13, 2.47 |
| Slope of time | ||||||
| Intercept | B10 | 0.022 | 0.007 | .003 | 1.02 | 1.01, 1.04 |
| ELA×Time | B11 | −0.009 | 0.009 | .327 | 0.991 | 0.975, 1.01 |
| Slope social stress | ||||||
| Intercept | B20 | 0.127 | 0.366 | .729 | 1.14 | 0.552, 2.34 |
| ELA×Social Stress Slope | B21 | −0.684 | 0.424 | .109 | 0.505 | 0.219, 1.17 |
Note: The results from the multilevel model predicting inflammatory cluster membership at study entry and over the follow-up from early life adversity (ELA) exposure and social stress over the follow-up are presented. Time (months since study entry) was entered uncentered at Level 1, social stress was entered group centered at Level 1, and early life adversity was entered grand mean centered at Level 2.
As shown, the patterns above remained significant when social stress was included in the models, suggesting that greater social stress over the study follow-up does not mediate links between early life adversity and inflammatory cluster membership. In these models, early life adversity continued to be associated with greater probability of being in the more inflammatory cluster at baseline (β01 = 0.514, SE 0.198, p = 011), independent of fluctuations in social stress over the follow-up. (As before, early life adversity did not predict change in inflammatory cluster membership over the follow-up, β11 = −0.009, SE = 0.009, p = 327.) For every one-unit increase in early life adversity, there was a 67% increase in the probability of being in the more inflammatory cluster.
Social stress over the follow-up, however, did not independently predict inflammatory cluster membership (β20 = 0.127, SE = 0.366, p = .729). In addition, social stress over the follow-up did not significantly interact with early life adversity to predict inflammatory cluster membership (β21 = −0.684, SE = 0.424, p = .11), a result that is inconsistent with the hypothesis that current social stress accentuates or attenuates the negative effects associated with early life adversity.
Again, this model was followed by entering participant demographics (age and race) at Level 2. Neither age nor race predicted inflammatory cluster membership. Early life adversity continued to predict more inflammatory cluster membership at study entry but not over the follow-up, and social stress over the follow-up did not predict inflammatory cluster membership, either at baseline or over the follow-up.
Discussion
According to the biological embedding model (Miller et al., 2011), individuals who experience early life adversity (e.g., through exposure to poverty, family stress, or maltreatment) will show evidence of a proinflammatory phenotype, characterized by both larger cytokine responses to threats and lower sensitivity to anti-inflammatory signals from cortisol. To date, however, researchers have examined these inflammatory characteristics in isolation, ignoring the coupling of these features that is thought to be detrimental for long-term health. This trend is perhaps not surprising; even in the present sample, the two indices are only significantly correlated at one out of six time points. Nevertheless, our findings suggest that these indices of the proinflammatory phenotype do aggregate, and participants can be classified based on how much IL-6 their cells produce after stimulation with bacterial products and how effectively those cells respond to cortisol’s anti-inflammatory properties. In many ways, the proinflammatory phenotype can be compared to a car that has a healthy accelerator and sluggish brakes. Although this phenotype is likely to confer advantages within certain contexts (e.g., when there is infection or wounding that require acute, intense responses), the biological embedding model suggests that if sustained, this proinflammatory phenotype would give rise to low-level, nonresolving inflammation. Acting in concert with genetic liabilities and other exposures, this inflammation could increase vulnerability to a variety of mental and physical health problems, including anxiety, depression, substance use, diabetes, heart disease, and autoimmune conditions (see Nusslock & Miller, 2016).
The present study provides the most direct test of the biological embedding model to date. In the present study, we found that we could successfully categorize adolescent girls on the basis of these two indices of inflammatory activity. In addition, as expected, we found considerable (although not complete) stability in cluster membership across the 2.5-year period. This stability is consistent with the hypothesis that early experiences calibrate monocytes in such a way that they consistently demonstrate altered response patterns in middle to late adolescence. At the same time, however, there was some evidence of phenotypic plasticity, as some individuals switched clusters over the course of the study. In addition, we found that exposure to early life adversity predicted increased odds of being categorized in the more inflammatory cluster, and this finding did not change over the six laboratory visits. One question that remains is whether these effects would be evident if participants were sampled many years later, well into adulthood and at a time when health problems have started to emerge. How sustained are the effects of a difficult childhood? How reversible are these effects as life circumstances change? These questions remain some of the central unresolved issues that underlie many studies in developmental psychopathology, and it is an open question as to whether individuals can change trajectories of their inflammatory phenotypes based on subsequent life experiences.
We did not find evidence for the role of ongoing social stress as a mediator of the link between early adversity and inflammatory cluster membership. This finding is inconsistent with the large body of literature linking current social stress with inflammatory outcomes (see reviews by Kiecolt-Glaser, Gouin, & Hantsoo, 2010; Slavich & Irwin, 2014). However, previous studies have focused largely on predicting inflammatory measures in isolation rather than aggregated clusters. This difference is important because it may be that social stressors modulate certain aspects of inflammatory activity while setting into motion other processes, which offset these effects. This kind of allostasis (McEwen & Seeman, 1999) might be especially likely to occur in otherwise healthy young people, such as those in our sample. However, with aging, these counterregulatory processes might break down, leading to more widespread immune modulation by social stress. Much more research in this area is needed to help identify the relative roles of early experience and current stress on the proinflammatory phenotype.
We also did not find evidence that early adversity sensitized girls’ responses to social stress. This finding contrasts with John-Henderson et al. (2016), who found support for this hypothesis in midlife adults. Perhaps a lack of social support is sufficient to disrupt various aspects of the inflammatory response, but it may not be sufficient to alter the coupling of inflammatory measures that was the focus of the present study. The pattern of results also did not suggest that positive social conditions could offset or reverse the proinflammatory traits associated with early adversity. It may be that other social experiences that were not measured in the present study, such as more nuanced aspects of close relationships, or outside influences, such as helpful community mentors, could change the risk for being categorized in the more inflammatory cluster. Another possibility is that a more powerful change to ongoing social experiences is needed in order to alter the inflammatory trajectories set in motion early in life. Some emerging evidence using family-based interventions holds promise in this regard (Miller, Brody, Yu, & Chen, 2014). In this study, Miller et al. found that African American youth who participated in a psychosocial intervention focused on improving parenting, strengthening family relationships, and building youth competencies had lower levels of six cytokines at age 19 compared to peers in the control group. Not all youth who experience early adversity will be able to benefit from supportive interventions, however, so it will be important to consider other naturally occurring protective factors that could be bolstered easily without the need for large-scale interventions.
These present study also highlights the utility of incorporating person-centered analyses, such as cluster analysis, into tests of connections between social stressors and inflammatory characteristics. The overwhelming majority of studies on psychosocial links to inflammation and health rely on variable-centered analytical approaches (e.g., regression-based approaches). By taking a person-centered approach, we were able to identify groups of adolescents who would not have been identified with a variable-centered analytic model. We encourage other researchers to consider how person-centered analytic methods might yield new insights into the ways in which social experiences relate to health outcomes. For instance, many researchers assess a variety of positive and negative psychosocial experiences, and a cluster analysis approach might reveal subgroups of individuals who differ in their levels of positive and negative social experience. It might be the unique patterning of positive and negative experience that best predicts indicators of health.
Limitations and future directions
Although this study adds to our understanding of the ways in which early life adversity might set the stage for a proinflammatory phenotype, several limitations should be addressed in future research. First, our study included only healthy adolescent girls, mostly of White and Asian ethnicities, and we do not know whether the findings would generalize to samples with adolescent boys or with adolescents of other ethnicities. Although we have no reason to believe that the ability to form clusters on the basis of inflammatory measures would differ as a function of race or gender, we are cautious about generalizing our findings to all adolescents until we can replicate the patterns in a more diverse sample. Similarly, we focused on a narrow window of childhood (recruiting adolescents who were 15 to 19 years old at study entry), and we will be interested to see if similar inflammatory clusters emerge in younger children, or if additional time is needed in order to distinguish children by inflammatory phenotypes.
Second, our measure of early life adversity was obtained at the baseline laboratory visit and does not allow us to comment fully on the prospective nature of early life adversity and later inflammatory phenotypes. That said, we selected indices of adversity that would be less likely to suffer from memory biases (e.g., renting vs. owning a home and parental education). Nevertheless, an ideal study design would include measures of early adversity that are collected closer to the early childhood period to alleviate any concerns about reporting biases.
It is also important to note that our sample was drawn from a community of adolescents living in the Vancouver, British Columbia, area. This sample characteristic bears noting for two reasons. First, the resulting sample is relatively more homogenous than many adolescent samples that would be drawn from cities in the United States, particularly with regard to ethnicity. Second, adolescent experiences of early life adversity in Canada are likely to be more limited than what many adolescents experience in the United States. Canadian and US cities differ dramatically in economic and social inequality, with less pronounced inequality in Canadian cities compared to those in the United States, and these differences could have real implications for health and mortality (e.g., Ross et al., 2000). Out of the five indicators of early life adversity used in the present study, no adolescent experienced more than three exposures to adversity, and approximately 40% of adolescents experienced no adversities at all. In contrast, evidence from US samples suggests that the experience of early adversity is quite common. In one study, nearly 75% of children experienced at least one major adversity by the age of 16, and nearly 50% of children in the United States have experienced multiple adversities (Kessler, Davis, & Kendler, 1997). Further, almost half of children in the United States live in poverty or low-income households (Children’s Defense Fund, 2012). We believe it will be especially important to test our hypothesis about the proinflammatory phenotype and early life adversity in samples with a greater range of exposures to early adversity.
Our study focused on two specific measures of inflammatory activity, production of IL-6 following exposure to LPS and glucocorticoid insensitivity, because these indices reflect the features described by the biological embedding model. Nevertheless, we would argue that before definitive conclusions are made, a larger battery of inflammatory measures should be included in the construction of inflammatory phenotypes. Other stimuli must be used to evoke cytokine production (e.g., viral particles, debris and lipids, and necrosis signals) and other molecules (e.g., IL-10 and transforming growth factor-β) to inhibit it. Nevertheless, we believe this issue will be a complicated one to sort out and will require large sample sizes with repeated assessments of multiple indices of inflammatory measures. When considering which measures should be used to construct inflammatory phenotypes, researchers will need to take into account the sample characteristics and developmental stage of their participants. For example, in childhood and adolescent samples, it may not be realistic to include measures of low-grade inflammatory markers like C-reactive protein (which tend to be undetectable in many children unless they are obese or have major health problems). However, these markers may aggregate with other characteristics of inflammatory activity in adult samples. The biological embedding model would predict that by adulthood, the clusters in the present study would expand to incorporate biomarkers of nonresolving inflammation. Long-term longitudinal studies will be especially important for this research, in part because the onset of chronic illness will become evident over longer spans of time, and it will be possible to evaluate whether a proinflammatory phenotype confers increased risk for developing health problems of clinical relevance (e.g., atherosclerosis, diabetes, and depression).
In summary, the findings from the present study suggest that even in adolescence, when children are still relatively healthy, characteristics of inflammatory activity can be clustered to form inflammatory phenotypes, and individuals in these phenotypes differ in their experience of early life adversity. Although in the present study we did not find evidence for accentuating or mitigating effects of ongoing social stress, there may be other factors that could offset the predicted trajectories formed as a result of exposure to early adversity.
Acknowledgments
This research was supported by grants from the Canadian Institutes of Health Research (67191) and the National Institute of Child Health and Human Development HD058502 (to G.E.M.) and HD076563 (to K.B.E).
References
- Appleyard K, Egeland B, van Dulmen MHM, Sroufe LA. When more is not better: The role of cumulative risk in child behavior outcomes. Journal of Child Psychology and Psychiatry. 2005;46:235–245. doi: 10.1111/j.1469-7610.2004.00351.x. [DOI] [PubMed] [Google Scholar]
- Azad MB, Lissitsyn Y, Miller GE, Becker AB, HayGlass KT, Kozyrskyj AL. Influence of socioeconomic status trajectories on innate immune responsiveness in children. PLOS ONE. 2012;7:e38669. doi: 10.1371/journal.pone.0038669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barker DJ. Fetal and infant origins of adult disease. London: BMJ Publishing Group; 1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Black JE, Jones TA, Nelson CA, Greenough WT. Neuronal plasticity and the developing brain. In: Alessi NE, Coyle JT, Harrison SI, Eth S, editors. Handbook of child and adolescent psychiatry: Vol 6 Basic psychiatric science and treatment. New York: Wiley; 1998. pp. 31–53. [Google Scholar]
- Blair C, Raver CC. Child development in the context of adversity: Experiential canalization of brain and behavior. American Psychologist. 2012;67:309–318. doi: 10.1037/a0027493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busillo JM, Cidlowski JA. The five Rs of glucocorticoid action during inflammation: Ready, reinforce, repress, resolve, and restore. Trends in Endocrinology & Metabolism. 2013;24:109–119. doi: 10.1016/j.tem.2012.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cameron NM, Champagne FA, Parent C, Fish EW, Ozaki-Kuroda K, Meaney MJ. The programming of individual differences in defensive responses and reproductive strategies in the rat through variations in maternal care. Neuroscience & Biobehavioral Reviews. 2005;29:843–865. doi: 10.1016/j.neubiorev.2005.03.022. [DOI] [PubMed] [Google Scholar]
- Chen E, Langer DA, Raphaelson YE, Matthews KA. Socioeconomic status and health in adolescents: The role of stress interpretations. Child Development. 2004;75:1039–1052. doi: 10.1111/j.1467-8624.2004.00724.x. [DOI] [PubMed] [Google Scholar]
- Children’s Defense Fund. The state of America’s children. Washington, DC: Author; 2012. [Google Scholar]
- Coe CL, Lubach GR. Mother–infant interactions and the development of immunity from conception through weaning. In: Ader R, editor. Psychoneuroimmunology. 4th. London: Academic Press; 2007. pp. 455–474. [Google Scholar]
- Csikszentmihalyi M, Larson R. Being adolescent: Conflict and growth in the teenage years. New York: Basic Books; 1984. [Google Scholar]
- Danesh J, Collins R, Appleby P, Peto R. Association of fibrinogen, C-reactive protein, albumin, or leukocyte count with coronary heart disease: Meta-analyses of prospective studies. Journal of the American Medical Association. 1998;279:1477–1482. doi: 10.1001/jama.279.18.1477. [DOI] [PubMed] [Google Scholar]
- Dixon D, Meng H, Goldberg R, Schneiderman N, Delamater A. Stress and body mass index each contributes independently to tumor necrosis factor-production in prepubescent Latino children. Journal of Pediatric Nursing. 2009;24:378–388. doi: 10.1016/j.pedn.2008.02.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodge KA, Pettit GS, Bates JE. Socialization mediators of the relation between socioeconomic status and child conduct problems. Child Development. 1994;65:649–665. [PubMed] [Google Scholar]
- Ehrlich KB, Miller GE, Chen E. Childhood adversity and adult physical health. In: Cicchetti D, editor. Developmental psychopathology. 3rd. Vol. 4. Hoboken, NJ: Wiley; 2016. pp. 1–42. [Google Scholar]
- Ehrlich KB, Miller GE, Rohleder N, Adam EK. Trajectories of relationship stress and inflammatory processes in adolescence. Development and Psychopathology. 2016;28:127–138. doi: 10.1017/S0954579415000334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuligni AJ, Telzer EH, Bower J, Cole SW, Kiang L, Irwin MR. A preliminary study of daily interpersonal stress and C-reactive protein levels among adolescents from Latin American and European backgrounds. Psychosomatic Medicine. 2009;71:329–333. doi: 10.1097/PSY.0b013e3181921b1f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furman W, Buhrmester D. Age and sex differences in the perceptions of networks of personal relationships. Child Development. 1992;63:103–115. doi: 10.1111/j.1467-8624.1992.tb03599.x. [DOI] [PubMed] [Google Scholar]
- Furman W, Shaffer L. The role of romantic relationships in adolescent development. In: Florsheim P, editor. Adolescent romantic relations and sexual behavior: Theory, research, and practical implications. Mahwah, NJ: Erlbaum; 2003. pp. 3–22. [Google Scholar]
- Gluckman PD, Hanson MA. The conceptual basis for developmental origins of health and disease. In: Gluckman P, Hanson M, editors. Developmental origins of health and disease. New York: Cambridge University Press; 2006. pp. 33–50. [Google Scholar]
- Graham J. Missing data analysis: Making it work in the real world. Annual Review of Psychology. 2009;60:549–576. doi: 10.1146/annurev.psych.58.110405.085530. [DOI] [PubMed] [Google Scholar]
- Hammen C. The generation of stress in the course of unipolar depression. Journal of Abnormal Psychology. 1991;100:555–561. doi: 10.1037//0021-843x.100.4.555. [DOI] [PubMed] [Google Scholar]
- Hertzman C. The biological embedding of early experience and its effects on health in adulthood. Annals of the New York Academy of Sciences. 1999;896:85–95. doi: 10.1111/j.1749-6632.1999.tb08107.x. [DOI] [PubMed] [Google Scholar]
- Hotamisligil GS. Inflammation and metabolic disorders. Nature. 2006;444:860–867. doi: 10.1038/nature05485. [DOI] [PubMed] [Google Scholar]
- Jelicic H, Phelps E, Lerner R. Use of missing data methods in longitudinal studies: The persistence of bad practices in developmental psychology. Developmental Psychology. 2009;45:1195–1199. doi: 10.1037/a0015665. [DOI] [PubMed] [Google Scholar]
- John-Henderson NA, Marsland AL, Kamarck TW, Muldoon MF, Manuck SB. Childhood socioeconomic status and the occurrence of recent negative life events as predictors of circulating and stimulated levels of IL-6. Psychosomatic Medicine. 2016;78:91–101. doi: 10.1097/PSY.0000000000000262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaptoge S, Di Angelantonio E, Lowe G, Pepys MB, Thompson SG, Collins R, et al. C-reactive protein concentration and risk of coronary heart disease, stroke, and mortality: An individual participant meta-analysis. Lancet. 2010;375:132–140. doi: 10.1016/S0140-6736(09)61717-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kessler RC, Davis CG, Kendler KS. Childhood adversity and adult psychiatry disorder in the U.S. National Comorbidity Survey. Psychological Medicine. 1997;27:1101–1119. doi: 10.1017/s0033291797005588. [DOI] [PubMed] [Google Scholar]
- Kessler RC, McLaughlin KA, Green JG, Gruber MJ, Sampson NA, Zaslavsky AM, et al. Childhood adversities and adult psy chopathology in the WHO World Mental Health Surveys. British Journal of Psychiatry. 2010;197:378–385. doi: 10.1192/bjp.bp.110.080499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiecolt-Glaser JK, Gouin JP, Hantsoo L. Close relationships, inflammation, and health. Neuroscience & Biobehavioral Reviews. 2010;35:33–38. doi: 10.1016/j.neubiorev.2009.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Libby P, Ridker PM, Hansson GK. Inflammation in atherosclerosis: From pathophysiology to practice. Journal of the American College of Cardiology. 2009;54:2129–2138. doi: 10.1016/j.jacc.2009.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Libby P, Theroux P. Pathophysiology of coronary artery disease. Circulation. 2005;111:3481–3488. doi: 10.1161/CIRCULATIONAHA.105.537878. [DOI] [PubMed] [Google Scholar]
- Lupien SJ, McEwen BS, Gunnar MR, Heim C. Effects of stress throughout the lifespan on the brain, behavior and cognition. Nature Reviews Neuroscience. 2009;10:434–445. doi: 10.1038/nrn2639. [DOI] [PubMed] [Google Scholar]
- Lynch J, Smith GD. A life course approach to chronic disease epidemiology. Annual Review of Public Health. 2005;26:1–35. doi: 10.1146/annurev.publhealth.26.021304.144505. [DOI] [PubMed] [Google Scholar]
- Markham JA, Koenig JI. Prenatal stress: Role in psychotic and depressive diseases. Psychopharmacology. 2011;214:89–106. doi: 10.1007/s00213-010-2035-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marques AH, Silverman MN, Sternberg EM. Glucocorticoid dysregulations and their clinical correlates: From receptors to therapeutics. Annals of the New York Academy of Sciences. 2009;1179:1–18. doi: 10.1111/j.1749-6632.2009.04987.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McEwen BS, Seeman T. Protective and damaging effects of mediators of stress: Elaborating and testing the concepts of allostasis and allostatic load. Annals of the New York Academy of Sciences. 1999;896:30–47. doi: 10.1111/j.1749-6632.1999.tb08103.x. [DOI] [PubMed] [Google Scholar]
- Meaney MJ. Maternal care, gene expression, and the transmission of individual differences in stress reactivity across generations. Annual Review of Neuroscience. 2001;24:1161–1192. doi: 10.1146/annurev.neuro.24.1.1161. [DOI] [PubMed] [Google Scholar]
- Meyer U, Feldon J, Dammann O. Schizophrenia and autism: Both shared and disorder-specific pathogenesis via perinatal inflammation? Pediatric Research. 2011;69:26R–33R. doi: 10.1203/PDR.0b013e318212c196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller GE, Cole SW. Clustering of depression and inflammation in adolescents previously exposed to childhood adversity. Biological Psychiatry. 2012;72:34–40. doi: 10.1016/j.biopsych.2012.02.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller GE, Brody GH, Yu T, Chen E. A family oriented psychosocial intervention reduces inflammation in low-SES African American youth. Proceedings of the National Academy of Sciences. 2014;111:11287–11292. doi: 10.1073/pnas.1406578111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller GE, Chen E. The biological residue of childhood poverty. Child Development Perspectives. 2013;7:67–73. doi: 10.1111/cdep.12021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller GE, Chen E, Fok AK, Walker H, Lim A, Nicholls EF, et al. Low early-life social class leaves a biological residue manifested by decreased glucocorticoid and increased proinflammatory signaling. Proceedings of the National Academy of Sciences. 2009;106:14716–14721. doi: 10.1073/pnas.0902971106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller GE, Chen E, Parker KJ. Psychological stress in childhood and susceptibility to the chronic diseases of aging: Moving toward a model of behavioral and biological mechanisms. Psychological Bulletin. 2011;137:959–997. doi: 10.1037/a0024768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller GE, Chen E, Sze J, Marin T, Arevalo JMG, Doll R, et al. A genomic fingerprint of chronic stress in humans: Blunted glucocorticoid and increased NF-κB signaling. Biological Psychiatry. 2008;64:266–272. doi: 10.1016/j.biopsych.2008.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller GE, Chen E, Zhou ES. If it goes up, must it come down? Chronic stress and the hypothalamic–pituitary–adrenocortical axis in humans. Psychological Bulletin. 2007;133:25–45. doi: 10.1037/0033-2909.133.1.25. [DOI] [PubMed] [Google Scholar]
- Miller GE, Rohleder N, Cole SW. Chronic interpersonal stress predicts activation of pro-and anti-inflammatory signaling pathways six months later. Psychosomatic Medicine. 2009;71:57–62. doi: 10.1097/PSY.0b013e318190d7de. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy MLM, Slavich GM, Chen E, Miller GE. Targeted rejection predicts decreased anti-inflammatory gene expression and increased symptom severity in youth with asthma. Psychological Science. 2015;26:111–121. doi: 10.1177/0956797614556320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy MLM, Slavich GM, Rohleder N, Miller GE. Targeted rejection triggers differential pro-and anti-inflammatory gene expression in adolescents as a function of social status. Clinical Psychological Science. 2013;1:30–40. doi: 10.1177/2167702612455743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nathan C, Ding A. Nonresolving inflammation. Cell. 2010;140:871–882. doi: 10.1016/j.cell.2010.02.029. [DOI] [PubMed] [Google Scholar]
- Nusslock R, Miller GE. Early-life adversity and physical and emotional health across the lifespan: A neuroimmune network hypothesis. Biological Psychiatry. 2016;80:23–32. doi: 10.1016/j.biopsych.2015.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poulton R, Caspi A, Milne BJ, Thomson WM, Taylor A, Sears MR, et al. Association between children’s experience of socioeconomic disadvantage and adult health: A life-course study. Lancet. 2002;360:1640–1645. doi: 10.1016/S0140-6736(02)11602-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raison CL, Miller AH. When not enough is too much: The role of insufficient glucocorticoid signaling in the pathophysiology of stressrelated disorders. American Journal of Psychiatry. 2003;160:1554–1565. doi: 10.1176/appi.ajp.160.9.1554. [DOI] [PubMed] [Google Scholar]
- Raudenbush SW. Toward a coherent framework for comparing trajectories of individual change. In: Collins LM, Sayer AG, editors. New methods for the analysis of change: Decade of behavior. Washington, DC: APA; 2001. pp. 35–64. [Google Scholar]
- Repetti RL, Robles TF, Reynolds BR. Allostatic processes in the family. Development and Psychopathology. 2011;23:921–938. doi: 10.1017/S095457941100040X. [DOI] [PubMed] [Google Scholar]
- Repetti RL, Taylor SE, Seeman TE. Risky families: Family social environments and the mental and physical health of offspring. Psychological Bulletin. 2002;128:330–366. [PubMed] [Google Scholar]
- Ridker PM. Inflammatory biomarkers and risks of myocardial infarction, stroke, diabetes, and total mortality: Implications for longevity. Nutrition Reviews. 2007;65:S253–S259. doi: 10.1111/j.1753-4887.2007.tb00372.x. [DOI] [PubMed] [Google Scholar]
- Rivest S. Chapter 4—Interactions between the immune and neuroendocrine systems. Progress in Brain Research. 2010;181:43–53. doi: 10.1016/S0079-6123(08)81004-7. [DOI] [PubMed] [Google Scholar]
- Ross NA, Wolfson MC, Dunn JR, Berthelot J, Kaplan GA, Lynch JW. Relation between income inequality and mortality in Canada and in the United States: Cross sectional assessment using census data and vital statistics. British Medical Journal. 2000;320:898–902. doi: 10.1136/bmj.320.7239.898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudolph KD, Hammen C. Age and gender as determinants of stress exposure, generation, and reactions in youngsters: A transactional perspective. Child Development. 1999;70:660–677. doi: 10.1111/1467-8624.00048. [DOI] [PubMed] [Google Scholar]
- Schreier HMC, Roy LB, Frimer LT, Chen E. Family chaos and adolescent inflammatory profiles: The moderating role of socioeconomic status. Psychosomatic Medicine. 2014;76:460–467. doi: 10.1097/PSY.0000000000000078. [DOI] [PubMed] [Google Scholar]
- Shih JH, Eberhart NK, Hammen CL, Brennan PA. Differential exposure and reactivity to interpersonal stress predict sex differences in adolescent depression. Journal of Clinical Child and Adolescent Psychology. 2006;35:103–115. doi: 10.1207/s15374424jccp3501_9. [DOI] [PubMed] [Google Scholar]
- Shonkoff JP, Boyce WT, McEwen BS. Neuroscience, molecular biology, and the childhood roots of health disparities: Building a new framework for health promotion and disease prevention. Journal of the American Medical Association. 2009;301:2252–2259. doi: 10.1001/jama.2009.754. [DOI] [PubMed] [Google Scholar]
- Shonkoff JP, Garner AS, The Committee on Psychosocial Aspects of Child and Family Health, Committee on Early Childhood, Adoption, and Dependent Care, and Section on Developmental and Behavioral Pediatrics. Siegel BS, et al. The lifelong effects of early childhood adversity and toxic stress. Pediatrics. 2012;129:e232–e246. doi: 10.1542/peds.2011-2663. [DOI] [PubMed] [Google Scholar]
- Slavich GM, Irwin MR. From stress to inflammation and major depressive disorder: A social signal transduction theory of depression. Psychological Bulletin. 2014;140:774–815. doi: 10.1037/a0035302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smetana JG, Campione-Barr N, Metzger A. Adolescent development in interpersonal and societal contexts. Annual Review of Psychology. 2006;57:255–284. doi: 10.1146/annurev.psych.57.102904.190124. [DOI] [PubMed] [Google Scholar]
- Tolan PH. Community violence exposure and developmental psychopathology. In: Cicchetti D, editor. Developmental psychopathology. 3rd. Vol. 4. Hoboken, NJ: Wiley; 2016. pp. 43–85. [Google Scholar]
- Weiss B, Dodge KA, Bates JE, Pettit GS. Some consequences of early harsh discipline: Child aggression and a maladaptive social information processing style. Child Development. 1992;63:1321–1335. doi: 10.1111/j.1467-8624.1992.tb01697.x. [DOI] [PubMed] [Google Scholar]
- West SG, Ryu E, Kwok OM, Cham H. Multilevel modeling: Current and future applications in personality research. Journal of Personality. 2011;79:2–50. doi: 10.1111/j.1467-6494.2010.00681.x. [DOI] [PubMed] [Google Scholar]
- Yeh ETH, Willerson JT. Coming of age of C-reactive protein: Using inflammation markers in cardiology. Circulation. 2003;107:370–371. doi: 10.1161/01.cir.0000053731.05365.5a. [DOI] [PubMed] [Google Scholar]
- Zhang X, Mosser DM. Macrophage activation by endogenous danger signals. Journal of Pathology. 2008;214:161–178. doi: 10.1002/path.2284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zunszain PA, Hepgul N, Pariante CM. Inflammation and depression. In: Cohen PJ, Sharp T, Lau JYF, editors. Behavioral neurobiology of depression and its treatment. New York: Springer; 2013. pp. 135–151. [Google Scholar]