

Abstract
Natural Killer cells (NK cells) are one of the main effector immune cells involved in Antibody-Dependent Cell-mediated Cytotoxicity (ADCC). Upon recognition of cell bound IgG antibodies, which occurs through Fc gamma receptors (FCGRs) expressed on the cell surface of NK cells, NK cells become activated and lyse target tumor or infected cells. The FCGRs, FCGR3A and FCGR2C, expressed on the surface of NK cells have single nucleotide polymorphisms (SNPs) that result in differential activity of NK cells. In addition to SNP genetic variation within each of these genes, the FCGRs are subject to copy number variation (CNV), which leads to variable protein expression levels on the cell surface. Studies have found that FCGR genotype for FCGR3A and FCGR2C is associated with variation in the response to immunotherapy.
Due to high sequence homology within FCGR3 and FCGR2 families, there are difficulties associated with genotyping these specific receptors related to cross-amplification of non-targeted FCGRs. To improve specificity for both FCGR3A and FCGR2C, Rnase-H primers (RH primers) were designed to amplify specifically FCGR3A (while not co-amplifying FCGR3B) and FCGR2C (while not co-amplifying FCGR2B). In addition, fluorescently labeled Locked Nucleic Acid (LNA) probes provide additional precision for determination of the SNPs within both FCGR3A and FCGR2C. For CNV determination, separate fluorescently labeled probes for FCGR3A, and for FCGR2C, can be used with the same RH primers for each gene. These probes can be combined in the same well with control primers/probe for a known diploid gene and used to calculate the copy number of both FCGR3A and FCGR2C. Here we provide new detailed methodology that allows for the specific amplification of these FCGRs in a single PCR reaction, allowing for genotyping of both the SNPs and CNVs using Real Time PCR.
Keywords: FCGR3A, FCGR2C, NK cells, ADCC, SNP, immunotherapy, cancer immunology, CNV
1. INTRODUCTION
The innate immune effector cells, Natural Killer cells (NK cells), are major contributors in the fight against certain diseases and infections (viral infections, various forms of cancer, etc.). NK cells can be targeted to “damaged” cells via antibody recognition through Fc gamma receptors [FCGR3A (CD16A) and FCGR2C (CD32C)] expressed on their cell surface, which recognize the Fc portion of the IgG antibodies [1]. Engagement of the FCGRs expressed on NK cells activates the NK cell to kill the target cell, a process termed Antibody-Dependent Cell-mediated Cytotoxicity (ADCC) [2,3].
The FCGRs are either activating or inhibitory, and are expressed on a variety of immune cells. They include variants such as FCGR1, FCGR2A, FCGR2B, FCGR2C, FCGR3A and FCGR3B. Both FCGR2C and FCGR3A are expressed on NK cells, and both have been shown to be subject to genetic variability within the population [4]. Along with some of the other FCGRs, both FCGR2C and FCGR3A have been found to be copy number variable (CNV), and, depending on the individual, can deviate from the “standard” 2 copies (inheriting one from maternal genome, one from paternal genome), with 2 copies occurring the most commonly among the population [5–8]. Additionally, FCGR2C and FCGR3A each have single nucleotide polymorphisms (SNPs); dependent upon the SNP that a person has for each of these genes, the different SNPs create variability in the ability of the FCGRs to interact with the Fc portion of the IgG antibodies.
FCGR3A has a SNP at amino acid position 158 that encodes for either a valine (V) or a phenylalanine (F), resulting in altered affinity of the receptor for IgG (rs396691) [1,9,10]. Alternatively, FCGR2C has a SNP in Exon 3 (rs759550223) that results in either expression, or non-expression, of the protein on the cell surface [11,12]. FCGR2C with a “C” at nucleotide position 202 results in an open reading frame (ORF) and expression on the cell surface. In contrast, a “T” at nucleotide position 202 results in a stop codon and the protein is not expressed on the cell surface [13,12,11]. Determining the SNP variant for each FCGR expressed on NK cells may help to predict the function of that individual’s NK cells in ADCC, and thus potentially indicate how a person may respond to ADCC-mediating antibody therapies that aim to target NK cells toward cells targeted for destruction.
For such genotyping, amplifying the area of DNA containing the SNP or CNV region has been difficult as the FCGRs have high sequence homology. In particular, FCGR3A and FCGR3B have very few nucleotide differences in the region that surrounds the amino acids that encode the FCGR3A-158-V/F SNP. In fact, the nucleotides at position FCGR3A-559-G/T translate to the amino acid SNP of FCGR3A-158-V/F SNP. Typing for these is complicated by the fact that the gene for FCGR3B has a “G” nucleotide at position 559 within its sequence. Since both FCGR3A and FCGR3B can have a “G” nucleotide at the region of this SNP, if FCGR3B is incorrectly amplified it will further confound the accuracy of the SNP determination for FCGR3A. Several studies have addressed the difficulties associated with accurately determining the FCGR3A-158-V/F SNP [14–18]. Designing unique methodology to specifically amplify FCGR3A, allowing for accurate determination of its SNP and as well as CNV associated with this gene, is of great importance when trying to associate clinical outcome with the SNP and CNV status for FCGR3A.
We developed Rnase-H primers (RH primers) to specifically amplify FCGR3A, while not co-amplifying FCGR3B [19]. We used Locked Nucleic Acid (LNA)-labeled probes to increase both the sensitivity and the specificity of the probe sequences, and also allow for improved precision of SNP determination [20,21]. The use of Rnase-H-dependent PCR (rhPCR) [Integrated DNA Technologies (IDTDNA)] allows for improved specificity of the intended PCR product (in this case FCGR3A). RH primers include an RNA base that specifically matches a unique nucleotide of FCGR3A, but not the sequence in FCGR3B (Figure 1) [19]. By designing the Real Time PCR reaction to include, within one of the primers, an RNA base that matches FCGR3A, but does not match FCGR3B, we were able to specifically amplify FCGR3A while not co-amplifying FCGR3B as confirmed by Real Time PCR in a single PCR reaction (data not shown). Likewise, FCGR2C was created from the unequal crossover of FCGR2A and FCGR2B, and thus shares high sequence homology to each of those FCGR variants, with sequence homology surrounding the region of the SNP to FCGR2B [11,13,12]. Thus, in a similar manner to FCGR3A, RH primers were designed to specifically amplify FCGR2C while not co-amplifying FCGR2B, with FCGR2C LNA-SNP probes used to determine the genetic variability of FCGR2C within individuals. Furthermore, separate CNV-probes were created for both FCGR3A and FCGR2C, to be used in combination with the same RH primers for both FCGR3A and FCGR2C, respectively. Both the FCGR3A RH primer/CNV-probe and the FCGR2C RH-primer/CNV-probe, can also be used in combination with primers/CNV-probe of a control gene that is known to be diploid within the human genome (RNASE P). By combining the query gene CNV reaction in the same tube as a control gene that is diploid, normalization to the control gene can be done for each sample to accurately analyze the amplification reaction.
Figure 1.
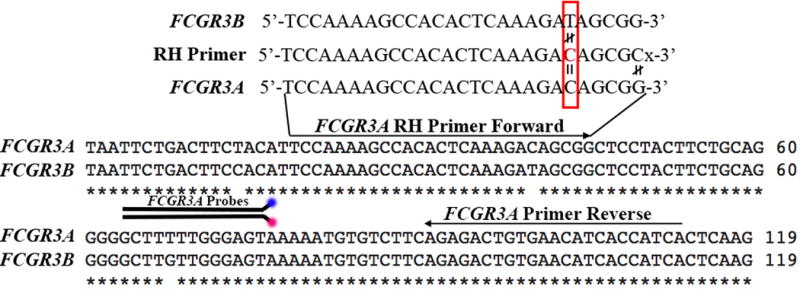
Sequence similarities between FCGR3A and FCGR3B can be resolved for specific amplification using an RNA base (red font) at the nucleotide position where the sequences differ (red box). The RNA base matches the sequence of FCGR3A, but does not match the sequence of FCGR3B. When Rnase Enzyme is included in the PCR reaction, the mismatch of the RNA base from FCGR3B will result in cleavage of the PCR product of FCGR3B not allowing it to amplify. Alternatively, as FCGR3A matches the RNA base, the cleavage will not occur and PCR product will continue to amplify. The 3′ of the RH primer has a mismatched base to FCGR3A followed by a spacer to ensure maximum efficiency of the end block.
By improving the specificity of the PCR reaction for each FCGR expressed on NK cells (FCGR3A and FCGR2C) through the use of RH primers for each gene, greater accuracy should be obtainable for the genotyping results. This improved genotyping accuracy should enable FCGR genotyping to be evaluated with respect to a variety of in vivo settings where the function of distinct FCGR SNP-variants might influence disease susceptibility, severity, or response to therapy.
2. MATERIALS
All work should be conducted in sterile conditions. If possible, prepare reagents in a PCR hood using nuclease-free materials (filter tips, sterile water, sterile microfuge tubes, sterile strip tubes and/or Real Time PCR plates).
2.1. Premade Reagents
Genotyping Master Mix (Applied Biosystems/Life Technologies). Refer to Note 1.
Express qPCR Master Mix (Invitrogen/Life Technologies). Refer to Note 1.
Rnase-H Enzyme and Rnase-H Buffer [Integrated DNA Technologies (IDTDNA)]. Dilute enzyme to 25mU/μL: add 2mL Rnase H2 Enzyme Dilution Buffer to 50U of Rnase-H Enzyme. Aliquot into 0.5mL sterile tubes at 100μL/tube.
2.2. Instrument requirements and Software
Real Time PCR machine with settings capable of assessing SNP allelic discrimination, such as “Genotyping” analysis found in the StepOnePlus Real Time PCR system (Applied Biosystems/Life Technologies). Refer to Note 2.
CNV analysis software, such as CopyCaller® Software (Applied Biosystems/Life Technologies)
2.3. Primers and Probes
All primers used in this protocol were ordered from IDTDNA (Table 1). Refer to Note 2. Resuspend stock primers and probes at 100μM. Separately, create a working solution of each primer and probe by diluting each primer and probe to 10μM (90μL of sterile DNase/Rnase-free water + 10μL of the 100μM stock primer or probe). Store all primers and probes at −20C.
All Gene Block (gBlock) control primers were ordered from IDTDNA. Resuspend gBlock control sequences (Table 2) and dilute to serve as controls for each SNP. Resuspend control sequences to a concentration of 1ng/μL with nuclease free water. Perform serial dilutions (add 1μL of 1ng/μL gBlock DNA to 99μL sterile water, vortex to mix well = 10−2 ng/μL; add 1μL of 10−2 ng/μL gBlock DNA to 99μL sterile water, vortex to mix well = 10−4 ng/μL; add 1μL of 10−4 ng/μL gBlock DNA to 99μL sterile water, vortex to mix well = 10−6 ng/μL = 1fg/μL; add 1μL of 1fg/μL gBlock DNA to 99μL sterile water, vortex to mix well = 10−2 fg/μL) in nuclease free water to a final concentration of 10−2 fg/μL. Refer to Note 4. For “heterozygous” controls, create a mixture of 1:1 FCGR3A-F-CNT with FCGR3A-V-CNT, and create a mixture of 1:1 FCGR2C-C-CNT with FCGR2C-T-CNT.
Table 1. Sequences of primers and probes as ordered through IDTDNA.
All primers are shaded in light gray, SNP probes are shaded in white, and CNV probes are shaded in dark grey. Unmodified/standard primers were purified by standard desalting, Rnase H primers were purified by HPLC. For Rnase H primers, at least 10 base pairs precede the RNA base (bolded). The RNA base is then followed by 5 more base pairs, which are specific for the intended PCR product (FCGR3A or FCGR2C), and then a C3 spacer was added. All probes include a fluorescent probe on the 5′ end of the DNA sequence (Hexachlorofluorescein (HEX), FAM, or ATTO™532), refer to Note 10. LNA modified bases are indicated by the “+” symbol within the DNA sequence. The ZEN® modified CNV probes include “/ZEN/” within the DNA sequence.
| FCGR3A Rnase-H Forward Primer | 5′ TCCAAAAGCCACACTCAAAGArCAGCGC - C3 spacer-3′ |
| FCGR3A Reverse Primer | 5′ GATGGTGATGTTCACAGTCTCT 3′ |
| FCGR3A-G Probe | 5′ FAM TCC+CA+A+C+AA+G+CC 3′ Iowa Black FQ Quencher |
| FCGR3A-T Probe | 5′ ATTO532N TCC+CA+A+A+AA+GC+CC 3′ Iowa Black FQ Quencher |
| FCGR3A-CNV Probe | 5′ FAM AAGCCCCCT/ZEN/GCAGAAGTAGGA 3′ Iowa Black FQ Quencher |
| FCGR2C Forward | 5′ TATTCCTGGCTCCTGTTGC 3′ |
| FCGR2C Rnase-H Reverse | 5′ TGTCAGAGTCACAGAGTCCTCrUTGGAC - C3 spacer-3′ |
| FCGR2C-C Probe | 5′ ATTO532N CAC+T+G+GGG+CT 3′ Iowa Black FQ Quencher |
| FCGR2C-T Probe | 5′ FAM TCCAC+T+A+GGG+CT 3′ Iowa Black FQ Quencher |
| FCGR2C CNV-Probe | 5′ FAM AGCC+C+C+AGTGG/3IABkFQ/ |
| RNASE P Forward | 5′ AGATTTGGACCTGCGAGCG 3′ |
| RNASE P Reverse | 5′ GAGCGGCTGTCTCCACAAGT 3′ |
| RNASE P CNV Probe | 5′ HEX TTCTGACCT/ZEN/GAAGGCTCTGCGCG 3′ Iowa Black FQ Quencher |
Table 2. Control sequences for FCGR3A and FCGR2C. SNP assays.
The sequences below can be ordered as “Gene Blocks” from IDTDNA to serve as positive controls for SNP assays.
| Geneblock Control | Sequence |
|---|---|
| FCGR3A-V-CNT | 5′- GACTTCTACATTCCAAAAGCCACACTCAAAGACAGCGGCTCCTACTTCTGCAGGGGGCTTGTTGGGAGTAAAAATGTGTCTTCAGAGACTGTGAACATCACCATCACTCAAGGTGAGACATGTGCCACCCT-3′ |
| FCGR3A-F-CNT | 5′- GACTTCTACATTCCAAAAGCCACACTCAAAGACAGCGGCTCCTACTTCTGCAGGGGGCTTTTTGGGAGTAAAAATGTGTCTTCAGAGACTGTGAACATCACCATCACTCAAGGTGAGACATGTGCCACCCT-3′ |
| FCGR2C-C-CNT | 5′- CAGCAGCTCCCCCAAAGGCTGTGCTGAAACTCGAGCCCCAGTGGATCAACGTGCTCCAAGAGGACTCTGTGACTCTGACATGCCGGGGGACTCACAGCCCTGAGAGCGACTCCATTCCGTGGTTCCACAATGGGAATCTCATTCCCACCCACACGCAGCCCAGCTACAGGTTCAAGGCCAACAACAATGACAGCGGGGAGTACACGTGCCAGACTGGCCAGACCAGCCTCAGCGACCCTGTGCATCTGACTGTGCTTTCTGGTCAGTGGAGGAAGGCCCCAGGGTGGACCTGGGAGGGCCAGGATGGATGAAATCTGCTTTCAGGCAG-3′ |
| FCGR2C-T-CNT | 5′- CAGCAGCTCCCCCAAAGGCTGTGCTGAAACTCGAGCCCTAGTGGATCAACGTGCTCCAAGAGGACTCTGTGACTCTGACATGCCGGGGGACTCACAGCCCTGAGAGCGACTCCATTCCGTGGTTCCACAATGGGAATCTCATTCCCACCCACACGCAGCCCAGCTACAGGTTCAAGGCCAACAACAATGACAGCGGGGAGTACACGTGCCAGACTGGCCAGATCAGCCTCAGCGACCCTGTGCATCTGACTGTGCTTTCTGGTCAGTGGAGGAAGGCCCCAGGGTGGACCTGGGAGGGCCAGGATGGATGAAATCTGCTTTCAGGCAG-3′ |
3. METHODS
All work should be conducted in sterile conditions, taking care to not cross-contaminate samples or reagents. If possible, prepare all reactions in a PCR hood, using sterile, nuclease-free materials (filter tips, sterile water, sterile microfuge tubes, sterile strip tubes and/or Real Time PCR plates). Keep all reagents on ice during assay setup procedures.
3.1. SNP Genotyping Methodology—FCGR3A-V158F and FCGR2C-C/T
Prepare the following reaction master mix as shown in Table 3. Refer to Note 3. The final concentration of the primers should be 0.3μM of each primer and 0.25μM of each probe included in the master mix of each reaction. Refer to Note 5 for reaction volumes; refer to Note 6 for Rnase H Enzyme concentrations; and refer to Note 14 for master mix tips.
Pipette 4μL of the reaction master mix into each well or tube.
Dilute all DNA samples, including the positive controls, to 5ng/μL. Add 1μL of 5ng/μL DNA sample to the appropriate well per the predesigned template. Refer to Note 7.
Add 1μL of sterile water to the no template well. Refer to Note 8. If known sample control genotypes are available for each SNP being analyzed, load 1μL of 5ng/μL DNA for each known sample control. Refer to Note 9.
Seal/cover the tubes or plate securely.
Place the sample in the Real Time PCR machine. Follow the Real Time PCR machine’s manufacturer’s settings for SNP genotyping settings.
Adjust the reaction tube volume to read 5μL. Set the cycling conditions to those as shown in Table 5. Refer to Note 15.
If known sample control genotypes are available, indicate which samples are known in the sample setup.
After the run is complete, analyze for genotype determinations according to the Real Time PCR machine’s manufacturer settings, or refer to Figure 2A (FCGR3A) and Figure 2B (FCGR2C) for example of expected qPCR results. Refer to Notes 9 and 10.
Table 3. Master Mix for FCGR3A SNP and FCGR2C SNP.
Multiply the number of reactions (# of unknowns= samples * # replicates/sample + # of no template samples + # control samples) * 1.1. Refer to Note 13 for each component by the reaction volume for 1 reaction (RXN).
| FCGR3A | FCGR2C | ||||
|---|---|---|---|---|---|
| Volume (μL) 1 Reaction | Final Concentration | Volume (μL) 1 Reaction | Final Concentration | ||
| 2X Genotyping Master Mix (ABI) | 2.5 | 1X | 2X Express Genotyping Master Mix (Invitrogen) | 2.5 | 1X |
| RH FCGR3A Forward Primer (10uM) | 0.15 | 0.3uM | FCGR2C Forward Primer (10uM) | 0.15 | 0.3uM |
| FCGR3A Reverse Primer (10uM) | 0.15 | 0.3uM | RH FCGR2C Reverse Primer (10uM) | 0.15 | 0.3uM |
| FCGR3A-G Probe (10uM) | 0.125 | 0.25uM | FCGR2C-C Probe (10uM) | 0.125 | 0.25uM |
| FCGR3A-T-Probe (10uM) | 0.125 | 0.25uM | FCGR2C-T-Probe (10uM) | 0.125 | 0.25uM |
| RH Enzyme in Buffer (25U/uL) | 0.2 | 1U/uL | RH Enzyme in Buffer (25U/uL) | 0.2 | 1U/uL |
| DNA (5ng/uL) | 1 | N/A | DNA (5ng/uL) | 1 | N/A |
| Sterile Water | 0.75 | N/A | Sterile Water | 0.75 | N/A |
Table 5. Real Time PCR machine cycling conditions for FCGR3A (SNP and CNV) and FCGR2C (SNP and CNV) reactions.
| FCGR3A | FCGR2C | ||||
|---|---|---|---|---|---|
|
| |||||
| Temperature | Temperature | ||||
| Cycle | ('C) | Time | Cycle | ('C) | Time |
|
| |||||
| 1 | 95 | 10 min | 1 | 95 | 3 min |
| 2 | 95 | 30 sec | 2 | 95 | 5 sec |
| 3 | 60 | 1 min | 3 | 63 | 15 sec |
| Repeat Cycles 2–3 × 40–50 more times | Repeat Cycles 2–3 × 40–50 more times | ||||
Figure 2. Example of Allelic Discrimination Plot results for FCGR3A and FCGR2C.
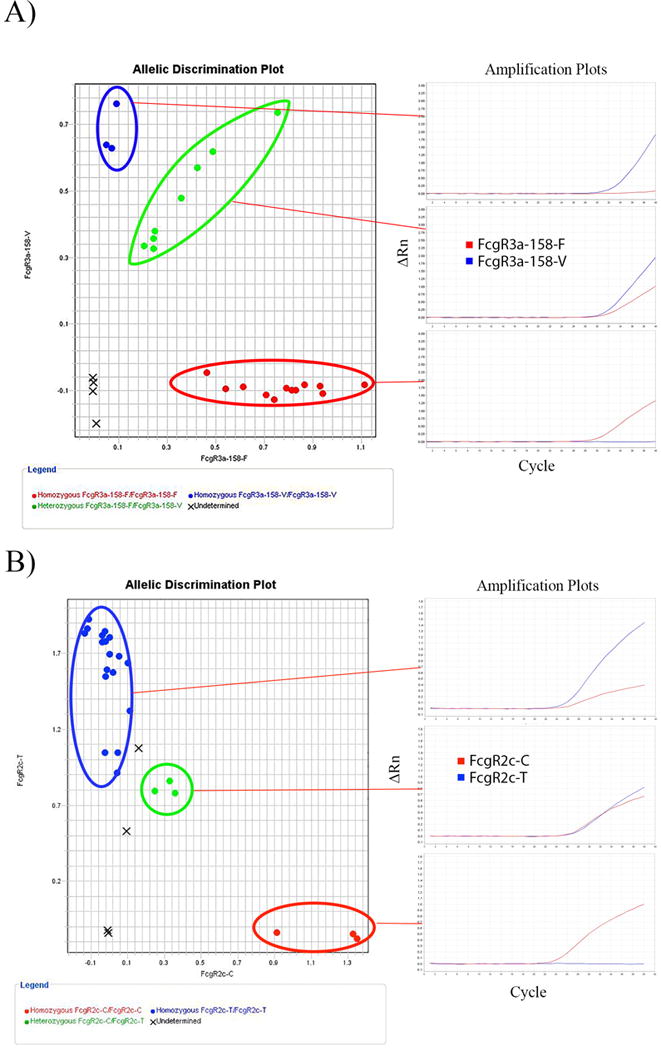
(A) Each allelic variant is separated into quadrants of the plot. Amplification of the ATTO532-T probe, but not of the FAM-G probe, indicates a V/V genotype (blue). Amplification of the FAM-G probe, but not of the ATTO532-T probe, indicates a F/F genotype (red). Amplification of both FAM-G probe and ATTO532-T probe, indicates a V/F genotype (green). No template controls are noted by an “X”. Additionally, samples for which the genotype could not be determined based on the algorithm within the software are also labeled as “X” in the center of the plot. (B) Each allelic variant is separated into quadrants of the plot. Amplification of the FAM-T probe, but not of the ATTO532-C probe, indicates a T/T genotype (blue). Amplification of the ATTO532-C probe, but not of the FAM-T probe, indicates a C/C genotype (red). Amplification of both FAM-T probe and ATTO532-C probe, indicates a C/T genotype (green).
3.2. CNV Genotyping Methodology— FCGR3A and FCGR2C
Prepare the following reaction master mix as shown in Table 4. Refer to Note 3. The final concentration of the primers should be 0.3μM of each primer and 0.25μM of each probe included in the master mix of each reaction. Refer to Note 12 for template suggestions; refer to Note 5 for reaction volumes; and refer to Note 14 for master mix tips.
Pipette 4μL of the reaction master mix into each well or tube.
Dilute all DNA samples, including the positive controls, to 5ng/μL. Add 1μL of 5ng/μL DNA sample to the appropriate well. Plate samples in triplicate per the predesigned template. Refer to Note 13.
Add 1μL of sterile water to the no template well. Refer to Note 8. If known sample control genotypes are available for each CNV being analyzed, load 1μL of 5ng/μL DNA for each known sample control. Refer to Note 12.
Seal/cover the tubes or plate securely.
Place the sample in the Real Time PCR machine. Follow the Real Time PCR machine’s manufacturer’s settings using standard cycling conditions settings.
Adjust the reaction tube volume to read 5μL. Set the cycling conditions to those as shown in Table 5. If known sample control genotypes are available, indicate which samples are known in the sample setup.
After the run is complete, analyze CN according to the Real Time PCR machine’s manufacturer settings. For a StepOnePlus, refer to the CopyCaller software manual for analysis. Adjust the threshold for each curve according to the settings as suggested in the software protocol, export results, and analyze in the CNV analysis software.
Table 4. Master Mix for FCGR3A CNV and FCGR2C CNV.
Multiply the number of reactions (# of unknowns= samples * # replicates/sample + # of no template samples + # control samples) * 1.1 Refer to Notes 11 and 12 for each component by the reaction volume for 1 reaction (RXN).
| FCGR3A | FCGR2C | ||||
|---|---|---|---|---|---|
| Volume (μL) 1 Reaction | Final Concentration | Volume (μL) 1 Reaction | Final Concentration | ||
| 2X Genotyping Master Mix (ABI) | 2.5 | 1X | 2X Express Genotyping Master Mix (Invitrogen) | 2.5 | 1X |
| RH FCGR3A Forward Primer (10uM) | 0.15 | 0.3uM | FCGR2C Forward Primer (10uM) | 0.15 | 0.3uM |
| FCGR3A Reverse Primer (10uM) | 0.15 | 0.3uM | RH FCGR2C Reverse Primer (10uM) | 0.15 | 0.3uM |
| FCGR3A CNV Probe (10uM) | 0.125 | 0.25uM | FCGR2C CNV Probe (10uM) | 0.125 | 0.25uM |
| RNASE P Forward Primer (10uM) | 0.15 | 0.3uM | RNASE P Forward Primer (10uM) | 0.15 | 0.3uM |
| RNASE P Reverse Primer (10uM) | 0.15 | 0.3uM | RNASE P Reverse Primer (10uM) | 0.15 | 0.3uM |
| RNASE P CNV Probe (10uM) | 0.125 | 0.25uM | RNASE P CNV Probe (10uM) | 0.125 | 0.25uM |
| RH Enzyme in Buffer (25U/uL) | 0.2 | 1U/uL | RH Enzyme in Buffer (25U/uL) | 0.2 | 1U/uL |
| DNA (5ng/uL) | 1 | N/A | DNA (5ng/uL) | 1 | N/A |
| Sterile Water | 0.45 | N/A | Sterile Water | 0.45 | N/A |
Acknowledgments
This work was supported, in part, by National Institutes of Health Grants CA032685, CA87025, CA166105, CA14520, a Stand Up To Cancer – St. Baldrick’s Pediatric Dream Team Translational Research Grant (SU2C-AACR-DT1113), The University of Wisconsin-Madison Institute for Clinical and Translational Research Grant 1TL1RR025013-01, and grants from the Midwest Athletes for Childhood Cancer Fund, The Crawdaddy Foundation, and Hyundai Hope on Wheels.
We would also like to thank the technical support staff at IDTDNA, in particular Dr. Elisabeth Wagner, for their assistance in RH primer and probe designs.
Footnotes
It is important to store reagents properly and avoid multiple freeze thaws in order prevent reagent degradation. As reagents become degraded, it is difficult to discriminate between genotype variations (both SNP and CNV).
The Real Time PCR instrument should be capable of duplex reads of, at the minimum, VIC and fluorescein (FAM). ATTO™532 is an N-Hydroxysuccinimide (NHS) Ester that can be substituted for VIC and, depending on the Real Time PCR machine, may require calibration. Hexachlorofluorescein (HEX) is another substitute for VIC, and calibration may be necessary depending on the instrument. HEX and ATTO™532 calibration mixes are each available from IDTDNA.
The Real Time PCR master mix used for the FCGR3A RH primer/probe reaction mix worked well with the Genotyping Master Mix from ABI. When using the same FCGR3A RH primer/probe reaction with the Express qPCR Master Mix from Invitrogen, the results were more difficult to interpret. Alternatively, the Real Time PCR master mix used for the FCGR2C RH primer/probe reaction mix worked well with the Express qPCR Master Mix from Invitrogen, but when using the same FCGR2C RH primer/probe reaction with the Genotyping Master Mix from ABI, the amplification of the separate alleles was not distinguishable. Thus, the Real Time PCR master mix used may affect the interpretability of the results.
It may be necessary to dilute the control gBlock DNA sequences for each gene (FCGR3A and FCGR2C). The goal is to have the control gBlock sequences amplify around the same cycle as the genomic unknown DNA sequences. After initial dilutions are made, run a small test plate to verify the amplification of the gBlock DNA sequences (Table 2) with a small subset of genomic DNA samples, that are each diluted to 5ng/μL, to determine if the samples amplify at the same cycle. Optimize the gBlock DNA concentration further if necessary by performing further 1:10 serial dilutions.
The total reaction volume per tube as prepared in this protocol is 5μL. This total reaction volume can be increased if desired by adjusting all reactions components accordingly to maintain the correct final concentration of the reaction components. If the reaction volume is increased, the concentration of DNA, however, can remain 5ng of DNA/reaction.
The amount of Rnase H Enzyme and Buffer may vary depending on the Real Time PCR master mix used, and may require optimization. The amount of Rnase H Enzyme/Buffer were optimized for the PCR master mix and primer/probe combinations. Depending on the dilution/lot number of the Rnase H Enzyme, optimization may be required.
It is highly recommended to predesign a template for the PCR reaction in a template table, such as in Excel. Run samples at least in duplicate if possible. Include at least one no template negative control. Also, if possible include ≥1 known sample controls for each genotype (e.g. DNA from a sample known to encode FCGR3A-158-F/F; DNA from a sample known to encode FCGR3A-158-V/F; and DNA from a sample known to encode FCGR3A-158-V/V).
The negative control samples should not show amplification.
The known sample controls then can be selected during assay setup allowing for the analysis software to calibrate each reaction read. Also, the user can compare the amplification curves of unknown samples to known controls if the software is unable to differentiate the genotype (Figure 2A and Figure 2B). If known sample genotypes are unavailable, refer to Note 4 and Table 2.
SNP determinants can be made by referring to the amplification plot of the amplified SNPs without allelic discrimination software. When using Allelic Discrimination Software, select samples that serve as positive controls for each allele that is amplified (each homozygote and heterozygotes). Be sure when selecting the positive control samples that the amplification of each allele is associated with the correct probe (for example, if “Allele 1” is associated with the genotype for the “FAM”-labeled probe, then the positive control “Allele1/Allele 1” should be “FAM”-labeled probe. Also, if the allelic discrimination plot yields genotype results that the software algorithm cannot interpret (Figure 2A and 2B “X” labeled “Undetermined”), it may be necessary to refer to the amplification plot to determine the genotype of undeterminable samples. This can be done by referencing the amplification of known genotypes.
It is crucial to have good positive controls (Table 2). The FCGR2C-T probe, although specific for T, does non-specifically amplify FCGR2C-C even when there is no FCGR2C-C present in the reaction mixture. Due to this, in order to determine the allelic discrimination of unknown samples, it is important to refer to control primer reaction (in particular for FCGR2C-T). Depending on the Real Time PCR instrument, it may be helpful to optimize the PCR reaction by performing an annealing temperature gradient. It may be necessary to decrease the annealing temperature slightly (to 58C or 59C) to improve the specificity of this reaction.
Again, it is highly recommended to predesign a template for the PCR reaction assay setup in a template table, refer to Note 7 above. The confidence in the data will be improved by including more DNA samples. During the analysis of CNV, if a known control for the number of copies of each gene (FCGR3a or FCGR2c) exists, it should be included in each run for use during copy number analysis. For these analyses, without a proper copy number control, the expected number of copies for both FCGR3A and FCGR2C can be set as 2 copies per gene. If possible, using healthy donor samples to standardize this assay for future studies can help to standardize the assay. The number of copies for this analysis can then be based on the expected copy number for such healthy donor samples. As such, it is best to use at least 10 different DNA samples for this type of analysis on one plate. By including an adequate number of individual DNA samples, more reliable results can be obtained.
The CNV assay is more reliable if each sample is repeated at least twice, on two separate plates. Each time that a sample is tested in a CNV assay, it should be prepared in triplicate sub-replicates on each plate.
When making the master mix for each reaction, always included additional reaction components to account for pipetting errors. Multiply the total number of samples [including: (unknown samples) × (the number of replicates), the number of no templates, and the number of positive control samples] by at least 1.1. For example, if analyzing 10 samples, prepare master mix for 11; if analyzing 96 samples, prepare master mix for 106 samples.
If desired for SNP genotyping assays, extend the cycling conditions out to 50 cycles instead of 40 cycles.
References
- 1.Bruhns P, Iannascoli B, England P, Mancardi DA, Fernandez N, Jorieux S, Daeron M. Specificity and affinity of human Fcgamma receptors and their polymorphic variants for human IgG subclasses. Blood. 2009;113(16):3716–3725. doi: 10.1182/blood-2008-09-179754. [DOI] [PubMed] [Google Scholar]
- 2.Metes D, Manciulea M, Pretrusca D, Rabinowich H, Ernst LK, Popescu I, Calugaru A, Sulica A, Chambers WH, Herberman RB, Morel PA. Ligand binding specificities and signal transduction pathways of Fc gamma receptor IIc isoforms: the CD32 isoforms expressed by human NK cells. European journal of immunology. 1999;29(9):2842–2852. doi: 10.1002/(SICI)1521-4141(199909)29:09<2842::AID-IMMU2842>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 3.Musolino A, Naldi N, Bortesi B, Pezzuolo D, Capelletti M, Missale G, Laccabue D, Zerbini A, Camisa R, Bisagni G, Neri TM, Ardizzoni A. Immunoglobulin G fragment C receptor polymorphisms and clinical efficacy of trastuzumab-based therapy in patients with HER-2/neu-positive metastatic breast cancer. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2008;26(11):1789–1796. doi: 10.1200/JCO.2007.14.8957. [DOI] [PubMed] [Google Scholar]
- 4.Bournazos S, Woof JM, Hart SP, Dransfield I. Functional and clinical consequences of Fc receptor polymorphic and copy number variants. Clin Exp Immunol. 2009;157(2):244–254. doi: 10.1111/j.1365-2249.2009.03980.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Henrichsen CN, Chaignat E, Reymond A. Copy number variants, diseases and gene expression. Human molecular genetics. 2009;18(R1):R1–8. doi: 10.1093/hmg/ddp011. [DOI] [PubMed] [Google Scholar]
- 6.Niederer HA, Willcocks LC, Rayner TF, Yang W, Lau YL, Williams TN, Scott JA, Urban BC, Peshu N, Dunstan SJ, Hien TT, Phu NH, Padyukov L, Gunnarsson I, Svenungsson E, Savage CO, Watts RA, Lyons PA, Clayton DG, Smith KG. Copy number, linkage disequilibrium and disease association in the FCGR locus. Human molecular genetics. 2010;19(16):3282–3294. doi: 10.1093/hmg/ddq216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hollox EJ, Detering JC, Dehnugara T. An integrated approach for measuring copy number variation at the FCGR3 (CD16) locus. Human mutation. 2009;30(3):477–484. doi: 10.1002/humu.20911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Breunis WB, van Mirre E, Geissler J, Laddach N, Wolbink G, van der Schoot E, de Haas M, de Boer M, Roos D, Kuijpers TW. Copy number variation at the FCGR locus includes FCGR3A, FCGR2C and FCGR3B but not FCGR2A and FCGR2B. Human mutation. 2009;30(5):E640–650. doi: 10.1002/humu.20997. [DOI] [PubMed] [Google Scholar]
- 9.Kim DH, Jung HD, Kim JG, Lee JJ, Yang DH, Park YH, Do YR, Shin HJ, Kim MK, Hyun MS, Sohn SK. FCGR3A gene polymorphisms may correlate with response to frontline R-CHOP therapy for diffuse large B-cell lymphoma. Blood. 2006;108(8):2720–2725. doi: 10.1182/blood-2006-01-009480. [DOI] [PubMed] [Google Scholar]
- 10.Koene HR, Kleijer M, Algra J, Roos D, von dem Borne AE, de Haas M. Fc gammaRIIIa-158V/F polymorphism influences the binding of IgG by natural killer cell Fc gammaRIIIa, independently of the Fc gammaRIIIa-48L/R/H phenotype. Blood. 1997;90(3):1109–1114. [PubMed] [Google Scholar]
- 11.Metes D, Ernst LK, Chambers WH, Sulica A, Herberman RB, Morel PA. Expression of functional CD32 molecules on human NK cells is determined by an allelic polymorphism of the FcgammaRIIC gene. Blood. 1998;91(7):2369–2380. [PubMed] [Google Scholar]
- 12.Su K, Wu J, Edberg JC, McKenzie SE, Kimberly RP. Genomic organization of classical human low-affinity Fcgamma receptor genes. Genes and immunity. 2002;3(Suppl 1):S51–56. doi: 10.1038/sj.gene.6363879. [DOI] [PubMed] [Google Scholar]
- 13.Metes D, Gambotto AA, Nellis J, Ruscin A, Stewart-Akers AM, Morel PA, Rao AS. Identification of the CD32/FcgammaRIIc-Q13/STP13 polymorphism using an allele-specific restriction enzyme digestion assay. Journal of immunological methods. 2001;258(1–2):85–95. doi: 10.1016/s0022-1759(01)00472-0. [DOI] [PubMed] [Google Scholar]
- 14.Burchard PR, Malhotra S, Kaur P, Tsongalis GJ. Detection of the FCGR3a polymorphism using a real-time polymerase chain reaction assay. Cancer Genet. 2013;206(4):130–134. doi: 10.1016/j.cancergen.2013.03.002. [DOI] [PubMed] [Google Scholar]
- 15.Haridan US, Mokhtar U, Machado LR, Abdul Aziz AT, Shueb RH, Zaid M, Sim B, Mustafa M, Nik Yusof NK, Lee CK, Abu Bakar S, AbuBakar S, Hollox EJ, Boon Peng H. A comparison of assays for accurate copy number measurement of the low-affinity Fc gamma receptor genes FCGR3A and FCGR3B. PLoS One. 2015;10(1):e0116791. doi: 10.1371/journal.pone.0116791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kim HK, Hwang HL, Park SY, Lee KM, Park WC, Kim HS, Um TH, Hong YJ, Lee JK, Joo SY, Seoh JY, Song YW, Kim SY, Kim YN, Hong KM. Simple and versatile molecular method of copy-number measurement using cloned competitors. PLoS One. 2013;8(7):e69414. doi: 10.1371/journal.pone.0069414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Matlawska-Wasowska K, Gale JM, Nickl CK, Khalili P, Shirley B, Wilson BS, Vasef MA, Winter SS. Pyrosequencing for classification of human FcgammaRIIIA allotypes: a comparison with PCR-based techniques. Mol Diagn Ther. 2014;18(6):665–673. doi: 10.1007/s40291-014-0120-5. [DOI] [PubMed] [Google Scholar]
- 18.van der Straaten T, Martijn R, el Hajoui T, Baak-Pablo R, Guchelaar HJ. A novel specific pyrosequencing method for genotyping FCGR3A rs396991 without coamplification of homologous gene FCGR3B. Pharmacogenet Genomics. 2013;23(11):631–635. doi: 10.1097/FPC.0b013e328365a4f2. [DOI] [PubMed] [Google Scholar]
- 19.Dobosy JR, Rose SD, Beltz KR, Rupp SM, Powers KM, Behlke MA, Walder JA. RNase H-dependent PCR (rhPCR): improved specificity and single nucleotide polymorphism detection using blocked cleavable primers. BMC Biotechnol. 2011;11:80. doi: 10.1186/1472-6750-11-80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Latorra D, Campbell K, Wolter A, Hurley JM. Enhanced allele-specific PCR discrimination in SNP genotyping using 3′ locked nucleic acid (LNA) primers. Hum Mutat. 2003;22(1):79–85. doi: 10.1002/humu.10228. [DOI] [PubMed] [Google Scholar]
- 21.Latorra D, Hopkins D, Campbell K, Hurley JM. Multiplex allele-specific PCR with optimized locked nucleic acid primers. Biotechniques. 2003;34(6):1150–1152. 1154, 1158. doi: 10.2144/03346bm06. [DOI] [PubMed] [Google Scholar]