

Abstract
Background
Animal models are an important tool to understand intestinal biology. Our lab previously generated C57BL/6-Tg(Car1-cre)5Flt transgenic mice (CAC) with large intestine specific Cre recombinase (Cre) expression as a model to study colon health.
Aims
To expand the utility of the CAC mouse model by determining the impact of chemically induced colitis on CAC transgene expression.
Methods
CAC mice were crossed to Rosa reporter mice (Rosa26Rflox/flox) with a lox-STOP-lox signal controlling β-galactosidase (βgal) expression and then further crossed with ApcCKO/CKO mice in some experiments to delete Apc alleles (ApcΔ580) Initially, 8-wk-old CACTg/WT;Rosa26Rflox/WT;ApcΔ580/WT mice were treated with dextran sulfate sodium (DSS) in drinking water (5 d, 0, 0.65%, 1.35%, or 2.0%). Colon tissue damage and βgal labelling were analyzed 10-d after stopping DSS. Next, 8-wk-old CACTg/WT;Rosa26Rflox/flox mice were treated with 0 or 1.35% DSS, and colonic β-gal labeling was assessed at 30-d post DSS treatment. Finally, 10-wk-old CACTg/WT;ApcΔ580/WT mice were treated with DSS (0 or 2%) for 5 d and colonic tumors were analyzed at 20-wk.
Results
CACTg/WT;Rosa26Rflox/WT;ApcΔ580/WT mice had a DSS dose-dependent increase in colon epithelial damage that correlated with increased epithelial β-gal labeling at 10-d (r2 = 0.9, β = 0.75). The β-gal labeling in CACTg/WT;Rosa26Rflox/flox mice colon remained high at 30-d, especially in the crypts of the healed ulcer. DSS also increased colon tumor incidence and multiplicity in CACTg/WT;ApcΔ580/WT mice.
Conclusions
DSS-mediated epithelial damage induces a persistent, Cre-mediated recombination of floxed alleles in CAC mice. This enables the examination of gene function in colon epithelium during experimental colitis and colitis-induced colon cancer.
Keywords: Colorectal cancer, Transgenic animal model, Cre recombinase, Colitis
Introduction
Colorectal cancer is a multi-factorial disease and its development can be influenced by genetics, diet, inflammation, environment and other factors [1-7]. Unfortunately, the etiology of colorectal cancer remains unclear, thus barriers exist to develop approaches to prevent and treat the disease [8-10]. Controlled animal studies are an important approach to understand colorectal cancer biology and to test promising strategies for disease prevention and treatment. As such, an optimal animal model of colorectal cancer should capture the pathological changes, molecular mechanisms, and clinical symptoms presented in the human disease. In addition, the cancer effect should be limited to colon to limit any confounding response from other tissues.
Many animal models have been developed for colorectal cancer research, and they have been reviewed in several recent comprehensive reviews [11-13]. Our lab previously generated a transgenic mouse model for colorectal cancer research with large intestine-specific Cre recombinase (Cre) transgene expression, i.e. the C57BL/6-Tg(Car1-cre)5Flt or CAC mouse [14]. In colon epithelial cells of CAC mice, Cre is expressed in approximately 10% of the epithelium. Here we report that Cre expression in colon epithelial cells of CAC mice can be induced during dextran sulfate sodium (DSS) induced colitis. In addition, our data suggest that these recombination events induce a persistent effect that can be used to study genes involved in the epithelial response to colitis or in colitis-associated colon cancer.
Materials and Methods
Animals
C57BL/6-Tg(Car1-cre)5Flt mice were created by our group [14] and maintained as heterozygotes (CACTg/WT). The Rosa26R strain, B6.129S4-Gt(ROSA)26Sortm1Sor/J, was obtained from The Jackson Laboratory (Bar Harbor, ME) and maintained as homozygotes (ROSAflox/flox). Rosa26R mice contain a STOP sequence that is flanked by LoxP sites upstream from the E. coli β-galactosidase (βgal) gene within a gene trap of the Rosa26 allele [15]. The ApcCKO mouse carrying a floxed exon 14 of the Apc gene (ApcCKO/CKO) was obtained from the Mouse Models of Human Cancer Consortium (National Cancer Institute, Frederick, MD). In cells expressing Cre, exon 14 is removed resulting in a truncated 580 amino acid long protein produced from one (ApcΔ580/WT) or both (ApcΔ580/Δ580) Apc alleles. Genotypes were determined by standard PCR methods as previously described for the CAC [14], Rosa26R [15], and floxed Apc alleles [14]. Each strain of mouse was maintained on a C57BL/6J background. Mice were bred to create experimental animals with specific combinations of genotypes. All mouse breeding colonies were housed individually, given a standard chow diet and water ad libitum, and exposed to a 12 hours light/12 hours dark cycle. The experiments were conducted with approval from the Purdue University Animal Care and Use Committee.
Experimental Design
Experiment 1
CACTg/WT;Rosa26Rflox/flox mice were crossed to ApcCKO/CKO mice to generate CACTg/WT;Rosa26Rflox/WT;ApcΔ580/WT mice. Experimental mice were weaned at 3 weeks of age and fed the AIN93G diet [16]. Mice were randomly assigned into one of four dextran sulfate sodium (DSS) treatment groups (0%, 0.65%, 1.35% and 2%, n=16 per group, 8 male and 8 female). DSS (MW = 36,000-50,000 d, MP Biomedicals, LLC, Solon, OH)) was diluted in deionized water (w/v) to the appropriate concentrations. At 8 weeks of age, mice received the DSS solution in place of water ad libitum for 5 consecutive days. Afterwards, DSS was replaced with deionized water and this was provided until the day of sacrifice. Half of the mice in each group were sacrificed 2 days after ending the DSS treatment and half were sacrificed 10 days after ending DSS treatment. All mice were fasted overnight before harvest. At necropsy, the proximal and distal colon were prepared as Swiss rolls and examined for β-galactosidase levels by immunohistochemistry.
Experiment 2
24 CACTg/WT;Rosa26Rflox/flox mice (12 female and 12 male) were weaned at 3 weeks of age and fed AIN93G diet. At 8 weeks of age, 16 mice (8 male and 8 female) were treated with 1.35% DSS for 5 days and the other 8 mice (4 male and 4 female) were used as vehicle controls (0% DSS). Half of the DSS treated mice and all the vehicle control mice were sacrificed 10 days after completing the DSS treatment. The other mice in the DSS treatment group were sacrificed 30 days after completing the DSS treatment. All mice were fasted overnight before harvest. At necropsy, the proximal and distal colon were prepared as Swiss rolls and examined for β-galactosidase levels by immunohistochemistry. Small intestine, kidney, spleen, liver and lung were also collected for detection of β-galactosidase enzymatic activity.
Experiment 3
13 CACTg/WT;Rosa26Rflox/WT;ApcΔ580/Δ580 mice with two recombined Apc alleles were generated by crossing CACTg/WT;Rosa26Rflox/WT;ApcΔ580/WT mice with ApcCKO/CKO mice. Mice were sacrificed at 4 weeks of age after an overnight fast. At necropsy, the proximal and distal colon were prepared as Swiss rolls and examined for β-galactosidase levels by immunohistochemistry.
Experiment 4
81 CACTg/WT;ApcΔ580/WT mice were generated by crossing CACTg/WT mice with ApcCKO/CKO mice. CACTg/WT;ApcΔ580/WT pups were weaned at 3 weeks of age and fed an AIN93G diet. CACTg/WT;ApcΔ580/WT mice were treated with either 2% DSS (n=38) or deionized water (n=43) at 10 weeks of age for 5 days, then the DSS was replaced with water. Mice were sacrificed at 20 weeks of age after an overnight fast. The colon was removed, opened longitudinally, and cleaned with PBS. Digital images were collected and analyzed to determine the number, location, and size of each tumor.
Detection of β-galactosidase Activity in Organs
Tissues were dissected from the mice, rinsed briefly in cold PBS, and fixed for 1-2 hours in ice cold 4% paraformaldehyde. After rinsing the tissue for 15 minutes in PBS, tissues were incubated for 4 hours in 1 mg/mL 5-bromo-4-chloro-3-indoyl-β-D-galactopyranoside (X-gal; Invitrogen; B-1690) solution (pH 7.6) at 37°C as previously described [14]. In tissues expressing βgal (indicative of Cre-mediated recombination of the Rosa26R locus), the enzyme converted the X-gal substrate into a dark blue crystalline precipitate that is visible upon gross examination.
Tissue Preparation and Immunohistochemical Detection of β-galactosidase
The colon was opened longitudinally, rinsed with cold PBS, and transected at its midpoint to separate the proximal and distal colon. Each section was split longitudinally and Swiss-rolled [17], resulting in 2 rolls of proximal colon and 2 rolls of distal colon per mouse. Tissues were immersion fixed for 48-72 hours in 10% neutral buffered formalin at 4°C before being transferred to 70% ethanol. Tissues were processed and embedded in paraffin within 7 days of collection at the Purdue Histology and Phenotyping Laboratory.
Four micron tissue sections were cut, de-paraffinized, and rehydrated through xylene, ethanol, and water by standard methods. For antigen retrieval, slides were submerged in 0.01 mol/L sodium citrate (pH 6.0) and heated to 96°C for 20 minutes in a laboratory microwave (PELCO, Edmond, OK). Immunohistochemistry was performed on a Dako Autostainer (Dako, Denmark). Slides were incubated with 0.03% hydrogen peroxide (5 minutes) and serum-free protein block (30 minutes; Dako; Denmark), followed by a 120 minute incubation with rabbit polyclonal anti–β-galactosidase (β gal) antibody (1:400 dilution; Novus; NB100-65209). Antigen binding was detected with a horseradish peroxidase–labeled anti-rabbit IgG polymer (Vector laboratories; MP-7410) and 3, 3'-diaminobenzidine (DAB; Vector laboratories; SK-4100) as the chromogen. Slides were counterstained with Harris hematoxylin (EK Industries, Juliet, IL). The digital images were collected with an Aperio ScanScope slide scanner (Leica Technologies, Buffalo Grove, IL) at 20X magnification.
Image Analysis
Mucosal transgene expression was determined by quantifying the percent βgal positive pixels after immunohistochemistry. The colonic mucosa was differentiated from the tunica muscularis using the Genie histology pattern recognition software (Aperio, Leica Biosystem) trained at 5X magnification and 1000 iterations to a mean training accuracy of 96%. A positive pixel count algorithm was then used to analyze DAB labeling within the mucosa. This analysis was done by a board certified veterinary pathologist (RLJ) and confirmed independently (by FW).
The area of βgal positive crypts was determined by manually measuring the length of the mucosal surface containing βgal labeled crypts. βgal positive crypts were included only if the entire crypt axis, from base to lumen, was within the plane of section or if the crypt base could be seen immediately adjacent to the muscularis mucosa. The percentage of area represented by βgal positive crypts was calculated by dividing the length of the βgal positive area by the total length of the mucosa surface.
The damaged tissue surface area included the areas of ulceration and areas undergoing active healing. Ulceration was defined as sections of the colon mucosa devoid of crypts. This phenotype is obvious at day 2 after DSS termination and increased with DSS dose. Tissue healing included restitution (a single layer of epithelial cells covering an ulcer bed), crypt fission, and crypt regeneration - two events that repopulate the organized crypt structure within an ulcer. These phenotypes were observed at day 10 after DSS termination and continued through day 30 after DSS termination. The percentage of damaged surface was defined as the length of damaged mucosa divided by the total length of the mucosal surface.
Statistical Analysis
Statistical analysis was performed with SAS Enterprise 5.1 software (SAS Institute, Cary, NC). All data were reported as the mean ± standard error of the mean (SEM). Normality of the data was determined by evaluation of its histogram and a Shapiro-Wilk normality test (p>0.05). All of the data were normally distributed. Statistical comparisons were made by two-tailed student's t-tests, one-way ANOVA or Chi-square test as appropriate. Following one-way ANOVA, the Bonferroni multiple comparison test or planned orthogonal contrasts were used to compare groups. Differences were considered significant at p ≤ 0.05 for all the statistical comparisons.
Results
Cre-mediated recombination is increased by DSS treatment in CACTg/WT;Rosa26Rflox/WT; ApcΔ580/WT mice
Our group has previously reported that CAC mice have recombination of the Rosa allele that is restricted to the large intestine epithelial cells [14]. In our experiments, β-gal labeling is used as an indicator of Cre-mediated transgene recombination. In experiment 1, we examined the proximal and distal colon of CACTg/WT;Rosa26Rflox/WT;ApcΔ580/WT mice without DSS treatment (Fig 1A-D). The proximal colon under the basal, non-DSS condition had significantly more β-gal positive cells than the distal colon (Fig. 1E, p=0.0023). However, β-gal labeling in proximal colon was mostly superficial, while entire crypts were labeled in distal colon (Fig. 1B, D). As a result, 5-fold more β-gal positive crypts were observed in the distal colon than the proximal colon, (Fig. 1F, p=0.0011).
Figure 1. Baseline βgal expression patterns are different between proximal and distal colon in CACTg/WT;Rosa26Rflox/WT;ApcΔ580/WT mice.
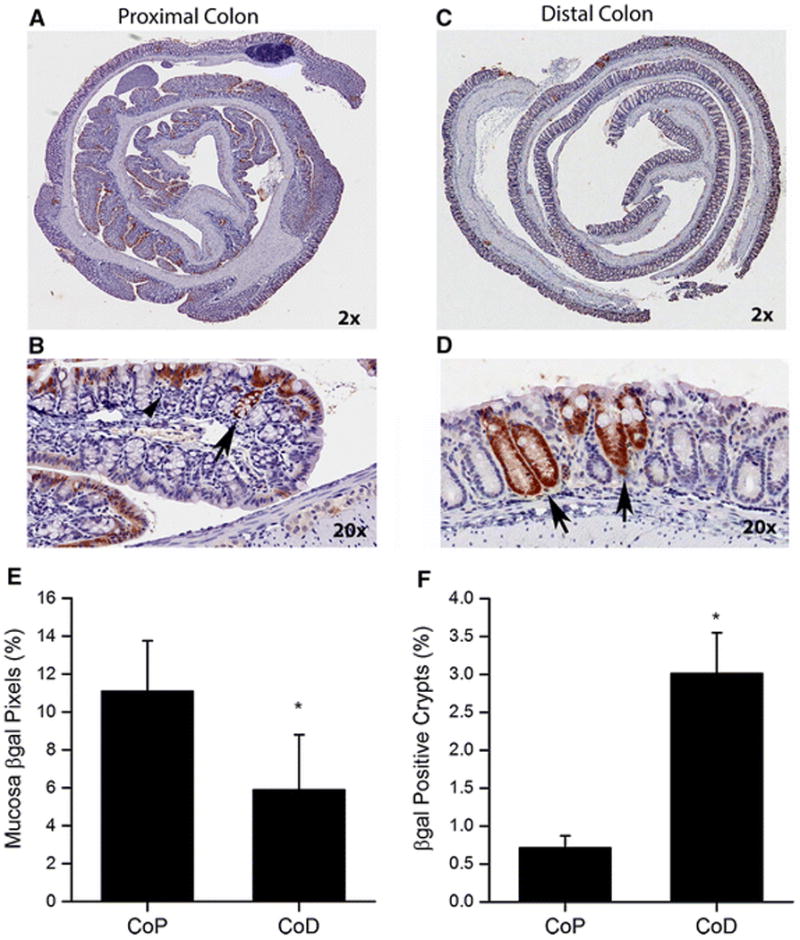
Colons from 10 weeks old mice were prepared as Swiss rolls, processed, and stained for β-gal expression by immunohistochemistry. Representative images were generated using an Aperio ScanScope digital slide scanner and are presented for proximal colon (A, B) and distal colon (C, D), The top pictures are at 2X magnification and the bottom pictures are at 20X magnification. (B) β-gal labeling in proximal colon Swiss rolls was predominantly superficial (arrow head) with occasional labeling to the crypt base (arrow). (D) β-gal labeling in distal colon Swiss rolls was seen from the crypt base to the luminal surface (arrow). (E) Total mucosal β-gal positive pixels and (F) β-gal positive crypts were quantified. The bars represent the means ± SEM of 8 animals for data from the proximal colon (CoP) and distal colon (CoD). * p< 0.05 vs CoP.
Ten days after DSS treatment β-gal labeling was significantly increased at all three DSS doses (Fig. 2A, vehicle vs DSS, Proximal colon: p<0.0001; Distal colon: p=0.0007). In both the proximal and distal colon, a significant increase in β-gal labeling was observed as more DSS was used, although the labeling plateaued in the 2% DSS treatment group (Fig. 2A). In addition, the increased β-gal labeling was present from the surface to the crypt base in both colonic segments (Fig. 3). The impact of DSS treatment on β-gal activity was also examined in the small intestine, liver, spleen, lung and kidney. As reported previously [14], β-gal activity was not observed in the small intestine, even after DSS induction (data not shown). Both liver and lung had low level baseline transgene recombination but this was not increased by DSS treatment. Kidney had non-specific β-gal activity in non-transgenic mice but this did not increase in either the CAC mouse or the CAC mouse treated with DSS. In the spleen there was no transgene expression either at baseline or after DSS treatment (Supplemental Fig. S1).
Figure 2. Colon β-gal expression is positively correlated with epithelial damage in CACTg/WT;Rosa26Rflox/WT;ApcΔ580/WT mice following DSS treatment.
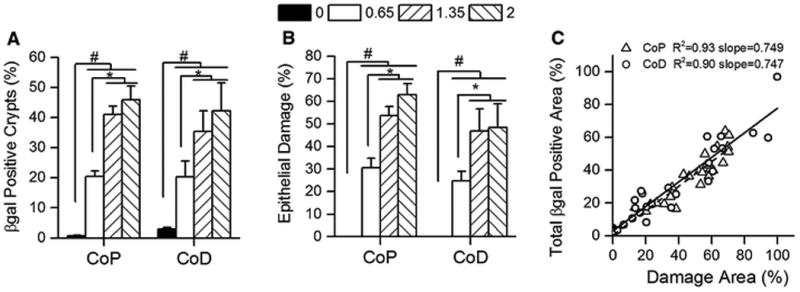
Mice were given increasing amounts of DSS for 5 days then examined histologically 10 days later. (A) The percentage of β-gal positive crypts in the proximal (CoP) or distal (CoD) colon. (B) The percentage of damaged epithelium (includes colon epithelial ulceration, restitution, crypt fission and other abnormal crypt structures observed during the healing process). Bars represent the mean ± SEM of 8 animals per group. Planned orthogonal comparisons were performed between no DSS vs the DSS treatment groups (# p<0.05), the low DSS vs high DSS groups (* p<0.05), and the two high DSS groups (not significant). (C) The relationship between the percentage epithelial damage and β-gal positive area in colon. The regression line for the proximal colon is shown as a dashed line. The regression line for the distal colon is shown as a solid line.
Figure 3. Immunohistochemical labeling for βgal in proximal and distal colon of DSS-treated CACTg/WT;Rosa26Rflox/WT;ApcΔ580/WT mice.
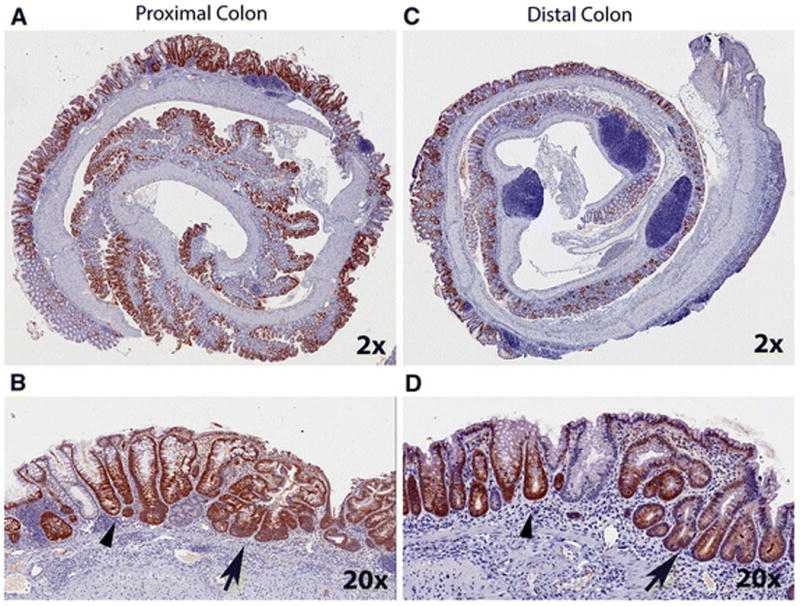
Animals were treated with 2% DSS for 5 days, and the tissues were collected at 10 days after stopping DSS treatment. Representative images were generated using an Aperio ScanScope digital slide scanner and are presented for proximal colon (A, B) and distal colon (C, D). The top pictures are at 2X magnification and the bottom pictures are at 20X magnification. Arrow head = normal crypt structure adjacent to a healing area. Arrow = crypt fission phenotype within a healing area.
As expected, the percentage of damaged epithelial surface was higher with increasing DSS dose in both proximal and distal colon (Fig. 2B). In addition, there was a direct, positive correlation between epithelial damage and β-gal expression in both colonic segments (r2 >0.9, slope = 0.75, Fig. 2C). Also, regardless of the DSS dose, approximately 60% of the abnormal crypts in the areas of epithelial damage were β-gal positive (data not shown). This indicates that Cre-mediated recombination of the LacZ locus has occurred in the epithelium that expanded to heal the DSS induced ulcers.
Increased β-gal expression resulting from DSS induced colon damage is sustained after tissue recovery
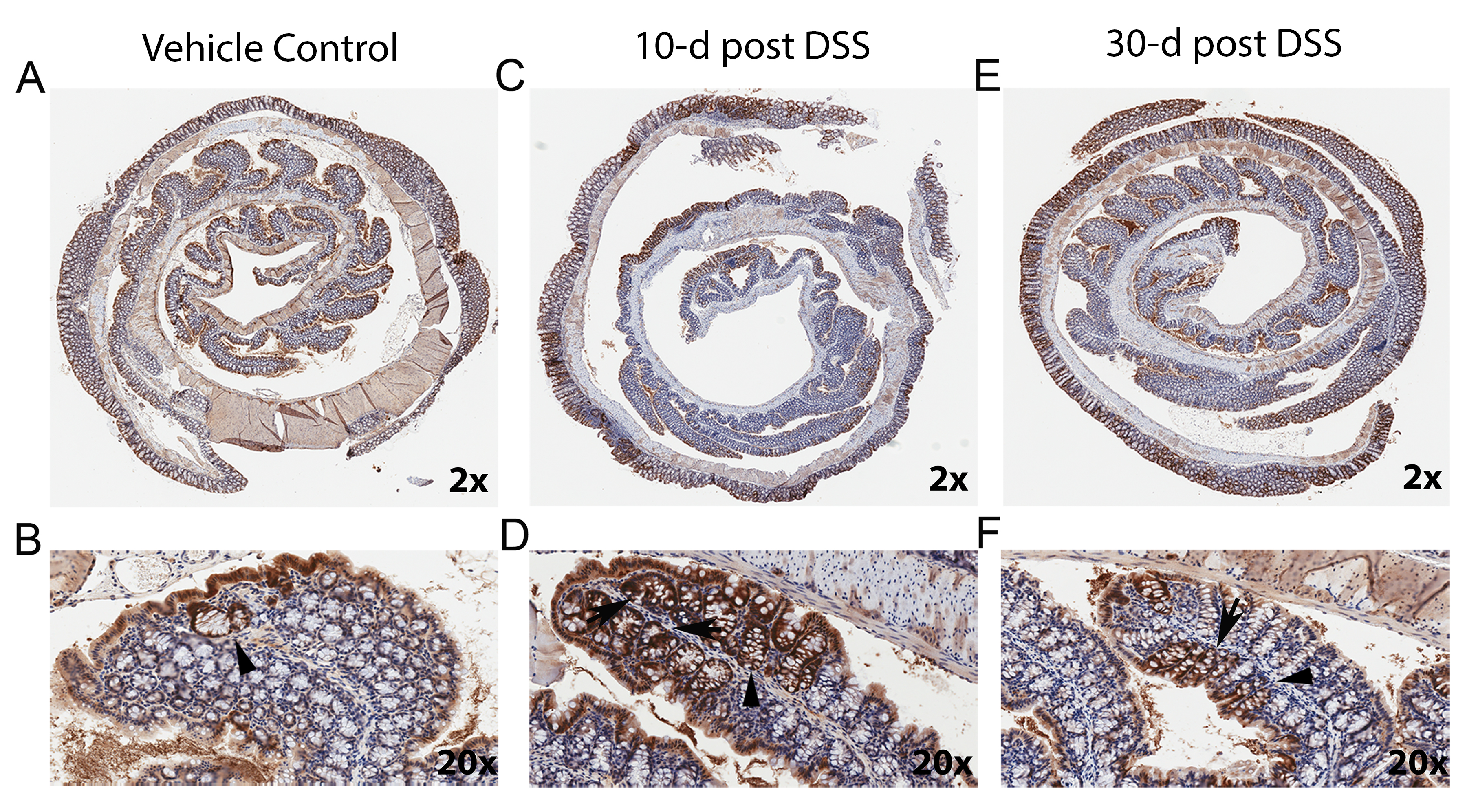
In normal colon mucosa, the crypts are well organized and separated by very little lamina propria (Fig 4A,B). 10 days after DSS treatment, the ulcerated mucosa was undergoing active healing, characterized by epithelial restitution, crypt fission, and crypt regeneration (Fig 4C, D). By 30 days after DSS treatment, the ulcerated mucosa was filled with normal-appearing crypts that were separated by slightly increased amounts of lamina propria (Fig 4E, F). As we observed in the first study, the total percentage of β-gal positive crypts was significantly higher in the DSS treatment groups than the vehicle controls at 10-day post DSS (proximal colon in Fig. 5A, p<0.0001, distal colon in Fig. 5D, p=0.0024) and greater than 60% of the crypts in the healing ulcer were β-gal positive (Fig. 5B, E). This increased β-gal staining remained 30 days after termination of DSS treatment (Fig. 4, Supplemental Fig. S2, and Fig. 5). In fact, the percentage of β-gal positive crypts in the healing/healed ulcer was higher at 30-day post DSS than 10-day post DSS (e.g. 75.2±4.4 % vs 57.9±6.3 % in proximal colon). The DSS treatment group also had significantly more β-gal positive crypts in areas of normal-appearing crypts that were adjacent to healing mucosa (Fig. 5C, F). This indicated that the transgene expression induced by DSS was sustained in the tissue after recovery of the damaged epithelium.
Figure 4. After DSS treatment transgene expression is increased in regenerating crypts and is sustained after healing of ulcers in distal colon.
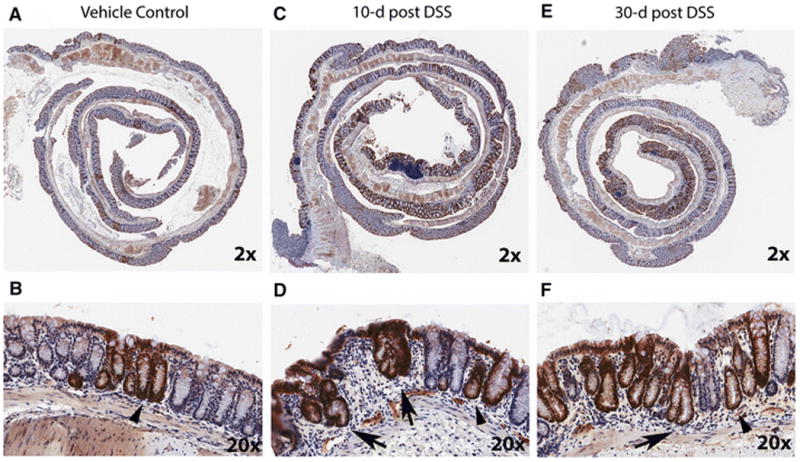
(A, B) Representative images of βgal expression level in the distal colon of a CACTg/WT;Rosa26Rflox/flox mouse treated with vehicle under 2X (A) and 20X (B) magnification. Crypt base labeling is highlighted with arrow head. (C, D) Images of βgal labeling in a CACTg/WT;Rosa26Rflox/flox mouse 10 days after the end of treatment with 1.35% DSS under 2X (C) and 20X (D) magnification. (E, F) Images of βgal labeling in a CACTg/WT;Rosa26Rflox/flox mouse 30 days after the end of treatment with 1.35% DSS under 2X (E) and 20X (F) magnification. In (C,D, E, F), Arrow = βgal positive crypts undergoing regeneration. Arrow head = β-gal positive crypts with normal phenotype adjacent to a healing area. All the images were generated using an Aperio ScanScope digital slide scanner.
Figure 5. The percentage of β-gal positive crypts increases after DSS treatment and is sustained after regeneration in the colon of CACTg/WT;Rosa26Rflox/flox mice.
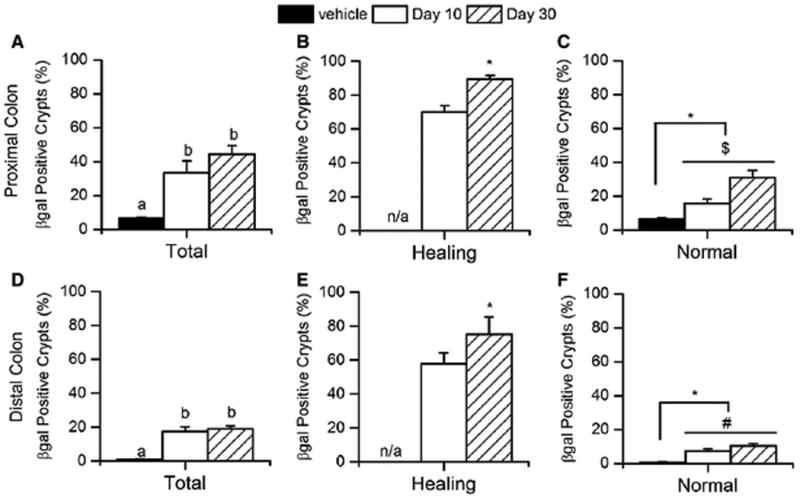
(A, D) The percentage of β-gal positive crypts was measured in proximal (A) and distal (D) colon at 10 or 30 days after ending DSS treatment. Means with different letters are significantly different (Bonferroni, p< 0.05). (B, E) The percentage of β-gal positive crypts in mucosa that exhibits a healing phenotype in the proximal (B) and distal (E) colon. Student t-tests were used to compare values between day 30 and day 10 (* p<0.05). (C, F) The percentage of βgal positive crypts in normal mucosa was measured in proximal (C) and distal (F) colon. Planned orthogonal comparisons were performed (* p<0.05, day 0 vs post-treatment values; $ p<0.05, day 10 vs day 30; # p<0.10, day 10 vs day 30). Bars represent the mean ± SEM of 8 animals per group.
DSS induced transgene recombination increases tumor formation
We first examined whether β-gal staining occurs coincident with adenomatous phenotypes that result from deletion of two floxed Apc alleles. In CACTg/WT;Rosa26Rflox/WT;ApcΔ580/Δ580 mice, 30% of the distal colon exhibited an adenomatous phenotype consistent with recombination of both Apc alleles (i.e. increased cytoplasmic basophilia, increased nuclear to cytoplasmic ratio, loss of nuclear basal polarity, disruption of crypt architecture). While only 2.4% of crypts in normal mucosa were β-gal positive (Fig. 6A), within the adenomatous regions 88.8±1.5% of the epithelial cells were positive for β-gal (Fig. 6B). The co-occurrence of β-gal labeling (indicative of recombination at the ROSA26R locus) and the adenomatous phenotype (indicative of recombination of both floxed Apc alleles) suggests that Cre mediated recombination of floxed alleles is very efficient.
Figure 6. Colon tissue with an adenomatous phenotype show high βgal expression level in 4-week-old CACTg/WT; Rosa26Rflox/WT; ApcΔ580/Δ580 mice.
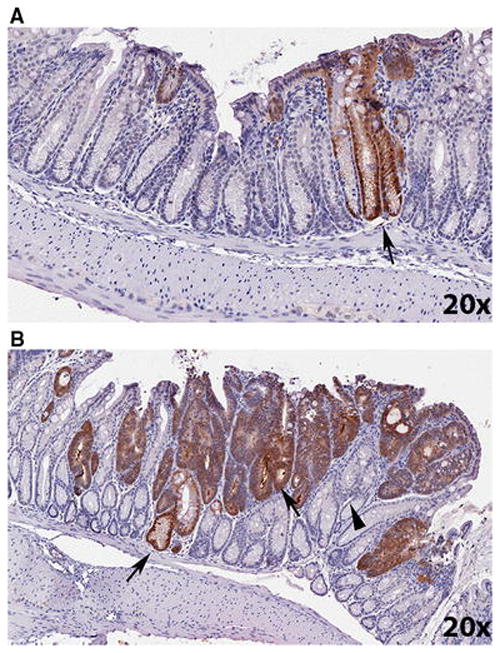
(A) βgal expression in a normal area of the distal colon. Arrow = βgal labeling in a normal crypt. (B) βgal expression in distal colon adenomatous phenotype. Arrow = adenomatous phenotype with βgal positive labeling. Arrow head = adenomatous phenotype with βgal negative labeling. Images were generated using an Aperio ScanScope digital slide scanner. Magnification: 20X.
Since DSS treatment induces Cre-mediated recombination in crypts, we hypothesized that DSS would increase tumor incidence in CACTg/WT;ApcΔ580/WT mice by expanding the colon-specific inactivation of a single Apc allele, coupled with the well-established tumor-promoting effects of inflammation [18]. In the vehicle treated group, 18.6% of the mice had a colonic tumor and the tumors were all in the distal colon (Table 1, 1.25 per mouse). In contrast, DSS treatment significantly increased the percentage of mice with colonic tumors to 78.9% (DSS vs Vehicle, p<0.0001) and this included the appearance of tumors in the proximal colon in 34% of mice. The number of colonic tumors developed per mouse was also significantly higher in the DSS treated mice compared to the vehicle controls (4.20 vs 1.25 per mouse, p<0.0001). The DSS treatment resulted in twice as many mice with tumors in the distal colon compared to the proximal colon (Table 1, 74% vs 34%, respectively) and twice as many tumors in the distal versus the proximal colon (Table 1, 3.71 vs 1.47 per mouse, respectively). Thus the CAC mediated recombination of Apc alleles induced by DSS along with the pro-inflammatory environment within DSS-induced ulcers was a potent promoter of colon cancer.
Table 1. DSS Increases Tumor Incidence in CACTg/WT;ApcΔ580/WT Mice.
| Vehicle | DSS | |||||||
|---|---|---|---|---|---|---|---|---|
|
| ||||||||
| n | # Tumors | Incidence (%) | # per mousea | n | # Tumors | Incidence (%) | # per mousea | |
| Any Tumor | 9 | 12 | 20.9 | 1.33±0.24 | 37 | 186 | 97.4* | 5.03±0.56# |
| Cecum | 2 | 2 | 4.7 | 1.00±0.00 | 30 | 60 | 81.6* | 2.00±0.32# |
| Proximal colon | 0 | 0 | 0.0 | 0.00 | 15 | 22 | 34.2* | 1.47±0.29# |
| Distal colon | 8 | 10 | 18.6 | 1.25±0.25 | 28 | 104 | 73.7* | 3.71±0.35# |
| Proximal + Distal | 8 | 10 | 18.6 | 1.25±0.25 | 30 | 126 | 78.9* | 4.20±0.40# |
| No Tumor | 34 | 0 | 79.1 | 1 | 0 | 2.6 | ||
| Total Mice | 43 | 38 | ||||||
Notes: Significantly different from the vehicle group
Chi-square test, p<0.05;
Student's T-tests, p<0.05.
Average number of tumors per tumor-bearing mouse
Discussion
Our results expand the utility of the CAC mouse model by demonstrating that recombination of floxed alleles driven by the Cre recombinase transgene is increased during DSS-colitis. This effect was restricted to large intestinal epithelial cells, was tightly correlated with DSS induced epithelial damage, was visible during the healing of ulcers, and persisted in crypts within the healed epithelium. However, since DSS causes colon epithelial damage that is accompanied by local inflammation, it is not clear whether induction of transgene activity is a result of the epithelial damage, healing, inflammation, or the factors combined. While the mechanism for this effect is not known, two lines of evidence suggest that the colonic stem cell has been affected. First, recombination is evident in epithelial cells from the luminal surface to the crypt base where colon stem cells reside, and second, the effect is persistent in crypts and lasts after the damaged epithelium has healed.
In addition to the CAC mouse we developed and used in these experiments [14], several other mouse models exist for colon research and each have advantages and disadvantages [12]. A 12.4 kb region of the mouse villin promoter drives Cre recombinase expression uniformly throughout the epithelial cells of the intestine [19]. However, villin-Cre transgene expression extends into the small intestine [19] and this can confound interpretation of events occurring in the colon. An improvement on the villin-Cre mouse was accomplished by using a 9 kb villin promoter to drive a CreERT2 transgene that is tamoxifen inducible and which can cause gene deletions that are spatially and temporally controlled. However, 60 days after treating villin-CreERT2 mice with tamoxifen, the persistence of recombination in small intestine was seen in 80% of the epithelium while in the colon it was just 40% [20]. Moreover, villin promoter driven Cre or Cre-ERT2 transgene expression level is lower in the crypt base where stem cells are found [19,20] and this suggests that Cre activity in the colon stem cell compartment may not be sufficient for optimal recombination.
Deletion of floxed alleles directly in intestinal stem cells can be accomplished using the Lgr5 promoter-CreERT2 mouse [21] and the Lrig1 promoter-CreERT2 mouse [22]. Unfortunately, inducible Cre-ERT2 transgene activity is very low in the large intestine of Lgr5 promoter-CreERT2 mice [23] and so, like the villin promoter models, the Lgr5 promoter-CreERT2 mouse is best for inducing small intestinal cancer. In contrast, the Lrig1 promoter-CreERT2 mouse forms multiple adenomas in the distal colon when it is used to delete an Apc allele; however, many tumors are also seen in the small intestine [22].
An improvement on these models is the CDX2 promoter-Cre mouse whose transgene expression is seen only in cecum, colon and distal ileum of adult mice. When it is used to delete a floxed Apc allele, tumors form in the ileum and distal colon [24]. Unfortunately, transient expression of the CDX2-Cre transgene during embryonic development causes recombination of floxed alleles throughout the distal half of the body. This could reduce the utility of the CDX2-Cre model for many genes whose functions are present outside the colon. A tamoxifen-inducible version of this model, CDX2-CreERT2, avoids the problem of embryonic transgene recombination [23]. However, the induced transgene activity is much higher in proximal colon than the distal colon so its utility may be limited.
Our original CAC model [14] has a major advantage over the other models just described -intestinal transgene expression is specific to the large intestine. This makes the model useful to study the role of specific genes in basic colonic biology and in cancer. A weakness of the CAC model is that CAC transgene expression affects just a small portion of the colonic epithelium and extends to the crypt base mostly in the distal colon. Here we report a major advance for the CAC mouse – that Cre-mediated recombination is induced during DSS-mediated epithelial damage in the colon. The effect of DSS-induced colitis on CAC transgene activity occurs to a comparable degree in both the proximal and distal colon. Thus, the DSS/CAC model will allow researchers to evaluate the role of epithelial cell specific genes in the development and recovery of colitis as well as in colitis-associated colon cancer. The DSS/CAC model also has the advantage that the increase in transgene activation is directly related to the dose of DSS used but that increase does not involve the entire mucosa. For example, we found that only 40-50% of the epithelial surface in the distal colon became transgene positive after treating mice with 2% DSS for 5 days. Thus, in this model both normal tissue (without transgene expression) and tissue with recombined alleles will be adjacent to one another. This will reduce the biological variation caused by intra-animal differences by providing an internal control for paired analysis of treatment effects.
As a preliminary test of the DSS/CAC model, we used it to examine the impact of Apc allele deletion on the development of colon cancer. We found that in CAC mice with one floxed Apc allele, DSS increased colonic tumor incidence 4-fold (to 78.9%) and tumor number per mouse 3-fold (to 4.5 colonic tumors per mouse). However, despite the fact that DSS-induces recombination of floxed alleles equally in both proximal and distal colon, tumors formed predominantly in the distal colon. This suggests that the different environmental influences interact with inflammation and Apc allele loss to affect cellular transformation and tumor development in the proximal and distal colon.
In summary, we observed a novel phenotype for the CAC model - that the Cre transgene mediated recombination is induced following DSS treatment. This opens the opportunity to study gene function at the time of colitis development as well as after the recovery of colitis. We believe the DSS/CAC model is an efficient, reliable model for research on the epithelial cell biology of colitis and inflammation-induced colon cancer. This will make the CAC/DSS model a powerful tool to study the mechanisms by which inflammation promotes transformation as well as to assess interventions to minimize the impact of inflammation on carcinogenesis.
Supplementary Material
{kind=link}
{kind=link}
Acknowledgments
Financial support: The work was supported by awards from the American Institute for Cancer Research (Award # 09A098, JCF) and the National Cancer Institute (NCI) (CA156240, JCF), a pilot grant from the Purdue University Center for Cancer Research, an NCI designated basic science cancer center (P30 CA02316, JCF), fellowship support from the Purdue Interdisciplinary Cancer Prevention Internship Program from National Institutes of Health (R25 CA128770, FW, MLD), and an Abbott Laboratories Pathology Research Fellowship (RLJ).
Abbreviations
- APC
adenomatosis polyposis coli
- CA1
carbonic anhydrase I
- CDX2
caudal type homeobox 2
- DSS
dextran sulfate sodium
- WT
wild-type
Footnotes
Conflict of interest: Dr. Fleet is on the Scientific Advisory Board for Innophos, Inc.
Ms. Fa Wang and Dr. DeSmet have no conflicts to report.
Dr. Johnson is currently an employee of Eli Lilly and Company.
Dr. Synder is currently an employee of EPL, Inc.
Ethical approval: All procedures performed in studies involving animals were in accordance with the ethical standards of Purdue University Animal Care and Use Committee at which the studies were conducted.
This article does not contain any studies with human participants performed by any of the authors.
Authors contributions: Conception and design: JCF; Development of methodology: FW and RLJ; Acquisition of data: FW, RLJ and MLD; Analysis and interpretation of data: FW, RLJ and JCF; Administrative, technical or material support: JCF and PWS; Study supervision: JCF.
References
- 1.Al-Sukhni W, Aronson M, Gallinger S. Hereditary colorectal cancer syndromes: familial adenomatous polyposis and lynch syndrome. Surg Clin North Am. 2008;88:819–844. vii. doi: 10.1016/j.suc.2008.04.012. [DOI] [PubMed] [Google Scholar]
- 2.Vargas AJ, Thompson PA. Diet and nutrient factors in colorectal cancer risk. Nutr Clin Pract. 2012;27:613–623. doi: 10.1177/0884533612454885. [DOI] [PubMed] [Google Scholar]
- 3.Monteleone G, Pallone F, Stolfi C. The dual role of inflammation in colon carcinogenesis. Int J Mol Sci. 2012;13:11071–11084. doi: 10.3390/ijms130911071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fleet JC, DeSmet M, Johnson R, Li Y. Vitamin D and cancer: a review of molecular mechanisms. Biochem J. 2012;441:61–76. doi: 10.1042/BJ20110744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Salaspuro M. Interactions of alcohol and tobacco in gastrointestinal cancer. J Gastroenterol Hepatol. 2012;27(Suppl 2):135–139. doi: 10.1111/j.1440-1746.2012.07017.x. [DOI] [PubMed] [Google Scholar]
- 6.Stone WL, Krishnan K, Campbell SE, Palau VE. The role of antioxidants and pro-oxidants in colon cancer. World J Gastrointest Oncol. 2014;6:55–66. doi: 10.4251/wjgo.v6.i3.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rezaei-Tavirani M, Safaei A, Zali MR. The Association between Polymorphismsin Insulin and Obesity Related Genesand Risk of Colorectal Cancer. Iran J Cancer Prev. 2013;6:179–185. [PMC free article] [PubMed] [Google Scholar]
- 8.Campos FG, Logullo Waitzberg AG, Kiss DR, Waitzberg DL, Habr-Gama A, Gama-Rodrigues J. Diet and colorectal cancer: current evidence for etiology and prevention. Nutr Hosp. 2005;20:18–25. [PubMed] [Google Scholar]
- 9.Tarraga Lopez PJ, Albero JS, Rodriguez-Montes JA. Primary and secondary prevention of colorectal cancer. Clin Med Insights Gastroenterol. 2014;7:33–46. doi: 10.4137/CGast.S14039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mundade R, Imperiale TF, Prabhu L, Loehrer PJ, Lu T. Genetic pathways, prevention, and treatment of sporadic colorectal cancer. Oncoscience. 2014;1:400–406. doi: 10.18632/oncoscience.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Johnson RL, Fleet JC. Animal models of colorectal cancer. Cancer Metastasis Rev. 2013;32:39–61. doi: 10.1007/s10555-012-9404-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fleet JC. Animal models of gastrointestinal and liver diseases. New mouse models for studying dietary prevention of colorectal cancer Am J Physiol Gastrointest Liver Physiol. 2014;307:G249–259. doi: 10.1152/ajpgi.00019.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sussman DA, Santaolalla R, Strobel S, Dheer R, Abreu MT. Cancer in inflammatory bowel disease: lessons from animal models. Curr Opin Gastroenterol. 2012;28:327–333. doi: 10.1097/MOG.0b013e328354cc36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Xue Y, Johnson R, DeSmet M, Snyder PW, Fleet JC. Generation of a transgenic mouse for colorectal cancer research with intestinal cre expression limited to the large intestine. Mol Cancer Res. 2010;8:1095–1104. doi: 10.1158/1541-7786.MCR-10-0195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Soriano P. Generalized lacZ expression with the ROSA26 Cre reporter strain. Nat Genet. 1999;21:70–71. doi: 10.1038/5007. [DOI] [PubMed] [Google Scholar]
- 16.Reeves PG, Nielsen FH, Fahey GC. AIN-93 purified diets for laboratory rodents: Final report of the american institute of nutrition Ad Hoc writing committee on the reformulation of the AIN-76A rodent diet. J Nutr. 1993;123:1939–1951. doi: 10.1093/jn/123.11.1939. [DOI] [PubMed] [Google Scholar]
- 17.Moolenbeek C, Ruitenberg EJ. The “Swiss roll”: a simple technique for histological studies of the rodent intestine. Lab Anim. 1981;15:57–59. doi: 10.1258/002367781780958577. [DOI] [PubMed] [Google Scholar]
- 18.Clapper ML, Cooper HS, Chang WC. Dextran sulfate sodium-induced colitis-associated neoplasia: a promising model for the development of chemopreventive interventions. Acta Pharmacol Sin. 2007;28:1450–1459. doi: 10.1111/j.1745-7254.2007.00695.x. [DOI] [PubMed] [Google Scholar]
- 19.Madison BB, Dunbar L, Qiao XT, Braunstein K, Braunstein E, Gumucio DL. Cis elements of the villin gene control expression in restricted domains of the vertical (crypt) and horizontal (duodenum, cecum) axes of the intestine. J Biol Chem. 2002;277:33275–33283. doi: 10.1074/jbc.M204935200. [DOI] [PubMed] [Google Scholar]
- 20.El Marjou F, Janssen KP, Chang BH, et al. Tissue-specific and inducible Cre-mediated recombination in the gut epithelium. Genesis. 2004;39:186–193. doi: 10.1002/gene.20042. [DOI] [PubMed] [Google Scholar]
- 21.Barker N, Ridgway RA, van Es JH, et al. Crypt stem cells as the cells-of-origin of intestinal cancer. Nature. 2009;457:608–611. doi: 10.1038/nature07602. [DOI] [PubMed] [Google Scholar]
- 22.Powell AE, Wang Y, Li Y, et al. The pan-ErbB negative regulator Lrig1 is an intestinal stem cell marker that functions as a tumor suppressor. Cell. 2012;149:146–158. doi: 10.1016/j.cell.2012.02.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Feng Y, Sentani K, Wiese A, et al. Sox9 induction, ectopic Paneth cells, and mitotic spindle axis defects in mouse colon adenomatous epithelium arising from conditional biallelic Apc inactivation. Am J Pathol. 2013;183:493–503. doi: 10.1016/j.ajpath.2013.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hinoi T, Akyol A, Theisen BK, et al. Mouse model of colonic adenoma-carcinoma progression based on somatic Apc inactivation. Cancer Res. 2007;67:9721–9730. doi: 10.1158/0008-5472.CAN-07-2735. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.