

Abstract
Background
Several consortia have pursued genome-wide association studies for identifying novel genetic loci for blood pressure (BP), lipids, hypertension, etc. They demonstrated the power of collaborative research through meta-analysis of study-specific results.
Methods and Results
The Gene-Lifestyle Interactions Working Group was formed to facilitate the first large, concerted, multi-ancestry study to systematically evaluate gene-lifestyle interactions. In Stage 1, genome-wide interaction analysis is carried out in 53 cohorts with a total of 149,684 individuals from multiple ancestries. In Stage 2 involving an additional 71 cohorts with 460,791 individuals from multiple ancestries, focused analysis is carried out for a subset of the most promising variants from Stage 1. In all, the study involves up to 610,475 individuals. Current focus is on cardiovascular traits including blood pressure and lipids, and lifestyle factors including smoking, alcohol, education (as a surrogate for socio-economic status), physical activity, psychosocial variables, and sleep. The total sample sizes vary among projects due to missing data. Large scale gene-lifestyle or more generally gene-environment interaction (G×E) meta-analysis studies can be cumbersome and challenging. This paper describes the design and some of the approaches pursued in the interaction projects.
Conclusions
The Gene-Lifestyle Interactions Working Group provides an excellent framework for understanding the lifestyle context of genetic effects and to identify novel trait loci through analysis of interactions. An important and novel feature of our study is that the gene-lifestyle interaction (G×E) results may improve our knowledge about the underlying mechanisms for novel as well as already known trait loci.
Keywords: meta-analysis, lifestyle, genetic epidemiology, Genome Wide Association Study, methodology, Gene-Lifestyle Interactions, GWAS, G×E
Journal Subject Terms: Genetic, Association Studies, Cardiovascular Disease, Risk Factors
Introduction
Remarkable advances in genomics, including the Human Genome Project (HGP) and 1000 Genomes (1000G) Project, have revolutionized methods for genetic dissection of common complex diseases and disease traits. Using Genome-Wide Association Studies (GWAS), large consortia such as CHARGE (Cohorts for Heart and Aging Research in Genomic Epidemiology1, ICBP (International Consortium of Blood Pressure), AGEN Asian Genetic Epidemiology Network), GLGC (Global Lipids Genetics Consortium), and DIAGRAM (Diabetes Genetics Replication and Meta-Analysis) have identified hundreds of common genetic variants associated with many common complex disease traits (https://www.genome.gov/26525384/catalog-of-published-genomewide-association-studies/). However, most of the identified genetic variants explain small proportions of the trait heritability, mostly through small main effects of common variants. It has been recognized that this focus on main effects may have become a barrier to further progress2,3.
Hypertension and dyslipidemia are common complex disorders that contribute to two of the leading causes of death (cardiovascular and cerebrovascular disease) and exhibit significant patterns of health disparity among racial/ancestral groups in the US4,5. While lifestyle factors have long been recognized as risk factors, modulation of the effects of genetic variants by lifestyle factors, and the underlying candidate pathobiological mechanisms have not received much attention. Understanding these genetic modifiers is important because it may provide valuable clues for lifestyle-based interventions which may result in a more successful management of these health conditions through personalized therapies, and may explain part of the “missing heritability”2,6.
The Gene-Lifestyle Interactions Working Group (hereafter referred to as “this study”) investigates gene-lifestyle interactions for uncovering more of the unexplained genetic variance in BP and lipids and for gaining insights into the biological mechanisms influencing these important morbid conditions. We will do this by leveraging the CHARGE infrastructure and the extensive resources of existing studies in multiple ancestries that have data on phenotypes, lifestyle factors, and dense genotype data.
Research involving gene-environment (G×E) interactions is now being reported7–9. These demonstrated the promise of G×E interactions for identifying genetic variants with large effects10–13. For example, mean triglyceride levels are 23 mg/dL lower in physically active versus sedentary individuals (88 vs 111 md/dL) who carry a C-allele at rs2070744 in NOS3, but there is little difference by physical activity status in TT homozygotes11. This shows the utility of G×E interactions for using genetic information to identify subpopulations in whom modifying the environmental factors is beneficial14–16, and that the main effect (of the genetic variant) alone is inadequate to inform lifestyle interventions that need to be personalized based on genotype17,18. In addition, G×E interactions may provide additional insight into biological mechanisms and pathways.
This is the first large, concerted, multi-ancestry study to systematically evaluate gene-lifestyle interactions using data from 610,475 individuals. Large scale G×E meta-analysis studies can be cumbersome and challenging. This paper describes the design and some of the approaches pursued in our ongoing Gene-Lifestyle Interaction projects.
Study Design
The CHARGE Consortium
This study leverages the infrastructure created by the CHARGE consortium1 which created several Working Groups (WGs), an internal wiki site, guidelines for collaboration and authorship, and periodic CHARGE meetings where WGs meet in person.
The Gene-Lifestyle Interactions WG
A new WG has been established for pursuing the major goals of this study. The WG includes investigators and analysts from the large group of studies participating in Stage 1 (Genome-Wide Discovery) as discussed later. Another large group of studies participates in Stage 2 (Focused Discovery/Replication). The WG is assisted by a Coordinating Center (CC) at Washington University in St. Louis.
This study operates through the Working Group (WG), an Analysis Committee, a Harmonization Committee, and multiple Project Teams. The WG meets twice a year at CHARGE meetings and meets by conference call twice a month. Research direction and priorities are set by the WG. The analysis and harmonization committees meet together once a year and by conference calls twice a month. All harmonization and analytical issues are resolved by these committees. There are multiple Project Teams, each leading interaction analyses for a combination of the phenotypes (BP or Lipids) and lifestyle domains (smoking, alcohol, education, PA, Psychosocial, Sleep). Finally, IRB approval has been obtained for the study.
Mission and Aim
The overall mission of the WG is to promote and facilitate large collaborative analysis of gene-lifestyle interactions on disease traits across a large number of cohorts from multiple ancestries. Primarily, the WG aims to better understand the lifestyle context of genetic effects and to discover new trait loci through analysis of interactions thereby explaining part of the missing heritability2 in the disease traits. An important and novel feature of our study is that the gene-lifestyle interaction (G×E) results may improve our knowledge about the underlying mechanisms for novel as well as already known trait loci.
Primary Hypothesis
We hypothesize that lifestyle (environment) variables modulate some of the genetic effects on cardiovascular traits and that accounting for lifestyle factors and gene-lifestyle interactions in genome-wide scans will identify multiple novel genetic variants.
Phenotypes and Lifestyle Variables
The primary phenotypes include BP and lipids. An analysis plan in the supplemental material discusses data definitions and adjustments. Future initiatives may consider other cardio-metabolic traits in collaboration with other working groups.
The primary BP phenotypes are resting/sitting Systolic Blood Pressure (SBP) (mmHg) and Diastolic Blood Pressure (DBP) (mmHg). For individuals taking any anti-hypertensive (BP lowering) medications, their SBP and DBP values are first adjusted by adding 15 mmHg to SBP and adding 10 mmHg to DBP. Mean Arterial Pressure (MAP) and Pulse Pressure (PP) are also derived, using the adjusted SBP and DBP values:
MAP = DBP + (SBP – DBP)/3, and
PP = SBP – DBP
The primary lipids phenotypes are High-density lipoprotein cholesterol (HDL, mg/dL), Triglycerides (TG, mg/dL) and Low-density lipoprotein cholesterol (LDL, mg/dL), either directly assayed (LDLda) or derived using the Friedewald equation (LDLF). For individuals with TG > 400 mg/dL, only directly assayed LDL (LDLda) is used. When using non-fasting samples or fasting < 8 hours, only LDLda and HDL are used (not LDLF or TG). Log transformations are used for HDL and TG, and LDL is adjusted for statin use (see the analysis plan in the supplementary materials).
The initial set of dichotomized lifestyle are: Smoking (current smoking and ever smoking), Alcohol Consumption (Current Drinking, Current Regular Drinking, and Quantity of Drinks (>7 drinks per week)), Education (as a measure of socioeconomic status, SES; Some College, and Graduated College), Physical Activity (Physically Inactive), Psychosocial Attributes (Depression, Trait Anxiety, and Social Support), and Sleep Duration (Short Sleep and Long Sleep). Future initiatives may consider other domains such as diet.
GWAS Data
Dosages derived from 1000 Genomes (1000G) imputation are the primary resource for GWAS analysis. 1000G imputations are based on the ALL ancestry panel from 1000G Phase I Integrated Release Version 3 Haplotypes (2010–11 data freeze, 2012-03-14 haplotypes) that contains haplotypes of 1,092 individuals of all ancestral backgrounds. Dosages based on HapMap Phase II/III reference panel is used if 1000G imputations are not available for a specific study. In general, rare variants (mean allele frequency (MAF) < 1%) and poorly imputed variants (Rsq <0.1) are excluded. Variants mapping to sex chromosomes or mitochondria have also been excluded. Although we refer to SNP (Single Nucleotide Polymorphism) variants, the imputed data also include indels (insertions and deletions).
Participating Studies and Ancestry Groups
Five ancestry groups are represented: European (EA), African (AA), Hispanic (HA), Asian (AS), and Brazilian admixed (BR). Men and women between the ages of 18–80 are included in the analyses. Although the participating studies are based on different study designs and populations, most of them have data on BP and lipid traits, a range of lifestyle variables, and genotypes across the genome. In total, this study comprises up to 610,475 individuals.
Stage 1 (Genome-Wide Discovery)
A total of 32 studies with data on 53 cohorts (see Table 1 in Supplemental Material) participate in the discovery phase (Stage 1), which involves genome-wide interaction analyses. In total, this stage includes up to 95,911 EA, 27,116 AA, 8,805 HA, 13,438 AS, and 4,414 BR individuals, to an overall total of 149,684 individuals in Stage 1.
Stage 2 (Focused Discovery/Replication)
A total of 46 studies with data on 71 cohorts (see Table 2 in Supplemental Material) participate in Stage 2, which involves analyses of small sets of variants that were identified in Stage 1 as either genome-wide significant (with p < 10−8) or suggestive (with p < 10−6). In total, this stage includes up to 290,552 EA, 7,785 AA, 13,522 HA, and 148,932 AS individuals, to a total of 460,791 individuals in Stage 2. There are no BR cohorts in Stage 2.
Analysis Models
The participating studies have considerable prior experience contributing to GWAS-based consortia studying the main effects of common variants (without interactions). For G×E work, existing analysis pipelines had to be modified. Based on extensive discussions with the Analysis Committee and the Working Group, an Analysis Plan was developed, addressing critical issues including: data preparation, analysis models, analysis methods, and software packages. Individual project teams made appropriate modifications to the analysis plan as needed. The most critical elements are summarized below. An example of a full analysis plan (Education-Lipids) is provided in the supplemental material.
We consider three different analysis models, each with slightly different purposes.
Joint model (Model 1)
This is our primary model which features joint analysis of the effects of the SNP, lifestyle, and their interaction. For each combination of phenotype (Y) and lifestyle exposure variable (E), each study fits the following linear model, separately by ancestry:
Y ~ E + SNP + E * SNP + C, or more formally,
E(Y) = β0 + βE E + βG SNP + βGE E * SNP + βC C
where SNP is the dosage of the genetic variant and C is the set of covariates (age, sex, principal components for controlling population stratification effects as needed, and other study-specific covariates), and therefore βC is a vector; body mass index (BMI) was specifically excluded as a covariate so that lifestyle interactions with related pathway genes (such as inflammation genes) can be identified). Participating studies provide estimates of βG and βGE along with their covariance matrix. If E is dichotomous (E= 0 or 1), the SNP effect (βG) represents the SNP effect in those who are unexposed (environmental variable E=0), and thus needs to be interpreted with caution. If E is continuous, it is often desirable to center it on its sample mean, so that βG approximates the overall effect of the SNP on Y (as is estimated by Model 2). In either case, the SNP effect is context-dependent and therefore should not be interpreted as the “main effect”. Model 1 was used by all studies in both stages. In addition to model 1, each study in stage 1 (only) uses at least one of two additional models presented below, depending on the specific needs of the respective project.
Main effects model (Model 2)
Analysis of the main effect only: For each Phenotype (Y), each study fits the following linear model, separately by ancestry:
Y ~ SNP + C, or more formally,
E(Y) = λ0 + λG SNP + λCC
Model 2 is used as a benchmark to identify which of our discoveries from the joint model would be found using analysis of main effects alone. Some projects also fit this model separately in the exposed and unexposed groups (i.e. they performed stratified analysis) and provide a 1 degree of freedom (df) test of the interaction term as well as a 2 df joint test of the SNP and interaction effects19,20. For each analysis, participating studies provide estimates of λG and its standard error. Stratified analysis and the joint analysis using model 1 in stage 1 cohorts have been shown to yield largely similar results21. Stratified analysis can help reduce inflation of type I error rates by fitting separate covariate effects and error variances by strata22–24.
Refined main effects model (Model 3)
Analysis of the SNP and lifestyle effects, without interaction. For each Phenotype (Y) and lifestyle exposure variable (E), each study fits the following linear model, separately by ancestry:
Y ~ E + SNP + C, or more formally,
E(Y) = γ0 + γE E + γG SNP + γCC
Model 3 is used to identify which of our discoveries from the joint model would be missed when the interaction term is not used. For each analysis, participating studies provide estimates of γG and and its standard error.
Analysis Methods
Analysis Methods for Low Frequency and Common Variants
We identify novel loci through SNP*E interaction effects alone or jointly with the SNP effects (or only through SNP effects in models 2 and 3). For continuous traits, the joint test of the SNP and SNP*E interaction effects is known to be powerful for this aim20, 25,26. Since our interaction projects involve many studies, we rely on existing methods and software, such as ProbABEL, Sandwich, and MMAP (see the analysis plan in the supplemental material), or those that are straightforward to implement using these tools.
Testing the significance of the SNP and the SNP*E interaction effects
In Model 1, the focus is on the test of the interaction effect and the joint effects of the SNP and the interaction. The interaction effect (βGE) is evaluated using a 1 degree of freedom (df) Wald test. The effects of both SNP (βG) and interaction (βGE) are tested jointly, using a 2 df Wald test25. In model 2, which does not include E or SNP*E terms, λG is the familiar main effect of the SNP which is tested using a 1-df Wald test. A 1 df Wald test is also used in model 3 for evaluating the SNP effect (γG) in the presence of E, which may be referred to as the refined SNP effect or context-dependent SNP effect. In all cases, we will use the “robust” Wald tests by using robust estimates of the standard errors (SEs) and covariances to protect against misspecification of the mean model27,28. When the SNP effect is weak and the SNP*E interaction effect is moderate, the joint 2 df test has been shown to be more powerful than either the 1 df test of the SNP effect or the 1 df test of the interaction effect alone25. The increase in power for the 2 df over either 1 df test can be particularly dramatic, especially when the type I error rate is controlled at very low levels (e.g., 5×10−8) as in this project29.
Analyses needed from each cohort
Each cohort carries out a genome-wide analysis of the SNP and SNP*E interaction effects, separately within each ancestry by accounting for possible population stratification (see the Analysis Plan in the supplemental material), and provides estimates of regressions coefficients and robust estimates of the corresponding standard errors (SEs) and covariance. Because the model is based on a standard regression framework, software to compute the relevant statistics is widely available. For studies of unrelated individuals, standard commands and the R sandwich package30 implement bivariate robust covariance estimates for SNP-specific analyses. To implement the analyses for all SNPs, the R interface in PLINK31 may be used; ProbABEL32 also provides appropriate utilities. For family studies in which relatedness must be taken into account, programs such as GenABEL/MixABEL33 and MMAP (O’Connell, unpublished; personal communication) implement mixed models that allow for relatedness. All cohorts analyze their data using these methods/software following a detailed Analysis Plan and upload results to a secure server.
Meta-analysis for combining results across studies
To combine estimates of the regression coefficients and their corresponding 2×2 covariance matrix provided by each cohort, we use the joint meta-analysis method developed by Manning et al26 who modified METAL34 to handle this joint 2 df meta-analysis. The joint meta-analysis provides inference on the SNP and SNP*E interaction effects pooled across all cohorts. Manning7 used this approach and demonstrated power enhancement for detecting interactions. We use the modified METAL for the joint meta-analysis and use METAL for carrying out meta-analysis of the 1 df analyses (interaction effect in model 1, main effect in model 2, and refined SNP effect in model 3). We use a genome-wide significance threshold of 5×10−8 for identifying significant results and use 10−6 for identifying suggestive results. Heterogeneity chi-square test is used to test for differences in any of the regression coefficients among the contributing studies. Early results indicate minimal differences even in the interaction coefficient.
Quality Control (See supplementary material for details)
Quality assurance is emphasized by preparing very detailed analysis plans with step by step instructions for preparing and analyzing data, and formatting results for uploading (see the Education-Lipids analysis plan in the supplementary materials). Extensive QC measures are used for processing all study-specific results centrally by each project team, at two levels. “Study-level” QC involves reviewing and harmonization of each individual result file separately. “Meta-level” QC involves reviewing and harmonizing results files across all available cohorts for a single analysis (e.g., comparing summary statistics across all SBP-Current Smoking-Model1 discovery cohorts). QC was performed using customized EasyQC scripts that provide a wide variety of QC checks for GWAS results35.
Analysis of interactions involving rare variants
Power of the joint test of SNP and SNP*E for testing individual rare variants is limited primarily due to their low frequency. Burden tests36–39 collapse all rare variants in a genomic region (typically a gene) into a single burden variable (essentially a “mega variant”, giving each subjects’ total dosage across a gene) and regress the phenotype on the burden variable to test for the combined effects of all rare variants in the region/gene. We apply the 2 df test directly to each burden variable. Each cohort creates burden variables by collapsing variants within the genomic regions using a MAF (pooled across studies) threshold (e.g., MAF < 0.01). We then perform meta-analysis of these results, similar to the meta-analysis described earlier but now with as many burden variables as the number of genomic regions. To assess the significance for the analysis of rare variants, we will use a Bonferroni-corrected significance threshold (α = 0.05/Nb where Nb= number of burden variables). The CHARGE consortium has provided detailed analysis guidelines for exome chip data and the CC has used some of these rare variant methods40–43.
Analysis of Stage 1 and Stage 2 Results
Primary publications resulting from the various analyses in stages 1 and 2 are pursuing two approaches shown in Figure 1: combined analysis of Stages 1 and 2 and traditional discovery/replication.
Figure 1.
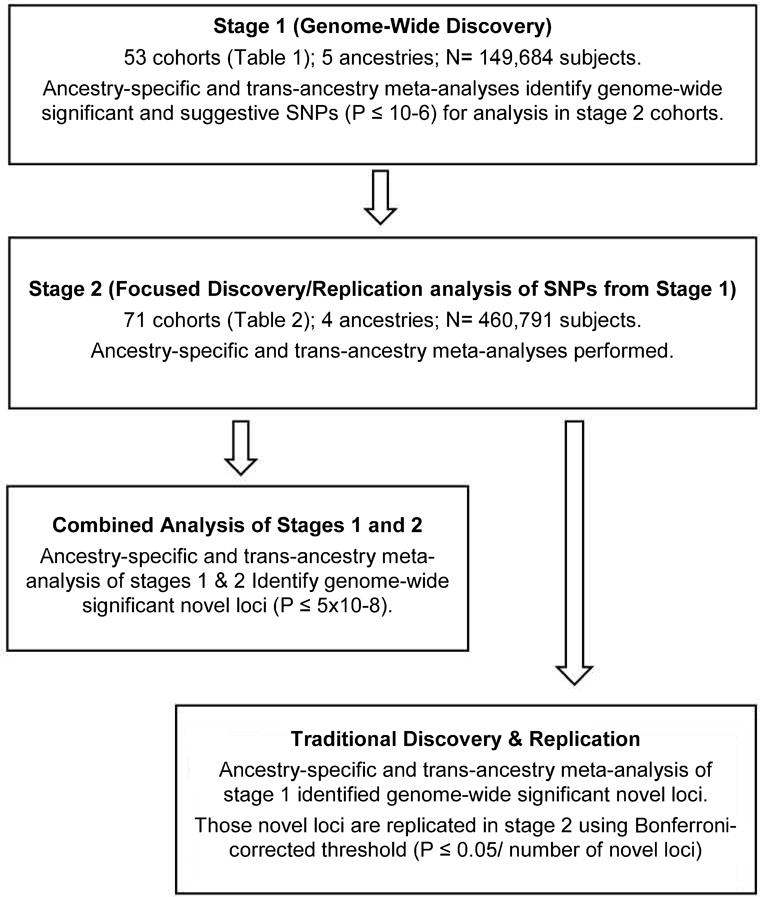
Overall flow of analyses. Combined analysis leverages the full power of Stages 1 and 2. The traditional discovery and replication approach identifies additional loci missed by the combined approach. Both approaches can be used for maximizing discovery.
Combined analysis of stages 1 and 2
This approach can be more powerful than other approaches44. For a given combination of phenotype and lifestyle, all significant and suggestive results (with α = 10−6) from stage 1 cohorts and the corresponding results from stage 2 cohorts are pooled through meta-analysis (first within each stage and then meta-analyzing the two stage-specific meta-analyses) separately by ancestry. A significance threshold of α = 5×10−8 is used to identify significant results from the combined meta-analysis results. Finally, all ancestry-specific meta-analyses are meta-analyzed as an approximate trans-ancestry analysis for identifying additional associations (if any) that are missed by ancestry-specific analyses.
Traditional Discovery/Replication Analysis
In this approach, all genome-wide significant results are identified from stage 1 results only, separately by ancestry, using a significance threshold of α = 5×10−8. Stage 2 results are then used to formally replicate the stage 1 findings, using appropriate Bonferroni correction such as 0.05 divided by the number of independent novel loci discovered in stage 1. Variants that are suggestive but not significant in Stage 1 are only considered in the combined analysis approach.
The combined approach is more powerful than the traditional approach. However, the traditional approach can identify additional novel validated loci missed by the combined approach (as shown most recently using a slight variation of this approach45). This justifies using both approaches. If only one approach were to be used, the combined one is the method of choice.
Statistical power for detecting associations
With the overall sample size used, this study is well powered for identifying novel discoveries even with moderately small effect sizes. To demonstrate this, we illustrate the sample sizes required to achieve at least 80% power to identify the genetic (G) effect and the G×E interaction effect using the 2 df joint test for a range of model parameters. We used QUANTO46, which computes power and sample size for both disease and quantitative trait studies of genes (G), environment factors (E), and G×E interactions. For our study of quantitative traits, the required sample sizes depend on the proportions of variance explained by the G (R2G), the lifestyle factor (R2E) and their interaction effect (R2GE). A wide range of R2E values yielded similar results, and therefore we fixed R2E = 0.1% and examined the effect of varying the other 2 parameters. Although low frequency variants explain large proportions of variance in some cases47, we limited this investigation to lower R2G values of 0.01%, 0.02%, 0.05%, and 0.1% because most variants identified through GWAS have much smaller effect sizes. Figure 2 shows the sample sizes required for a range of R2GE values corresponding to each of the four values for R2G using a significance threshold of 5×10−8. These values are smaller than what we found in our preliminary studies (not reported), suggesting that our power estimates may be conservative.
Figure 2.
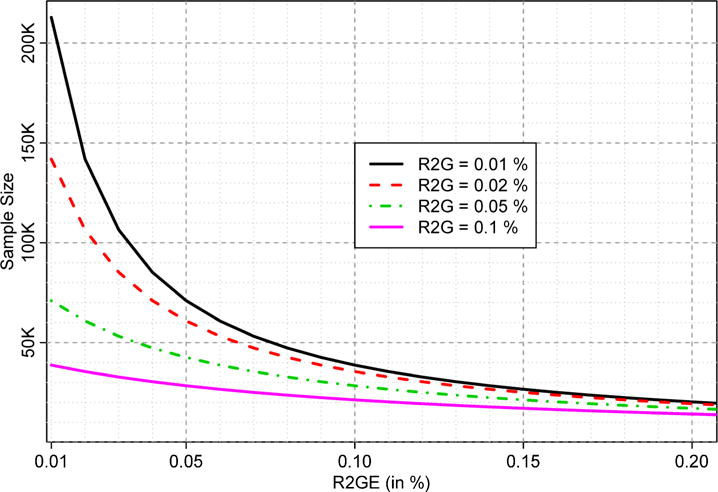
Sample sizes needed for 80% power using the 2 df joint test. Sample size (Y-axis) is plotted as a function of the percent variance explained by the interaction (R2GE; X-axis), for each of 4 different values of the percent variance explained by the genetic effect (R2G); that due to the lifestyle factor (R2E) is fixed at 0.1% (see the text).
The sample sizes should be more than adequate for 80% power in EA and AA using Stage 1 samples alone, so long as the SNP effect is not very small (e.g., R2G > 0.05%). In fact, for R2G = 0.05%, significance level of 5×10−8, and the stage 1 sample sizes shown in Supplemental Table 1, the minimum detectable R2GE at 80% power are < 0.01%, 0.11%, 0.44%, and 0.27% for EA, AA, HA, and AS, respectively. When Stages 1 and 2 are combined, even smaller effect sizes are detectable (although the exact calculations are complex since Stage 2 studies did not carry out genome-wide interaction analyses). In any case, the combined sample size of stages 1 and 2 appears well poised for powerful discoveries even with smaller effect sizes than assumed in these estimates.
Discussion
Current Status and Anticipated Benefits
Our study has made considerable progress to date. Four projects have completed all analyses in stages 1 and 2 and are processing the final results for publications (Smoking-BP, Smoking-Lipids, Alcohol-BP, and Alcohol-Lipids). In addition, three other projects (Education-BP, Education-Lipids, and PA-Lipids) have completed stage 1 analyses for which stage 2 analyses are in progress, and two projects (Psychosocial-BP and Psychosocial-Lipids) have completed stage 1 analyses and are preparing for stage 2 analysis. More projects are getting underway. We believe that these projects will make major contributions to the genetic dissection of cardiovascular traits and that the G×E analysis can help improve understanding of the mechanisms underlying the novel as well as known loci which have been identified previously through main effects.
What are the unique benefits of our approach? How critical is the consideration of lifestyle and interactions (models 1 and 3)?
Emerging results indicate that a large proportion of novel findings originate from models 1 and 3, i.e. results that would be missed by limiting analyses to main effects (model 2). This suggests that inclusion of the lifestyle context and/or gene-lifestyle interaction is important for identifying novel signals.
Collaboration levels are unprecedented
In an area where direct competition among study groups was the norm until about a decade ago, collaborative GWAS-based consortia such as CHARGE represent an innovative model for research. Through working together, the contributing studies have achieved much more than they could have working alone. The Gene-Lifestyle Interactions Working Group takes this model further, assembling 610,475 subjects in 124 cohorts. Other studies with appropriate data are welcome to join. While the collaborative nature of the work requires some compromises (e.g. using standard software, and meta-analysis of relatively simple analyses) the results will hopefully deepen what has already been learned from GWAS.
Supplementary Material
Clinical Perspective.
Gene-lifestyle interaction studies provide an excellent framework for understanding the lifestyle context of genetic effects on disease traits. Understanding these genetic modifiers is important because it may provide valuable clues for lifestyle-based interventions which may result in a more successful management of these health conditions through personalized therapies. An important feature of our study is that such interaction results may improve our knowledge about the underlying mechanisms for novel as well as already known trait loci.
Acknowledgments
The authors wish to thank several members of the Working Group (WG) for their overall contributions to the WG, notably Hugues Aschard, Sharon Kardia, Ruth Loos, Alisa Manning, Jeff O’Connell, Michael Province, Patricia Peyser, Jerome Rotter, Xiaofeng Zhu, among others. The full list of the WG members can be found at: http://depts.washington.edu/chargeco/wiki/Gene-Lifestyle_Interactions. The authors also wish to thank Matthew Brown for his critical help in preparing some of the materials for this publication. Study descriptions and study-specific acknowledgments are included in the Supplemental Materials along with an example analysis plan. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Sources of Funding: This multi-ancestry study of gene-lifestyle interactions is sponsored by R01HL118305 from the National Heart, Lung, and Blood Institute (NHLBI), NIH. The CHARGE infrastructure on which this study is based is also sponsored by another NHLBI grant HL105756.
Dr. Psaty serves on the DSMB of a clinical trial funded by Zoll LifeCor and on the Steering Committee of the Yale Open Data Access Project funded by Johnson & Johnson.
Footnotes
Disclosures
All others have none.
References
- 1.Psaty BM, O’Donnell CJ, Gudnason V, Lunetta KL, Folsom AR, Rotter JI, et al. Cohorts for Heart and Aging Research in Genomic Epidemiology (CHARGE) Consortium: Design of prospective meta-analyses of genome-wide association studies from 5 cohorts. Circ Cardiovasc Genet. 2009;108:73–80. doi: 10.1161/CIRCGENETICS.108.829747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Manolio TA, Collins FS, Cox NJ, Goldstein DB, Hindorff LA, Hunter DJ, et al. Finding the missing heritability of complex diseases. Nature. 2009:747–753. doi: 10.1038/nature08494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Visscher PM, Brown MA, McCarthy MI, Yang J. Five years of GWAS discovery. Am J Hum Genet. 2012;90:7–24. doi: 10.1016/j.ajhg.2011.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Murphy SL, Xu J, Kochanek KD. Deaths: Preliminary Data for 2010, in National Vital Statistics Reports. National Center for Health Statistics; Hyattsville, MD: 2012. [Google Scholar]
- 5.Roger VL, Go AS, Lloyd-Jones DM, Benjamin EJ, Berry JD, Borden WB, et al. Heart disease and stroke statistics–2012 update: a report from the American Heart Association. Circulation. 2012;125:e2–e220. doi: 10.1161/CIR.0b013e31823ac046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zheng JS, Arnett DK, Lee YC, Shen J, Parnell LD, Smith CE, et al. Genome-wide contribution of genotype by environment interaction to variation of diabetes-related traits. PloS One. 2013;8:e77442. doi: 10.1371/journal.pone.0077442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Manning AK, Hivert MF, Scott RA, Grimsby JL, Bouatia-Naji N, Chen H, et al. A genome-wide approach accounting for body mass index identifies genetic variants influencing fasting glycemic traits and insulin resistance. Nat Genet. 2012;44:659–669. doi: 10.1038/ng.2274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hutter CM, Mechanic LE, Chatterjee N, Kraft P, Gillanders EM, NCI Gene-Environment Think Tank Gene-environment interactions in cancer epidemiology: a National Cancer Institute Think Tank report. Genet Epidemiol. 2013;37:643–657. doi: 10.1002/gepi.21756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sung YJ, de las Fuentes L, Schwander KL, Simino J, Rao DC. Gene-smoking interactions identify several novel blood pressure loci in the Framingham Heart Study. Am J Hypertens. 2015;28:343–354. doi: 10.1093/ajh/hpu149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Montasser ME, Shimmin LC, Hanis CL, Boerwinkle E, Hixson JE. Gene by smoking interaction in hypertension: identification of a major quantitative trait locus on chromosome 15q for systolic blood pressure in Mexican-Americans. J Hypertens. 2009;27:491–501. doi: 10.1097/hjh.0b013e32831ef54f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Higashibata T, Hamajima N, Naito M, Kawai S, Yin G, Suzuki S, et al. eNOS genotype modifies the effect of leisure-time physical activity on serum triglyceride levels in a Japanese population. Lipids Health Dis. 2012;11:150. doi: 10.1186/1476-511X-11-150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Grarup N, Andreasen CH, Andersen MK, Albrechtsen A, Sandbaek A, Lauritzen T, et al. The -250G>A promoter variant in hepatic lipase associates with elevated fasting serum high-density lipoprotein cholesterol modulated by interaction with physical activity in a study of 16,156 Danish subjects. J Clin Endocrinol Metab. 2008;93:2294–2299. doi: 10.1210/jc.2007-2815. [DOI] [PubMed] [Google Scholar]
- 13.Parnell LD, Blokker BA, Dashti HS, Nesbeth PD, Cooper BE, Ma Y, et al. CardioGxE, a catalog of gene-environment interactions for cardiometabolic traits. BioData Min. 2014;7:21. doi: 10.1186/1756-0381-7-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hunter DJ. Gene-environment interactions in human diseases. Nat Rev Genet. 2005;6:287–298. doi: 10.1038/nrg1578. [DOI] [PubMed] [Google Scholar]
- 15.Murcray CE, Lewinger JP, Gauderman WJ. Gene-Environment Interaction in Genome-Wide Association Studies. Am J Epidemiol. 2009;169:219–226. doi: 10.1093/aje/kwn353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Thomas D. Gene-environment-wide association studies: emerging approaches. Nat Rev Genet. 2010;11:259–272. doi: 10.1038/nrg2764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Taylor JY, Maddox R, Wu CY. Genetic and Environmental Risks for High Blood Pressure Among African American Mothers and Daughters. Biol Res Nurs. 2009;11:53–65. doi: 10.1177/1099800409334817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Green ED, Guyer MS. Charting a course for genomic medicine from base pairs to bedside. Nature. 2011;470:204–213. doi: 10.1038/nature09764. [DOI] [PubMed] [Google Scholar]
- 19.Randall JC, Winkler TW, Kutalik Z, Berndt SI, Jackson AU, Monda KL, et al. Sex-stratified genome-wide association studies including 270,000 individuals show sexual dimorphism in genetic loci for anthropometric traits. PLoS Genet. 2013;9:e1003500. doi: 10.1371/journal.pgen.1003500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Aschard H, Hancock DB, London SJ, Kraft P. Genome-wide meta-analysis of joint tests for genetic and gene-environment interaction effects. Hum Hered. 2010;70:292–300. doi: 10.1159/000323318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sung YJ, Winkler TW, Manning AK, Aschard H, Gudnason V, Harris TB, et al. An Empirical Comparison of Joint and Stratified Frameworks for Studying G × E Interactions: Systolic Blood Pressure and Smoking in the CHARGE Gene-Lifestyle Interactions Working Group. Genet Epidemiol. 2016;40:404–415. doi: 10.1002/gepi.21978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.VanderWeele TJ, Ko Yi-An, Mukherjee Bhramar. Environmental Confounding in Gene-Environment Interaction Studies. Am J Epidemiol. 2013;178:144–152. doi: 10.1093/aje/kws439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dudbridge F, Fletcher O. Gene-Environment Dependence Creates Spurious Gene-Environment Interaction. Amer J Hum Genet. 2014;95:301–307. doi: 10.1016/j.ajhg.2014.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Keller MC. Gene × Environment Interaction Studies Have Not Properly Controlled for Potential Confounders: The Problem and the (Simple) Solution. Biol Psychiatry. 2014;75:18–24. doi: 10.1016/j.biopsych.2013.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kraft P, Yen YC, Stram DO, Morrison J, Gauderman WJ. Exploiting gene-environment interaction to detect genetic associations. Hum Hered. 2007;63:111–119. doi: 10.1159/000099183. [DOI] [PubMed] [Google Scholar]
- 26.Manning AK, LaValley M, Liu CT, Rice K, An P, Liu Y, et al. Meta-analysis of gene-environment interaction: joint estimation of SNP and SNP × environment regression coefficients. Genet Epidemiol. 2011;35:11–18. doi: 10.1002/gepi.20546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tchetgen EJ, Kraft P. On the robustness of tests of genetic associations incorporating gene-environment interaction when the environmental exposure is misspecified. Epidemiology. 2011;22:257–261. doi: 10.1097/EDE.0b013e31820877c5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Voorman A, Lumley T, McKnight B, Rice K. Behavior of QQ-plots and genomic control in studies of gene-environment interaction. PLoS One. 2011;6:e19416. doi: 10.1371/journal.pone.0019416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Morris N, Elston R. A Note on Comparing the Power of Test Statistics at Low Significance Levels. Am Stat. 2011;65:164–166. doi: 10.1198/tast.2011.10117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zeileis A. Object-oriented computation of sandwich estimators. J Stat Softw. 2006;16 [Google Scholar]
- 31.Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Aulchenko YS, Struchalin MV, van Duijn CM. ProbABEL package for genome-wide association analysis of imputed data. BMC Bioinformatics. 2010;11:134. doi: 10.1186/1471-2105-11-134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Aulchenko YS, Ripke S, Isaacs A, van Duijn CM. GenABEL: an R library for genome-wide association analysis. Bioinformatics. 2007;23:294–296. doi: 10.1093/bioinformatics/btm108. [DOI] [PubMed] [Google Scholar]
- 34.Willer CJ, Li Y, Abecasis GR. METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics. 2010;26:2190–2191. doi: 10.1093/bioinformatics/btq340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Winkler TW, Day FR, Croteau-Chonka DC, Wood AR, Locke AE, Magi R, et al. Quality control and conduct of genome-wide association meta-analyses. Nat Protoc. 2014;9:1192–1212. doi: 10.1038/nprot.2014.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Morgenthaler S, Thilly WG. A strategy to discover genes that carry multi-allelic or mono-allelic risk for common diseases: a cohort allelic sums test (CAST) Mutat Res. 2007;615:28–56. doi: 10.1016/j.mrfmmm.2006.09.003. [DOI] [PubMed] [Google Scholar]
- 37.Li B, Leal SM. Methods for detecting associations with rare variants for common diseases: application to analysis of sequence data. Am J Hum Genet. 2008;83:311–321. doi: 10.1016/j.ajhg.2008.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Madsen BE, Browning SR. A groupwise association test for rare mutations using a weighted sum statistic. PLoS Genet. 2009;5:e1000384. doi: 10.1371/journal.pgen.1000384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Morris AP, Zeggini E. An evaluation of statistical approaches to rare variant analysis in genetic association studies. Genet Epidemiol. 2010;34:188–193. doi: 10.1002/gepi.20450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sung YJ, Rice TK, Rao DC. Application of collapsing methods for continuous traits to the Genetic Analysis Workshop 17 exome sequence data. BMC Proc. 2011;5(Suppl 9):S121. doi: 10.1186/1753-6561-5-S9-S121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sun YV, Sung YJ, Tintle N, Ziegler A. Identification of genetic association of multiple rare variants using collapsing methods. Genet Epidemiol. 2011;35(Suppl 1):S101–106. doi: 10.1002/gepi.20658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mallaney C, Sung YJ. Rare variant analysis of blood pressure phenotypes in the Genetic Analysis Workshop 18 whole genome sequencing data using SKAT. BMC Proc. 2014 Jun 17;8:S10. doi: 10.1186/1753-6561-8-S1-S10. Suppl 1 Genetic Analysis Workshop 18 Vanessa Olmo. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sung YJ, Basson J, Rao DC. Whole genome sequence analysis of the simulated SBP in Genetic Analysis Workshop 18 family data: Long term average and collapsing methods. BMC Proc. 2014 Jun 17;8:S12. doi: 10.1186/1753-6561-8-S1-S12. Suppl 1 Genetic Analysis Workshop 18 Vanessa Olmo. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Skol AD, Scott LJ, Abecasis GR, Boehnke M. Joint analysis is more efficient than replication-based analysis for two-stage genome-wide association studies. Nat Genet. 2006;38:209–213. doi: 10.1038/ng1706. [DOI] [PubMed] [Google Scholar]
- 45.Surendran P, Drenos F, Young R, Warren H, Cook JP, Manning AK, et al. Trans-ancestry meta-analyses identify rare and common variants associated with blood pressure and hypertension. Nat Genet. 2016;48:1151–1161. doi: 10.1038/ng.3654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gauderman W, Morrison J. QUANTO 1:2: A computer program for power and sample size calculations for genetic-epidemiology studies. 2009 http://biostats.usc.edu/software.
- 47.Bowden DW, An SS, Palmer ND, Brown WM, Norris JM, Haffner SM, et al. Molecular basis of a linkage peak: exome sequencing and family-based analysis identify a rare genetic variant in the ADIPOQ gene in the IRAS Family Study. Hum Mol Genet. 2010;19:4112–4120. doi: 10.1093/hmg/ddq327. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.