

Abstract
Human breast tumors contain significant amounts of stromal cells. There exists strong evidence that these stromal cells support cancer development and progression by altering various pathways (e.g. downregulation of tumor suppressor genes or autocrine signaling loops). Here, we suggest that stromal carcinoma-associated fibroblasts (CAFs), shown to be generated from bone marrow-derived mesenchymal stem cells, may (i) recycle tumor-derived lactate for their own energetic requirements, thereby sparing glucose for neighboring glycolytic tumor cells, and (ii) subsequently secrete surplus energetically and biosynthetically valuable metabolites of lactate oxidation, such as pyruvate, to support tumor growth. Lactate, taken up by stromal CAFs, is converted to pyruvate, which is then utilized by CAFs for energy needs as well as excreted and shared with tumor cells. We have interrogated lactate oxidation in CAFs to determine what metabolites may be secreted, and how they may affect the metabolism and growth of MDA-MB-231 breast cancer cells. We found that CAFs secrete pyruvate as a metabolite of lactate oxidation. Further, we show that pyruvate is converted to lactate to promote glycolysis in MDA-MB-231 cells and helps to control elevated ROS levels in these tumor cells. Finally, we found that inhibiting or interfering with ROS management, using the naturally occurring flavonoid phloretin (found in apple tree leaves), adds to the cytotoxicity of the conventional chemotherapeutic agent doxorubicin. Our work demonstrates that a lactate-pyruvate, reciprocally-supportive metabolic relationship may be operative within the tumor microenvironment (TME) to support tumor growth, and may be a useful drug target.
Keywords: monocarboxylate, stroma, metabolism, MR spectroscopy
Graphical abstract
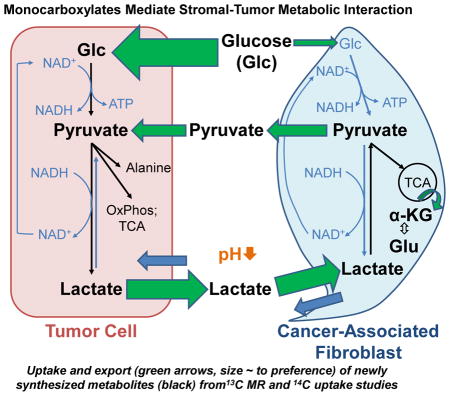
Introduction
The Warburg effect, which describes the dependence of tumors on glucose and constitutive aerobic glycolysis, results in the accumulation of cellular lactate as a metabolic byproduct [1, 2]. The excess lactate is evacuated from the tumor cell to prevent intracellular acidosis and subsequent apoptosis [3–5]. To facilitate the trafficking of lactate and other monocarboxylic acids, tumor cells and normal tissue express a family of proton-coupled monocarboxylate transporters (MCTs) [6, 7]. Specifically, it has been shown that lactate, secreted via low-affinity MCT4 in white, fast-twitch glycolytic myocytes, is taken up through high-affinity MCT1 in neighboring red, slow-twitch oxidative muscle fibers, wherein the metabolite is used to drive energy production [7]. These studies were first evidence of the existence of lactate shuttles and have collectively characterized MCT1 and MCT4 proteins as respective net importers and exporters of lactate [8]. A compelling finding of these investigations is that MCT4/MCT1-mediated lactate shuttles allow oxidative muscles to utilize lactate preferentially over glucose, thereby sparing it for the more glycolytic myocytes [7, 8]. Subsequent studies demonstrated the existence of such cell-cell lactate shuttles in astrocytes and neurons [9] and in sperm [10]. Investigations of intercellular metabolic complementarity in pathological systems have suggested that such lactate shuttles may be operative in tumors. Indeed, a recent report indicates that glycolytic triple negative basal breast adenocarcinoma MDA-MB-231 cells secrete lactate that is then taken up by oxidative luminal breast adenocarcinoma MCF-7 cells (MCFs) for conversion to pyruvate [11]. Other recent data suggest that the interplay between tumor and carcinoma-associated fibroblasts (CAFs) is more complex and that tumor cells may induce aerobic glycolysis in CAFs as a result of oxidative stress through secretion of hydrogen peroxide [12]. Tumor and tumor-associated stromal cells have the ability to adapt to oxidative stress, induced by increased production of cellular reactive oxygen species (ROS), by shifting oxidative mitochondrial metabolism toward glycolytic metabolism [13, 14].
Human breast tumors have been shown to contain a significant number of stromal cells [15] and extensive evidence indicates that these stromal cells support tumor growth [16]. Although numerous mechanisms have been suggested [12, 17–19], the mechanisms underpinning this relationship remain unclear. As tumors secrete significant amounts of lactate into the stroma, we had previously hypothesized that, in addition to preventing intracellular acidosis, this excreted lactate may serve to help recruit stromal components for tumor development [20]. We have demonstrated that lactate is taken up by stromal CAFs, and subsequently utilized for energy needs [20]. We now extend our original hypothesis to propose that stromal CAFs may (i) recycle tumor-derived lactate for their own energetic needs, thereby sparing glucose for the neighboring glycolytic tumor cells, and (ii) subsequently secrete energetically and biosynthetically valuable metabolites of lactate oxidation, such as pyruvate, to support tumor cell growth. As ROS modulation is linked to a shift in cellular metabolic pathways [13, 14], we hypothesize that (i) metabolic cooperation between tumor and stromal cells also modulates ROS levels and (ii) modulating ROS production by inhibitors acting across several pathways has the ability to attenuate ROS levels and glycolysis in tumor cells. Using ROS inhibitors with broad activity may also promote future clinical translation as an adjuvant to chemotherapy.
The current study interrogated lactate oxidation in CAFs to identify secreted metabolites and determine how they may affect the metabolism and growth of MDA-MB-231 cells. We found that CAFs secrete pyruvate as a metabolite of lactate oxidation. Further, we show that pyruvate is converted to lactate to promote glycolysis in MDA-MB-231 cells, and helps to control elevated reactive oxygen species (ROS) levels in these tumor cells. Finally, we report that flavonoid-mediated inhibition of or interference with MCT function and ROS management adds to the cytotoxicity of doxorubicin, a conventional chemotherapeutic agent widely used in treatment of breast cancer [21]. Our work demonstrates that a lactate-pyruvate, reciprocally-supportive metabolic relationship may be operative within the tumor microenvironment (TME) to support tumor growth.
Materials and Methods
Cell culture
The breast cancer cell line MDA-MB-231 was purchased from ATCC (Manassas, VA). Bone marrow-derived mesenchymal stem cells (MSCs) were purchased from Lonza Walkersville, Inc. (Walkersville, MD; Lonza Group Ltd.). All cells were maintained in Dulbecco’s Modified Essential Medium (DMEM, Sigma-Aldrich, St. Louis, MO), supplemented with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin, at 37°C and 5% CO2 in a humidified atmosphere. The complete culture medium contained 25 mM glucose, 4 mM glutamine and no pyruvate. Trace amounts of lactate are present in culture medium from glucose. The pyruvate concentrations used for the experimental studies ranged from 0 mM to 5 mM, based on the study published by Diers et al. [22].
CAF production
Medium from MDA-MB-231 cells growing at 70%–80% confluence was collected and centrifuged at 200×g for 7 minutes. The supernatant was transferred to a new tube, centrifuged as before, and subsequently filtered through a 0.2 μm membrane. For the purposes of this study, this filtrate was termed tumor-conditioned medium (TCM). The medium of MSCs growing at 60%–70% confluence was replaced with TCM every 12 hours for 10 days or every 24 hours for 20–30 days to induce the CAF-like state [23]. For the purposes of this study MSCs and CAFs are collectively termed stromal cells.
Extracellular acidification rate (ECAR) and basal oxygen consumption rate (OCR) measurements
The ECAR and OCR of CAFs and MDA-MB-231 cells (Fig. 1A, Supplementary Figure) were determined using a Seahorse XF96 Analyzer as per manufacturer’s instructions. Briefly, 20,000 cells/well (for cell number titration experiments 10,000 (10k), 20,000 (20k), and 40,000 (40k) cells/well respectively) were seeded into XF96 PET cell culture microplates (Part #101104-004, Seahorse Bioscience, North Billerica, MA). On the same day, the sensor cartridge was pre-incubated in XF96 calibrant solution (Par #100840-0000, Seahorse Bioscience, North Billerica, MA) in an XF Prep Station. Twenty-four hours later media in the XF96 PET plates were aspirated. Cells were washed one time with 100 μL phosphate buffered saline (PBS)/well and subsequently treated with 150 μL/well DMEM deficient in FBS and sodium bicarbonate, containing 1% penicillin/streptomycin, pH 7.4, in an XF Prep Station for 20 minutes. The ECAR and OCR of cells were then monitored for approximately 30–60 minutes at intervals of approximately 5–8 minutes.
Figure 1.

Glycolytic flux and lactate metabolism. (A) The extracellular acidification rate (ECAR; mean±SD) of MDA-MB-231 cells and CAFs, measured by Seahorse analyzer, reveals that MDA-MB-231 cells are more glycolytic than CAFs. The extracellular acidification rate ECAR measures proton excretion (representing cellular glycolysis) over time in units mpH/min where 1 mpH = 4.3 pmole excreted H+. (B) Extracellular glucose consumption and lactate production of MDA-MB-231 cells and stromal cells confirm the higher glycolytic activity of MDA-MB-231 cells compared to CAFs observed by Seahorse Analyzer analysis. Data are displayed as mean±SD (n = 3). (C) The glucose uptake is significantly higher in MDA-MB-231 cells (MDAs) than in MSCs or CAFs, in good agreement with the higher aerobic glycolysis observed in the cancer cells. Data are displayed as mean±SD (n = 4). (D) CAFs take up and metabolize lactate as well as secrete lactate oxidation metabolites, as shown by 13C MR spectroscopy on cell extracts and CCM. Signal assignments are: α-KG – α-ketoglutarate, Glu – glutamate; Ala – alanine; Lac – lactate; Pyr – pyruvate; α-Glc & β-Glc – α-glucose and β-glucose; “-C” followed by number – position of 13C labeling as a result of metabolic conversion of exogenous 10 mM 3-13C-L-lactate. ** p<0.005, *** p<0.0005 by two-tailed, unpaired, unequal variance Student’s T-test; Abbreviations: MDAs – MDA-MB-231 cells; MSCs: human mesenchymal stem cells; CAFs: cancer-associated fibroblasts; CCM – CAF-conditioned medium;
Lactate concentration in cell-conditioned media
Three hundred thousand CAFs or MDA-MB-231 cells per well were seeded into 24-well plates in 10 mM glucose-containing complete medium. Conditioned media were collected from each well twenty-four hours after growth and centrifuged at 200 × g RCF for 5 minutes. Twenty microliters of each sample were then applied to a lactate test strip and the lactate concentration determined, using a lactate analyzer (Hoffman-La Roche Ltd., Roche Diagnostics, USA) in accordance to the manufacturer’s instructions. The concentration was normalized to 1•105 cells to account for differences in the number of cells conditioning the medium.
Glucose concentration in cell-conditioned media
Three hundred thousand CAFs or MDA-MB-231 cells per well were seeded into 24-well plates in 10 mM glucose-containing complete medium. Conditioned media were collected from each well twenty-four hours after growth and centrifuged at 200 × g RCF for 5 minutes. Three microliters of each sample were then applied to a glucose test strip and the glucose concentration measured, using the GlucCell Glucose Monitoring System (Chemglass Life Sciences, Vineland, NJ) according to manufacturer’s instructions. The concentration was normalized to 1•105 cells to account for differences in the number of cells conditioning the medium.
Glucose uptake assay
Seventy-five thousand MSCs, CAFs, or MDA-MB-231 cells per well were seeded into 12-well plates. After attachment and growth for 24 h (Fig. 1C) to 48 h (Fig. 2C and 6E), cells were washed once with 1 mL PBS and incubated for 1 h with 0.025 μCi [U-14C]-D-glucose in 200 μL glucose uptake buffer (GUB: 120 mM NaCl, 25 mM NaHCO3, 4 mM KCl, 1.2 mM KH2PO4, 2.5 mM MgSO4-7 hydrate, 70 μM CaCl2-dihydrate, pH 7.4) per well at 37°C and 5% CO2. For experiments, studying the effects of a flavonoid on glucose uptake (Fig. 6E), 100 μM phloretin was added to GUB. All cells were subsequently washed with 500 μL ice-cold GUB to stop transport. Cells were then exposed to 200 μL 0.25% Trypsin / 0.53 mM EDTA per well for 15 minutes at 37°C and 5% CO2. The entire content of each well was placed into scintillation liquid and counted in a scintillation counter to generate counts per minute (CPM). Where appropriate (Fig. 1C), CPM was normalized to 100,000 cells. In co-culture experiments (Fig. 4B), 5•104 MDA-MB-231 cells were seeded into the chambers of a Boyden chamber plate and 1•105 MDA-MB-231 cells or CAFs were seeded into the inserts. Cells were co-cultured for 2 weeks, after which MDA-MB-231 cells in chambers were assayed to determine glucose uptake as described above.
Figure 2.

Lactate oxidation metabolite (LOM)-mediated growth of MDA-MB-231 cells – Modulation of MDA-MB-231 cell growth in the absence or presence of 2.5 mM glucose (Gluc) by 2.5 mM of (A) α-alanine (α-Ala), β-alanine (β-Ala), GABA, acetate (Acet) or (B) by 2.5 mM pyruvate (sodium pyruvate). Compared to control cells exposed to culture medium without glucose and added nutrients (Dep), neither GABA, alanine or acetate produced a significant growth benefit for MDA-MB-231 cells; While α-alanine appears to increase cell growth in the presence of 2.5 mM glucose, GABA, β-alanine, and acetate abrogate the growth benefit of 2.5 mM glucose, as seen in (A, B). Glucose alone (B) and pyruvate alone (B) enhanced MDA-MB-231 cell growth similarly, while both together enhanced growth synergistically (B). (C) Pyruvate does not modulate glucose uptake in MDA-MB-231 cells (over a range of 1.25 mM to 5 mM). Data are displayed as mean±SD (n = 3 for A, B and n = 4 for C); * p<0.05, ** p<0.005, *** p<0.0005 by two-tailed, unpaired, unequal variance Student’s T-test (compared to Dep or as indicated by lines between subgroups).
Figure 6.

Phloretin as a potential inhibitor of tumor-TME metabolic coupling – (A) Left panel: Electrostatic potential map of solvent accessible surface area of MCT1. Site A = positive electrostatic potential, Site B = negative electrostatic potential; Right panel: 3D model of Phloretin binding to Site B (Eint = −50.7 kcal/mol). (B) Growth inhibitory profile of flavonoids in MDA-MB-231 cells in response to 24 h exposure to 100 μM of indicated drug. (C) Phloretin inhibits 14C-lactate uptake in MSC and CAFs. (D) Phloretin inhibits 14C-pyruvate uptake in MDA-MB-231 cells. (E) Phloretin inhibits 14C-D-glucose uptake in MDA-MB-231 cells. 14C-L-glucose was used as a negative control. (F) Phloretin decreases ROS accumulation in MDA-MB-231 cells and protects from the ROS accumulation effects of H2O2exposure. (G) Phloretin attenuates MDA-MB-231 cell growth in CCM. The glucose concentrations at the start of the experiments were 21 mM and 11 mM for complete medium (Comp Media) and CCM, respectively. In B (n=3), C (n=4), D (n=4), E (n=4), F (n=6), and G (n=3) data are displayed as mean±SD; * p<0.05, ** p≤0.005, *** p<0.0005, **** p<0.0001 by two-tailed, unpaired, unequal variance Student’s T-test for the comparison to corresponding controls (“Untreated”, no line) and comparisons as indicated by line.
Figure 4.

Conditioned medium from CAFs (CCM) influences 14C-pyruvate (A) and 14C-glucose uptake (B) in MDA-MB-231 cells (MDAs). Data are displayed as mean±SD (n=4 for A and n=3 for B); ** p=0.005, *** p<0.0005 by two-tailed, unpaired, unequal variance Student’s T-test (compared to MDAs).
Lactate/pyruvate uptake assay
For measuring lactate uptake in stromal cells (Fig. 6C), 5•104 MSCs or CAFs were seeded per well into 24-well plates. For measuring pyruvate uptake in tumor cells (Fig. 6D), 7.5•104 MDA-MB-231 cells were seeded per well into 12-well plates. For both assays, all cells were washed 24 h post seeding once with 1 mL PBS and incubated for 1 h with 0.05 μCi [U-14C]-L-lactate or 0.05 μCi [U-14C]-pyruvate in 200 μL lactate uptake buffer (LUB: 10 mM HEPES, 5 mM KCl, 1 mM MgCl2, 150 mM NaCl, 1 mM CaCl2, pH 7.5) per well at 37 °C and 5% CO2. All cells were subsequently washed with 500 μL ice-cold LUB to stop transport. Cells were then exposed to 200 μL 0.025% Trypsin / 0.53 mM EDTA per well for 15 minutes at 37 °C and 5% CO2. The entire content of each well was placed into scintillation liquid and counted in a scintillation counter to generate CPM. Where appropriate, CPM was normalized to 100,000 cells. In co-culture experiments (Fig. 4A), 5•104 MDA-MB-231 cells were seeded into the chambers of a Boyden chamber plate and 105 MDA-MB-231 cells or CAFs were seeded into the inserts. Cells were co-cultured for 4 days, after which MDA-MB-231 cells in chambers were assayed to determine pyruvate uptake as described above.
Reverse transcriptase polymerase chain reaction (RT-PCR)
The expression of several genes involved in the production or management of reactive oxygen species (ROS) was evaluated by RT-PCR. To assess glucose 6-phosphate dehydrogenase (G6PD) mRNA expression (Fig. 5C), 7.5•104 MDA-MB-231 cells were grown with 2.5 mM of glucose (Gluc), pyruvate (Pyr), both (Gluc+Pyr), in DMEM medium for 5 days followed by RT-PCR analysis for G6PD with β-Actin as loading control, as per protocol below. In co-culture experiments (Fig. 5E), 5•104 MDA-MB-231 cells were seeded into the chambers of a Boyden chamber plate and 105 MDA-MB-231 cells or CAFs were seeded into the inserts. Cells were co-cultured for 4 days, after which MDA-MB-231 cells in chambers were assayed to determine manganese super oxide dismutase (Mn-SOD), G6PD, and nicotinamide adenine dinucleotide phosphate (NADPH) oxidase-1 (Nox-1) mRNA expression with glyceraldehyde-3-phosphate dehydrogenase (GAPDH) as loading control, as described below. Total RNA from cells was extracted using Trizol as per manufacturer’s protocol (Invitrogen™, Life Technologies, Thermo Fisher Scientific, Grand Island, NY). Fifty nanograms of total RNA per sample were reverse transcribed and subjected to PCR using SuperScript One-Step RT-PCR System with Platinum® Taq DNA Polymerase (Invitrogen™). Seven to 15 μL of each reaction were run on 1% agarose gels to resolve DNA bands of interest. Bands were visualized using ChemiDoc (Bio-Rad, Hercules, CA) and, where indicated, quantified using Quantity One 1-D Analysis Software version 4.6.9 (Bio-Rad).
Figure 5.

Pyruvate and CCM modulate ROS in MDA-MB-231 cells (MDAs) – Pyruvate decreases ROS accumulation after 5 hours (A) or 5 days (top panel full-scale data, bottom panel zoomed) (B) of exposure. (C) Pyruvate increases G6PD mRNA per se or in presence of glucose. (D) ROS accumulation in MDA-MB-231 cells (top panel full-scale data, bottom panel zoomed; statistical results are reported in Supplementary Table S1 and S2). In MDA-MB-231 cells, CCM and H2O2 increase synergistically ROS accumulation, while either condition alone does not increase significantly ROS levels (Supplementary Table S1). Over time, significant accumulation of ROS can be detected in all groups besides the MDAs+MDAs control group (Supplementary Table S2), with ROS accumulation below 120 min significantly increasing by the addition of H2O2 (Supplementary Table S2). (E) The gene expression of the ROS producers and scavengers Mn-SOD, G6PD and Nox-1 increases in MDA-MB-231 cells in response to CCM. (F) Intracellular reduced glutathione (GSH) levels are increased in MDA-MB-231 cells in response to CCM, as determined from MDA-MB-231 / CAF co-culture experiments (Normalized absorbance: OD relative to OD of MDAs). In A, B, and G data are displayed as mean±SD (n=3); * p≤0.05, ** p<0.005, *** p<0.0005 by two-tailed, unpaired, unequal variance Student’s T-test for comparison to Dep (no line) and comparisons as indicated by line. In B only, one outlier in the Dep group (included in the plot) has been excluded for the statistical analysis. In D, data are displayed as mean±SD (n=3); significance of ROS accumulation for (i) differences between groups for each time point (Supplementary Table S1) and (ii) changes over time within each group (Supplementary Table S2) was evaluated by two-way ANOVA with multiple comparison.
Assessing Cellular Metabolic Fate of 3-13C-L-lactate or 3-13C-pyruvate by high-resolution magnetic resonance spectroscopy (MRS)
We used high-resolution 13C MRS to assess in vitro the metabolic fate of lactate and pyruvate in CAFs, MSCs and MDA-MB-231 cells respectively, based on the 13C label incorporation into downstream metabolites (Fig. 1D, 3). CAFs or MSCs were allowed to expand to 90% confluence in T150 flasks. Two flasks each of MSCs or CAFs were incubated for 24 h with cell culture media, supplemented with 10 mM sodium 3-13C-L-lactate (ISOTEC®, Sigma Aldrich, Miamisburg, OH). Two T225 flasks of MDA-MB-231 cells were expanded to 70–90% confluence and subsequently incubated for 24 h with culture medium, containing 5 mM 3-13C-pyruvate (ISOTEC®, Sigma Aldrich, Miamisburg, OH). Following 24 h incubation, cells, irrespective of nutrient used, were harvested and extracted using perchloric acid, based on previously published methods [20, 24]. For some samples and prior to cell extraction, TCM or CAF-conditioned media (CCM) were collected for magnetic resonance (MR) analysis. All MR measurements were performed in the NMR Analytical Core Facility at Memorial Sloan Kettering Cancer Center on a 600 MHz Bruker-Avance III MR spectrometer with a 13C/1H cryoprobe. One-dimensional 1H-decoupled 13C MR spectra were acquired using a 30° flip angle, a waltz-16 decoupling scheme, 67072 acquisition size, and a 2 s relaxation delay. Additional acquisition parameters are as follows: (i) with nuclear Overhauser enhancement (NOE), 42016.87 Hz sweep width (SW), 2 dummy scans (DS), 3072 averages (NA), and 1.5964 s acquisition time for cell extract samples of CAFs and MSCs; (ii) without NOE, 52631.6 Hz SW, 2 DS, 1536 NA, and 1.2744 s acquisition time for CCM; (iii) with NOE, 52631.6 Hz SW, 8 DS, 1024 NA, and 1.2744 s acquisition time for samples of MDA-MB-231 cell extracts, TCMs, and control media. After applying a 1 Hz exponential line-broadening filter, the free induction decays were Fourier transformed, zero filled, phase corrected, and baseline corrected by Bernstein polynomial fit. The reference standard sodium 2,2-dimethyl 2-silapentane-5-sulfonate (DSS) was set to 0.015 parts per million (ppm). Signals were assigned to metabolites based on their 13C chemical shift. The number behind “-C” depicts the position of the 13C-labeled carbon in each metabolite.
Figure 3.

One-dimensional 1H-decoupled 13C MR spectra, acquired with nuclear Overhauser enhancement (NOE) of (A) MDA-MB-231 breast cancer cells extracted with perchloric acid after 24 h growth in DMEM containing either 0 mM, 5 mM, or 25 mM glucose and 5 mM 3-13C-pyruvate; (B) MDA-MB-231 cell-conditioned media (TCM), and (C) culture media control samples (DMEM containing either 0 mM, 5 mM, or 25 mM glucose and 5 mM 3-13C-pyruvate prior the addition to tumor cells) – In (B) and (C), the left panel depicts the spectral region from 210 ppm to 160 ppm and the middle panel from 100 ppm to −2 ppm, both zoomed with the same relative factor to enhance visibility of signals with lower signal intensity. The right panel (framed in grey) depicts the spectral region from 30 ppm to 22 ppm, zoomed out to fully appreciate the full signal height of the 5 mM 3-13C-pyruvate and 3-13C-lactate respectively. The glucose concentration in the respective culture medium at the start of the 24 h exposure (A–C) and the number of cells at extraction (A, B) are depicted on the spectra. The spectra in A and B were normalized to the DSS signal (0.05 ppm to −0.05 ppm), accounting for differences in sample volume (3 mm and 5 mm tubes) and shim quality, followed by scaling with the depicted factor (beside cell number) to account for differences in cell number between samples. Signal assignments with the number following “-C” depicting the position of the incorporated 13C carbon labelare: Lac – lactate; Glc – glucose (natural abundance); Glu – glutamate; Gln – glutamine; Pyr – pyruvate; ParaPyr – parapyruvate; Ace – acetate; Ala – alanine; Orange * - Pyr-C3(OH); ** Possibly Acetoacetate-C2; ? Unassigned signal. Note: The Ace-C2 signal may overlap with a GABA-C3 signal, and thus, these two may not be distinguishable.
Cell Growth Assay
For the cell growth studies displayed in Fig. 2A–B, fifty thousand MDA-MB-231 cells per well were seeded into 12-well plates in triplicate. Twenty-four hours later, cells were washed with 1X PBS and exposed for 5 days to 5 mM of either α-alanine, β-alanine, GABA, acetate or pyruvate (sodium pyruvate) in the absence or presence of 2.5 mM glucose, after which cells were harvested by trypsinization and counted via Trypan Blue exclusion assay, using a ViCell Cell Viability Analyzer (Beckman Coulter, Indiananpolis, IN). The total number of viable cells for each condition was normalized to the number viable cells for the depleted condition (glucose-depleted medium with no added alanine, GABA, acetate or pyruvate). Pyruvate was added essentially as done by Diers et al. [22]. To assess cell growth inhibitory effects of various flavonoids on MDA-MB-231 cells (Fig. 6B), 1.5×105 cells were seeded into 12-well plates in triplicate. Twenty-four hours later, cells were treated with 100 μM Phloretin, Phloredzin, Quercetin, Isoquercetin, or p-Phenylenediacetic acid. Cells were harvested 6 days later for cell counting via Trypan blue exclusion (ViCell). To investigate the combined cytotoxicity of the chemotherapy drug doxorubicin and Phloretin, 3×105 cells were seeded into 12-well plates in triplicate and treated after 24 h attachment with 100 μM Phloretin, 5 nM doxorubicin each alone or both combined in the presence of absence of CCM for 6 days, followed by harvest and cell counting (Fig. 6G).
Reactive oxygen species detection assay
Fifty thousand cells/well were seeded into black, clear-bottom, 96-well plates. After 24 h, ROS were quantified, using a 2′,7′–dichlorofluorescein diacetate (DCFDA) Cellular ROS Detection Kit (Abcam®, Cambridge, MA) as per manufacturer’s protocol. Briefly, cells were washed once with 200 μL/well of 1X assay buffer solution and subsequently incubated with 100 μL/well of 25 μM DCFDA in 1X assay buffer at 37 °C and 5% CO2 for 45 minutes. Afterwards, all wells were washed with 1X assay buffer as before, followed by treatment with 100 μL 1X assay buffer/well with or without indicated drug or nutrient for indicated duration. At each time point, plates were read in a fluorometer at an excitation wavelength of 485 nm and at an emission wavelength of 535 nm. Statistically significant change in fluorescence relative to controls was considered to be a significant change in amount of intracellular ROS. In co-culture experiments (Fig. 5D), 5•104 MDA-MB-231 cells were seeded into the chambers of a Boyden chamber plate and 105 MDA-MB-231 cells or CAFs were seeded into the inserts. Cells were co-cultured for 96 h, after which MDA-MB-231 cells in chambers were assayed intracellular ROS accumulation over 180 min. Viable MDA-MB-231 cell number was determined by Trypan Blue exclusion assay and fluorescence levels were normalized to 1•105 MDA-MB-231 cells. In experiments evaluating the effects of Phloretin on ROS production, 5•104 MDA-MB-231 cells were seeded in 96-well plates. After 24 h attachment period, cells were treated for 90 minutes with 100 μM Phloretin in the absence or presence of 100 μM tert-butyl hydrogen peroxide (H2O2).
Glutathione Assay
In co-culture experiments (Fig. 5F), 5•104 MDA-MB-231 cells were seeded into the chambers of a Boyden chamber plate and 105 MDA-MB-231 cells or CAFs were seeded into the inserts. Cells were co-cultured for 4 days, after which MDA-MB-231 cells in chambers were assayed for intracellular, reduced glutathione (GSH) levels using a Glutathione Colorimetric Assay Kit (# K261-100, BioVision Inc., Milpitas, CA) as per manufacturer’s protocol. Briefly, cells were washed once with 1X PBS and centrifuged at 600 × g RCF for 5 minutes and lysates prepared as per manufacturer’s protocol. Two to 10 μL of lysate supernatant of each sample were used to determine the GSH detection limit of the kit in a 96-well plate. The volume of lysate corresponding to the linear detection range of the GSH standard curve was used for downstream data transformation. Sample absorbance values (optical density, OD) were determined at 405 nm. Statistically significant change in OD relative to controls (P ≤ 0.05, unpaired t-Test) was considered to be a significant change in amount of GSH.
Results
CAFs are functionally less glycolytic than MDA-MB-231 cells
In this study, we used metastatic, triple negative (null for estrogen receptor, progesterone receptor, and human epidermal growth factor receptor) MDA-MB-231 breast cancer cells. These cells are dependent on stromal support for in vivo growth [16], implicating the presence of a metabolic relationship between MDA-MB-231 cells and their TME. Further, our hypothesis regarding metabolic complementarity between tumor cells and CAFs indicate that the latter are important constituents of the TME, and predict that the former are more glycolytic than the latter [20]. Thus, it was of interest to directly compare the glycolytic profiles and mitochondrial respiration of the two cell types.
We measured the ECAR of MDA-MB-231 cells and CAFs via a Seahorse XF96 Analyzer, which enables real-time monitoring of the rate of cellular proton excretion (mpH/min), and therefore, provides a measure of glycolysis. As indicated in Figure 1A and the Supplementary Figure (Top), the ECAR values of MDA-MB-231 cells were consistently higher over the 35 minutes of monitoring than the ECAR values of CAFs. This is consistent with the greater extracellular acidity of MDA-MB-231 cells as predicted by their higher metastatic potential relative to that of cell lines of lesser metastatic potential [25]. Further, the consistently higher basal OCR values in MDA-MB-231 cells compared to CAFs at equal cell numbers indicates higher mitochondrial respiration in MDA-MB-231 cells than in CAFs (Supplementary Figure (Bottom)). These results indicate that MDA-MB-231 cells are more metabolically active than CAFs. It has been shown that MDA-MB-231 cells secrete more lactate than MCF-7 cells under both normoxic and hypoxic conditions [11], thereby confirming reports of MDA-MB-231 cells’ high glycolytic metabolism [2, 26]. We compared the lactate production of CAFs and of MDA-MB-231 cells to gain insight into the relative oxidative or glycolytic metabolism of each cell type. We found that the lactate output of MDA-MB-231 cells is approximately 2-fold greater than that of CAFs (Fig. 1B and [20]). To more accurately profile differences in glucose consumption between MDA-MB-231 cells and CAFs, a glucose analyzer was used to quantify the remaining glucose in the media of cells after they had been incubated for 24 h in glucose-replete, complete medium. The glucose concentration of CAF media was almost unchanged relative to its initial concentration of 10 mM, while the glucose concentration of MDA-MB-231 medium dropped by more than 2 fold (Fig. 1B), suggesting that MDA-MB-231 cells consume more glucose than CAFs. In agreement with such glycolytic flux differences, MDA-MB-231 cells show significantly higher glucose transport than the stromal cells (Fig. 1C). Coupled with the ECAR results, these data indicate that MDA-MB-231 cells are significantly more glycolytic than CAFs, and implicate a potential cross-supportive lactate source - lactate sink (MDA-MB-231 cells - CAFs) relationship.
CAFs secrete several lactate-oxidation metabolites (LOMs)
We have shown that CAFs and MSCs take up lactate and oxidize it for energetic purposes (Fig. 1D and [20]). Using 1H-decoupled 13C MRS of CCM collected from CAFs grown for 24 h in medium containing 10 mM 3-13C-L-lactate, we identified signals specific to alanine, γ-aminobutyric acid (GABA), acetate, and pyruvate (Fig. 1D). The presence of acetate and alanine is not unexpected, as these are immediate downstream metabolites of pyruvate, which is an immediate metabolite of lactate oxidation. As the signal of GABA labeled in C3 may overlap with acetate-C2, there is the possibility that lactate may be converted to butyric acid in CAFs. The latter may occur via the cycling of lactate-derived pyruvate through the tricarboxylic acid cycle (TCA), yielding α-ketoglutarate (α-KG), an intermediate that is transaminated to glutamate as evidenced by the labeling of glutamate in the C4 position (Glu-C4), and which subsequently may be metabolized to GABA. Although studies have shown that bone marrow stromal cells have the potential to synthesize GABA de novo when appropriately differentiated [27], the data in Figure 1D may be the first indication of the presence of metabolically-derived GABA in stromal cells. Collectively, these results support our previous finding that lactate is metabolized oxidatively in CAFs. Additionally, these data suggest that CAFs secrete LOMs that may be of energetic and/or biosynthetic value to neighboring tumor cells or other cell types (e.g. immune cells) in the TME.
Pyruvate synergistically increases MDA-MB-231 cell growth in the presence of glucose
The presence of LOMs in the medium of 3-13C-L-lactate-treated CAFs suggests that lactate shuttling from MDA-MB-231 cells to CAFs may lead to reciprocal metabolic support of the former by the latter. To determine the functional value of LOMs to MDA-MB-231 cells, we profiled their growth in the presence or absence of α-alanine, β-alanine, GABA, acetate, or pyruvate. As glucose is a limiting nutrient in the TME, it was of interest to determine the contribution of exogenous LOMs to MDA-MB-231 cell growth within or without the context of minimal glucose (2.5 mM). Interestingly, alanine, GABA, and acetate had no effect on MDA-MB-231 cell growth in the absence of glucose, while only α-alanine appeared to show a modest growth benefit in the presence of glucose (Fig. 2A). Pyruvate (2.5 mM) increased the growth of MDA-MB-231 cells by approximately 1.7 fold, and in the presence of 2.5 mM glucose increased their growth synergistically (Fig. 2B). To ensure that pyruvate-mediated growth of MDA-MB-231 cells in the presence of glucose was not simply due to an increase in glucose uptake, we tested the uptake of 14C-D-glucose with or without pyruvate. We found that pyruvate did not affect glucose uptake in MDA-MB-231 cells (Fig. 2C). This is consistent with our observation that simply increasing the glucose concentration in the culture medium does not affect MDA-MB-231 cell growth kinetics (data not shown). That pyruvate per se moderately increased MDA-MB-231 growth was expected, as the monocarboxylate may serve as a potential energetic and biosynthetic source of carbon. However, its synergy, rather than simple additivity, with glucose in promoting MDA-MB-231 cell growth indicates that exogenous pyruvate may partially relieve the requirement of MDA-MB-231 cells to route glucose to produce energy via canonical glycolysis. This may increase the bioavailability of glucose to biosynthetic routes, such as anaplerosis or the pentose phosphate pathway. Thus, CAF-secreted pyruvate may help to balance energetic and biosynthetic demands of the tumor cells, especially when glucose is limited, to robustly increase MDA-MB-231 cell growth.
MDA-MB-231 cells take up pyruvate for glycolytic and biosynthetic needs
The increase in MDA-MB-231 cell growth by pyruvate suggested that the monocarboxylate was internalized to support the metabolic needs of the tumor cells. To directly test the fate of pyruvate in MDA-MB-231 cells, we exposed them to 5 mM 13C-pyruvate for 24 hours in the presence of varying glucose concentrations and subsequently subjected cellular extracts and culture media (TCM and control) to 13C proton-decoupled magnetic resonance (MR) analysis. The incorporation of the 13C label into specific carbon positions of downstream metabolites permits one to identify which metabolic pathways define the metabolic fate of pyruvate [28, 29]. Low levels of intracellular glucose were detected in extracts from MDA-MB-231 cells exposed for 24 h to medium containing glucose (Fig. 3A pink shaded area marked with Glc, natural abundance). Intracellular glucose also leads to the metabolic conversion of unlabeled glucose-derived pyruvate in the TCA cycle, as evidenced by the appearance of intracellular lactate derived from 13C-labeled pyruvate, which is not visible when glucose is absent or limited (Fig. 3A). The pyruvate-derived 13C labeling of glutamate and glutamine in all medium conditions demonstrates the uptake and metabolic conversion of pyruvate in the TCA cycle (Fig. 3A, B). The presence of α-KG in MDA-MB-231 cell extracts (Fig. 3A) indicates that some pyruvate may have entered the TCA cycle for anaplerotic purposes. Although a small portion of pyruvate does shunt into the TCA cycle in MDA-MB-231 cells, most is converted to lactate and excreted extracellularly. Accordingly, the signal intensity of pyruvate-derived lactate is high in the TCMs (Fig. 3B). The labeling of alanine in the C3 position is higher in glucose-free TCM than in TCM supplemented with glucose (Fig. 3B). The 13C labeling of lactate in the C2 and C1 position did not change significantly in response to glucose deprivation (Fig. 3B).
Increased use of pyruvate for the TCA cycle in glucose-free medium compared to glucose-containing media is supported by (i) the increased ratio of 13C-labeled glucose and glutamine to lactate-C3 and (ii) the lower pyruvate-derived lactate-C3 and higher acetate-C2, relative to 3-13C-pyruvate (Fig. 3B). These data are supported by control media sample data (Fig. 3C), which show no spectral differences aside from glucose signal levels. In addition to the strong 3-13C-labeled pyruvate signal in the control media, several natural abundance 13C signals of medium components can be identified; of note is that control media typically contain low levels of lactate (< 1 mM, Fig. 3C). The presence of parapyruvate in control media samples is from the non-enzymatic conversion of pyruvate to parapyruvate in aqueous solutions [30]. Parapyruvate disappears as the cells metabolize pyruvate, indicating the reversion of the non-enzymatic reaction that produced it in the control media (Fig. 3B, C). Exogenous pyruvate (5 mM, which is higher than the 0–1 mM typically contained in culture media) does not fully recapitulate the MDA-MB-231 growth potential of glucose, as evidenced by the decrease in cell number and concomitant with the decrease in glucose concentration in the medium (Fig. 3A) and consistent with results in Figure 2B. As indicated by the absence of glucose signals, MDA-MB-231 cells metabolize 5 mM glucose within the 24 h incubation period (Fig. 3B), suggesting that MDA-MB-231 cells depend significantly on glucose to support their metabolic and energetic needs.
Further, exposure of MDA-MB-231 cells to CCM from CAFs in a Boyden chamber co-culture system increased pyruvate uptake (Fig. 4A), the expression of glucose transporter 1 (GLUT1), glucose transporter 3 (GLUT3), lactate exporter MCT4 (not shown), and glucose uptake (Fig. 4B) in these tumor cells. These data collectively indicate that pyruvate and other CCM factors may support and amplify glycolysis in MDA-MB-231 cells.
In MDA-MB-231 cells, alanine and glutamate are most commonly synthesized by exogenous glutamine via the TCA. These cells are reliant on exogenous glutamine as they do not express glutamine synthetase and therefore do not undergo do novo glutamine synthesis [31]. Glutamine is the major contributor to anaplerotic flux in the TCA of these cells [32] and when deprived of this amino acid, MDA-MB-231 cells are forced to use pyruvate carboxylase to form TCA intermediates necessary for biosynthesis [33, 34]. Pyruvate carboxylase shunts pyruvate into the anaplerotic, non-oxidative route of the TCA [35]. Although we did not detect 13C-labeled alanine in cell extracts of MDA-MB-231 cells treated with 13C-pyruvate alone (Fig. 3A), alanine was present in the TCM (Fig. 3B). This is consistent with the observation that cancer cells secrete high levels of alanine as a means to dispose of excess nitrogen [36]. Some of the pyruvate produced from glucose may be shunted into anaplerotic reactions, thereby producing α-KG, glutamate, and alanine via alanine transaminase [37]. Thus, 13C-labeled species of α-KG, glutamate, and alanine may have the potential to arise by exogenous pyruvate contributing to this reaction in a similar manner. Relatedly, the extra pyruvate provisioned to cells exposed to glucose and 13C-pyruvate may enable MDA-MB-231 cells to better balance their energetic and biosynthetic needs, allowing for more robust growth (Fig. 2B).
These data suggest that CAFs may secrete pyruvate to provide MDA-MB-231 cells with important glycolytic and biosynthetic substrates, potentially releasing paracrine factors into the CCM that may modulate expression of glycolysis-promoting genes. Given the role of tumor-conditioned medium in differentiating MSCs [23], the data presented in Figure 3 demonstrate that factors in CCM and CAF-derived pyruvate may help complete a lactate-pyruvate metabolic loop between tumor cells and the TME.
CAFs modulate ROS in tumor cells
Tumors increase reactive oxygen species (ROS) production to promote pro-proliferative/pro-survival pathways and genetic mutagenesis important in adaptive growth [38]. However, excessive intracellular ROS causes irreparable genetic damage and induces cell death; accordingly, studies have shown that tumor cells may upregulate expression of antioxidant enzymes to combat excess ROS accumulation [39]. Thus, tumors balance the pro-growth and anti-survival effects of ROS in a seemingly paradoxical context in which high ROS production is maintained in the presence of high antioxidant levels.
Dysfunctional oxidative phosphorylation in glycolytic tumors results in the production of mitochondrial ROS, while the pentose phosphate arm and anaplerotic shunt of glucose metabolism produce reducing equivalents that power antioxidant systems [40]. As our data collectively indicate that pyruvate and other CCM factors may support and amplify glycolysis in MDA-MB-231 cells (Fig. 1–4), we investigated in preliminary studies the role that the monocarboxylate or CCM may play in ROS modulation in these tumor cells. Figure 5A shows the effect of exposing MDA-MB-231 cells for 5 h to 5 mM pyruvate and/or 5 mM glucose on ROS accumulation, in the presence or absence of 25 μM tert-butyl hydrogen peroxide (H2O2). Relative to nutrient depletion (Dep), this short-term exposure to pyruvate per se resulted in a moderate but statistically insignificant decrease in basal ROS levels, and significantly decreased ROS accumulation in the presence of glucose. The greatest pyruvate-mediated reduction in ROS levels was observed in the presence of H2O2, irrespective of the presence of glucose, indicating that short-term exposure to pyruvate may protect against ROS accumulation above basal levels. Exposure of pyruvate per se for 5 days markedly decreased ROS accumulation relative to Dep, and significantly attenuated ROS accumulation in the presence of glucose relative to pyruvate or glucose alone (Fig. 5B). Glucose deprivation (Dep) is known to increase ROS production [41], which is in agreement with the high ROS levels observed in this and the glucose-alone conditions (5 mM glucose was exhausted almost immediately relative to the 5 total days of the experiment [data not shown]). Collectively, these data are in accordance with aforementioned studies, which indicate that glucose metabolism attenuates ROS accumulation, and are consistent with investigations showing that pyruvate inactivates H2O2 by directly decarboxylating the oxidant [42]. Relatedly, exogenous pyruvate may function in concert with glucose to increase the mRNA expression of glucose-6-phosphosphate dehydrogenase (G6PD) (Fig. 5C), the enzyme that catalyzes the first and rate-liming reaction that shunts glucose from the canonical glycolysis pathway to the pentose phosphate pathway [43]. Thus, stroma-derived pyruvate may act as an antioxidant while promoting glycolytic and biosynthetic functions in MDA-MB-231 cells to robustly increase their proliferation.
To determine if the ROS-attenuation effects of pyruvate can be observed in MDA-MB-231 cells exposed to CCM, we co-cultured the tumor cells with themselves or CAFs in transwell chambers for 4 days and subsequently interrogated for intracellular ROS levels. Interestingly, MDA-MB-231s co-cultured with CAFs showed an increase in ROS accumulation (Fig. 5D), concurrent with an increase in nicotinamide adenine dinucleotide phosphate (NADPH) oxidase-1 (Nox-1) and manganese super oxide dismutase (Mn-SOD) gene expression (Fig. 5E). While Nox-1 produces super oxide (SO), Mn-SOD scavenges SO radicals to produce H2O2, making Mn-SOD both a ROS producer and scavenger [44, 45]. Increased G6PD gene expression in MDA-MB-231 cells in response to CCM (Fig. 5E) implies an increase in the reducing equivalent NADPH. This may play an important role in powering Nox-1 function [44] and in the reduction of glutathione sulfide to glutathione (GSH), a critical antioxidant that inactivates H2O2 by reducing it to water [40]. Relatedly, GSH levels increased in MDA-MB-231 cells after co-culture with CAFs (Fig. 5F). Consistent with the role of the Warburg effect in the reported nuanced management of ROS in tumor cells [45, 46], our results indicate that CAFs may support this function by modulating the redox balance and enabling increased, stroma-mediated glycolysis in MDA-MB-231 cells.
Phloretin as a potential inhibitor of tumor-TME metabolic coupling
Our investigation into tumor-TME metabolic coupling identifies monocarboxylate transport, glycolysis, and ROS regulation as potential therapeutic targets. Flavonoids, a large group of polyphenols commonly found in the human diet, have been shown to inhibit lactate and pyruvate transport, glucose transport, and ROS accumulation [47, 48]. To determine which flavonoids would be appropriate for our studies, we first conducted in silico modeling studies of MCT1 to define likely binding pockets. As no crystal structure of human MCTs exist, a homology model of human MCT1 was built using the crystal structure of the E. coli glycerol-3-phosphate transporter (PDB ID 1PW4) [49]. The homology model was built via the single template approach using the MODELLER program2 [50]. The model was subsequently used for in silico screening of small molecules. Electrostatic potential was mapped to the solvent-accessible surface area of MCT1. These sites, marked A and B in Fig. 6A - left panel, are distinguished by the difference in electrostatic potential. Site A is dominated by positive potential (colored blue), whereas site B is dominated by negative electrostatic potential (colored red). Site A is the region recognized for binding lactate. Subsequently, an interaction energy (Eint) analysis using the UCSF DOCK program3 [51] was conducted for known compounds that bind site B, and thus, would limit stereotactic binding of lactate to Site A (Fig. 6A - right panel). We calculated binding of the flavonoids quercetin, isoquercetin, p-phenylenediacetic acid, phloredzin, and phloretin; we found phloretin to have the lowest Eint (−50 kcal/mol), indicating that this flavonoid had the tightest binding potential of these 5 drugs to Site B.
To determine the relative cytotoxicity of these drugs, we tested their potency in attenuating MDA-MB-231 cell growth. After 6 days of exposure, phloretin and quercetin respectively decreased MDA-MB-231 cell growth by 20% and 60% (Fig. 6B). Given phloretin’s low cytotoxicity in MDA-MB-231 cells (relative to quercetin) and based on its favorable pharmacokinetics, its previous use in animal studies, and its low Eint values from MCT1 docking studies, we chose to use phloretin for subsequent studies.
To determine if phloretin affected monocarboxylate transport, glucose transport, and modulation of ROS levels, we treated stromal cells and MDA-MB-231 cells with the flavonoid. Phloretin exposure resulted in approximately 80% and 60% respective decrease in lactate uptake in MSCs and CAFs (Fig. 6C), and approximately 50% and 95% respective decrease in pyruvate uptake (Fig. 6D) and glucose uptake (Fig. 6E) in MDA-MB-231 cells. Additionally, basal ROS levels and H2O2-mediated ROS accumulation were inhibited by phloretin in MDA-MB-231 cells (Fig. 6F). These data suggest that phloretin has the potential to significantly attenuate glycolysis and ROS accumulation in tumors, while disrupting monocarboxylate exchange between the tumor cells and the TME.
Given that metabolic and paracrine interplay may underlie TME-mediated support of tumor growth, determining the potency of phloretin in the context of the TME is important. To that end, we have initiated preliminary investigations into the effect of phloretin on MDA-MB-231 cell growth in the absence or presence of CCM. As presented in Figure 6G, phloretin was significantly more cytotoxic to MDA-MB-231 cells in CCM than in controls. As monocarboxylate transport and glucose uptake is important for tumor cell metabolism [52, 53], treatment with phloretin in combination with other drugs may allow for the use of this flavonoid at sub-IC50 concentrations to minimize potential toxicity to host cells. As such, we queried the potential for phloretin to potentiate the toxicity of doxorubicin, a conventional chemotherapeutic. Phloretin cytotoxicity did not contribute to that of doxorubicin in complete medium but was additive in CCM (Fig. 6G). We predict that in the poorly vascularized TME, glucose is limiting and tumors may therefore be more reliant on exogenous nutrients to maintain their proliferative potential. As such, the potency of conventional anti-cancer drugs may be increased in the presence of adjuvants, such as phloretin, that antagonize primary (glucose uptake) and secondary (monocarboxylate exchange) metabolic dependencies of tumors.
Discussion
Cross talk between tumor cells and other cellular components of their TME has been well studied in the context of cytokines, growth factors, extracellular matrix proteins, and other modulators of signal transduction [54–57]. The recent resurgence in cancer metabolism research has focused investigations on how such factors may modulate glycolysis in tumors [58]. Using reports of intercellular nutrient cycling in muscles and brain [59, 60] as a guide to study potential lactate metabolite shuttling from the TME to tumor cells, our work reports that lactate oxidation in CAFs may provide pyruvate to MDA-MB-231 cells to support their biosynthetic and energetic needs. We also observed evidence of pyruvate-fueled anabolism in the presence of glucose as 13C-labeled TCA intermediates were detected. The pathways involved in the metabolic fate of lactate and pyruvate were identified via the 13C labeling of downstream metabolites using steady state measurements. The limitation of this approach is that a single time point measurement does not permit the calculation of synthesis or consumption rates or to model the relative contribution of the different metabolic pathways. These limitations can be overcome in future studies beyond the scope of this manuscript by using an MR-compatible cell perfusion bioreactor to measure the 13C-labeling pattern time dependent in live cells [28]. While serial 1H-decoupled 13C MRS of live cells assesses cycling through Krebs cycle and associated pathways [28], the addition of hyperpolarized 13C MRS (a clinically applicable imaging modality) with its 10,000-fold boost in detection sensitivity would enable the direct measurement of the exchange reaction of lactate to pyruvate conversion in real time [61–64]. Despite its advantages, hyperpolarized 13C MRS also pose some challenges including the limited number of substrates that can be hyperpolarized, the expensive equipment and long time needed to hyperpolarize a substrate, and the quick loss of hyperpolarization during 13C MRS at physiological conditions, requiring fast MRS acquisition methods [61–64].
In our study, MDA-MB-231 cells, a representative model of TNBC, were more glycolytic and demonstrated higher oxidative metabolism than the CAFs. Further, CAFs metabolized lactate – extruded by glycolytic epithelial cancer cells in tumors – and extrude pyruvate into the environment for use as energy source by other cells. Contrary to our results and termed the “reverse Warburg effect”, recent data suggest that tumor cells may induce aerobic glycolysis in CAFs as a result of oxidative stress through secretion of hydrogen peroxide [12]. Metabolites, excreted by CAFs in response to cancer-cell-induced oxidative stress, may include lactate, ketone bodies, and glutamine which can serve in turn as fuel for cancer cells [12, 65–67]. Further, Sonveaux et al. found that the human cervix squamous carcinoma cell line SiHa preferentially uses lactate, taken up via MCT-1, for oxidative metabolism [68]. However consistent with our results, Giatromanolaki et al. found based on the analysis of LDH isoenzymes in patient prostate cancer samples that tumor-associated fibroblasts use secreted lactate from glycolytic cancer cells to produce pyruvate to be used as continuous energy supply by the prostate cancer cells [69]. Further, in breast cancer patient samples, Choi et al. found that TNBC was predominantly associated with a Warburg-type (tumor: GLUT-1- and/or CA-IX-positive; stroma: Glut-1 and CAIX-negative) or mixed-type (tumor and stroma: GLUT-1- and/or CA-IX-positive) classification, while luminal breast cancer associated mainly with the reverse Warburg-type (tumor: Glut-1 and CAIX-negative; stroma: GLUT-1- and/or CA-IX-positive) and null-type (tumor and stroma: Glut-1 and CAIX-negative) classification [70]. These findings translated into the separation of disease-free and overall survival based on the metabolic tumor phenotype, with Warburg-type tumors associated with the worst and null-type tumors with the best outcome [70]. Our results are consistent with the findings by Choi et al. [70], as MDA-MB-231 cells represent TNBC, the most aggressive breast cancer type associated with the worst outcome.
Taken together, these data demonstrate the complex nature of the interplay between tumor and stromal cells and tumor metabolic heterogeneity, where no single cancer model covers all instances.
Pyruvate may also support the intracellular antioxidant arsenal used to manage elevated ROS levels, which correlated with increased CCM-mediated glycolysis. This study offers evidence of a reciprocal metabolic relationship between tumors and the TME that may be disturbed by phloretin, a flavonoid that inhibits monocarboxylate transport, glucose transport, ROS accumulation, and MDA-MB-231 cell growth.
Consistent with reports indicating that tumors upregulate ROS production in the context of elevated expression of antioxidants [35, 38, 39], we have found that pyruvate decreased oxidant burden in MDA-MB-231 cells, while CCM increased glutathione (anti-ROS) levels and increased the expression of Nox-1 (pro-ROS), Mn-SOD (pro/anti-ROS), and G6PD (anti-ROS). The myriad of signaling pathways affecting ROS accumulation [71] and glycolysis [72] however undoubtedly confound determination of the causal versus correlative relationship between tumor metabolism and oxidant regulation.
In healthy mammalian cells, including hMSCs, utilizing oxygen results in the production of ROS which are kept at low intracellular levels by antioxidants realizing a highly reducing intracellular environment [13, 14]. As mitochondria consume most of cellular oxygen, they are considered a major cellular source of ROS [13], although, the NOX enzyme family and other pathways, such as peroxisomal metabolism, cytochrome 450 metabolism, lysyl oxidases metabolism, and inflammation, may also contribute to cellular ROS [13]. Healthy cells undergoing oxidative stress may be damaged beyond repair and undergo apoptosis [13].
Cancer cells tend to be more metabolically active than healthy cells and also demonstrate higher ROS production than their healthy counterpart [13]. As oxidative stress outweighs the capacity for repair, healthy cells may adapt by ramping up their antioxidant defense system or, if the damage is beyond repair, undergoing apoptosis [13]. Oxidative stress may mediate DNA damage, leading to genetic mutations, including mutations affecting the mitochondrial electron transport chain, that benefit cancer development [13, 14]. During cancer development, cells adapt to increased ROS by shifting oxidative mitochondrial metabolism toward glycolytic metabolism [13].
Although the precise mechanism of the interplay between ROS and glycolysis modulation in breast cancer cells by CAFs is unclear at present, this study indicates that substrate availability is a growth-limiting constraint for glycolytic tumors. Our findings further indicate that Intercellular metabolic coupling between tumor cells and stromal cells may be a means by which tumors secure auxiliary sources of energetic and biosynthetic metabolites, which may synergize with paracrine signaling to robustly promote tumor growth. Indeed, a recent study by Huang, et al. [73] supports our contention that pyruvate confers a growth and survival advantage to tumor cells in the nutrient-depleted TME. Additionally, an investigation of the expression of monocarboxylates in lung cancer and associated tumor stroma indicated an expression profile in which cancer cells express high levels of the lactate exporter MCT4 and the glucose importer GLUT1, while stromal cells show high expression of the lactate importer MCT1. Importantly, the study showed that MCT1 was negatively prognostic for disease-specific survival [74]. Such studies underscore the importance of the lactate-pyruvate shuttle as an attractive target for tumor therapy.
Supplementary Material
Supplementary Figure – Extracellular acidification rate (ECAR; mean±SD) (Top) and basal oxygen consumption rate (OCR; mean±SD) (Bottom) measured by Seahorse Analyzer for cell number titrations of MDA-MB-231 (MDA) and CAFs (CAF) respectively. −10k, −20k, and −40k depict the respective cell number for each group. For equal cell numbers, the ECAR and OCR were always higher in MDA-MB-231 cells than in CAFs. Further, for each group, ECAR and basal OCR remained unchanged over the time course of the experiment. For the lowest cell number, OCR for CAFs was at the detection limit of the instrument. The ECAR results indicate that MDA-MB-231 cells are more glycolytic than CAFs, while the OCR results indicate higher mitochondrial respiration in MDA-MB-231 cells than in CAFs.
Acknowledgments
This work was carried out with grant support from: New Jersey Commission on Cancer Research NJCCR-10-1964-CCR-EO (DB), The Graduate School of Biomedical Sciences, Robert Wood Johnson Medical School, Rutgers Biomedical and Health Sciences, Rutgers University (DB), the Jack and Mimi Diamond Foundation (DB), Medical Director’s Breast Cancer Research Fund of the Evelyn H. Lauder Breast Center (JAK), R01 CA172846 (RGB, JAK), and P30 CA008748 (Memorial Sloan Kettering Cancer Center Support Grant). The authors gratefully acknowledge technical support by Dr. George D. Sukenick (Nuclear Magnetic Resonance Analytical Core Facility, Memorial Sloan-Kettering Cancer Center) and Ms. Natalia Kruchevsky. This work has been presented in part as a poster at the Stem Cell Biology Symposium at Cold Spring Harbor Laboratory, Cold Spring Harbor, NY on Oct. 7–11, 2015.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Warburg O. On respiratory impairment in cancer cells. Science. 1956;124:269–270. [PubMed] [Google Scholar]
- 2.Gatenby RA, Gillies RJ. Why do cancers have high aerobic glycolysis? Nat Rev Cancer. 2004;4:891–899. doi: 10.1038/nrc1478. [DOI] [PubMed] [Google Scholar]
- 3.Fang J, Quinones QJ, Holman TL, Morowitz MJ, Wang Q, Zhao H, Sivo F, Maris JM, Wahl ML. The H+-linked monocarboxylate transporter (MCT1/SLC16A1): a potential therapeutic target for high-risk neuroblastoma. Molecular pharmacology. 2006;70:2108–2115. doi: 10.1124/mol.106.026245. [DOI] [PubMed] [Google Scholar]
- 4.Schornack PA, Gillies RJ. Contributions of cell metabolism, H+ diffusion to the acidic pH of tumors. Neoplasia (New York, NY. 2003;5:135–145. doi: 10.1016/s1476-5586(03)80005-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Griffiths JR, McIntyre DJ, Howe FA, Stubbs M. Why are cancers acidic? A carrier-mediated diffusion model for H+ transport in the interstitial fluid. Novartis Found Symp. 2001;240:46–62. doi: 10.1002/0470868716.ch4. discussion 62–47, 152–153. [DOI] [PubMed] [Google Scholar]
- 6.Halestrap AP, Wilson MC. The monocarboxylate transporter family--role and regulation. IUBMB life. 2012;64:109–119. doi: 10.1002/iub.572. [DOI] [PubMed] [Google Scholar]
- 7.Halestrap AP, Meredith D. The SLC16 gene family-from monocarboxylate transporters (MCTs) to aromatic amino acid transporters and beyond. Pflugers Archiv : European journal of physiology. 2004;447:619–628. doi: 10.1007/s00424-003-1067-2. [DOI] [PubMed] [Google Scholar]
- 8.Ullah MS, Davies AJ, Halestrap AP. The plasma membrane lactate transporter MCT4, but not MCT1, is up-regulated by hypoxia through a HIF-1alpha-dependent mechanism. J Biol Chem. 2006;281:9030–9037. doi: 10.1074/jbc.M511397200. [DOI] [PubMed] [Google Scholar]
- 9.Bouzier-Sore AK, Voisin P, Canioni P, Magistretti PJ, Pellerin L. Lactate is a preferential oxidative energy substrate over glucose for neurons in culture. J Cereb Blood Flow Metab. 2003;23:1298–1306. doi: 10.1097/01.WCB.0000091761.61714.25. [DOI] [PubMed] [Google Scholar]
- 10.Storey BT, Kayne FJ. Energy metabolism of spermatozoa. VI. Direct intramitochondrial lactate oxidation by rabbit sperm mitochondria. Biol Reprod. 1977;16:549–556. [PubMed] [Google Scholar]
- 11.Kennedy KM, Scarbrough PM, Ribeiro A, Richardson R, Yuan H, Sonveaux P, Landon CD, Chi JT, Pizzo S, Schroeder T, Dewhirst MW. Catabolism of exogenous lactate reveals it as a legitimate metabolic substrate in breast cancer. PLoS One. 2013;8:e75154. doi: 10.1371/journal.pone.0075154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pavlides S, Vera I, Gandara R, Sneddon S, Pestell RG, Mercier I, Martinez-Outschoorn UE, Whitaker-Menezes D, Howell A, Sotgia F, Lisanti MP. Warburg meets autophagy: cancer-associated fibroblasts accelerate tumor growth, metastasis via oxidative stress, mitophagy and aerobic glycolysis. Antioxid Redox Signal. 2012;16:1264–1284. doi: 10.1089/ars.2011.4243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhou D, Shao L, Spitz DR. Reactive oxygen species in normal and tumor stem cells. Advances in cancer research. 2014;122:1–67. doi: 10.1016/B978-0-12-420117-0.00001-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fridovich I. Oxygen: how do we stand it? Medical principles and practice : international journal of the Kuwait University. Health Science Centre. 2013;22:131–137. doi: 10.1159/000339212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Moorman AM, Vink R, Heijmans HJ, van der Palen J, Kouwenhoven EA. The prognostic value of tumour-stroma ratio in triple-negative breast cancer. European journal of surgical oncology : the journal of the European Society of Surgical Oncology and the British Association of Surgical Oncology. 2012;38:307–313. doi: 10.1016/j.ejso.2012.01.002. [DOI] [PubMed] [Google Scholar]
- 16.Aboussekhra A. Role of cancer-associated fibroblasts in breast cancer development and prognosis. The International journal of developmental biology. 2011;55:841–849. doi: 10.1387/ijdb.113362aa. [DOI] [PubMed] [Google Scholar]
- 17.Mao Y, Keller ET, Garfield DH, Shen K, Wang J. Stromal cells in tumor microenvironment and breast cancer. Cancer metastasis reviews. 2013;32:303–315. doi: 10.1007/s10555-012-9415-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bremnes RM, Donnem T, Al-Saad S, Al-Shibli K, Andersen S, Sirera R, Camps C, Marinez I, Busund LT. The role of tumor stroma in cancer progression and prognosis: emphasis on carcinoma-associated fibroblasts and non-small cell lung cancer. Journal of thoracic oncology : official publication of the International Association for the Study of Lung Cancer. 2011;6:209–217. doi: 10.1097/JTO.0b013e3181f8a1bd. [DOI] [PubMed] [Google Scholar]
- 19.Pietras K, Ostman A. Hallmarks of cancer: interactions with the tumor stroma. Exp Cell Res. 2010;316:1324–1331. doi: 10.1016/j.yexcr.2010.02.045. [DOI] [PubMed] [Google Scholar]
- 20.Rattigan YI, Patel BB, Ackerstaff E, Sukenick G, Koutcher JA, Glod JW, Banerjee D. Lactate is a mediator of metabolic cooperation between stromal carcinoma associated fibroblasts and glycolytic tumor cells in the tumor microenvironment. Exp Cell Res. 2012;318:326–335. doi: 10.1016/j.yexcr.2011.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.O’Shaughnessy JA. Pegylated liposomal doxorubicin in the treatment of breast cancer. Clin Breast Cancer. 2003;4:318–328. doi: 10.3816/cbc.2003.n.037. [DOI] [PubMed] [Google Scholar]
- 22.Diers AR, Broniowska KA, Chang CF, Hogg N. Pyruvate fuels mitochondrial respiration and proliferation of breast cancer cells: effect of monocarboxylate transporter inhibition. The Biochemical journal. 2012;444:561–571. doi: 10.1042/BJ20120294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mishra PJ, Mishra PJ, Humeniuk R, Medina DJ, Alexe G, Mesirov JP, Ganesan S, Glod JW, Banerjee D. Carcinoma-associated fibroblast-like differentiation of human mesenchymal stem cells. Cancer research. 2008;68:4331–4339. doi: 10.1158/0008-5472.CAN-08-0943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Street JC, Mahmood U, Ballon D, Alfieri AA, Koutcher JA. 13C and 31P NMR investigation of effect of 6-aminonicotinamide on metabolism of RIF-1 tumor cells in vitro. J Biol Chem. 1996;271:4113–4119. doi: 10.1074/jbc.271.8.4113. [DOI] [PubMed] [Google Scholar]
- 25.Montcourrier P, Silver I, Farnoud R, Bird I, Rochefort H. Breast cancer cells have a high capacity to acidify extracellular milieu by a dual mechanism. Clin Exp Metastasis. 1997;15:382–392. doi: 10.1023/a:1018446104071. [DOI] [PubMed] [Google Scholar]
- 26.Robey IF, Lien AD, Welsh SJ, Baggett BK, Gillies RJ. Hypoxia-inducible factor-1alpha, the glycolytic phenotype in tumors. Neoplasia (New York, NY. 2005;7:324–330. doi: 10.1593/neo.04430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mohammad-Gharibani P, Tiraihi T, Mesbah-Namin SA, Arabkheradmand J, Kazemi H. Induction of bone marrow stromal cells into GABAergic neuronal phenotype using creatine as inducer. Restorative neurology and neuroscience. 2012;30:511–525. doi: 10.3233/RNN-2012-100155. [DOI] [PubMed] [Google Scholar]
- 28.Simoes RV, Serganova IS, Kruchevsky N, Leftin A, Shestov AA, Thaler HT, Sukenick G, Locasale JW, Blasberg RG, Koutcher JA, Ackerstaff E. Metabolic Plasticity of Metastatic Breast Cancer Cells: Adaptation to Changes in the Microenvironment. Neoplasia (New York, NY. 2015;17:671–684. doi: 10.1016/j.neo.2015.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Martin G, Chauvin MF, Dugelay S, Baverel G. Non-steady state model applicable to NMR studies for calculating flux rates in glycolysis, gluconeogenesis and citric acid cycle. J Biol Chem. 1994;269:26034–26039. 24. [PubMed] [Google Scholar]
- 30.Willems JL, de Kort AF, Vree TB, Trijbels JM, Veerkamp JH, Monnens LA. Non-enzymic conversion of pyruvate in aqueous solution to 2,4-dihydroxy-2-methylglutaric acid. FEBS letters. 1978;86:42–44. doi: 10.1016/0014-5793(78)80094-5. [DOI] [PubMed] [Google Scholar]
- 31.Kung HN, Marks JR, Chi JT. Glutamine synthetase is a genetic determinant of cell type-specific glutamine independence in breast epithelia. PLoS genetics. 2011;7:e1002229. doi: 10.1371/journal.pgen.1002229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gaglio D, Metallo CM, Gameiro PA, Hiller K, Danna LS, Balestrieri C, Alberghina L, Stephanopoulos G, Chiaradonna F. Oncogenic K-Ras decouples glucose and glutamine metabolism to support cancer cell growth. Molecular systems biology. 2011;7:523. doi: 10.1038/msb.2011.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Phannasil P, Thuwajit C, Warnnissorn M, Wallace JC, MacDonald MJ, Jitrapakdee S. Pyruvate Carboxylase Is Up-Regulated in Breast Cancer and Essential to Support Growth and Invasion of MDA-MB-231 Cells. PLoS One. 2015;10:e0129848. doi: 10.1371/journal.pone.0129848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cheng T, Sudderth J, Yang C, Mullen AR, Jin ES, Mates JM, DeBerardinis RJ. Pyruvate carboxylase is required for glutamine-independent growth of tumor cells. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:8674–8679. doi: 10.1073/pnas.1016627108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tennant DA, Duran RV, Boulahbel H, Gottlieb E. Metabolic transformation in cancer. Carcinogenesis. 2009;30:1269–1280. doi: 10.1093/carcin/bgp070. [DOI] [PubMed] [Google Scholar]
- 36.Ahn CS, Metallo CM. Mitochondria as biosynthetic factories for cancer proliferation. Cancer & metabolism. 2015;3:1. doi: 10.1186/s40170-015-0128-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ochoa-Ruiz E, Diaz-Ruiz R. Anaplerosis in cancer: Another step beyond the Warburg effect. American Journal of Molecular Biology. 2012;2:291–303. [Google Scholar]
- 38.Giannoni E, Buricchi F, Raugei G, Ramponi G, Chiarugi P. Intracellular reactive oxygen species activate Src tyrosine kinase during cell adhesion and anchorage-dependent cell growth. Molecular and cellular biology. 2005;25:6391–6403. doi: 10.1128/MCB.25.15.6391-6403.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Aykin-Burns N, Ahmad IM, Zhu Y, Oberley LW, Spitz DR. Increased levels of superoxide and H2O2 mediate the differential susceptibility of cancer cells versus normal cells to glucose deprivation. The Biochemical journal. 2009;418:29–37. doi: 10.1042/BJ20081258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schafer ZT, Grassian AR, Song L, Jiang Z, Gerhart-Hines Z, Irie HY, Gao S, Puigserver P, Brugge JS. Antioxidant and oncogene rescue of metabolic defects caused by loss of matrix attachment. Nature. 2009;461:109–113. doi: 10.1038/nature08268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Graham NA, Tahmasian M, Kohli B, Komisopoulou E, Zhu M, Vivanco I, Teitell MA, Wu H, Ribas A, Lo RS, Mellinghoff IK, Mischel PS, Graeber TG. Glucose deprivation activates a metabolic and signaling amplification loop leading to cell death. Molecular systems biology. 2012;8:589. doi: 10.1038/msb.2012.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Desagher S, Glowinski J, Premont J. Pyruvate protects neurons against hydrogen peroxide-induced toxicity. J Neurosci. 1997;17:9060–9067. doi: 10.1523/JNEUROSCI.17-23-09060.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Frederiks WM, Vizan P, Bosch KS, Vreeling-Sindelarova H, Boren J, Cascante M. Elevated activity of the oxidative and non-oxidative pentose phosphate pathway in (pre)neoplastic lesions in rat liver. International journal of experimental pathology. 2008;89:232–240. doi: 10.1111/j.1365-2613.2008.00582.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kleniewska P, Piechota A, Skibska B, Goraca A. The NADPH oxidase family and its inhibitors. Archivum immunologiae et therapiae experimentalis. 2012;60:277–294. doi: 10.1007/s00005-012-0176-z. [DOI] [PubMed] [Google Scholar]
- 45.Holley AK, Dhar SK, St Clair DK. Curbing cancer’s sweet tooth: is there a role for MnSOD in regulation of the Warburg effect? Mitochondrion. 2013;13:170–188. doi: 10.1016/j.mito.2012.07.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hamanaka RB, Chandel NS. Cell biology. Warburg effect and redox balance. Science. 2011;334:1219–1220. doi: 10.1126/science.1215637. [DOI] [PubMed] [Google Scholar]
- 47.Brunetti C, Di Ferdinando M, Fini A, Pollastri S, Tattini M. Flavonoids as antioxidants and developmental regulators: relative significance in plants and humans. International journal of molecular sciences. 2013;14:3540–3555. doi: 10.3390/ijms14023540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nijveldt RJ, van Nood E, van Hoorn DE, Boelens PG, van Norren K, van Leeuwen PA. Flavonoids: a review of probable mechanisms of action and potential applications. The American journal of clinical nutrition. 2001;74:418–425. doi: 10.1093/ajcn/74.4.418. [DOI] [PubMed] [Google Scholar]
- 49.Huang Y, Lemieux MJ, Song J, Auer M, Wang DN. Structure and mechanism of the glycerol-3-phosphate transporter from Escherichia coli. Science. 2003;301:616–620. doi: 10.1126/science.1087619. [DOI] [PubMed] [Google Scholar]
- 50.Eswar N, Webb B, Marti-Renom MA, Madhusudhan MS, Eramian D, Shen MY, Pieper U, Sali A. Comparative protein structure modeling using Modeller. Curr Protoc Bioinformatics. 2006;Chapter 5(Unit 5):6. doi: 10.1002/0471250953.bi0506s15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Meng EC, Shoichet BK, Kuntz ID. Automated Docking with Grid-Based Energy Evaluation. J Comput Chem. 1992;13:505–524. [Google Scholar]
- 52.Sanita P, Capulli M, Teti A, Galatioto GP, Vicentini C, Chiarugi P, Bologna M, Angelucci A. Tumor-stroma metabolic relationship based on lactate shuttle can sustain prostate cancer progression. BMC cancer. 2014;14:154. doi: 10.1186/1471-2407-14-154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lu W, Logsdon CD, Abbruzzese JL. Cancer Metabolism and Its Therapeutic Implications. Journal of Cell Science & Therapy. 2013;4:143–152. [Google Scholar]
- 54.Mishra P, Banerjee D, Ben-Baruch A. Chemokines at the crossroads of tumor-fibroblast interactions that promote malignancy. Journal of leukocyte biology. 2011;89:31–39. doi: 10.1189/jlb.0310182. [DOI] [PubMed] [Google Scholar]
- 55.Chekhun VF. Stroma -- regulator of cancer cell progression. Experimental oncology. 2009;31:126. [PubMed] [Google Scholar]
- 56.Raman D, Baugher PJ, Thu YM, Richmond A. Role of chemokines in tumor growth. Cancer letters. 2007;256:137–165. doi: 10.1016/j.canlet.2007.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Orimo A, Weinberg RA. Stromal fibroblasts in cancer: a novel tumor-promoting cell type. Cell cycle (Georgetown, Tex. 2006;5:1597–1601. doi: 10.4161/cc.5.15.3112. [DOI] [PubMed] [Google Scholar]
- 58.Burgess DJ. Metabolism: Unravelling metabolic dependencies. Nat Rev Cancer. 2012;12:321. doi: 10.1038/nrc3273. [DOI] [PubMed] [Google Scholar]
- 59.Gladden LB. Lactate metabolism: a new paradigm for the third millennium. J Physiol. 2004;558:5–30. doi: 10.1113/jphysiol.2003.058701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Brooks GA. Lactate shuttles in nature. Biochemical Society transactions. 2002;30:258–264. doi: 10.1042/. [DOI] [PubMed] [Google Scholar]
- 61.Daniels CJ, McLean MA, Schulte RF, Robb FJ, Gill AB, McGlashan N, Graves MJ, Schwaiger M, Lomas DJ, Brindle KM, Gallagher FA. A comparison of quantitative methods for clinical imaging with hyperpolarized C-13-pyruvate. NMR in biomedicine. 2016;29:387–399. doi: 10.1002/nbm.3468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hu S, Lustig M, Balakrishnan A, Larson PE, Bok R, Kurhanewicz J, Nelson SJ, Goga A, Pauly JM, Vigneron DB. 3D compressed sensing for highly accelerated hyperpolarized (13)C MRSI with in vivo applications to transgenic mouse models of cancer. Magn Reson Med. 2010;63:312–321. doi: 10.1002/mrm.22233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.von Morze C, Larson PE, Hu S, Yoshihara HA, Bok RA, Goga A, Ardenkjaer-Larsen JH, Vigneron DB. Investigating tumor perfusion, metabolism using multiple hyperpolarized (13)C compounds: HP001 pyruvate and urea. Magnetic resonance imaging. 2012;30:305–311. doi: 10.1016/j.mri.2011.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bokacheva L, Ackerstaff E, LeKaye HC, Zakian K, Koutcher JA. High-field small animal magnetic resonance oncology studies. Phys Med Biol. 2014;59:R65–R127. doi: 10.1088/0031-9155/59/2/R65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Icard P, Kafara P, Steyaert JM, Schwartz L, Lincet H. The metabolic cooperation between cells in solid cancer tumors. Biochim Biophys Acta. 2014;1846:216–225. doi: 10.1016/j.bbcan.2014.06.002. [DOI] [PubMed] [Google Scholar]
- 66.Morandi A, Chiarugi P. Metabolic implication of tumor:stroma crosstalk in breast cancer. Journal of molecular medicine. 2014;92:117–126. doi: 10.1007/s00109-014-1124-7. [DOI] [PubMed] [Google Scholar]
- 67.Lisanti MP, Martinez-Outschoorn UE, Sotgia F. Oncogenes induce the cancer-associated fibroblast phenotype: metabolic symbiosis “fibroblast addiction” are new therapeutic targets for drug discovery. Cell cycle (Georgetown, Tex. 2013;12:2723–2732. doi: 10.4161/cc.25695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sonveaux P, Vegran F, Schroeder T, Wergin MC, Verrax J, Rabbani ZN, De Saedeleer CJ, Kennedy KM, Diepart C, Jordan BF, Kelley MJ, Gallez B, Wahl ML, Feron O, Dewhirst MW. Targeting lactate-fueled respiration selectively kills hypoxic tumor cells in mice. J Clin Invest. 2008;118:3930–3942. doi: 10.1172/JCI36843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Giatromanolaki A, Koukourakis MI, Koutsopoulos A, Mendrinos S, Sivridis E. The metabolic interactions between tumor cells and tumor-associated stroma (TAS) in prostatic cancer. Cancer Biol Ther. 2012;13:1284–1289. doi: 10.4161/cbt.21785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Choi J, Kim do H, Jung WH, Koo JS. Metabolic interaction between cancer cells and stromal cells according to breast cancer molecular subtype. Breast Cancer Res. 2013;15:R78. doi: 10.1186/bcr3472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Finkel T. Signal transduction by reactive oxygen species. J Cell Biol. 2011;194:7–15. doi: 10.1083/jcb.201102095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Cairns RA, Harris IS, Mak TW. Regulation of cancer cell metabolism. Nat Rev Cancer. 2011;11:85–95. doi: 10.1038/nrc2981. [DOI] [PubMed] [Google Scholar]
- 73.Huang CY, Kuo WT, Huang YC, Lee TC, Yu LC. Resistance to hypoxia-induced necroptosis is conferred by glycolytic pyruvate scavenging of mitochondrial superoxide in colorectal cancer cells. Cell death & disease. 2013;4:e622. doi: 10.1038/cddis.2013.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Eilertsen M, Andersen S, Al-Saad S, Kiselev Y, Donnem T, Stenvold H, Pettersen I, Al-Shibli K, Richardsen E, Busund LT, Bremnes RM. Monocarboxylate transporters 1–4 in NSCLC: MCT1 is an independent prognostic marker for survival. PLoS One. 2014;9:e105038. doi: 10.1371/journal.pone.0105038. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure – Extracellular acidification rate (ECAR; mean±SD) (Top) and basal oxygen consumption rate (OCR; mean±SD) (Bottom) measured by Seahorse Analyzer for cell number titrations of MDA-MB-231 (MDA) and CAFs (CAF) respectively. −10k, −20k, and −40k depict the respective cell number for each group. For equal cell numbers, the ECAR and OCR were always higher in MDA-MB-231 cells than in CAFs. Further, for each group, ECAR and basal OCR remained unchanged over the time course of the experiment. For the lowest cell number, OCR for CAFs was at the detection limit of the instrument. The ECAR results indicate that MDA-MB-231 cells are more glycolytic than CAFs, while the OCR results indicate higher mitochondrial respiration in MDA-MB-231 cells than in CAFs.