

Abstract
There are currently no disease-modifying therapies for the treatment of tauopathies, a group of progressive neurodegenerative disorders that are pathologically defined by the presence of tau protein aggregates in the brain. Current challenges for the treatment of tauopathy include the inability to diagnose early and to confidently discriminate between distinct tauopathies in patients, alongside an incomplete understanding of the cellular mechanisms involved in pathogenic tau-induced neuronal death and dysfunction. In this review, we describe current diagnostic and therapeutic strategies, known drivers of pathogenic tau formation, recent contributions to our current mechanistic understanding of how pathogenic tau induces neuronal death, and potential diagnostic and therapeutic approaches.
Keywords: tauopathy, tau, neurodegeneration, Alzheimer's disease, traumatic brain injury, metabolic syndrome
Tau and Tauopathies
In 1975, Weingarten and colleagues identified a protein contaminant that co-purified with microtubules. Upon further investigation, this contaminant, now known as the “microtubule-associated protein tau,” or “tau,” was found to be critical for microtubule stability [1]. Later studies identified the tau protein as the major protein constituent of neurofibrillary tangles [2, 3], one of the two major pathological hallmarks of human Alzheimer's disease. Subsequent studies found that tau aggregates are the primary pathological feature of clinically heterogeneous neurodegenerative disorders that are now collectively termed “tauopathies.” Tauopathies, including but not limited to Alzheimer's disease, progressive supranuclear palsy, corticobasal syndrome, some frontotemporal dementias, and chronic traumatic encephalopathy, are progressive neurodegenerative disorders that are pathologically defined by tau-positive deposits in the brain.
Current Strategies for Diagnosis and Treatment
Diagnosis
Current clinical diagnosis of distinct tauopathies relies upon clinical history, including symptom onset, progression, and overall course. Though there are numerous neurodegenerative diseases with tau inclusions, clinical research is arguably most attentive to Alzheimer's disease, progressive supranuclear palsy, and corticobasal syndrome. Each clinical syndrome presents with a unique constellation of symptoms thought to represent anatomical regions of vulnerability. Patients with “prototypical” Alzheimer's disease experience an insidious pattern of forgetfulness as pathology builds in the hippocampus and entorhinal cortex [4]. Frequent falls and impaired ocular movements highlight pathology within subcortical structures, suggestive of progressive supranuclear palsy or corticobasal syndrome [4]. The earliest observed changes in cognition and/or behavior occur decades after pathophysiological changes. This extensive prodromal stage provides a wide window for potential therapeutic intervention.
Neuropsychological evaluation
Comprehensive neuropsychological evaluations are instrumental in detecting the earliest signs of neurodegenerative disease. These lengthy assessments evaluate aspects of cognition, behavior and movement, often utilizing demographically-adjusted norms. Distinct cognitive profiles are interpreted within the context of presenting symptoms and reported history, helping to distinguish among competing clinical syndromes. Typical Alzheimer's disease patients show impairments in memory, with rapid forgetting, some degree of anomia, poor visuoconstruction, and impaired category fluency, with preserved phonemic fluency [4]. Subcortical dementias reveal inefficient recall, but preserved recognition memory. Patients with progressive supranuclear palsy often have low letter-cued fluency, varying degrees of executive dysfunction, and difficulty with motorized sequences, while those with suspected corticobasal syndrome often present with asymmetric motor abnormalities and profound apraxia [5].
Neurological evaluation
In conjunction with neuropsychological findings, medical and neurological examination further inform diagnosis, as abnormalities, or the lack thereof, provide additional insight regarding suspected pathology. Axial rigidity, frequent falls, and disturbed eye movements all suggest supranuclear palsy, while asymmetric motor disturbance, alien limb syndrome, and ocular apraxia are associated with corticobasal syndrome [5]. Neurological exam for those with Alzheimer's disease remains largely normal until advanced stages.
Neuroimaging
Neuroimaging is useful for identifying tau-mediated changes in brain structure and function. Magnetic resonance imaging (MRI) captures structural changes of both gray and white matter, while positron emission tomography (PET) imaging utilizes a radioactive tracer measuring brain function [6]. On MRI, Alzheimer's disease patients have hippocampal and parietal atrophy [4]. Patients with progressive supranuclear palsy have notable atrophy of the midbrain, while those with corticobasal syndrome most reliably show changes of the basal ganglia and bilateral frontal lobes [5].
Treatment
Treatment of tau-related neurodegenerative syndromes is limited and largely symptomatic. Currently, there are two classes of medication approved for cognition: cholinesterase inhibitors and an N-methyl-D-aspartate (NMDA) receptor antagonist, each demonstrating modest effects [4] (Table 1). Motor symptoms may respond to dopaminergic regimens [5], speech therapy has shown some efficacy with aphasia syndromes, and physical therapy can prove helpful in prolonging motor function. Symptoms of apathy and depression are quite common and often respond to therapy or pharmacologic intervention, including selective serotonin reuptake inhibitors [5]. There are currently no disease-modifying therapies for tauopathies, including Alzheimer's disease.
Table 1.
Current pharmacological strategies for treatment of cognitive deficits in tauopathies.
| Year Approved | Drug | Brand Name | Disease Stage | Mechanism of Action | Disease Modifying | Delay Disease Progression |
|---|---|---|---|---|---|---|
| 1996 | Donepezil | Aricept | All stages | Cholinesterase inhibitor | No | No |
| 2000 | Rivastigmine | Exelon | All stages | Cholinesterase inhibitor | No | No |
| 2001 | Galantamine | Razadyne | Mild to moderate | Cholinesterase inhibitor | No | No |
| 2003 | Memantine | Namenda | Moderate to Severe | NMDA receptor antagonist | No | No |
| 2014 | Donepezil+ Memantine | NAMZARIC | Moderate to Severe | Cholinesterase inhibitor + NMDA receptor antagonist | No | No |
Stimulators of Pathogenic Tau Formation
What is “pathogenic” tau?
The most well-described role for physiological tau is that of a microtubule-associated protein that binds to and stabilizes microtubules, particularly microtubules of neuronal axons [7]. In disease states, tau assumes a number of features, including A) Aberrant phosphorylation [8-10] and other posttranslational modifications [11], B) Truncation [2], and C) Aggregation into oligomers and larger insoluble filaments [7] (for a comprehensive review, see Arendt et. al [12]) (Figure 1). In this section, we will discuss stimulators of pathogenic tau formation other than amyloid β and α-synuclein, which have been reviewed extensively [13, 14][15].
Figure 1.
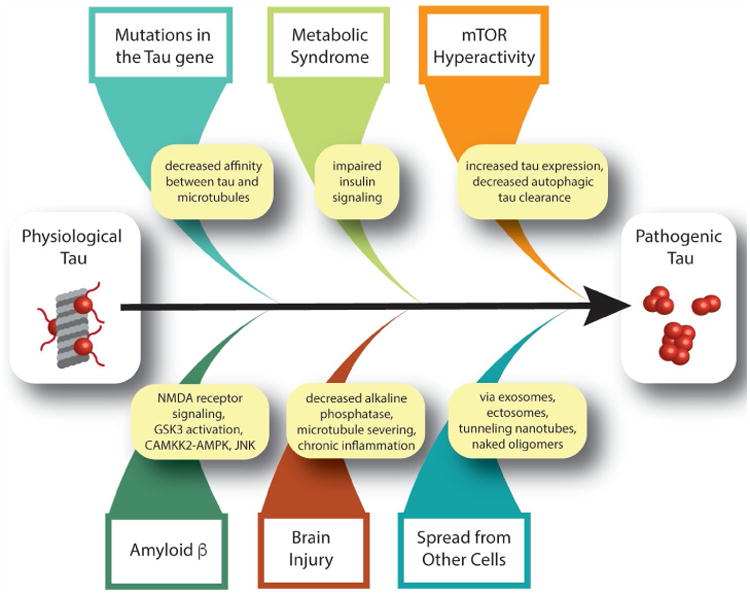
Known stimulators of pathogenic tau formation.
Diagnostic potential
Significant research effort has been devoted to developing diagnostic tools based on detection of pathogenic tau in body fluids and brain. A recent meta-analysis analyzing thirty years of published biomarker data from patients with mild cognitive impairment and Alzheimer's disease indicates that levels of total tau (cerebrospinal fluid and plasma) and total phosphotau (plasma) are strongly associated with Alzheimer's disease and mild cognitive impairment due to Alzheimer's disease [16] (available in an interactive format at www.alzforum.org/alzbiomarker). Recently, a mass spectrometry-based assay called “FLEXITau” (Full-Length EXpressed stable Isotope-labeled Tau) was developed for unbiased quantification of post-translational tau modifications in vitro and in vivo. The FLEXITau technology allows a comprehensive view of the tau post-translational landscape, and has the potential to be adapted for diagnostic purposes [10]. PET-based imaging of pathogenic tau in living humans using 18F-AV-1451 [17, 18] has recently spurred a wealth of studies demonstrating a significant correlation between tau-positive PET, disease severity and post mortem histopathological data for Alzheimer's disease [18, 19]. Tau-based PET provides information regarding the anatomical distribution of pathogenic tau within the brain, which, with the development of better tau tracers, may eventually allow one to differentiate between non-Alzheimer's disease tauopathies. While initial costs and limited accessibility may hinder the use of tau-based imaging in most clinical settings, the use of tau PET in specialized dementia centers will likely transform disease identification, progression, and treatment.
Therapeutic potential
Therapeutic efforts focused on targeting any one of the aforementioned features of pathogenic tau have been recently reviewed [20], and include but are not limited to inhibitors of aberrant tau post-translational modifications, proteolytic processing, and aggregation. A strategy utilizing antisense oligonucleotides to reduce tau mRNA has recently been reported to decrease pathogenic tau, halt neuronal loss, and extend lifespan of mice transgenically expressing human tau harboring the disease-associated P301S mutation (PS19 mouse model), even when administered well after tau deposition [21]. While antisense oligonucleotides targeting tau must undergo further toxicological studies before entering human clinical trials, they were found to be well tolerated in cynomolgus monkeys, and significantly reduced tau mRNA and protein in the brain, spinal cord, and cerebrospinal fluid [21]. Encouragingly, antisense oligonucleotides targeting mutant survival motor neuron protein 1 recently received FDA approval for treatment of spinal muscular atrophy [22], a disease caused by degeneration of lower motor neurons.
Mutations in the tau gene
Mutations in the microtubule-associated protein tau (MAPT) gene have been identified in over 150 families with frontotemporal lobar degeneration (termed “FTLD-tau with MAPT mutation,” previously known as “FTDP-17”) [23-25]. Clinical manifestations of FTLD-tau with MAPT mutation are syndromes of disordered movement, behavioral and personality changes, and cognitive deterioration. Disease phenotypes, age of onset and severity can differ significantly among families with the same mutation, suggesting that other factors (genetic modifiers and/or environment) are likely involved. Of the 57 pathogenic MAPT mutations identified, most either occur within the microtubule binding domains or affect the number of microtubule binding domains present in the mature tau protein. Familial tauopathies are associated with aberrantly phosphorylated, inclusion-forming tau protein, clearly demonstrating that mutations in the MAPT gene are sufficient to induce formation of pathogenic tau [26].
Brain injury
Chronic traumatic encephalopathy, previously known as “dementia pugilistica,” is a progressive neurodegenerative tauopathy caused by multiple traumatic brain injuries. In humans, post mortem pathological tau staining [27], tau PET [28], and presence of pathogenic tau in plasma exosomes [29] demonstrate an association between chronic traumatic encephalopathy and tauopathy. The defining neuropathological feature of chronic traumatic encephalopathy, an irregular pattern of accumulation of aberrantly phosphorylated tau in neurons and astroglia around small blood vessels at sulcal depths, is distinct from Alzheimer's disease-related tauopathy [27]. In animal models, brain injury is sufficient to induce tau cleavage [30], acute and sustained aberrant tau phosphorylation and aggregation [31, 32] and cis-trans isomerization of tau, termed “cistauosis” [33, 34]. In mice, traumatic brain injury-induced cistauosis occurs prior to overt tau oligomer or aggregate formation, disrupts axonal microtubule networks and mitochondrial transport, spreads to other neurons, and induces apoptosis [34]. Evidence supports several non-mutually exclusive mechanisms of traumatic brain injury-mediated stimulation of pathogenic tau formation. First, alkaline phosphatase, which dephosphorylates tau, decreases in response to brain injury in rats, causing accumulation of phosphotau [31]. Second, injury-induced severing of neuronal axons may cause tau to dissociate from microtubules, which could facilitate subsequent tau aggregation [35]. Third, the chronic inflammation that results from brain injuries likely exacerbates and sustains the acute tauopathy that occurs shortly after injury [36].
Tau-based PET indicates that the temporal neocortex of most individuals over the age of 65 contain pathogenic tau [37]. Curiously, the presence of pathogenic tau in this context, which is termed “Primary Age-Related Tauopathy” is not always associated with deficits in cognition. Basal ganglia and neocortical neurons of cynomolgus monkeys also exhibit an age-associated accumulation of tau deposits, suggesting that aging-associated tau accumulation may be a general feature of primate aging [38]. A current hypothesis posits that Primary Age-Related Tauopathy, while not a brain injury in the classical sense, is a result of the mild “wear and tear” on the brain that inevitably accumulates over a lifetime [39].
Diagnostic potential
Similar to findings in patients with frontotemporal dementia and Alzheimer's disease [40], a recent study reports higher levels of plasma-isolated exosomal tau in former professional American football players, a population especially vulnerable to chronic traumatic encephalopathy, versus controls [29]. These studies suggest that increased exosomal tau in plasma may be an indicator of tauopathy, but cannot currently be used as a diagnostic to discriminate between distinct tauopathies.
Therapeutic potential
Treating mice subject to a single severe traumatic brain injury with an antibody recognizing cis-phosphotau (cistau) prevents formation of tau oligomers and tangles, prevents cell-to-cell spread of pathogenic tau, reduces brain atrophy and restores long term potentiation [34]. Since cistauosis appears to be an early change in the course of both traumatic brain injury [34]- and Alzheimer's disease-associated tauopathy [33], detecting cistau may be useful for early diagnosis, and targeting cistau may be useful for prevention and therapeutic treatment of various tauopathies. In addition, therapeutic benefits of lithium [41], via inhibition of glycogen synthase kinase-3β, and a peptide inhibitor (D-JNKi1) of c-Jun N-terminal kinase [42], both of which are kinases associated with aberrant tau phosphorylation, have been reported in mouse models of traumatic brain injury.
Metabolic syndrome
Metabolic syndrome is a clinical diagnosis that includes obesity, hyperglycemia, hypertension and dyslipidemia. Affected individuals are at higher risk for developing more serious health conditions including diabetes and Alzheimer's disease [43]. Insulin resistance, a condition in which the body does not respond sufficiently to physiological levels of insulin, is central to metabolic syndrome. Postmortem brains from patients with tauopathies including Alzheimer's disease, Pick's disease, corticobasal degeneration and progressive supranuclear palsy have increased levels of abnormally phosphorylated insulin receptor substrate 1, a marker of insulin resistance [44]. Within tauopathy brains, neurons with elevated levels of abnormally phosphorylated insulin receptor substrate 1 also harbor pathogenic tau, suggesting a link between impaired insulin signaling and pathogenic tau [44]. Genetic induction of type II diabetes in mice causes disease-associated tau phosphorylation and cleavage [45]. Similarly, streptozotocin, a drug used to artificially induce insulin resistance and a type I diabetes-like phenotype, increases disease-associated tau phosphorylation in wild type mice, but, interestingly, not tau cleavage [45]. An increase in disease-associated phosphotau is also observed in streptozotocin-induced diabetic vervet monkeys [46]. Streptozotocin has also been shown to exacerbate tau phosphorylation and cognitive decline in mice transgenically expressing human tau harboring the P301L mutation alongside proteins associated with familial Alzheimer's disease (3xTg-AD) [47]. The onset of tauopathy in 3xTg-AD mice can also be accelerated by diet-induced diabetes [48]. In mice, streptozotocin-induced cognitive decline is blocked by knocking out the mouse endogenous MAPT gene [49], suggesting that tau is the key link between dysregulated insulin signaling and cognitive decline. Taken together, these studies suggest that diabetes is sufficient to drive formation of pathogenic tau. Mechanistically, evidence from cell culture suggests that insulin resistance promotes formation of pathogenic tau through reduced PI3K-Akt activity and subsequent activation of GSK3β, a kinase known to aberrantly phosphorylate tau in disease settings [50-52].
Therapeutic potential
Based on the link between tauopathy and insulin resistance, several FDA approved therapies developed for managing diabetes, including insulin, liraglutide, linagliptin, and metformin, are being tested in preclinical and clinical settings in the context of tauopathy. Pilot studies in patients with amnestic mild cognitive impairment or Alzheimer's disease have reported cognitive benefits of intranasally-administered insulin [53, 54] and inspired the ongoing clinical trial: The Study of Nasal Insulin in the Fight Against Forgetfulness (SNIFF, NCT01767909). The SNIFF study is examining effects of intranasally-administered insulin on cognition and brain atrophy. While it is currently unknown how intranasal insulin affects tau specifically, the SNIFF study will measure fluid biomarkers, including tau, in cerebrospinal fluid. An advantage of nasally-administered insulin is that insulin will not enter the peripheral bloodstream, which could cause hypoglycemia or insulin resistance.
Liraglutide, a glucagon-like peptide 1 (GLP-1) analog that stimulates insulin secretion, prevents aberrant tau phosphorylation in a mouse model of type II diabetes [55] and significantly reduces aberrantly phosphorylated tau and improves motor function in a mouse model of tauopathy (hTauP301L) [56]. Similarly, linagliptin has also recently been reported to reduce aberrant tau phosphorylation and improve cognition in 3xTg-AD mice [57]. A recent clinical trial reports that liraglutide prevents the decline of brain glucose metabolism that signifies cognitive impairment in Alzheimer's disease patients, but no improvement in cognitive performance was detected over the short six month study [58].
Metformin, a drug that suppresses hepatic glucose production, is commonly prescribed for management of type II diabetes. A small pilot study investigating the effects of metformin treatment in patients with amnestic mild cognitive impairment at risk for developing type II diabetes reports an improvement in one, but not all, measures of cognition [59]. A larger study investigating the effects of lifestyle intervention and metformin on brain glucose metabolism and cognition is currently in phase 4 clinical trials (NCT02409238). In the context of pathogenic tau specifically, metformin has been shown to prevent aberrant tau phosphorylation in a neuronal cell model of insulin resistance [60]. While studies in tau transgenic mice also report a reduction in disease-associated phosphotau in response to metformin treatment, the authors also report a metformin-induced increase in tau cleavage, aggregation, synaptic disruption, and hind limb atrophy. Based on these findings, the authors caution that treatment of diabetic patients with metformin may place them at higher risk for tauopathy [61].
Currently, the best supported and most efficacious intervention to prevent tauopathy associated with metabolic syndrome is a healthy lifestyle. For example, cognitive decline, hippocampal volume and metabolic measures improve in patients participating in the metabolic enhancement for neurodegeneration (MEND) protocol, which focuses on personalized therapeutic needs of the individual using a combination of treatment strategies including exercise and diet [62].
Spread of pathogenic tau from other cells
While not infectious in a classical sense, evidence clearly demonstrates that cells can release pathogenic tau, which “infects” neighboring and synaptically-connected cells in culture and in mice [63-66]. When taken in by neighboring cells, these pathogenic tau seeds stimulate aggregation of natively folded tau within the naïve cell [63, 67]. Studies utilizing recombinant, in vitro prepared tau aggregates as well as tau aggregates purified from postmortem Alzheimer's disease brain identify a tau trimer as the minimal unit necessary for cellular uptake of tau aggregates and subsequent seeding of intracellular tau aggregation in culture [67]. Tau may also spread from cell to cell in extracellular secreted microvesicles called “exosomes.” Tau-filled exosomes are present in cerebrospinal fluid and plasma of patients with mild Alzheimer's disease and frontotemporal dementia [40, 68]. Exosomal tau is phosphorylated at threonine 181, a phosphoepitope associated with pathogenic tau in the brain [68]. While exosomal tau has been reported to be secreted by both neurons [68] and microglia [69], blocking exosome formation in mouse microglia stops the spread of pathogenic tau from the entorhinal cortex to the hippocampus [69], suggesting that microglia-derived exosomes could be a vehicle for tau propagation. Studies in cultured cells and a rat model of tauopathy indicate that tau is also packaged into ectosomes, larger vesicles that are shed from cells via plasma membrane budding. When comparing exosomal versus ectosomal tau packaging, the authors report that ectosomal tau packaging occurs when tau is expressed at physiological levels, while exosomal tau packaging only occurs when tau is overexpressed [70]. Tunneling nanotubes, membranous structures that facilitate exchange of factors between cells, could also facilitate transfer of physiological and pathogenic tau between cells [71]. Interestingly, recent studies suggest that neuronal secretion of tau may play a role in normal, physiological brain function, as healthy neurons secrete tau in response to neuronal activity in cultured cells [72] and in wild-type mice [73]. The potentially “normal” function of neuronal secretion of tau may exacerbate tau spread in pathological conditions, as neuronal activity has also been reported to increase the rate of pathogenic tau spread in mice [74]. It is currently unknown whether pathogenic tau released in response to neuronal activity is free or vesicle-bound. Based on the ever-expanding studies in the field of pathogenic tau spread, it is clear that cells likely have multiple mechanisms to secrete physiological and pathogenic tau in vivo. The species of tau multimer and the packaging (or lack thereof) of tau multimer that is most efficient at propagating a misfolded state in vivo remains to be determined.
Therapeutic potential
Multiple companies and academic laboratories are working to develop immunotherapy strategies to clear pathogenic tau species from the brain. These active vaccines and anti-tau antibodies, several of which are currently in clinical trials [75], function by binding and clearing pathogenic tau from the brain, thereby preventing intercellular tau spread. Results from phase I trials for AADvac1, a tau vaccine, report favorable safety and immunogenicity outcomes [76]. Anti-tau antibodies are reported to be actively taken up by neurons in ex vivo cultured brain slices as well as in vivo [77-80], and tau-antibody complexes are proposed to be cleared via the lysosome [78, 79]. In addition to reducing levels of pathogenic tau, the tau antisense oligonucleotide described above also reduces tau seeding activity in tau transgenic mice [21].
Mechanistic target of rapamycin (mTOR) hyperactivity
mTOR is a serine/threonine protein kinase critical for cell survival and growth among cell types, including neurons. Through signal transduction pathways, mTOR integrates messages from the intra- and extracellular environment to modulate cellular functions including but not limited to transcription, translation, autophagy, cell signaling, metabolism, cytoskeletal dynamics, and memory formation [81]. Clinically, mTOR hyperactivity has been described in postmortem brain tissue collected from patients with progressive supranuclear palsy, corticobasal degeneration and Alzheimer's disease [82, 83]. Studies using cultured cells, tau transgenic mice and tau transgenic Drosophila have linked mTOR activation to elevated tau protein expression [48, 84, 85], aberrant tau phosphorylation [84, 86] decreased autophagic clearance of pathogenic tau [87-89], initiation of tau secretion [90], and tau-induced activation of the cell cycle in post-mitotic neurons [91]. Taken together, these studies indicate that mTOR activation is a potential driver of pathogenic tau formation, and suggest its relevance as a therapeutic target.
Therapeutic potential
Several mTOR inhibitors are FDA approved for treating cancer, are used as immunosuppressants for organ transplant, and extend lifespan in model organisms ranging from yeast to mammals [92]. The mTOR inhibitor rapamycin reduces pathogenic tau and improves cognition in numerous mouse models by upregulating autophagy [87-89, 93], and prevents neuronal death in tau transgenic Drosophila [91]. While treatment of 3xTg-AD mice with rapamycin prior to tau tangle formation significantly reduces formation of tau tangles and cognitive deficits, tangles are not reduced when rapamycin is administered after tangle formation, suggesting that beneficial effects of rapamycin may require an early diagnosis [94]. On the other hand, treatment of human tau P301S transgenic mice with rapamycin reduces aberrant tau phosphorylation and aggregation when administered early or late within the disease course, suggesting that early treatment with rapamycin may not be required in all contexts [89]. Analogs of rapamycin, or “rapalogs,” are also under investigation for the therapeutic treatment of tauopathies. Temsirolimus, for example, has better stability and pharmacological properties than rapamycin. In two separate studies, temsilorimus-treated tau transgenic mice were reported to have increased autophagy, decreased phosphotau and neurofibrillary tangle load, and improved cognition [95, 96]. Wogonin, a natural compound that inhibits mTOR, also reduces phosphotau in cultured cells [97]. Since several mTOR inhibitors are FDA approved, repurposing them for the treatment of neurodegenerative tauopathies may expedite the discovery of therapeutics. A clinical trial (NCT02874924) is currently underway to investigate the effects of rapamycin in adults aged 70-95; results from this study will provide data for rapamycin's tolerability and efficacy in improving life functions in older individuals. These outcomes will certainly guide future clinical studies.
Cellular Processes That Link Pathogenic Tau to Neuronal Death and Dysfunction
The cellular processes that link pathogenic tau to neuronal death and dysfunction provide additional targets for therapeutic intervention. Studies combining genetic approaches in tau transgenic Drosophila melanogaster with comparative studies in post mortem human control versus Alzheimer's disease brain have delineated a detailed cellular cascade that begins with pathogenic tau-mediated over-stabilization of the actin cytoskeleton and ends with neuronal death. The interaction between tau and filamentous actin is thought to play a physiological role in synaptic plasticity [98]. In a pathological setting, however, tau promotes filamentous actin stabilization and formation of filamentous actin bundles [99]. Among the many repercussions that an overly rigid cytoskeleton may have, tau-induced stabilization of filamentous actin is known to impact mitochondrial dynamics, which causes oxidative stress [100]. In addition, excess filamentous actin causes the nuclear envelope to involute, which disrupts the underlying nucleoskeleton [101], a lamin-rich meshwork that lines the nuclear envelope and provides a scaffold for the attachment of highly condensed heterochromatic DNA. Pathogenic tau-induced oxidative stress and lamin depletion cause constitutive heterochromatin to relax, which allows genes that are normally silenced by constitutive heterochromatin to be transcribed [101, 102]. These events activate the cell cycle in neurons [101, 102], which are normally post-mitotic. Activation of the cell cycle in neurons triggers apoptotic cell death [91, 103].
Tau may also have a physiological role in the nucleus where it protects DNA and regulates DNA packaging. Tau binds directly to DNA in vitro [104], protects against peroxidation-induced DNA damage [105], binds directly to AT-rich α-satellite heterochromatin in non-neuronal cultured cells [106], and colocalizes with pericentromeric constitutive heterochromatin in primary neuronal cultured from wild-type mice [107]. Markers of constitutive heterochromatin are reduced in neurons of tau knockout mice [107]. Taken together, these studies, combined with studies demonstrating that pathogenic tau induces heterochromatin relaxation via altered nucleoskeletal coupling and dysfunction of the lamin nucleoskeleton, suggest that the observed loss of markers of constitutive heterochromatin in human Alzheimer's disease brains [102, 108], could be a combined result of both toxic gain of function and loss of normal tau function.
An interaction between physiological tau and actin also regulates the relocalization of tau from dendritic shafts to the postsynaptic density in response to synaptic activity, which is important for synaptic plasticity [98]. In mouse hippocampal slice cultures exposed to amyloid β [98], or when transgenically expressed in mice in a disease-associated acetylated form [109], tau has less dendritic-to-postsynaptic mobility in response to synaptic activity, which is thought to result from a decreased interaction between tau and filamentous actin. Taken together, these data suggest that the physiological interaction between tau and actin is important for synaptic plasticity, that a reduced interaction between tau and actin at the synapse disrupts synaptic plasticity, and that over-stabilization of filamentous actin by pathogenic tau can ignite a chain of events that leads to neuronal death. Additional mechanisms of tau-induced neurotoxicity, including synaptic loss/dysfunction, impaired axonal transport, and microtubule destabilization have also been investigated in the context of tauopathy, and have been reviewed elsewhere [12, 110].
Therapeutic potential
In Drosophila, genetic reversal of filamentous actin over-stabilization, nuclear envelope disruption, heterochromatin relaxation, neuronal cell cycle activation, and reduction of oxidative stress significantly reduce tau-induced neurotoxicity [91, 99-102, 111], suggesting that pharmacological approaches that target these pathways may yield therapeutic benefit. Pharmacological approaches to reduce oxidative stress suppress tau-induced toxicity in Drosophila [111] and mice [112], however clinical trials in humans with mild to moderate Alzheimer's disease fail to show a cognitive benefit [113].
Concluding Remarks and Future Perspectives
The phenotypic heterogeneity and long disease duration of tauopathies present considerable challenges for diagnosis and development of disease-modifying therapies (see Outstanding Questions). In recent years, however, the tau community has made unprecedented strides toward establishing tools and identifying cellular pathways for diagnostic and therapeutic targeting. Transformative achievements include 1) Entry of tau-based immunotherapy into clinical trials, 2) First reports of tau-targeting antisense oligonucleotides, 3) Development of PET-based in vivo tau imaging, 4) A greater understanding of avenues in which pathogenic tau may spread through the brain, and 5) Identification of cellular mechanisms, including significant disruption of nuclear and genomic architecture, that mediate pathogenic tau-induced neuronal death and dysfunction. These advances are rapidly changing the landscape of biological understanding, diagnosis, and treatment for tauopathies, and offer great hope for the future.
Outstanding Questions.
Will in vivo tau imaging be able to differentiate between distinct tauopathies?
Will tau immunotherapy be effective in slowing cognitive decline in tauopathies?
What species of tau multimer and what type of multimer packaging is most efficient at propagating a misfolded state in vivo?
Can pharmacologic restoration of genomic architecture be used to prevent or slow tauopathy?
Trends Box.
Current diagnostic strategies cannot detect prodromal tauopathy
Current therapeutic strategies for tauopathies are not disease-modifying
Mutations in the tau gene, brain injuries, metabolic syndrome, TOR hyperactivity, and spread from neighboring cells can stimulate formation of pathogenic tau
Pathogenic tau-induced alterations in genomic architecture cause neuronal death
Acknowledgments
This study was supported by a grant from the National Institute of Neurological Disorders and Stroke (R00NS088429).
Footnotes
The authors declare no conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Weingarten MD, et al. A protein factor essential for microtubule assembly. Proc Natl Acad Sci U S A. 1975;72(5):1858–62. doi: 10.1073/pnas.72.5.1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wischik CM, et al. Isolation of a fragment of tau derived from the core of the paired helical filament of Alzheimer disease. Proc Natl Acad Sci U S A. 1988;85(12):4506–10. doi: 10.1073/pnas.85.12.4506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Goedert M, et al. Cloning and sequencing of the cDNA encoding a core protein of the paired helical filament of Alzheimer disease: identification as the microtubule-associated protein tau. Proc Natl Acad Sci U S A. 1988;85(11):4051–5. doi: 10.1073/pnas.85.11.4051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Miller BL, Boeve BF. The behavioral neurology of dementia. Cambridge University Press; 2009. [Google Scholar]
- 5.Miller BL Oxford University Press. Frontotemporal dementia, Contemporary neurology series 85. Oxford University Press; New York: 2014. p. xiii.p. 180. 1 online resource. [Google Scholar]
- 6.Lebby PC. Brain imaging : a guide for clinicians. Oxford University Press; 2013. [Google Scholar]
- 7.Holtzman DM, et al. Tau: From research to clinical development. Alzheimers Dement. 2016;12(10):1033–1039. doi: 10.1016/j.jalz.2016.03.018. [DOI] [PubMed] [Google Scholar]
- 8.Morishima-Kawashima M, et al. Hyperphosphorylation of tau in PHF. Neurobiol Aging. 1995;16(3):365–71. doi: 10.1016/0197-4580(95)00027-c. discussion 371-80. [DOI] [PubMed] [Google Scholar]
- 9.Hanger DP, et al. New phosphorylation sites identified inhyperphosphorylated tau (paired helical filament-tau) from Alzheimer's disease brainusing nanoelectrospray mass spectrometry. J Neurochem. 1998;71(6):2465–76. doi: 10.1046/j.1471-4159.1998.71062465.x. [DOI] [PubMed] [Google Scholar]
- 10.Mair W, et al. FLEXITau: Quantifying Post-translational Modifications of Tau Protein in Vitro and in Human Disease. Anal Chem. 2016;88(7):3704–14. doi: 10.1021/acs.analchem.5b04509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang Y, Mandelkow E. Tau in physiology and pathology. Nat Rev Neurosci. 2016;17(1):5–21. doi: 10.1038/nrn.2015.1. [DOI] [PubMed] [Google Scholar]
- 12.Arendt T, et al. Tau and tauopathies. Brain Res Bull. 2016;126(Pt 3):238–292. doi: 10.1016/j.brainresbull.2016.08.018. [DOI] [PubMed] [Google Scholar]
- 13.Nisbet RM, et al. Tau aggregation and its interplay with amyloid-beta. Acta Neuropathol. 2015;129(2):207–20. doi: 10.1007/s00401-014-1371-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ittner LM, Gotz J. Amyloid-beta and tau--a toxic pas de deux in Alzheimer's disease. Nat Rev Neurosci. 2011;12(2):65–72. doi: 10.1038/nrn2967. [DOI] [PubMed] [Google Scholar]
- 15.Moussaud S, et al. Alpha-synuclein and tau: teammates in neurodegeneration? Mol Neurodegener. 2014;9 doi: 10.1186/1750-1326-9-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Olsson B, et al. CSF and blood biomarkers for the diagnosis of Alzheimer's disease: a systematic review and meta-analysis. Lancet Neurol. 2016;15(7):673–84. doi: 10.1016/S1474-4422(16)00070-3. [DOI] [PubMed] [Google Scholar]
- 17.Chien DT, et al. Early clinical PET imaging results with the novel PHF-tau radioligand [F-18]-T807. J Alzheimers Dis. 2013;34(2):457–68. doi: 10.3233/JAD-122059. [DOI] [PubMed] [Google Scholar]
- 18.Kolb HC, Andres JI. Tau Positron Emission Tomography Imaging. Cold Spring Harb Perspect Biol. 2016 doi: 10.1101/cshperspect.a023721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Johnson KA, et al. Tau positron emission tomographic imaging in aging and early Alzheimer disease. Ann Neurol. 2016;79(1):110–9. doi: 10.1002/ana.24546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Khanna MR, et al. Therapeutic strategies for the treatment of tauopathies: Hopes and challenges. Alzheimers Dement. 2016;12(10):1051–1065. doi: 10.1016/j.jalz.2016.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.DeVos SL, et al. Tau reduction prevents neuronal loss and reverses pathological tau deposition and seeding in mice with tauopathy. Sci Transl Med. 2017;9 doi: 10.1126/scitranslmed.aag0481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hua Y, et al. Enhancement of SMN2 exon 7 inclusion by antisense oligonucleotides targeting the exon. PLoS Biol. 2007;5(4):e73. doi: 10.1371/journal.pbio.0050073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Poorkaj P, et al. Tau is a candidate gene for chromosome 17 frontotemporal dementia. Ann Neurol. 1998;43(6):815–25. doi: 10.1002/ana.410430617. [DOI] [PubMed] [Google Scholar]
- 24.Hutton M, et al. Association of missense and 5'-splice-site mutations in tau with the inherited dementia FTDP-17. Nature. 1998;393(6686):702–5. doi: 10.1038/31508. [DOI] [PubMed] [Google Scholar]
- 25.Spillantini MG, et al. Mutation in the tau gene in familial multiple system tauopathy with presenile dementia. Proc Natl Acad Sci U S A. 1998;95(13):7737–41. doi: 10.1073/pnas.95.13.7737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ghetti B, et al. Invited review: Frontotemporal dementia caused by microtubule-associated protein tau gene (MAPT) mutations: a chameleon for neuropathology and neuroimaging. Neuropathol Appl Neurobiol. 2015;41(1):24–46. doi: 10.1111/nan.12213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.McKee AC, et al. The first NINDS/NIBIB consensus meeting to define neuropathological criteria for the diagnosis of chronic traumatic encephalopathy. Acta Neuropathol. 2016;131(1):75–86. doi: 10.1007/s00401-015-1515-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dickstein DL, et al. Cerebral [18 F]T807/AV1451 retention pattern in clinically probable CTE resembles pathognomonic distribution of CTE tauopathy. Transl Psychiatry. 2016;6(9):e900. doi: 10.1038/tp.2016.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Stern RA, et al. Preliminary Study of Plasma Exosomal Tau as a Potential Biomarker for Chronic Traumatic Encephalopathy. J Alzheimers Dis. 2016;51(4):1099–109. doi: 10.3233/JAD-151028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gabbita SP, et al. Cleaved-tau: a biomarker of neuronal damage after traumatic brain injury. J Neurotrauma. 2005;22(1):83–94. doi: 10.1089/neu.2005.22.83. [DOI] [PubMed] [Google Scholar]
- 31.Arun P, et al. Acute decrease in alkaline phosphatase after brain injury: A potential mechanism for tauopathy. Neurosci Lett. 2015;609:152–8. doi: 10.1016/j.neulet.2015.10.036. [DOI] [PubMed] [Google Scholar]
- 32.Thomsen GM, et al. A model of recurrent concussion that leads to long-term motor deficits, CTE-like tauopathy and exacerbation of an ALS phenotype. J Trauma Acute Care Surg. 2016;81(6):1070–1079. doi: 10.1097/TA.0000000000001248. [DOI] [PubMed] [Google Scholar]
- 33.Nakamura K, et al. Proline isomer-specific antibodies reveal the early pathogenic tau conformation in Alzheimer's disease. Cell. 2012;149(1):232–44. doi: 10.1016/j.cell.2012.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kondo A, et al. Antibody against early driver of neurodegeneration cis P-tau blocks brain injury and tauopathy. Nature. 2015;523(7561):431–6. doi: 10.1038/nature14658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Collins-Praino LE, Corrigan F. Does neuroinflammation drive the relationship between tau hyperphosphorylation and dementia development following traumatic brain injury? Brain Behav Immun. 2017;60:369–382. doi: 10.1016/j.bbi.2016.09.027. [DOI] [PubMed] [Google Scholar]
- 36.Cherry JD, et al. Microglial neuroinflammation contributes to tau accumulation in chronic traumatic encephalopathy. Acta Neuropathol Commun. 2016;4(1):112. doi: 10.1186/s40478-016-0382-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Scholl M, et al. PET Imaging of Tau Deposition in the Aging Human Brain. Neuron. 2016;89(5):971–82. doi: 10.1016/j.neuron.2016.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Uchihara T, et al. Tau pathology in aged cynomolgus monkeys is progressive supranuclear palsy/corticobasal degeneration- but not Alzheimer disease-like -Ultrastructural mapping of tau by EDX. Acta Neuropathol Commun. 2016;4(1):118. doi: 10.1186/s40478-016-0385-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Crary JF. Primary age-related tauopathy and the amyloid cascade hypothesis: the exception that proves the rule? J Neurol Neuromedicine. 2016;1(6):53–57. doi: 10.29245/2572.942x/2016/6.1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Goetzl EJ, et al. Cargo proteins of plasma astrocyte-derived exosomes in Alzheimer's disease. Faseb j. 2016;30(11):3853–3859. doi: 10.1096/fj.201600756R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yu F, et al. Lithium ameliorates neurodegeneration, suppresses neuroinflammation, and improves behavioral performance in a mouse model of traumatic brain injury. J Neurotrauma. 2012;29(2):362–74. doi: 10.1089/neu.2011.1942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tran HT, et al. Inhibition of JNK by a peptide inhibitor reduces traumatic brain injury-induced tauopathy in transgenic mice. J Neuropathol Exp Neurol. 2012;71(2):116–29. doi: 10.1097/NEN.0b013e3182456aed. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schrijvers E, et al. Insulin metabolism and the risk of Alzheimer disease: The Rotterdam Study. Neurology. 2010;75(22):1982–7. doi: 10.1212/WNL.0b013e3181ffe4f6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yarchoan M, et al. Abnormal serine phosphorylation of insulin receptor substrate 1 is associated with tau pathology in Alzheimer's disease and tauopathies. Acta Neuropathol. 2014;128(5):679–89. doi: 10.1007/s00401-014-1328-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kim B, et al. Increased tau phosphorylation and cleavage in mouse models of type 1 and type 2 diabetes. Endocrinology. 2009;150(12):5294–301. doi: 10.1210/en.2009-0695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Morales-Corraliza J, et al. Brain-Wide Insulin Resistance, Tau Phosphorylation Changes, and Hippocampal Neprilysin and Amyloid-beta Alterations in a Monkey Model of Type 1 Diabetes. J Neurosci. 2016;36(15):4248–58. doi: 10.1523/JNEUROSCI.4640-14.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chen Y, et al. Intracerebroventricular streptozotocin exacerbates Alzheimerlike changes of 3xTg-AD mice. Mol Neurobiol. 2014;49(1):547–62. doi: 10.1007/s12035-013-8539-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Orr ME, et al. Mammalian target of rapamycin hyperactivity mediates the detrimental effects of a high sucrose diet on Alzheimer's disease pathology. Neurobiol Aging. 2014;35(6):1233–42. doi: 10.1016/j.neurobiolaging.2013.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Abbondante S, et al. Genetic ablation of tau mitigates cognitive impairment induced by type 1 diabetes. Am J Pathol. 2014;184(3):819–26. doi: 10.1016/j.ajpath.2013.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hong M, Lee VM. Insulin and insulin-like growth factor-1 regulate tau phosphorylation in cultured human neurons. J Biol Chem. 1997;272(31):19547–53. doi: 10.1074/jbc.272.31.19547. [DOI] [PubMed] [Google Scholar]
- 51.Lesort M, et al. Insulin transiently increases tau phosphorylation: involvement of glycogen synthase kinase-3beta and Fyn tyrosine kinase. J Neurochem. 1999;72(2):576–84. doi: 10.1046/j.1471-4159.1999.0720576.x. [DOI] [PubMed] [Google Scholar]
- 52.Kim B, et al. Cortical neurons develop insulin resistance and blunted Akt signaling: a potential mechanism contributing to enhanced ischemic injury in diabetes. Antioxid Redox Signal. 2011;14(10):1829–39. doi: 10.1089/ars.2010.3816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Reger MA, et al. Intranasal insulin improves cognition and modulates beta-amyloid in early AD. Neurology. 2008;70(6):440–8. doi: 10.1212/01.WNL.0000265401.62434.36. [DOI] [PubMed] [Google Scholar]
- 54.Craft S, et al. Intranasal insulin therapy for Alzheimer disease and amnestic mild cognitive impairment: a pilot clinical trial. Arch Neurol. 2012;69(1):29–38. doi: 10.1001/archneurol.2011.233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ma DL, et al. Early intervention with glucagon-like peptide 1 analog liraglutide prevents tau hyperphosphorylation in diabetic db/db mice. J Neurochem. 2015;135(2):301–8. doi: 10.1111/jnc.13248. [DOI] [PubMed] [Google Scholar]
- 56.Hansen HH, et al. The GLP-1 receptor agonist liraglutide reduces pathology-specific tau phosphorylation and improves motor function in a transgenic hTauP301L mouse model of tauopathy. Brain Res. 2016;1634:158–70. doi: 10.1016/j.brainres.2015.12.052. [DOI] [PubMed] [Google Scholar]
- 57.Kosaraju J, et al. Linagliptin, a Dipeptidyl Peptidase-4 Inhibitor, Mitigates Cognitive Deficits and Pathology in the 3xTg-AD Mouse Model of Alzheimer's Disease. Mol Neurobiol. 2016 doi: 10.1007/s12035-016-0125-7. [DOI] [PubMed] [Google Scholar]
- 58.Gejl M, et al. In Alzheimer's Disease, 6-Month Treatment with GLP-1 Analog Prevents Decline of Brain Glucose Metabolism: Randomized, Placebo-Controlled, Double-Blind Clinical Trial. Front Aging Neurosci. 2016;8:108. doi: 10.3389/fnagi.2016.00108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Luchsinger JA, et al. Metformin in Amnestic Mild Cognitive Impairment: Results of a Pilot Randomized Placebo Controlled Clinical Trial. J Alzheimers Dis. 2016;51(2):501–14. doi: 10.3233/JAD-150493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gupta A, et al. Peripheral insulin-sensitizer drug metformin ameliorates neuronal insulin resistance and Alzheimer's-like changes. Neuropharmacology. 2011;60(6):910–20. doi: 10.1016/j.neuropharm.2011.01.033. [DOI] [PubMed] [Google Scholar]
- 61.Barini E, et al. Metformin promotes tau aggregation and exacerbates abnormal behavior in a mouse model of tauopathy. Mol Neurodegener. 2016;11:16. doi: 10.1186/s13024-016-0082-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bredesen DE, et al. Reversal of cognitive decline in Alzheimer's disease. Aging (Albany NY) 2016;8(6):1250–8. doi: 10.18632/aging.100981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Frost B, et al. Propagation of tau misfolding from the outside to the inside of a cell. J Biol Chem. 2009;284(19):12845–52. doi: 10.1074/jbc.M808759200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Clavaguera F, et al. Transmission and spreading of tauopathy in transgenic mouse brain. Nat Cell Biol. 2009;11(7):909–13. doi: 10.1038/ncb1901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Liu L, et al. Trans-synaptic spread of tau pathology in vivo. PLoS One. 2012;7(2):e31302. doi: 10.1371/journal.pone.0031302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.de Calignon A, et al. Propagation of tau pathology in a model of early Alzheimer's disease. Neuron. 2012;73(4):685–97. doi: 10.1016/j.neuron.2011.11.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Mirbaha H, et al. Tau Trimers Are the Minimal Propagation Unit Spontaneously Internalized to Seed Intracellular Aggregation. J Biol Chem. 2015;290(24):14893–903. doi: 10.1074/jbc.M115.652693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Saman S, et al. Exosome-associated tau is secreted in tauopathy models and is selectively phosphorylated in cerebrospinal fluid in early Alzheimer disease. J Biol Chem. 2012;287(6):3842–9. doi: 10.1074/jbc.M111.277061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Asai H, et al. Depletion of microglia and inhibition of exosome synthesis halt tau propagation. Nat Neurosci. 2015;18(11):1584–93. doi: 10.1038/nn.4132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Dujardin S, et al. Ectosomes: a new mechanism for non-exosomal secretion of tau protein. PLoS One. 2014;9(6):e100760. doi: 10.1371/journal.pone.0100760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Tardivel M, et al. Tunneling nanotube (TNT)-mediated neuron-to neuron transfer of pathological Tau protein assemblies. Acta Neuropathol Commun. 2016;4(1):117. doi: 10.1186/s40478-016-0386-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Pooler AM, et al. Physiological release of endogenous tau is stimulated by neuronal activity. EMBO Rep. 2013;14(4):389–94. doi: 10.1038/embor.2013.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Yamada K, et al. Neuronal activity regulates extracellular tau in vivo. J Exp Med. 2014;211(3):387–93. doi: 10.1084/jem.20131685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Wu JW, et al. Neuronal activity enhances tau propagation and tau pathology in vivo. Nat Neurosci. 2016;19(8):1085–92. doi: 10.1038/nn.4328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Pedersen JT, Sigurdsson EM. Tau immunotherapy for Alzheimer's disease. Trends Mol Med. 2015;21(6):394–402. doi: 10.1016/j.molmed.2015.03.003. [DOI] [PubMed] [Google Scholar]
- 76.Novak P, et al. Safety and immunogenicity of the tau vaccine AADvac1 in patients with Alzheimer's disease: a randomised, double-blind, placebo-controlled, phase 1 trial. Lancet Neurol. 2017;16(2):123–134. doi: 10.1016/S1474-4422(16)30331-3. [DOI] [PubMed] [Google Scholar]
- 77.Asuni AA, et al. Immunotherapy targeting pathological tau conformers in a tangle mouse model reduces brain pathology with associated functional improvements. J Neurosci. 2007;27(34):9115–29. doi: 10.1523/JNEUROSCI.2361-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Krishnamurthy PK, et al. Mechanistic Studies of Antibody-Mediated Clearance of Tau Aggregates Using an ex vivo Brain Slice Model. Front Psychiatry. 2011;2:59. doi: 10.3389/fpsyt.2011.00059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Collin L, et al. Neuronal uptake of tau/pS422 antibody and reduced progression of tau pathology in a mouse model of Alzheimer's disease. Brain. 2014;137(Pt 10):2834–46. doi: 10.1093/brain/awu213. [DOI] [PubMed] [Google Scholar]
- 80.Gu J, et al. Two novel Tau antibodies targeting the 396/404 region are primarily taken up by neurons and reduce Tau protein pathology. J Biol Chem. 2013;288(46):33081–95. doi: 10.1074/jbc.M113.494922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Switon K, et al. Molecular neurobiology of mTOR. Neuroscience. 2017;341:112–153. doi: 10.1016/j.neuroscience.2016.11.017. [DOI] [PubMed] [Google Scholar]
- 82.Gentry EG, et al. Rho Kinase Inhibition as a Therapeutic for Progressive Supranuclear Palsy and Corticobasal Degeneration. J Neurosci. 2016;36(4):1316–23. doi: 10.1523/JNEUROSCI.2336-15.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Tramutola A, et al. Alteration of mTOR signaling occurs early in the progression of Alzheimer disease (AD): analysis of brain from subjects with pre-clinical AD, amnestic mild cognitive impairment and late-stage AD. J Neurochem. 2015;133(5):739–49. doi: 10.1111/jnc.13037. [DOI] [PubMed] [Google Scholar]
- 84.An WL, et al. Up-regulation of phosphorylated/activated p70 S6 kinase and its relationship to neurofibrillary pathology in Alzheimer's disease. Am J Pathol. 2003;163(2):591–607. doi: 10.1016/S0002-9440(10)63687-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Morita T, Sobue K. Specification of neuronal polarity regulated by local translation of CRMP2 and Tau via the mTOR-p70S6K pathway. J Biol Chem. 2009;284(40):27734–45. doi: 10.1074/jbc.M109.008177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Meske V, et al. Coupling of mammalian target of rapamycin with phosphoinositide 3-kinase signaling pathway regulates protein phosphatase 2A- and glycogen synthase kinase-3 -dependent phosphorylation of Tau. J Biol Chem. 2008;283(1):100–9. doi: 10.1074/jbc.M704292200. [DOI] [PubMed] [Google Scholar]
- 87.Caccamo A, et al. Molecular interplay between mammalian target of rapamycin (mTOR), amyloid-beta, and Tau: effects on cognitive impairments. J Biol Chem. 2010;285(17):13107–20. doi: 10.1074/jbc.M110.100420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Caccamo A, et al. mTOR regulates tau phosphorylation and degradation: implications for Alzheimer's disease and other tauopathies. Aging Cell. 2013;12(3):370–80. doi: 10.1111/acel.12057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Ozcelik S, et al. Rapamycin attenuates the progression of tau pathology in P301S tau transgenic mice. PLoS One. 2013;8(5):e62459. doi: 10.1371/journal.pone.0062459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Tang Z, et al. mTor mediates tau localization and secretion: Implication for Alzheimer's disease. Biochim Biophys Acta. 2015;1853(7):1646–57. doi: 10.1016/j.bbamcr.2015.03.003. [DOI] [PubMed] [Google Scholar]
- 91.Khurana V, et al. TOR-mediated cell-cycle activation causes neurodegeneration in a Drosophila tauopathy model. Curr Biol. 2006;16(3):230–41. doi: 10.1016/j.cub.2005.12.042. [DOI] [PubMed] [Google Scholar]
- 92.Lamming DW. Inhibition of the Mechanistic Target of Rapamycin (mTOR)-Rapamycin and Beyond. Cold Spring Harb Perspect Med. 2016;6 doi: 10.1101/cshperspect.a025924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Siman R, et al. The mTOR Inhibitor Rapamycin Mitigates Perforant Pathway Neurodegeneration and Synapse Loss in a Mouse Model of Early-Stage Alzheimer-Type Tauopathy. PLoS One. 2015;10(11):e0142340. doi: 10.1371/journal.pone.0142340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Majumder S, et al. Inducing autophagy by rapamycin before, but not after, the formation of plaques and tangles ameliorates cognitive deficits. PLoS One. 2011;6(9):e25416. doi: 10.1371/journal.pone.0025416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Frederick C, et al. Rapamycin ester analog CCI-779/Temsirolimus alleviates tau pathology and improves motor deficit in mutant tau transgenic mice. J Alzheimers Dis. 2015;44(4):1145–56. doi: 10.3233/JAD-142097. [DOI] [PubMed] [Google Scholar]
- 96.Jiang T, et al. Temsirolimus attenuates tauopathy in vitro and in vivo by targeting tau hyperphosphorylation and autophagic clearance. Neuropharmacology. 2014;85:121–30. doi: 10.1016/j.neuropharm.2014.05.032. [DOI] [PubMed] [Google Scholar]
- 97.Zhu Y, Wang J. Wogonin increases beta-amyloid clearance and inhibits tau phosphorylation via inhibition of mammalian target of rapamycin: potential drug to treat Alzheimer's disease. Neurol Sci. 2015;36(7):1181–8. doi: 10.1007/s10072-015-2070-z. [DOI] [PubMed] [Google Scholar]
- 98.Frandemiche ML, et al. Activity-dependent tau protein translocation to excitatory synapse is disrupted by exposure to amyloid-beta oligomers. J Neurosci. 2014;34(17):6084–97. doi: 10.1523/JNEUROSCI.4261-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Fulga TA, et al. Abnormal bundling and accumulation of F-actin mediates tau-induced neuronal degeneration in vivo. Nat Cell Biol. 2007;9(2):139–48. doi: 10.1038/ncb1528. [DOI] [PubMed] [Google Scholar]
- 100.DuBoff B, et al. Tau promotes neurodegeneration via DRP1 mislocalization in vivo. Neuron. 2012;75(4):618–32. doi: 10.1016/j.neuron.2012.06.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Frost B, et al. Lamin Dysfunction Mediates Neurodegeneration in Tauopathies. Curr Biol. 2016;26(1):129–36. doi: 10.1016/j.cub.2015.11.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Frost B, et al. Tau promotes neurodegeneration through global chromatin relaxation. Nat Neurosci. 2014;17(3):357–66. doi: 10.1038/nn.3639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Arendt T, et al. Selective cell death of hyperploid neurons in Alzheimer's disease. Am J Pathol. 2010;177(1):15–20. doi: 10.2353/ajpath.2010.090955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Hua Q, et al. Microtubule associated protein tau binds to double-stranded but not single-stranded DNA. Cell Mol Life Sci. 2003;60(2):413–21. doi: 10.1007/s000180300034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Wei Y, et al. Binding to the minor groove of the double-strand, tau protein prevents DNA from damage by peroxidation. PLoS One. 2008;3(7):e2600. doi: 10.1371/journal.pone.0002600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Sjoberg MK, et al. Tau protein binds to pericentromeric DNA: a putative role for nuclear tau in nucleolar organization. J Cell Sci. 2006;119(Pt 10):2025–34. doi: 10.1242/jcs.02907. [DOI] [PubMed] [Google Scholar]
- 107.Mansuroglu Z, et al. Loss of Tau protein affects the structure, transcription and repair of neuronal pericentromeric heterochromatin. Sci Rep. 2016;6 doi: 10.1038/srep33047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Hernandez-Ortega K, et al. Altered Machinery of Protein Synthesis in Alzheimer's: From the Nucleolus to the Ribosome. Brain Pathol. 2016;26(5):593–605. doi: 10.1111/bpa.12335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Tracy TE, et al. Acetylated Tau Obstructs KIBRA-Mediated Signaling in Synaptic Plasticity and Promotes Tauopathy-Related Memory Loss. Neuron. 2016;90(2):245–60. doi: 10.1016/j.neuron.2016.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Frost B, et al. Connecting the dots between tau dysfunction and neurodegeneration. Trends Cell Biol. 2015;25(1):46–53. doi: 10.1016/j.tcb.2014.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Dias-Santagata D, et al. Oxidative stress mediates tau-induced neurodegeneration in Drosophila. J Clin Invest. 2007;117(1):236–45. doi: 10.1172/JCI28769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Nakashima H, et al. Effects of alpha-tocopherol on an animal model of tauopathies. Free Radic Biol Med. 2004;37(2):176–86. doi: 10.1016/j.freeradbiomed.2004.04.037. [DOI] [PubMed] [Google Scholar]
- 113.Polidori MC, Nelles G. Antioxidant clinical trials in mild cognitive impairment and Alzheimer's disease - challenges and perspectives. Curr Pharm Des. 2014;20(18):3083–92. doi: 10.2174/13816128113196660706. [DOI] [PubMed] [Google Scholar]