

Abstract
Like all positive strand RNA viruses, the picornaviruses replicate their genomes using a virally encoded RNA-dependent RNA polymerase enzyme known as 3Dpol. Over the past decade we have made tremendous advances in our understanding of 3Dpol structure and function, including the discovery of a novel mechanism for closing the active site that allows these viruses to easily fine tune replication fidelity and quasispecies distributions. This review summarizes current knowledge of picornaviral polymerase structure and how the enzyme interacts with RNA and other viral proteins to form stable and processive elongation complexes. The picornaviral RdRPs are among the smallest viral polymerases, but their fundamental molecular mechanism for catalysis appears to be generally applicable as a common feature of all positive strand RNA virus polymerases.
Keywords: picornavirus, polymerase, RNA-dependent RNA polymerase, structure, positive strand RNA virus
1. Introduction
Positive strand RNA viruses provide their own genome replication machinery via an RNA-dependent RNA polymerase (RdRP) protein, allowing them to replicate without a DNA intermediate. For the picornaviruses the RdRP is 3Dpol, a ≈460 residue protein found at the very C-terminal end of the ~250 kDa viral polyprotein. The core function of 3Dpol is genome replication, but the protein also functions as part of the 3CDpro precursor to modulate protease domain specificity and binding to RNA sequences that control virus replication. Notably, the picornaviral 3CDpro proteins have no polymerase activity, which is elegantly activated only upon proteolytic processing to generate the mature 3Dpol enzyme.
The first positive strand RNA virus RdRP structures were solved in the late 1990’s with a partial poliovirus polymerase structure (Hansen et al., 1997) that was followed by a pair of hepatitis C virus polymerase structures (Bressanelli et al., 1999; Lesburg et al., 1999), the caliciviral rabbit hemorrhagic disease virus polymerase structure (Ng et al., 2002), and then the essentially simultaneous publication of complete picornaviral polymerase structures from foot and mouth disease virus, three rhinovirus strains, and poliovirus (Ferrer-Orta et al., 2004; Love et al., 2004; Thompson and Peersen, 2004). Since that time, a plethora of RdRP structures have been solved as isolated proteins, in proteolytic precursor forms, and in complexes with other viral proteins, RNA, nucleotides. Together, these structures highlight the strong conservation of structural and functional elements needed for replicative polymerase activity. The picornaviral 3Dpols are the smallest positive strand RNA virus polymerases yet their highly conserved structure is found at the core of many larger viral RdRPs. The picornaviral polymerases are also proving to be excellent systems for studying the molecular mechanisms underlying catalysis and nucleotide selection, providing insights that pertain to most (+) strand RNA virus polymerases.
2. Genome Architecture and Polymerase Biochemistry
2.1 Genome Structure
The picornaviruses currently encompass 54 species grouped into 31 genera (www.picornaviridae.com) and have 7–8 Kb long single stranded RNA genomes. Using the enteroviruses as an example (Figure 1A), the viral genome begins with a 5′ UTR composed of a ~100 nt long RNA cloverleaf structure followed by a ~700 nt long IRES, the highly structured RNA that directly recruits ribosomes for viral protein translation (Fitzgerald and Semler, 2009). Next, a 6500–7200 nt open reading frame encodes for a single 240–260 kDa polyprotein that is ultimately cut into 11–13 different proteins by the viral 2Apro, 3Cpro, and 3CDpro proteases. Note that there are some differences in the genome organization and resulting proteomes among the different picornaviral genera (Palmenberg et al., 2010). The most common is replacing the 2A protease with a leader protease (L) found at the very beginning of the polyprotein, and some viruses have multiple tandem copies of the 3B (i.e. VPg) peptide (Gorbalenya and Lauber, 2010). The pathways for polyprotein processing are temporally complex and generate multiple intermediate species that provide functions and interactions somewhat different from the fully processed proteins, expanding the biochemical diversity arising from the rather small genome. These processing pathways will not be discussed here except for their role in activating 3Dpol function upon cleavage of the 3CDpro precursor protein. Finally, there is a short ~300 nt 3′ UTR that includes a pair of RNA stem-loops followed by a 25–100 nt long poly(A) tail.
Figure 1. Picornaviral Genome Structure and Polymerase Functions.
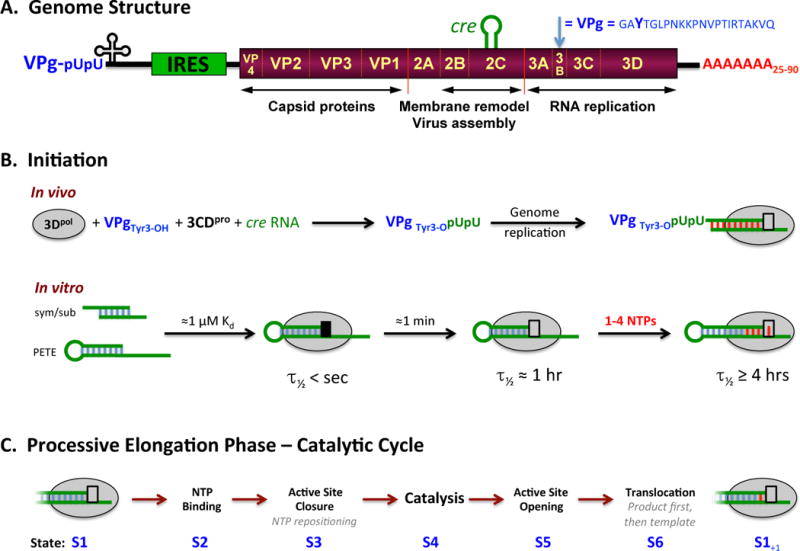
(A) Schematic representation of the poliovirus genome as a representative picornavirus. The genome encodes a single ≈250 KDa polyprotein that is translated from an internal ribosome entry site (IRES) and cleaved into about a dozen smaller proteins and functional intermediates by the viral 2Apro, 3Cpro, and 3CDpro proteases. The last part of the polyprotein is 3Dpol, a RNA-dependent RNA polymerase that is only active upon cleavage of the 3Cpro–3Dpol junction. Many picornaviruses replace 2Apro with a N-terminal leader (L) protease. (B) The native initiation pathway for 3Dpol uses the viral VPg protein (i.e. 3B) whose Tyr3 becomes doubly uridylylated via a cre RNA templated reaction in the context of a viral replication center. In vitro, however, 3Dpol will initiate using short RNA duplexes, such as the self-complementary sym/sub sequences used extensively by the Cameron lab or RNA hairpins “PETE” constructs used by the Peersen group. There is a stepwise assembly pathway whereby the initial 3Dpol-RNA complex needs to undergo a conformational transition to become catalytically competent, as indicated by the black versus grey box at the active site. (C) The full catalytic cycle that takes place repeatedly during processive elongation can be divided into six major structural states, S1–S6, as previously described (Gong and Peersen, 2010).
The picornaviral genomes also contain an important internal regulatory element, the RNA stem-loop structure known as cre (oriI). Cre templates the addition of two uracils onto the hydroxyl group of Tyr3 on the viral 3B protein, resulting in the VPg-pUpU primer used for all RNA synthesis by 3Dpol in vivo (Paul et al., 2000). The cre secondary structure, as a RNA stem with a 14-nt loop containing two key adenosines, is conserved among picornaviruses but its exact sequence and location within the genome vary greatly (Yang et al., 2002). Last, the poliovirus genome also contains a pair of RNA hairpin elements called α and β that are important for proper RNA synthesis during virus infection (Burrill et al., 2013; Song et al., 2012). These 75-nucleotide segments are located within the 3Dpol coding region, where α encompasses residues 341–356 within 3Dpol folding motif D and β encompasses residues 416–440 within the thumb domain. They are conserved among the group C enteroviruses, but their importance for replication beyond poliovirus is not yet known.
2.2 3Dpol Biochemistry
Initiation of RNA synthesis during normal virus replication always uses a tyrosine residue on the viral 3B (VPg) protein, but in an in vitro context with purified components 3Dpol is capable of initiation on RNA primer-template pairs and RNA hairpin structures (Figure 1B). This ability to efficiently initiate in vitro has enabled detailed studies of RNA binding, initiation kinetics, and subsequent RNA elongation steps. Overall, the data from these experiments have shown that 3Dpol forms a very stable and highly processive elongation complex with a well defined catalytic cycle. We now have a very good biochemical and structural understanding of the steps involved in each cycle of nucleotide incorporation (Figure 1B,C).
RNA binding takes on the order of a minute with a dissociation constant in the low micromolar range when studied as purified components in solution at moderate 75–100 mM monovalent salt concentrations (Arnold and Cameron, 2004; Gohara et al., 2000; Mestas et al., 2007). The primer can be either RNA or DNA in vitro, but the reaction is much less efficient with a DNA primer. This can be overcome by using terminal transferases to add 2–3 ribonucleotides to the 3′ end of the DNA primer (Svensen et al., 2016). Following initial RNA binding there is a conformational change in the 3Dpol–RNA complex that is necessary before the first nucleotide can be added to the RNA primer. This change occurs with a time constant of ≈13 seconds and it thus takes about a minute for an entire population to convert to the catalytically competent state (Arnold and Cameron, 2000). Notably, the 3Dpol-RNA complex becomes more stable as a result of this conformational change, resulting in a half-life of ≈1 hour. Incorporation of 1–4 nucleotides further stabilize the complex, increasing its half life to ≈4 hours or more and resulting in the highly processive elongation complex (EC) that is capable of replicating genome length RNAs without dissociating. Stalled ECs remain functional for hours on the bench (Arnold and Cameron, 2000), including in high salt concentrations that eliminate RNA rebinding (Hobdey et al., 2010), and they are stable for over a week at 16 °C as purified complexes for crystallization (Gong and Peersen, 2010). Notably, this high stability has allowed us to purify and crystallize multiple stalled elongation complexes, leading to structures that have provided tremendous structural insights into the 3Dpol catalytic cycle (Gong et al., 2013).
Replicative polymerases share a catalytic mechanism in which two magnesium ions are locked into the active site via interactions with aspartic acid residues on the protein, the priming nucleotide 3′ hydroxyl group, and the NTP triphosphate (Steitz, 1998). The magnesium ions stabilize the transition state and help deprotonate the primer 3′ hydroxyl group, setting it up for a nucleophilic attack on the NTP alpha phosphate. The picornaviral 3Dpol enzymes utilize this highly conserved mechanism and structural framework for catalysis, but notably they arrive at the final catalytic conformation by molecular motions that are distinct from those used by other single subunit polymerases (Gong and Peersen, 2010). The fine details of these different mechanisms are discussed in the Active Site Closure section below, and this alternate pathway appears to pertain to all (+) strand RNA virus polymerases. Importantly, this alternate mechanism is the molecular key to the relatively high error rates characteristic of viral RdRPs that in turn give rise to the quasispecies populations of virus variants (Lauring and Andino, 2010; Lauring et al., 2013).
3. Picornaviral Polymerase Structure
The remainder of this article will go into greater depth about our current understanding of the structures and molecular mechanisms underpinning a number of the biochemical functions described above. Specific structures are referenced by their 4-character Protein Data Bank (PDB, www.wwpdb.org) codes in brackets, e.g. [3OL6+], where a + sign indicates multiple related structures are available from the PDB, usually as part of the same publication. Residue numbering is based on poliovirus 3Dpol unless otherwise stated, and thus may differ slightly from other RdRPs and their prior publications. The structure superpositions presented are obtained using a maximum likelihood based algorithm that effectively downweights less conserved portions of the structures (www.theseus3d.org), producing more accurate results than the traditional least-squares approach (Theobald and Steindel, 2012; Theobald and Wuttke, 2008).
3.1 Overall 3Dpol Structure
The ≈460 residue picornaviral polymerase is a minimal viral RdRP structure with a three-dimensional fold resembling a cupped human right hand. This has led to the assignment of its major structural units as being the palm, fingers, and thumb domains (Figure 2A). This analogy was originally used to describe the DNA polymerase I Klenow fragment, and this core arrangement is proving to be a common feature of replicative DNA and RNA polymerases. The structural palm, fingers, and thumb domains are in turn composed of the classic motifs A–G arising from sequence conservation (Cerny et al., 2014). These motifs are shared among multiple classes of both DNA and RNA polymerases that utilize a two-metal catalytic mechanism with common active site geometry. The active site is built on a three-stranded β-sheet composed of one strand from motif A and two strands from motif C that are packed against a long α-helix from motif B.
Figure 2. Overview of picornaviral RdRP structures.
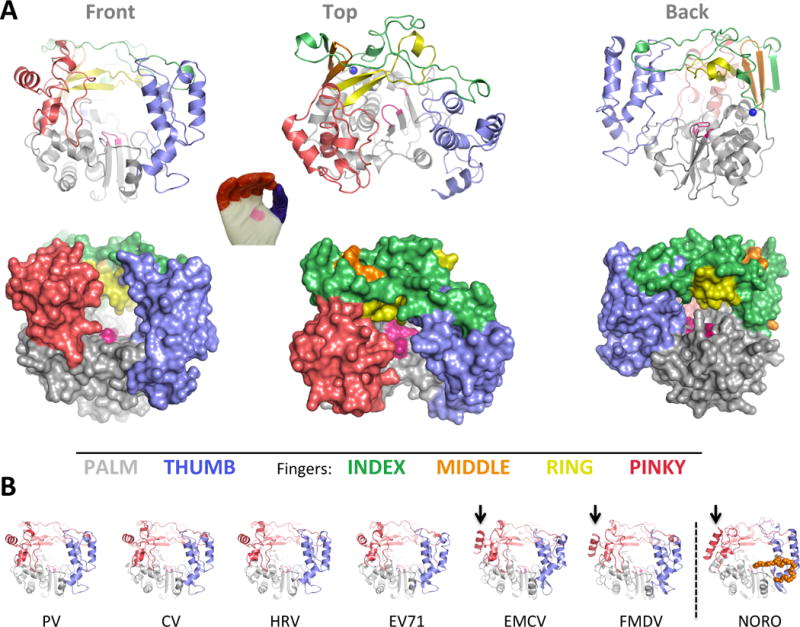
(A) Cartoon and surface representations of poliovirus 3Dpol in three different orientations. The structure resembles a cupped right hand composed of palm, fingers, and thumb domains. The fingers domain can be further divided into five distinct structures (per color key), and the active site in the palm domain is shown as a patch of magenta. Note that the index finger reaches across the palm to contact the top of the thumb, creating a channel at the back of the enzyme whereby NTPs access the active site. (B) All the picornaviral 3Dpol structures solved to date exhibit a very high degree of structural homology. Note that one helix on the pinky finger changes orientation between the enteroviruses and EMCV/FMDV groups, with the latter resembling the helix orientation seen in the non-picornaviral norovirus 3Dpol. Norovirus polymerase also has a C-terminal extension (orange) that reaches into the RNA exit channel.
Relating primary sequence to three-dimensional protein structure, the palm and fingers are interspersed with multiple crossovers between the two domains and the thumb is always the last 80–100 residues of the protein. Within the fingers domain there are four distinct sub-structures that are sometimes referred to by the anatomical analogy of index, middle, ring, and pinky fingers (Thompson and Peersen, 2004). Together, the index, middle, and ring finger form what is technically an extended 5-stranded β-sheet topology that is conserved among viral RdRPs (Lu and Gong, 2013).
The viral RdRPs share a folding interaction where the fingertips, i.e. the top of the fingers domain, reach across the active site to contact the top of the thumb. When the index finger contacts the thumb it traps the ring finger underneath it, resulting in the ring finger becoming the NTP entry channel roof and supplying multiple basic residues for making triphosphate contacts. This finger–thumb contact is absolutely conserved in all the positive strand RNA virus RdRP structures and it also plays an important role in maintaining structural integrity. Mutation of the poliovirus polymerase index finger-thumb interface reduces the protein thermal denaturation point by ≈5 °C while adding NTPs conversely stabilizes the structure by ≈5 °C (Thompson et al., 2007). As a class, the viral RdRPs are relatively small polymerases and they have likely evolved this fingers-thumb interaction to maintain proper folding at physiological temperatures.
This basic palm-fingers-thumb structure represents the complete sequence of smaller viral polymerases that are typified by the picornaviral 3Dpol proteins. There are now hundreds of viral RdRP structures in the PDB from more than twenty different (+) strand RNA viruses, and the picornaviruses being well represented within that set by 3Dpol structures from poliovirus (PV), coxsackievirus B3 (CV), enterovirus 71 (EV71), human rhinovirus (HRV), encephalomyocarditis virus (EMCV), and foot and mouth disease virus (FMDV) (Table I, Figure 2B). The RdRPs from larger RNA viruses generally have additional domains that provide other replication-linked activities and these proteins are often referred to as more encompassing “replicases”, as reviewed by Gong in this issue of Virus Research (Gong, 2017). Their added functionalities include N-terminal methyl transferase domains that provide RNA capping activity, extended loops on the thumb domain that protrude into the RNA duplex exit channel to provide a platform for primer-independent de novo initiation, and amphipathic helices that provide membrane anchoring functions.
Table 1.
Notable Picornaviral Polymerase Structures
| Virus | PDBa | Description |
|---|---|---|
| Poliovirus (PV) | ||
| 1RDR | First RdRP structure solved, but a partial structure with fingers missing | |
| 1RA6+ | Complete structure showing buried N-terminus | |
| 2ILY+ | Polymerase with bound NTPs in the absence of RNA | |
| 3OL6+ | Elongation complex structures showing palm based active site closure | |
| 4K4S+ | Additional stalled EC’s with in-crystal catalysis and translocation | |
| Coxsackievirus B3 (CV) | ||
| 3DDK | Polymerase | |
| 3CDU | Polymerase | |
| 3CDW | Polymerase with VPg at thumb/palm junction on back of 3Dpol | |
| 4K4X+ | Elongation complexes, including in-crystal translocation | |
| 4ZPA+ | Rearrangements of motif D and active site closure | |
| Rhinovirus (HRV) | ||
| 1XR5 | HRV 14 polymerase (originally 1TEB) | |
| 1XR6 | HRV 1B polymerase (originally 1TE8) with bound K+ near N-terminus pocket | |
| 1XR7 | HRV 16 polymerase (originally 1TE9) | |
| 1TP7 | HRV 16 polymerase | |
| 4K50 | HRV 16 elongation complex | |
| Enterovirus 71 (EV71) | ||
| 3N6L | Polymerase | |
| 3N6M | Polymerase with bound GTP | |
| 4IKA | Polymerase with the VPg at base of the thumb | |
| 5F8G+ | Elongation complexes with in-crystal catalysis and product strand translocation | |
| Encephalomyocarditis virus (EMCV) | ||
| 4NZ0 | Polymerase (6 independent copies, copy F distorted by crystal contact) | |
| 4NYZ | Polymerase with non-buried N-terminus and rearranged motif A | |
| Foot-and-mouth disease virus (FMDV) | ||
| 1U09 | Polymerase | |
| 1WNE | Polymerase with primer-template RNA | |
| 2D7S | Polymerase with VPg near active site | |
| 2F8E | Polymerase with VPg-pU near active site | |
| 2E9R+ | Polymerase with RNA and in-crystal elongation products | |
| Norovirus (not a picornavirus, but a similar and important structure) | ||
| 1SH0+ | Norwalk virus polymerase | |
| 3BS0+ | Polymerase co-crystallized with RNA and CTP, closed pre-catalysis active site | |
Structure coordinates and viewing tools are available via www.wwpdb.org, which provides links to member sites that provide different user interfaces and tools for examining a shared common PDB database of structures. A “+” after the PDB entry code indicates there are multiple related structures sharing the same primary publication. Direct links to these additional structures can be found in the “Literature” section of the primary entry on the RCSB version of the PDB (www.rcsb.org).
In fact, the structure of other picornaviral polymerases can now be predicted with fairly good accuracy using on-line modeling servers such as Phyre (Kelley et al., 2015) that require only the primary sequence as their input. Phyre first uses rigorous sequence database analysis to generate a hidden Markov model representation of the evolutionary profile of the protein of interest. This model is then matched with similar models derived from all the known protein structures in the PDB. Finally, the matched proteins from the PDB are used as structural frameworks for building multiple predicted structures of the protein of interest. This method is very effective, in large part because maintaining protein structure is a stronger evolutionary constraint than sequence conservation. It is also a fast way to structurally identify and visualize the boundaries of the classic conserved polymerase sequence motifs (motif A, B, C…) that map to key conserved structural elements.
3.2 Polyprotein Processing and Activation
The picornaviral polymerases are generally inactive when initially translated as part of the viral polyprotein and only gain polymerase activity upon 3C–3D junction cleavage to generate the fully processed 3Dpol enzyme (Harris et al., 1992). The structural basis for this activation was elucidated by the complete PV 3Dpol structure showing the newly created N-terminus being buried in a shallow pocket at the base of the fingers domain (Figure 3A) (Thompson and Peersen, 2004). The N-terminal glycine residue, resulting from cleavage at the glutamine–glycine polyprotein junction, is held tightly in this pocket by four hydrogen bonds, three to the terminal nitrogen atom and the fourth to the Gly1 carbonyl group. Two of these hydrogen bonds are between Gly1 and Gly64, an important interaction will be discussed later in the context of altering 3Dpol fidelity. The other two hydrogen bonds are with the backbone carbonyls of residues 239 and 241 (PV) located at the end of motif A. This is immediately adjacent to the Asp238 residue that helps close the active site for catalysis as a result of NTP recognition and repositioning. At a molecular level, the proper N-terminus burial likely stiffens the polymerase structure at the fingers–palm domain junction.
Figure 3. Proteolytic activation of 3Dpol.
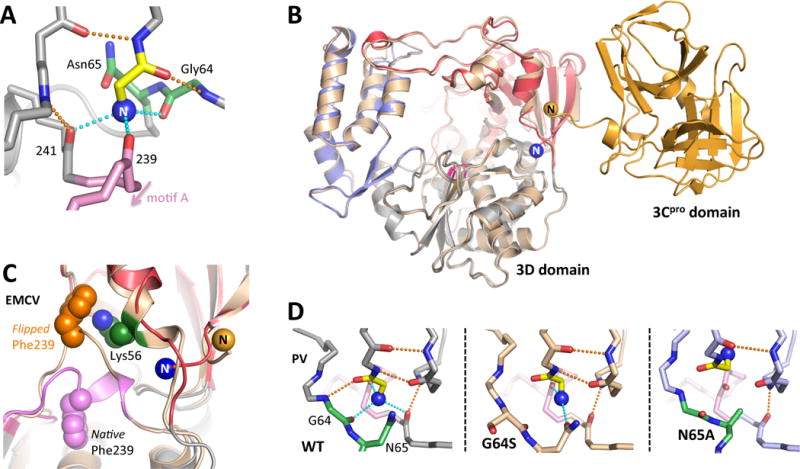
The very N-terminus (blue sphere N) of a picornaviral polymerase is buried in a pocket at the base of the fingers domain, resulting in activation of the enzyme through stabilization of the motif A movements needed for catalysis. (A) Structural interactions involving the PV 3Dpol N-terminus as viewed from the direction of the active site. (B) Backside view of the PV 3CDpro structure where the 3Dpol N-terminus does not exist because it is part of the flexible linker between the 3Cpro and 3Dpol domains. (C) In one EMCV 3Dpol structure [4YNZ] the N-terminus was no longer bound in its pocket and there is a major rearrangement of Phe239 within motif A. (D) Structural plasticity of the PV N-terminus binding pocket, where the native conformation is stabilized by six hydrogen bonds to residues Gly1 and Glu2. In the G64S structure one only of the native H-bonds to the N-terminus exists, but Asn65 has formed a new H-bond. Mutating Asn65 to alanine causes the pocket to collapse and the N-terminus folds up into the fingers domain.
This exact geometry for burying the N-terminus is also seen in the CV, HRV, EV71, EMCV, and FMDV polymerases, making it a universal feature of picornaviral 3Dpol structures thus far. Experiments with both PV and CV polymerase have demonstrated that adding or deleting even a single residue at the 3Dpol N-terminus totally abolishes polymerase activity, pointing out the critical dependence of polyprotein processing for polymerase activation (Campagnola et al., 2008; Thompson and Peersen, 2004). Two 3Dpol structures without a buried N-terminus have also been solved. The first is the PV 3CDpro precursor [2IJD] where the 3Dpol N-terminus does not exist (Marcotte et al., 2007). This structure is basically a fusion of the 3C and 3D domains, both of which retain the overall fold they have as separate proteins (Figure 3B). The polymerase Gly1 and Glu2 residues become part of a flexible 5-residue linker between the domains and the N-terminus binding pocket collapses slightly as a result of their removal, primarily through backbone movements of residues 61–66 that form the outside of the binding pocket (Figure 3D). Second, there is an EMCV 3Dpol structure [4NYZ] where, for unknown reasons, the very N-terminus moved out of its binding pocket (Vives-Adrian et al., 2014). In this structure there is a major rearrangement of residues 237–242, the structural link between motif A in the active site and the buried N-terminus, and Phe239 flips from being buried down in the palm domain to being tucked up into a shallow pocket in the fingers where it makes a cation–π interaction with Lys56 (Figure 3C).
The requirement for a glycine as the first residue in 3Dpol can be a potential problem for expression of recombinant polymerases because the N-terminus cannot be modified with an initiator methionine residue. Bacterial methionine aminopeptidases are fairly efficient at removing the Met1 residue, but they can be overloaded under very high-level protein expression. To circumvent this issue, the Cameron lab developed expression systems using N-terminal fusions with ubiquitin (Gohara et al., 1999) and SUMO (Arnold et al., 2006) that are cleaved in situ by co-expressed proteases, generating 3Dpol with a native N-terminal glycine residue for direct purification from bacterial lysates.
3.3 Polymerase-RNA Elongation Complexes
The core function of the RdRP is to carry out genome replication, and the picornaviral polymerases do so in a primer dependent manner. In vivo initiation uses the viral VPg protein, but for in vitro biochemical studies one can initiate with short (6–20 base pair) primers annealed to template RNA. The RNA path through the RdRP is well established from the plethora of RdRP-RNA complex structures that have been solved over the past decade, including structures that capture distinct conformational states at different points in the catalytic cycle and provide direct insights into molecular mechanisms of nucleotide selection and replication.
The first picornaviral RdRP–RNA structures were from FMDV, where polymerase was co-crystallized with a self-complementary 10-mer RNA that formed a six base pair duplex with four nucleotide 5′ overhangs [1WNE] (Ferrer-Orta et al., 2004). These crystals were also capable of in-crystal nucleotide incorporation upon soaking in NTP solutions [2E9R+], providing the initial structural insights into NTP binding and the RdRP catalytic cycle (Ferrer-Orta et al., 2007). Co-crystallization with RNA and NTPs has also worked for the very similar norovirus polymerase [3BSO] (Zamyatkin et al., 2008), but analogous structures of hepatitis C virus (HCV) polymerase were obtained by bathing crystals in RNA solutions [1NB7, 4E78, 4E7A] (Mosley et al., 2012; O’Farrell et al., 2003), utilizing an experiment affectionately known as “soak-and-pray” because a favorable outcome is by no means guaranteed. In our own work, we were unable to obtain poliovirus 3Dpol-RNA structures by either of these methods. We therefore embarked on a more laborious path of first forming 3Dpol-RNA complexes, initiating by adding the NTPs needed for 4–6 elongations steps, and then purifying these stalled elongation complexes prior to crystallization [3OL6+] (Gong and Peersen, 2010). The ECs were extremely stable and we used this same EC assembly and purification approach to also solve the structures of CV and HRV-16 3Dpol-RNA elongation complexes with multiple RNAs [4K4S+] (Gong et al., 2013). Conceptually identical methods can also be used to assemble stable HCV polymerase elongation complexes from dinucleotide primers, but no structures from these have been published yet (Jin et al., 2012). More recently, the Gong group has used this method to solve a series of enterovirus 71 EC structures [5F8G+] that are providing tremendous insights into RNA translocation (Shu and Gong, 2016). Together, the structures from all three approaches have complemented each other to yield a high-resolution atomic picture of RdRP structure and how it relates to catalysis and function.
3.4 RNA Path Through Polymerase
Based on multiple EC structures, 3Dpol directly contacts eight base pairs of upstream duplex product RNA and three nucleotides of downstream single stranded template RNA (Figure 4A–D) (Gong et al., 2013). The template-product helix emerges from the large opening between the thumb and pinky finger as a RNA duplex with a thumb domain α–helix lining the RNA major groove. The RNA is well ordered in electron density maps and there is no sign of any strand separation mechanism within any of the monomeric polymerase structures. However, mutations in the thumb domain can affect poly(A) tail length, suggesting some slippage between the two RNA strands may happen in vivo despite the lack of formal strand separation (Kempf et al., 2013).
Figure 4. RNA interactions with 3Dpol in the Elongation Complex.
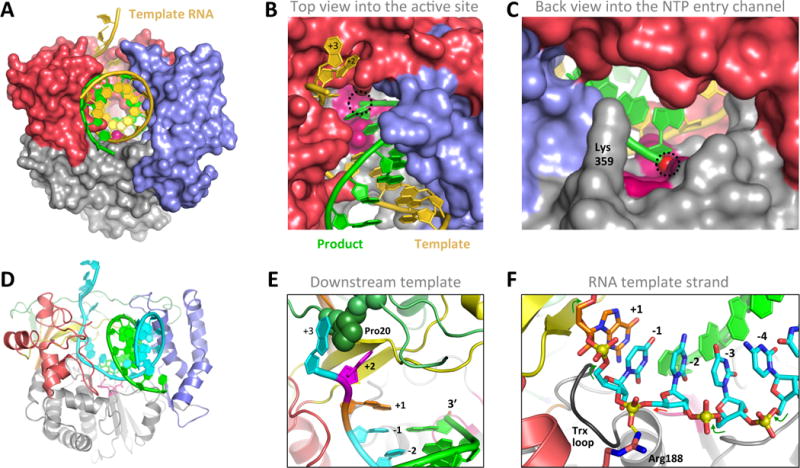
(A–C) Three views of the PV 3Dpol-RNA elongation complex oriented as in Figure 2A. The single stranded template RNA enters the polymerase from the top and takes a ≈90° turn as it contacts the palm at the active site, where it forms a duplex with the product strand and exits the polymerase via the wide front channel. The location of the priming 3′ hydroxyl group at the active site is marked with a dashed circle. (D) Cartoon representation of the elongation complex with the template strand in cyan and the fingers colored as in Figure 2A. (E) Details of key interaction within the 3Dpol-RNA complex, including Pro20 inserted between the +2 and +3 template strand bases, the binding pocket for the unstacked +2 nucleotide on the template strand, and the pre-positioning of the templating +1 nucleotide above the active site where it is poised for base pairing with an incoming NTP. (E) The conformation of the template RNA strand as it passes through the polymerase active site. Note the backbone linkage between the −1 and −2 nucleotides is not standard A-form (red arrow vs green arrows) due to a salt bridge interaction with Arg188.
Within the active site, the most unique aspect of the picornaviral RdRP complexes is that the templating nucleotide is fully pre-positioned for catalysis in the absence of a bound NTP [3OL6+] (Gong and Peersen, 2010). The templating base (+1 position) sits right above the active site and is fully stacked with the first base pair of the product helix (Figure 4E). As such, the polymerase-RNA complex is effectively poised for an incoming NTP that will be recognized by base pairing and trigger catalysis with very little movement of the RNA. This relatively simple nucleotide selection mechanism has major implications for RdRP replication fidelity, as discussed in greater detail below.
Downstream of the active site, the RdRPs only contact a few nucleotides of the incoming template. In the enteroviral polymerases there is a well defined binding pocket for the +2 base wherein the nucleotide becomes fully unstacked from both the upstream and downstream neighboring bases (Figure 4B) [4K4S+] (Gong et al., 2013). One side of the pocket has a proline that provides a hydrophobic interaction with the +2 nucleotide (Figure 4E) and the other side has a lysine that extends across the nucleotide to contact the phosphate backbone. The +3 base stacks on the other side of the proline and the downstream template then extends away from the polymerases surface without any further protein contacts. A similar RNA path is seen in the apthoviral FMDV polymerase structures, but the downstream +2 base is not nearly as well ordered in those electron density maps [1WNE, 2E9R+] (Ferrer-Orta et al., 2004; Ferrer-Orta et al., 2007). This heterogeneity may due to inherently weaker downstream RNA interactions in the FMDV 3Dpol complex because the proline seen in the enteroviral +2 binding pocket is replaced with a much more flexible arginine, or it may be due to induced fit effects where the enteroviral elongation complexes are more ordered because they have undergone several cycles of elongation prior to crystallization. An induced fit mechanism may also be suggested by recent structural [4WYL+] and biochemical work showing structural flexibility in the FMDV +2 pocket, including mutations of Lys18 and Lys20 that affect RNA binding, processivity, and nucleotide selection (Ferrer-Orta et al., 2015). Notably, the index finger residues making up the +2 pocket of FMDV 3Dpol also function as a nuclear localization signal (Sanchez-Aparicio et al., 2013), adding amino acid sequence constraints that may be in direct competition with establishing ideal RNA binding interactions. The +2 pocket region is thus important for virus replication via both direct effects on polymerase function and indirect effects on host cell interactions.
3.5 Template RNA Binding and Initiation
The exact structural changes associated with the biochemical transition to the long-lived 3Dpol–RNA complex are not readily apparent from the structures solved thus far. In fact, the 3Dpol protein structures observed in the absence or presence of RNA are essentially identical and there is not a clear conformational switch in the protein signifying a catalysis-ready complex. Thus, these necessary switches most likely have to do with the fine details of establishing proper interactions with the RNA phosphate backbone to fully position the template-primer RNA in the active site. For example, the +1 templating base needs to be locked into position above the active site, an unusual backbone conformation needs to be established between the −1 and −2 template strand nucleotides (Figure 4F), and the product strand must be properly oriented to set up the 3′ OH group for an efficient attack on the NTP phosphate. Establishing these interactions may require a few cycles of elongation to actively move the RNA through the complex, perhaps explaining why the RNA is generally more ordered in the stalled elongation complex structures than in the RNA soak structures.
The picornaviral polymerases can replicate through a duplex template in vitro (Cho et al., 1993) and 3Dpol must therefore have some inherent strand separation ability. The EC structures suggest this happens in a stepwise fashion (Gong et al., 2013); the +4 nucleotide can base pair in a downstream RNA duplex, the +3 nucleotide cannot base pair because index finger residues block access to the hydrogen bonding face of the base, the +2 nucleotide is fully unstacked from both neighboring bases, and the +1 base is stacked on the product duplex in the active site and awaiting the arrival of an NTP for catalysis. Strand separation between the +4 and +3 positions would direct a non-template strand on a path across the top of the middle finger, but we do not yet have a structure with a significant strand-separated sequence to show such interactions.
There are also subtle indications that we may not yet know the full story about downstream template strand interactions beyond the +2 nucleotide. Despite multiple attempts, none of the picornaviral ECs have crystallized with extensive single stranded RNA as the downstream sequence. However, inspection of the 3Dpol structure reveals a possible template entry channel with strong positive electrostatic surface potential that could contact three or four more bases using basic residues for phosphate contact and aromatic tyrosines for base stacking interactions. Mutations of these residues affect RNA binding and elongation kinetics in vitro, and when mutant polymerases are introduced into infectious virus there is strong selection for phenylalanine and tyrosine residues at many of these positions (Kortus et al., 2012). There is also the observation that residue 5, which is located near the exit of this putative RNA channel, is strongly favored to be a large hydrophobic amino acid despite being exposed on the polymerase surface. Mutating the native Phe5 in CV 3Dpol to smaller residues drastically reduces polymerase activity, as measured by in vitro product RNA formation, while replacing it with a larger tryptophan increases activity (Campagnola et al., 2008). More detailed studies of the corresponding Trp5 in PV 3Dpol have shown the primary effect of these mutations is to alter the temporal stability of the post-initiation elongation complex, which in turn greatly affects processivity and virus replication (Hobdey et al., 2010). Together, these structural and biochemical observations raise the possibility of a fingers domain refolding event associated with actively elongating ECs, but the structures relax back to the non-elongating conformation before crystals grow. Crystallization typically takes several days, making such as structure very difficult to capture by this technique.
4. Catalytic Cycle
The overall 3Dpol catalytic cycle that takes place repeatedly during elongation can be divided into six discrete steps, as outlined in Figure 1C. The rates of these steps have been worked out biochemically, X-ray crystallography has illustrated many of the associated structures, and NMR methods are providing key insights into the role of protein dynamics during nucleotide binding and recognition. For this cycle, nucleotides first enter the 3Dpol active site via the large opening at the back of the polymerase (Figure 4C) whose entrance is rimmed with lysine and arginine residues positioned to interact with the nucleotide triphosphates. The initial NTP association event is non-specific and non-cognate NTPs compete with weak millimolar inhibition constants (Karr and Peersen, 2016), but there is no structure of a 3Dpol-RNA in complex with a non-cognate NTP. Cognate NTPs, on the other hand, fully enter the active site and are recognized by basepairing to the templating base, and this event triggers molecular motions in the palm domain to close the active site for catalysis. Following catalysis, the active site opens again by reversing the movement of motif A in the palm, and this is followed by a distinct translocation step to complete the catalytic cycle and position the next templating base in the active site.
4.1 RdRP Active Site Closure
PV and EV71 EC structures show that picornaviral RdRPs close their active sites for catalysis using a seemingly subtle conformational change in the palm domain (Gong and Peersen, 2010; Shu and Gong, 2016). In essence, the triple β-strand motif that forms the highly conserved polymerase core structure only becomes fully structured for catalysis in the presence of a bound nucleotide. This novel mechanism for active site closure provides the RdRP with a fidelity control point that allows these viruses to evolutionarily fine tune quasispecies distributions for optimal growth (Campagnola et al., 2015). A comparison of RdRP palm domain structures suggests that all (+) strand RNA viruses use this palm-based active site closure mechanism, but the dsRNA and (−) strand RNA viruses most likely do not.
In contrast, single subunit polymerases, as exemplified by T7 RNA polymerase and Taq polymerase, utilize a pre-insertion site to first bind the incoming NTP via base pairing interactions with the templating base. This is followed by a major swinging movement of the fingers domain and its “O-helix” structure that repositions the template–NTP base pair, placing the triphosphate into the active site for catalysis. The key active site aspartic acid residues from motifs A and C are pre-positioned for catalysis and there are only minor structural changes within the palm domain during active site closure. In essence, proper template–NTP pairing is selected for at two different binding sites, increasing the fidelity of these enzymes as compared to a single site palm domain based mechanism used by the (+) strand RNA virus RdRPs.
The palm domain conformation change is illustrated in Figure 5A. It primarily involves motif A that has a β-strand conformation but is not fully hydrogen bonded with the adjacent motif C in either 3Dpol alone or 3Dpol-RNA EC structures. However, when the cognate nucleotide is present there is a subtle conformational change such that motif A forms a canonical antiparallel β-sheet with motif C. This is driven by a network of hydrogen bonds linking Asn297 in the motif B helix to the NTP 2′ hydroxyl to Ser288 in the fingers domain to a movement of Asp238 in motif A that triggers active site closure (Figure 5B) (Gong and Peersen, 2010). Importantly, Asp233 is now positioned to coordinate magnesium ions in the active site, setting up the classic geometry for polymerase catalysis. This involves two Mg2+ ions; metal A that is pre-bound to the polymerase and becomes located between Asp233 and the priming 3′ hydroxyl group, and metal B that is part of the incoming NTP-Mg2+ complex and becomes coordinate by Asp233 and the NTP α and β phosphate oxygens (Figure 5C). The detailed geometry of protein and nucleic acid groups, waters, and bound Mg2+ in the active site during catalysis have been visualized in both PV [3OL6+] and EV71 [5F8G+] EC structures (Gong and Peersen, 2010; Shu and Gong, 2016). All the closed state picornaviral ECs show a post-catalysis structure with UMP incorporation and bound pyrophosphate, but the structurally analogous pre-catalysis closed state has been captured in a norovirus polymerase complex [3BSO] (Zamyatkin et al., 2008), as also discussed in this issue of Virus Research (Jin et al., 2017). The initial PV elongation complex structures did not show the polymerase bound metal A magnesium, but it was later seen in CV and EV71 EC structures at a site ≈5.3 Å away from its catalytic position. The magnesium interacts with the active site Asp329 and Asp233 residues in this alternate site (Figure 5C) and is presumably brought into the active site via electrostatic interactions as both aspartates reposition during active site closure.
Figure 5. Active site closure via a unique palm domain based mechanism.
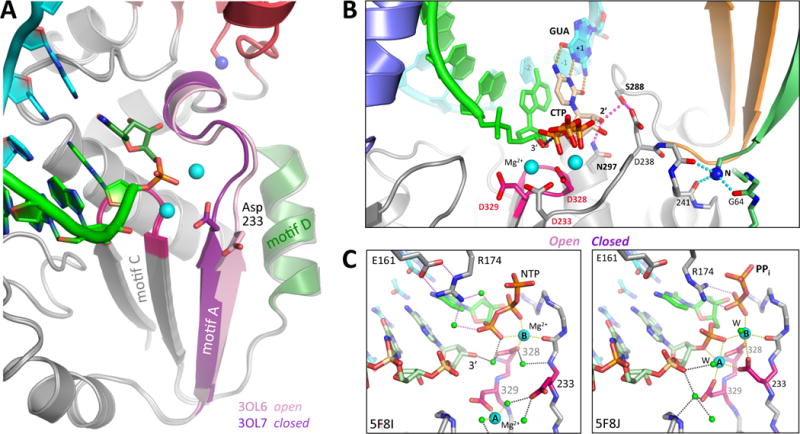
(A) The picornaviral RdRPs use the same two metal catalytic mechanism as other replicative polymerases, but close their active sites for catalysis by a novel movement of motif A in response to correct NTP binding. This moves the essential Asp233 into the active site to enable catalysis. (B) Structure of the closed PV 3Dpol active site following CTP binding and catalysis [3OL7], with the set of stabilizing hydrogen bonds directly linking the NTP 2′ hydroxyl to Asp238 in the repositioned motif A highlighted in magenta. (C) Structural details of the open and closed EV71 polymerase active sites where all twelve magnesium ion coordination ligands have been captured in a single structure. In the pre-catalysis open state the NTP phosphate interacts with Arg174 from the ring finger via two water molecules, and Arg174 is itself positioned by interactions with Glu161. The metal A magnesium ion is located ≈5 Å away from the active site in the open state, but is moved into the catalytic center during active site closure when both Asp233 and Asp329 reorient to support catalysis. In the closed state, the magnesiums are axially coordinated by the two Asp328 oxygens from below and by a pair of water molecules (W) from above.
4.2 Translocation
Another unique aspect of the picornaviral RdRP elongation complexes is that after catalysis their active sites return to the open state with the newly incorporated nucleotide remaining in the active site. This is also fundamentally different from what occurs in most other replicative polymerases, where the fingers domain swinging motion that opens the active site also drives translocation via a direct molecular contact between the “O-helix” and the RNA. In those polymerases, the opening step appears to be largely driven by a “power-stroke” mechanism where pyrophosphate release and loss of electrostatic interactions release the fingers domain, triggering a reversal of the swinging movement that previously closed that active site. In contrast, the viral RdRPs appear to translocate their RNA using more of a “Brownian ratchet” mechanism in which the RNA is moved via a series of smaller steps driven by thermal motions. In effect, the RNA can adopt a series of marginally stable binding registers with smaller energy barriers to entry and exit before finally dropping into a deeper energy minimum after the complete ≈2.4 Å base-step of the A-form RNA.
The strongest evidence for such a Brownian ratchet comes from a recent series of EV71 EC structures showing multiple conformational states in a crystal lattice where the product RNA extended into an open solvent channel without any direct molecular contact that could impeded its motion [5F8G+] (Shu and Gong, 2016). Figure 6 shows a comparison of the RNA structure seen at the very beginning of the catalytic cycle, before NTP entry, and that observed at the very end after incorporation of a new nucleotide and active site opening. From these structures, it is apparent that the RNA duplex is initially somewhat strained with the product strand being pulled toward its 3′ end in the active site such that the base pairs are not fully planar (Figure 6A). However, at the end of the cycle the priming nucleotide (−1, blue) has moved up out of the active site and the product strand RNA had slid toward its 5′ end, restoring planar base pairing and a lower energy state of the product duplex. Notice that the template RNA strand is essentially stationary during the movement of the product strand, and the shift in relative positions of the two strands can be appreciated by the change in the alignment of the base pairing hydrogen bonds (Figure 6B).
Figure 6. RNA Translocation Mechanism.
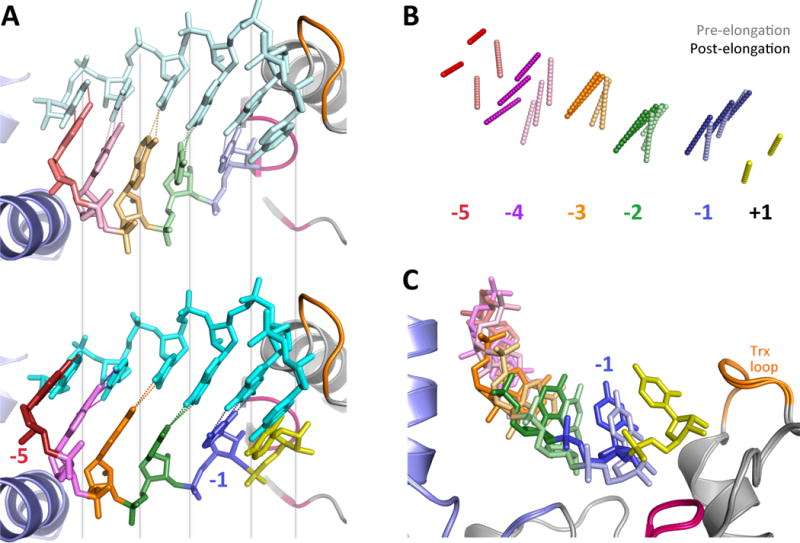
Following catalysis the 3Dpol active site opens back up without RNA translocation, unlike what seen in many other replicative polymerases where those two events are coupled. EV71 elongation complex structure suggest a Brownian ratchet mechanism for translocation. (A) Comparison of EV71 structures captured at the beginning state 1 [5F8L] and ending state 6 [5F8N] of a single nucleotide incorporation cycle show a sliding movement of the product RNA strand. In state 1 (top) the priming duplex is strained and its base pairs are not planar because the 3′ end of the product strand is being pulled toward the active site. After catalysis in state 6 (bottom) the product strand has moved toward the thumb such that the duplex now forms proper planar base pairs. Note that the template strand backbone does not move, as per the grey visual alignment lines. (B) Direct comparison of the base pair hydrogen bonding patterns at the beginning (lighter) and end (darker) of the catalytic cycle show how the product strand slides relative to the fixed template strand. (C) The movement toward the 5′ end is facilitate by the priming −1 nucleotide (blue) moving up and out the active site after the addition of the +1 nucleotide (yellow), releasing RNA-polymerase contacts and allowing the RNA to relax to the more favorable base pairing conformation.
These EV71 EC structures imply the existence of a final translocation step where the template strand moves by a single base register. This is likely to be a much higher energy transition that is not easily reversible, and thus effectively locks the RNA into position for the next round of catalysis. Two structural features of the template are likely involved in this step; the unique RNA backbone conformation between the −1 and −2 nucleotides, and the movement of the +2 base out of its well defined binding pocket and into the templating +1 site (Figure 4E,F). These two interactions are quite close in space and probably occur simultaneously, perhaps driven by a loss of the ionic contact between the −2 base phosphate group and Arg188; that would allow the template RNA to move away from direct contact with the polymerase and slide to re-establish the distorted interactions with the product strand that has already moved through its Brownian ratchet mechanism.
There is some structural evidence suggestive of a direct protein interaction during this final translocation step via a flexible loop in motif B that can interconvert between in and out conformations (Sholders and Peersen, 2014). The loop is in close contact with the RNA (Figure 6C) and its out conformation leads to a steric clash with the template strand, suggesting the loop may help move the templating nucleotide from the +1 position to the −1 position after catalysis (there is no “0” position). Mutations in the loop can result in translocation deficient polymerases that only incorporate one nucleotide, consistent with a role in translocation. However, the loop movements have only been observed in structures of 3Dpol alone, without bound RNA or NTPs, and it is possible that the loop flexibility serves some other purpose in the elongation or initiation complexes.
5. 3Dpol–VPg Complexes
In vivo initiation by 3Dpol always involves the 22–24 residue 3B protein, also known as VPg, wherein a tyrosine side chain serves as the primer for initial uridylylation reactions to generate a VPg-pUpU molecule. This reaction uses the normal 3Dpol active site in a macromolecular complex where the VPg tyrosine hydroxyl is presumably positioned in the active site to mimic a RNA 3′ hydroxyl group. In vitro uridylylation is possible using a mixture of 3Dpol, VPg, poly(A) RNA, and UTP, and for this reaction there is significant cross-reactivity between different viral 3Dpol and VPg proteins (Schein et al., 2015). However, uridylylation is much more efficient with cre RNA and 3CDpro as cofactors to form a complex with a significantly lower Km for UTP (Paul et al., 2000; Steil and Barton, 2008; Yin et al., 2003). Interestingly, the same adenosine base in cre templates both uracil addition steps, implicating slippage of the product relative to the cre to reposition the VPg-pU for the second uridylylation event (Paul et al., 2003). However, despite multiple structures there is not yet a clear consensus for how this is accomplished at a molecular level.
3Dpol–VPg complexes with CV, EV71, and FMDV polymerases have been solved, but they show divergent binding sites and none of the structures contain the poly(A) or cre RNA templates needed for uridylylation (Figure 7A). Based on solution NMR data, VPg is extended and disordered in solution [2BBL+] (Schein et al., 2006) while the singly uridylylated VPg-pU form adopts a more compact conformation with numerous medium and long range NOE cross-peaks (Schein et al., 2010). In the crystal structures, VPg makes a few discrete contacts with the polymerase, as opposed to forming a clearly defined and extended binding interaction. Density for CV VPg was observed in the motif E region at the junction of the palm and thumb domains on the backside of 3Dpol [3CDW], but only residues 7–15 were resolved (Gruez et al., 2008). The Tyr3 residue is not observed in the density maps and the VPg is oriented with its C-terminus pointing toward the 3Dpol active site, making it difficult to envision this as a correct binding site for a productive complex. This coxsackievirus VPg binding site is fairly close to a region identified as being important for the poliovirus 3Dpol-3AB interaction based on yeast two hybrid studies (Hope et al., 1997). In the FMDV system there are structures of both VPg [2D7S] and VPg-pU [2F8E] in a conformation where the peptide has an extended conformation and is cradled into the 3Dpol duplex RNA binding channel (Figure 7B) (Ferrer-Orta et al., 2006). The VPg shows strong electron density and its N-terminal extends out of the NTP entry channel, but then loops back to place Tyr3 and the Tyr3-pU residue just above the active site. However, the VPg is not positioned to be a substrate for the standard 3Dpol catalytic mechanism; the Tyr3 OH is ≈7 Å away from the normal RNA 3′ OH location and bound in a very different orientation, and the UMP in the VPg-UMP structure is not positioned for base pairing per the normal template RNA position (Figure 7C). Poly(A)10 RNA was present in the crystallization mixture and templated the VPg-pU product seen in the structure, but this happened prior to crystal growth and there is no electron density for bound RNA in the final crystal lattice.
Figure 7. Structures of 3Dpol–VPg complexes.
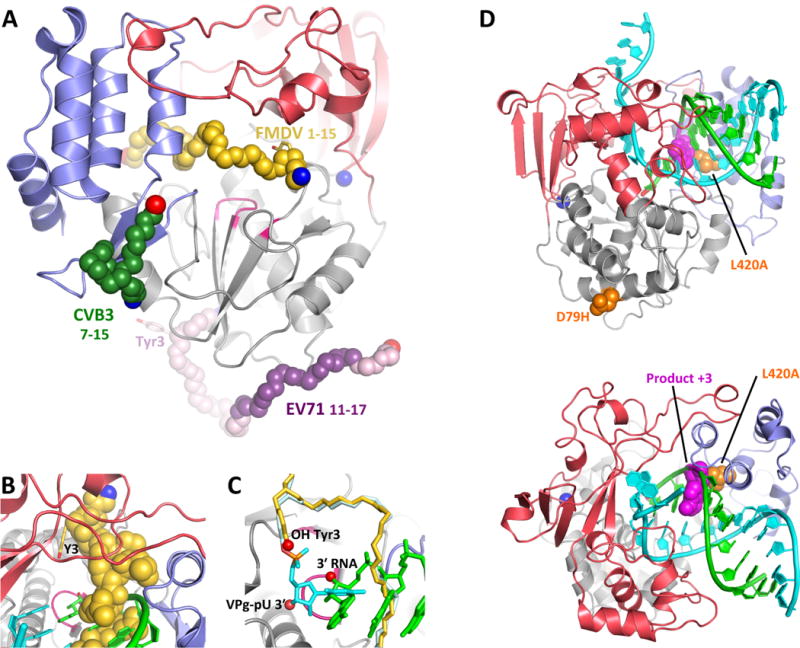
(A) The structure of CV 3Dpol onto which the backbone structures of CV, EV71, and FMDV VPg complexes have been superimposed. The N- and C- ends of the VPg peptides are indicated in blue and red, respectively, and the parts of the EV71 VPg structure in weak electron density are shown in lighter purple. Note that only the FMDV VPg is bound near the active site. (B,C) Detailed views of FMDV VPg (and VPg-pU bounds near the active site, with the product RNA strand from PV elongation complex superimposed for comparison. Note the VPg Tyr3 OH group is bound above and across the active site from the normal RNA 3′ OH group and the uracil base of the VPg-pU complex is perpendicular to the bases of the normal RNA binding orientation. (D) Recombination mutants in PV 3Dpol shown on the wildtype EC structure in two orientations. The D79H mutation is located on the exterior surface of the polymerase far from the RNA binding surface. L420A, on the other hand, makes a direct contact with the product strand RNA at the third nucleotide from the active site (magenta) and biochemical data indicate it reduces recombination by reducing the efficiency of re-initiation on a new template strand.
Provocative data concerning an alternative VPg interaction comes from the EV71 system where both biochemical and structural data implicate an in trans mechanism involving two 3Dpol molecules. Using 3Dpol surface mutagenesis, uridylylation assays, and pull-down experiments, Sun et al. identified an area at the base of the palm domain as a VPg binding site (Sun et al., 2012). Intriguingly, uridylylation did not require a functional active site on the 3Dpol binding to VPg, but instead required a second polymerase that must have a functional active site. This suggests a trans mechanism where higher order interactions between 3Dpol oligomers may be used to present the VPg into the active site for the uridylylation reaction. The EV71 3Dpol-VPg complex structure, solved by co-crystallization, showed residues ≈11–17 of VPg interacting with the bottom of the palm [4IKA] (Chen et al., 2013). This short stretch of VPg had electron density comparable to that of nearby polymerase residues, but an interaction with Tyr3 is not clearly defined because residues 1–10 showed very weak density and were modeled into a crystal solvent channel far away from any active site.
6. Polymerase and Recombination
Recombination is common in the picornaviruses, where it plays major roles in speciation and the development of viral subgroups, in part by allowing for the separate evolution of the capsid-forming structural proteins and the biochemical activities of the non-structural proteins (Lowry et al., 2014; McIntyre et al., 2013). Recombination can also be important for counteracting the effects of excessive deleterious mutations by providing a method for combining multiple beneficial mutations into a single genome. The dominant pathway is one in which abortive elongation results in partial genomes that can anneal to and re-initiate elongation on a new template strand, producing a full-length genome made from two different templates. The re-initiation of RNA synthesis happens via a RNA-RNA duplex that requires some amount of sequence homology/complementarity in order to properly fit into the 3Dpol active site, and the event is thus distinct from the VPg-based initiation mechanism used for native genome replication. The specific structural and molecular mechanisms by which recombination occurs in the context of a membrane bound viral replication center structure are not well understood, but two recently identified 3Dpol variants provide some insights into the process.
The first of these variants was obtained by isolating virus variants that retained a green fluorescent protein gene inserted into the viral genome, potentially because they were less efficient at removing it by recombination (Xiao et al., 2016). This led to the identification of an Asp79 to histidine (D79H) mutation in 3Dpol that was subsequently shown to have a ≈10-fold lower recombination rate without affecting the fitness of the virus. The D79H mutation is located on the 3Dpol surface at the bottom of the palm domain and it is far away from the RNA in the elongation complex (Figure 7D). From a structural viewpoint it is unlikely that the recombination effect is due to changes in direct 3Dpol–RNA contacts, and the location the middle of a long well ordered α-helix is not suggestive of effects on protein conformational dynamics. The mechanism of action for this recombination mutation is thus difficult to envision within the context of a single 3Dpol-RNA complex, and it is quite possible that the Asp79 residue plays a role in the higher order organization of the membrane associated viral replication centers.
The second recombination variant is a Leu420 to alanine mutation that was identified in directed assays looking for recombination effects arising from mutation of the 3Dpol-RNA interface in the elongation complex (Kempf et al., 2016). Leu420 is located on a thumb domain α-helix in the product RNA exit channel, and it interacts directly with the ribose group of the third nucleotide out from the active site on the product RNA strand (Figure 7D). Depending on cell type, recombination rates were reduced 16 to 60-fold in viruses with the L420A mutant polymerase. In vitro biochemical assays showed the L420A polymerase to have a wildtype elongation rate, but a reduced initiation rate and reduced elongation complex stability. These observations are indicative of altered interactions between 3Dpol and the RNA, which was to be expected given the direct contact of Leu420 with the product RNA strand. Interestingly, an analogous mutation of the adjacent Leu419 residue that makes a similar ribose interaction with the template RNA strand did not affect recombination, showing that the effect was strand specific. During recombination, the partial product strand becomes the primer for the re-initiation reaction, and the conclusion from this study was that the L420A mutation slightly alters the binding geometry of the primer strand. This subtly shifts the position and orientation of the 3′ hydroxyl group in the active site, which reduces re-initiation efficiency and recombination rates.
7. Fidelity Control in Picornaviral Polymerases
RNA viruses are generally known for having low fidelity polymerases that give rise to a heterogeneous genome population referred to as a quasispecies (Andino and Domingo, 2015). The breadth of this quasispecies population in terms of sequence space and genome diversity is linked to virus fitness by complex interaction networks that can differ greatly among even seemingly closely related viruses (Lauring and Andino, 2010; Lauring et al., 2013). Some viruses exist in a tightly confined sequence space centered on optimal fitness where few variants are seen in the population because most mutations are seriously compromised. Conversely, other viruses exist as a much broader distribution of genetic variants that all have comparable fitness and therefore persist in the population. These two populations will respond quite differently to changes in RdRP fidelity, with the highly optimized population rapidly losing fitness if RdRP fidelity is decreased, but the broader population can survive because it is inherently more tolerant of an increased mutation rate. In addition, there are effects from recombination that create hybrid viral genomes as a result of abortive replication events followed by a resumption of RNA elongation on a different template genome. Two recent papers have shown that such events can counter effects from low fidelity polymerases by providing a method of purging the genome of deleterious mutations (Kempf et al., 2016; Xiao et al., 2016).
The active site closure step elucidated from the PV elongation complex structure plays a key role in controlling polymerase function and can be used to modulate both replication rate and replication fidelity. Biochemical data show the conformational change step after NTP binding but before catalysis is a major fidelity checkpoint (Arnold et al., 2005). The closed active site structures show an extended network of hydrogen bonds emanating from the 2′ hydroxyl group of the base paired NTP, connecting through the motif B loop in the fingers domain, and ending at the top of motif A (Gong et al., 2013; Gong and Peersen, 2010; Shu and Gong, 2016). This network provides a direct link between the properly positioned NTP and a set of structural interactions that promote catalysis by stabilizing the closed active site.
Fidelity studies thus far have largely focused on three different areas of the polymerase that are all closely tied to these active site motions (Figure 8A). These are described in further detail below, and their origins reflect the different approaches being taken to discover or design polymerases with altered fidelity. First, resistance to a mutagenic antiviral drug led to the discovery of the high-fidelity PV G64S mutation. Second, a series of low-fidelity motif A mutants in CV were designed to alter fidelity based on perturbing specific interactions observed in the crystal structures of the various catalytic cycle states. Third, a recent focus on motif D has grown out of NMR based dynamics studies that showed protein motions in this region of the structure were very sensitive to nucleotide binding and active site closure. Notably, all three areas are near but not directly in the active site, stressing the importance of conformational dynamics for proper NTP selection and catalysis.
Figure 8. Fidelity modulation sites in 3Dpol.
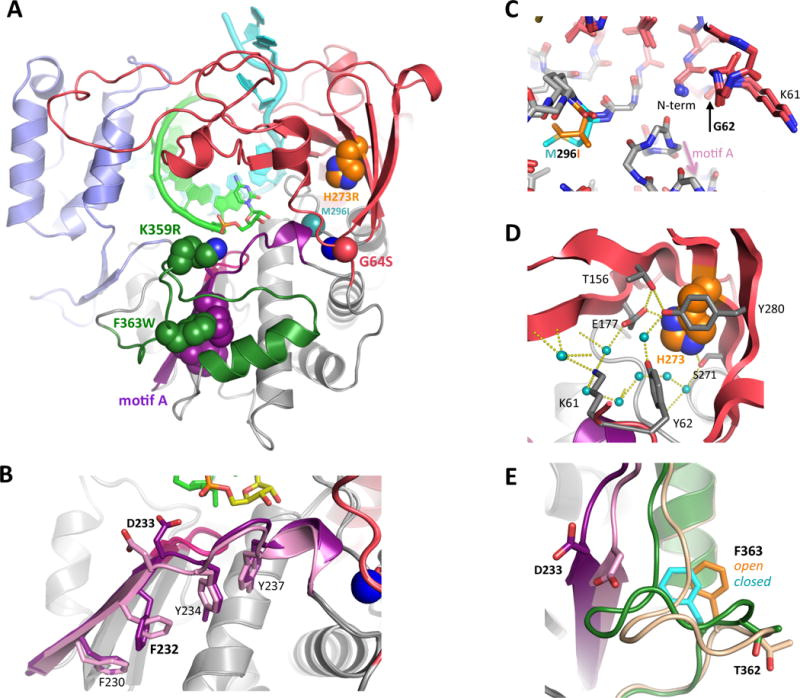
Work from multiple groups over the past decade has identified several 3Dpol regions where mutations can affect replication fidelity and virus growth. (A) Structure of the poliovirus elongation complex showing the three major regions for fidelity mutants; 1: G64S, M296I, and H273R near the N-terminus, 2: motif A in dark purple, and 3: motif D with K359 and F363 in dark green. (B) Structural changes on the underside of motif A as it changes form the open (light) to the closed (dark) conformation during active site closure. Note the rotamer switch of Phe232 that allows the backbone to move significantly with minimal movement of the side chain itself. (C) Comparison of the N-terminus binding pocket structures of wildtype and M296I FMDV 3Dpol. The M296I variant is a higher fidelity polymerase, much like G64S in PV, and it has similar effects on the structure of the N-terminus even though the mutation is in the protein interior. (D) The structure and dynamics of the extensive hydration and hydrogen bonding network within the fingers domain involving His273 are likely affected by the low fidelity PV H273R mutation. (E) The sliding movement of Phe363 atop the motif D helix during active site closure, where the motif D loop is pulled in toward the active site along with motif A (purple). Mutating Phe363 to tryptophan increased 3Dpol fidelity. The motif D loop also contains T362 that is mutated to an isoleucine in the Sabin type 1 oral poliovirus strain, yielding a lower fidelity polymerase.
7.1 N-terminus Binding Pocket
The interest in a molecular understanding of picornavirus RdRP fidelity largely began with the identification of a high-fidelity variant of PV 3Dpol in the form of a G64S point mutation (Pfeiffer and Kirkegaard, 2003; Vignuzzi et al., 2006). This poliovirus mutation was initially isolated by its resistance to ribavirin, an antiviral nucleoside analog that can basepair with either cytosine or uracil to cause transition mutations. Virus growth experiments quickly indicated G64S 3Dpol was a high fidelity polymerase based on reduced sensitivity to other nucleoside analogs and the reduced emergence of resistance to guanidine hydrochloride, an inhibitor of the viral 2CATPase protein. A more extensive biochemical analysis showed the G64S mutation slowed the pre-catalysis conformational change step, effectively changing the equilibrium constant for a nucleotide being bound into the correct geometry for chemistry to take place (Arnold et al., 2005). This is a rate limiting step in the 3Dpol catalytic cycle and as a result the overall 3Dpol elongation rate is reduced by 2- to 4-fold based on in vitro assays (Arnold et al., 2005; Campagnola et al., 2015). The reduced elongation rate is sufficient to support poliovirus growth in tissue culture with titers comparable to the wildtype virus, showing RNA replication is not a limiting factor for poliovirus production. However, the rate defect does become an issue upon co-infection with wildtype virus, at which point the faster replicating wildtype rapidly becomes dominant in the population even if initially present at less than one tenth of the inoculum (Pfeiffer and Kirkegaard, 2005). For this reason, the G64S mutation also shows rapid reversion to wildtype in the absence of selective pressure with ribavirin.
Structurally, glycine 64 plays an important role in stabilizing the buried N-terminus in its pocket, where it forms two of the four hydrogen bonds anchoring Gly1. The result is a backbone conformation for Gly64 that would be considered a strained Ramachandran plot outlier for a non-glycine amino acid. Consistent with this, the 3Dpol G64S structure (Figure 3D) shows a slight opening of the N-terminus binding pocket [2IJF] (Marcotte et al., 2007). This in turn changes the protein dynamics in not only the adjacent active site, but also throughout the fingers domain based on NMR studies (Yang et al., 2010). These dynamics changes likely make 3Dpol subtly more reliant on the stabilizing effects from ideal NTP base pairing in the active site, thus increasing replication fidelity.
Although Ser64 forms a weak hydrogen bond with Glu2 in the G64S PV 3Dpol structure, there is not a strong structural requirement for a serine at position 64 (Marcotte et al., 2007). Consistent with this, PV with Gly64 changed to alanine, threonine, valine, and leucine are also viable, but they have reduced growth rates and only alanine and threonine show similar ribavirin resistance to G64S (Vignuzzi et al., 2008). Similar testing of all 19 other amino acids in coxsackievirus 3Dpol showed that only alanine, serine, and glutamine were viable and genetically stable in that virus. The growth of these CV variants was too compromised to measure fidelity by next-generation sequencing analysis (Gnadig et al., 2012), but in vitro 3Dpol biochemistry indicates CV G64S does have slightly higher fidelity than the wildtype enzyme (Campagnola et al., 2015). Gly64 mutations in EV71 3Dpol have shown that arginine and threonine confer ribavirin resistance while asparagine does not, and substitutions to alanine, leucine, and proline were nonviable or not genetically stable (Sadeghipour et al., 2013).
In FMDV 3Dpol the structurally equivalent G62S mutation has similar effects on the N-terminus pocket structure, but the mutation does not confer increased ribavirin resistance in a virus context. Instead, FMDV growth on ribavirin selected for an M296I mutation located nearby at the end of motif B (Sierra et al., 2007). This is interesting because like FMDV G62S and PV G64S, this M296I mutation directly impacts the structure and interactions surrounding the buried N-terminus, and also increases 3Dpol fidelity (Figure 8C) (Arias et al., 2008; Ferrer-Orta et al., 2010). Furthermore, passaging M296I FMDV in the presence of ribavirin results in two additional mutations, P44S and P169S, both located at the top of the NTP entry channel (Agudo et al., 2010). These mutations are structurally very similar to the P48K, S164P, and K170E mutations observed in CVB3 3Dpol when low-fidelity motif A variants in that virus were passaged (Gnadig et al., 2012). Thus, there appear to be fidelity effects linking the N-terminus binding pocket to the roof of the NTP entry tunnel via a mechanism that is not yet understood.
One major lesson from the extensive Gly64 studies in multiple viruses is that the effect of mutations is highly context dependent in ways that are difficult to predict. Even though the primary effect of any mutation is likely through alterations in the backbone geometry that affect N-terminus binding, there are secondary effects on protein dynamics associated with NTP repositioning and active site closure. The dynamics changes make for a complicated set of intra- and intermolecular interactions. For example, PV H273R [4R0E] is a low fidelity mutation located in the interior of the fingers domain where the mutation likely disrupts the dynamics associated with an extensive network of hydrogen bonded buried water molecules (Figure 8D) (Moustafa et al., 2014). Efforts to model such effects using molecular dynamics (MD) calculations have identified sets of correlated motions and interactions within PV 3Dpol (Moustafa et al., 2011), but it is not clear if these effects can be directly translated into other viral RdRPs. The alternative of carrying out MD calculations of other polymerases is possible, but does represent a significant effort in terms of needed computational time, especially if multiple mutations will be analyzed.
7.2 Low and High Fidelity Variants via Motif A Mutations
The structural changes which take place during the catalytic cycle have been used to intentionally alter 3Dpol fidelity with mutations targeting interactions that change during the closure step (Figure 5A). This led to the identification of about a dozen CV variants whose fidelity was decreased as much as 3-fold based on sequencing of progeny virus genomes, and the strongest of these were mutations of Phe230 and Phe232 located within the motif A β-strand (Figure 8B) (Campagnola et al., 2015; Gnadig et al., 2012). Several of these low-fidelity variants were used to infect mice and examine tissue tropism, revealing that low fidelity variants are also attenuated for growth and the extent of attenuation increased with decreased fidelity.
In vitro biochemical assays of CV 3Dpol activity showed replication fidelity was correlated with elongation rates, which could be almost doubled from 22 to 38 nt/s, with faster polymerases being lower fidelity polymerases (pH 7, 75 mM NaCl, 37°C) (Campagnola et al., 2015). The fidelity effects also correlated with 3Dpol sensitivity to having a 2′ hydroxyl group on the NTP substrate, as determined by how well the enzyme can discriminate between CTP and 2′-deoxy-CTP. Wildtype CV 3Dpol has a 360-fold preference for CTP over dCTP, and this was reduced to ≈175-fold for the lowest fidelity motif A mutants. The 2′ OH sensitivity provides an in vitro biochemical assay for fidelity as an alternative to virus based studies, and importantly it allows for the study of drastically altered polymerases whose fidelity or rate are too low or too high to support virus growth. For example, in vitro assays showed the slow growing G64S is in fact a slight high-fidelity variant in CV 3Dpol and the nonviable F232L single and I230F+F232Y double mutants were both low fidelity variants. Extrapolating from the pool of viable viruses, these two mutants were estimated to generate 14–16 mutations per 10kb synthesized, which is likely too high to maintain genomic stability and support virus growth (Gnadig et al., 2012).
A second major finding from these studies was that motif A mutations can have either low or high fidelity phenotypes depending on virus background. Parallel biochemical experiments with PV and CV polymerases under identical conditions showed that these highly structurally homologous enzymes from closely related viruses differ greatly in their biochemical activities: CV 3Dpol is slow and has higher fidelity while PV 3Dpol is very fast and has lower fidelity (Campagnola et al., 2015). PV 3Dpol has maximal processive elongation rate of 88 nt/s, about 4-fold faster than the CV enzyme, and a CTP-vs-2′dCTP discrimination factor of only ≈120, one third that observed for CV 3Dpol. In terms of quasispecies populations, these data indicate that PV polymerase produces a broad population of divergent variants with rapid replication rates that are probably near their maximum. CV, on the other hand, has a more restricted quasispecies population due to higher fidelity polymerase and it may have close to the minimum elongation rate needed to support virus replication based on the slow growth of its G64S virus.
7.3 Motif D and NTP Delivery
The last region of 3Dpol that has been the focus of fidelity studies is motif D, a helix-loop structure that forms the outside bottom rim of the NTP entry channel (Figure 8A & 8E). The interest in motif D stemmed from lysine 359 and its possible role as a proton donor during catalysis (Castro et al., 2007). Poliovirus 3Dpol Lys359 was initially misidentified as the homolog of a highly conserved “O-helix” lysine in the fingers domain of DNA dependent polymerases, where it forms a salt bridge with the NTP triphosphate and acts as a proton donor during catalysis (Castro et al., 2009). However, the structures of viral RdRPs and reverse transcriptases indicate that for RNA-templated polymerases the more appropriate structural homolog of the “O-helix” lysine is an arginine in in the ring finger, e.g. Arg174 of PV 3Dpol (Gong and Peersen, 2010). This highly conserved arginine from conserved motif F interacts directly with the NTP phosphates in the PV EC structure and indirectly via two water molecules in EV71 EC structures (Figure 5C). Lys359, on the other hand, is located in motif D at the entrance to the NTP entry tunnel, placing it near the NTP γ phosphate but too far (4–5 Å) away for a direct contact based on the structures solved thus far. However, despite the different interaction geometry in the active site, biochemical data does support Lys359 being a functional homolog of the O-helix lysine; mutating Lys359 to leucine reduces PV 3Dpol catalytic rate 50-fold with only a 3-fold effect on the ATP Kd, and replacement with arginine (K359R) shifts the rate optimum to higher pH, leading to the conclusion that Lys359 plays a role in donating a proton to the pyrophosphate leaving group (Castro et al., 2009). In CV 3Dpol, an analogous K360R mutation similarly yields a slow and high fidelity enzyme based on biochemical data, but that polymerase does not support CV growth, presumably because it is too slow (Campagnola et al., 2015; Gnadig et al., 2012). Last, an alternative explanation for the importance of Lys359 has been put forth based on molecular dynamics calculations that suggest it plays a key role in delivering NTPs into the active site and transporting pyrophosphate out of the active site (Shen et al., 2012). In this model, Lys359 acts as a sensor that helps trigger active site closure and catalysis when the cognate NTP becomes fully bound at the catalytic center.
In addition to slowing the polymerase, the K359R mutation was found to increase fidelity by affecting rate limiting conformational change steps that occur both before and after chemistry (Yang et al., 2012). This study used 1H and 13C NMR with [methyl-13C] methionine to provide sparse labels located throughout the polymerase, an approach that can be used to directly measure protein dynamics in solution. 3Dpol is too large for full-blown NMR structure determination, but sparse labeling schemes have proven to be very effective, especially when applied to 3Dpol-RNA and 3Dpol-RNA-NTP complexes (Boehr et al., 2014; Yang et al., 2010). The NMR signal from Met355, located in motif D and only four residues away from Lys359, is sensitive to both NTP binding and the open versus closed state of the active site, providing observable differences between correct and incorrect nucleotides in the active site. Combined with a chain terminating deoxy ribose base on the 3′ end of the priming RNA, NMR experiments detect how protein dynamics within the 3Dpol structure are altered upon NTP binding and active site cycling, providing solution-based data to complement the EC crystal structures. Such NMR experiments have also been used to investigate a nearby Thr362 mutation to isoleucine, a variant in the motif D loop that is found in the Sabin 1 oral poliovirus vaccine strain (Liu et al., 2013). Although Thr362 is largely solvent exposed on the exterior face of the motif D loop and has minimal contact with other residues (Figure 8E), the mutation to isoleucine triggered long-range changes in protein dynamics that made the active site closure step more efficient. This is turn led to an increased tendency to misincorporate nucleotides, resulting in a mutator phenotype poliovirus with increased sensitivity to ribavirin.
We recently used structure based modeling to design a high-fidelity motif D variant of CV 3Dpol by mutating the highly conserved Phe364 to a tryptophan (i.e. residue 363 in PV) (McDonald et al., 2016). The idea came from a comparison of the open and closed active structures that revealed Phe364 undergoing a sliding movement atop the motif D helix and underneath a conserved proline in an interaction that likely helps the active site “lock” into the closed state for catalysis (Figure 8E). Molecular modeling showed that a slightly large tryptophan could nominally cover both locations of Phe364 with a smaller sliding movement, and subsequently alter replication fidelity by changing the protein dynamics associated with active site closure. This did prove to be the case, and the F364W mutation increased CTP-vs-dCTP discrimination factor in vitro, decreased mutation rates 2-fold in vivo based on sequencing progeny from infectious virus studies, and attenuated pathogenesis and tissue specific titers in mice. None of the experiments so far have resulted in compensatory mutations or reversion, likely due to several key features of this specific mutation design; 1) Trp364 is on the 3Dpol surface and has limited interactions with other 3Dpol residues, 2) the Pro357 interaction partner is unlikely to change because of its unique structure and backbone conformation, and 3) the tryptophan is encoded with a TGG codon where the more common transition mutations will results in stop codons or small non-planar amino acids that do not support virus growth (McDonald et al., 2016).
8. Protein Engineering for Live-attenuated Vaccine Development
The increased understanding of the RdRP catalytic cycle opens up the possibility of using protein engineering to generate live-attenuated vaccine strains by altering polymerase fidelity so as to limit virus growth and pathogenesis while maintaining immunogenicity (Lauring et al., 2010; Vignuzzi et al., 2008; Weeks et al., 2012). Quasispecies theory predicts that either increasing or decreasing replication fidelity should reduce fitness, and both effects have now been observed with multiple fidelity variant picornaviral polymerases, as discussed above. Extending this to infectious virus studies in animal models, poliovirus with the high-fidelity G64S mutation is strongly attenuated and has reduced pathogenicity in mice despite the mutation having only slight effects on growth in tissue culture. The conclusion from this work was that the high-fidelity virus has reduced genetic diversity and is therefore less able to adapt to the different tissue types it encounters in an animal host (Pfeiffer and Kirkegaard, 2005; Pfeiffer and Kirkegaard, 2006; Vignuzzi et al., 2006). In effect, it is unable to regenerate the quasispecies distribution “on the fly” as the virus replicates, limiting its ability to efficiently spread inside a host organism. At the other end of the spectrum, the protein engineering studies which led to the low fidelity CV variants showed that reducing fidelity can also attenuate virus growth and limit tissue tropism during infection of mice (Gnadig et al., 2012). The conclusion there was that the small viral populations present after inter-organ bottlenecking could not tolerate the high mutation frequency, leading to extinction. Similar results have also been obtained with PV polymerase, where the H273R mutation in the fingers domain reduces virulence and symptoms in mice, but notably the mutant virus provided enough of an infection to elicit a protective immune response (Korboukh et al., 2014).
These studies set the stage for using protein design methods to build variant polymerases that alter replication fidelity, and picornaviruses provide a good platform for such studies for multiple reasons. They grow rapidly, are amenable to both reverse genetics and animal pathogenesis studies, their RdRPs can be purified for in vitro biochemistry studies, and many high-resolution 3Dpol structures have now been solved. The issue of reversion can be a major concern, especially for low-fidelity variants. Picornaviruses replicate very rapidly and to very high titers, and in the process they explore an amazingly broad swath of sequence space. It is altogether not surprising that attenuated strains obtained by adaptation pathways also show reversal of adaption upon reintroduction into their native host. The power of protein engineering approaches is to minimize reversion potential by introducing mutations that alter function as desired, but are also drastic enough to be minimally prone to reversion.
One could introduce multiple mutations at locations that are far apart in the primary sequence but pack together into functional units in the folded protein structure. The palm domain movements during 3Dpol active site closure is a good example of a location where a mutation in motif A could be combined with changes in the adjacent motifs C and D to redesign a cluster of closely packed residues essential for polymerase function. Alternatively, such clusters could be transplanted between related viral polymerases, i.e. replace a group of four interacting residues in PV 3Dpol with their structural homologs in CV. Restoration of full wildtype activity would then be less likely because it requires multiple mutation events. This would be further minimized by codon usage that takes the effects of the more likely transition mutations (A ←→ G and C ←→ T) into account. For example, encoding a structurally desired leucine with CTG means that a proline (CCN) is the most likely mutation to occur, and this residue will likely debilitate polymerase function because it strongly distorts the peptide backbone geometry. Introducing tryptophan residues in place of tyrosine or phenylalanine, the other two planar amino acids, can also be very effective approach because this mutation is relatively unlikely to revert. Tryptophan has only one codon, TGG, from which single base changes result in stop codons (TGA, TAG), or arginine, cysteine, serine, leucine, and glycine. These alternative amino acids all differ drastically from tryptophan in terms of size and biochemical characteristic and are likely to affect polymerase folding, dynamics, and function. In effect, a tryptophan can act as a molecular poison pill to curtail the replication of pseudo-revertant viruses. This likely explains the genetic stability of F364W in CV 3Dpol (McDonald et al., 2016); the native phenylalanine is well conserved in the picornaviral 3Dpol enzymes and there is no precedent for a tryptophan, but that may in fact be because it is difficult to mutate to a Trp due the very same codon restrictions that make it difficult to mutate away from a Trp.
While the idea of using protein design principles to engineer stable fidelity variant polymerases is attractive, it is also important to recognize there are caveats in the approach. The finding that the fidelity variants, such as CV 3Dpol F364W, can be genetically stable without major reversion issues is very encouraging, but the tight interplay of RdRP error rates, recombination, quasispecies distributions, and fitness means that this may not hold for all viruses.
9. Conclusions
The past decade has been fruitful for the viral RNA-dependent RNA polymerase structure field and we have now solved the structures of representative enzymes from the major virus families. The small picornaviral 3Dpols are well represented in this group, and they are providing tremendous insights into the molecular mechanisms and structures responsible for nucleotide selection, catalysis, and translocation. There is every indication that these fundamental principles are used by nearly all positive-strand RNA virus polymerases even though they have larger replicase proteins that impart additional functionalities. Our increased understanding of polymerase structure, function, and fidelity determinants is enabling directed reverse genetics studies to address the importance of population diversity for virus growth, and probing ways in which protein engineering can be used to stably attenuate viruses for possible vaccine development.
Highlight.
Picornavirus 3Dpol structure and function review
Mechanistic origins of low fidelity and quasispecies distributions
General principles for all (+) strand RNA virus polymerases
Acknowledgments
I would like to thank the people who have passed through my research laboratory over the past fifteen years for their devotion to the bench, the graphics workstation, and the scientific discussion that have led to our current understanding of RdRP structure and function. Amongst this group, Peng Gong, Grace Campagnola, Aaron Thompson, Aaron Sholders, Seth McDonald, and Sarah Hobdey deserve special recognition. I am also very grateful for excellent collaborations with David Barton and Marco Vignuzzi that have allowed us to better explore how altering 3Dpol biochemistry affects virus replication in both tissue culture and animal systems. This writing of this review was supported by NIH award R21-AI124123 to OBP.
Abbreviations
- 3Dpol
picornavirus RNA-dependent RNA polymerase
- CTP
cytosine triphosphate
- CV
coxsackievirus B3
- EC
polymerase-RNA elongation complex
- EMCV
encephalomyocarditis virus
- EV71
enterovirus 71
- FMDV
foot-and-mouth disease virus
- HCV
hepatitis C virus
- HRV
human rhinovirus
- IRES
internal ribosome entry site
- NMR
nuclear magnetic resonance
- NTP
nucleotide triphosphate
- PDB
protein databank
- PV
poliovirus
- RdRP
RNA-dependent RNA polymerase
- UMP
uracil monophosphate
- UTR
untranslated region
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Agudo R, Ferrer-Orta C, Arias A, de la Higuera I, Perales C, Perez-Luque R, Verdaguer N, Domingo E. A multi-step process of viral adaptation to a mutagenic nucleoside analogue by modulation of transition types leads to extinction-escape. PLoS pathogens. 2010;6:e1001072. doi: 10.1371/journal.ppat.1001072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andino R, Domingo E. Viral quasispecies. Virology. 2015:479–480. doi: 10.1016/j.virol.2015.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arias A, Arnold JJ, Sierra M, Smidansky ED, Domingo E, Cameron CE. Determinants of RNA-dependent RNA polymerase (in)fidelity revealed by kinetic analysis of the polymerase encoded by a foot-and-mouth disease virus mutant with reduced sensitivity to ribavirin. J Virol. 2008;82:12346–12355. doi: 10.1128/JVI.01297-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnold JJ, Bernal A, Uche U, Sterner DE, Butt TR, Cameron CE, Mattern MR. Small ubiquitin-like modifying protein isopeptidase assay based on poliovirus RNA polymerase activity. Analytical biochemistry. 2006;350:214–221. doi: 10.1016/j.ab.2005.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnold JJ, Cameron CE. Poliovirus RNA-dependent RNA polymerase (3D(pol)). Assembly of stable, elongation-competent complexes by using a symmetrical primer-template substrate (sym/sub) J Biol Chem. 2000;275:5329–5336. doi: 10.1074/jbc.275.8.5329. [DOI] [PubMed] [Google Scholar]
- Arnold JJ, Cameron CE. Poliovirus RNA-dependent RNA polymerase (3Dpol): pre-steady-state kinetic analysis of ribonucleotide incorporation in the presence of Mg2+ Biochemistry. 2004;43:5126–5137. doi: 10.1021/bi035212y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnold JJ, Vignuzzi M, Stone JK, Andino R, Cameron CE. Remote site control of an active site fidelity checkpoint in a viral RNA-dependent RNA polymerase. J Biol Chem. 2005;280:25706–25716. doi: 10.1074/jbc.M503444200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boehr DD, Liu X, Yang X. Targeting structural dynamics of the RNA-dependent RNA polymerase for anti-viral strategies. Current opinion in virology. 2014;9:194–200. doi: 10.1016/j.coviro.2014.08.006. [DOI] [PubMed] [Google Scholar]
- Bressanelli S, Tomei L, Roussel A, Incitti I, Vitale RL, Mathieu M, De Francesco R, Rey FA. Crystal structure of the RNA-dependent RNA polymerase of hepatitis C virus. Proc Natl Acad Sci U S A. 1999;96:13034–13039. doi: 10.1073/pnas.96.23.13034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burrill CP, Westesson O, Schulte MB, Strings VR, Segal M, Andino R. Global RNA structure analysis of poliovirus identifies a conserved RNA structure involved in viral replication and infectivity. J Virol. 2013;87:11670–11683. doi: 10.1128/JVI.01560-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campagnola G, McDonald S, Beaucourt S, Vignuzzi M, Peersen OB. Structure-function relationships underlying the replication fidelity of viral RNA-dependent RNA polymerases. J Virol. 2015;89:275–286. doi: 10.1128/JVI.01574-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campagnola G, Weygandt M, Scoggin K, Peersen O. Crystal structure of coxsackievirus B3 3Dpol highlights the functional importance of residue 5 in picornavirus polymerases. J Virol. 2008;82:9458–9464. doi: 10.1128/JVI.00647-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castro C, Smidansky E, Maksimchuk KR, Arnold JJ, Korneeva VS, Gotte M, Konigsberg W, Cameron CE. Two proton transfers in the transition state for nucleotidyl transfer catalyzed by RNA- and DNA-dependent RNA and DNA polymerases. Proc Natl Acad Sci U S A. 2007;104:4267–4272. doi: 10.1073/pnas.0608952104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castro C, Smidansky ED, Arnold JJ, Maksimchuk KR, Moustafa I, Uchida A, Gotte M, Konigsberg W, Cameron CE. Nucleic acid polymerases use a general acid for nucleotidyl transfer. Nature structural & molecular biology. 2009;16:212–218. doi: 10.1038/nsmb.1540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerny J, Cerna Bolfikova B, Valdes JJ, Grubhoffer L, Ruzek D. Evolution of tertiary structure of viral RNA dependent polymerases. PLoS One. 2014;9:e96070. doi: 10.1371/journal.pone.0096070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C, Wang Y, Shan C, Sun Y, Xu P, Zhou H, Yang C, Shi PY, Rao Z, Zhang B, Lou Z. Crystal structure of enterovirus 71 RNA-dependent RNA polymerase complexed with its protein primer VPg: implication for a trans mechanism of VPg uridylylation. J Virol. 2013;87:5755–5768. doi: 10.1128/JVI.02733-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho MW, Richards OC, Dmitrieva TM, Agol V, Ehrenfeld E. RNA duplex unwinding activity of poliovirus RNA-dependent RNA polymerase 3Dpol. J Virol. 1993;67:3010–3018. doi: 10.1128/jvi.67.6.3010-3018.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrer-Orta C, Arias A, Agudo R, Perez-Luque R, Escarmis C, Domingo E, Verdaguer N. The structure of a protein primer-polymerase complex in the initiation of genome replication. Embo J. 2006;25:880–888. doi: 10.1038/sj.emboj.7600971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrer-Orta C, Arias A, Perez-Luque R, Escarmis C, Domingo E, Verdaguer N. Structure of foot-and-mouth disease virus RNA-dependent RNA polymerase and its complex with a template-primer RNA. J Biol Chem. 2004;279:47212–47221. doi: 10.1074/jbc.M405465200. [DOI] [PubMed] [Google Scholar]
- Ferrer-Orta C, Arias A, Perez-Luque R, Escarmis C, Domingo E, Verdaguer N. Sequential structures provide insights into the fidelity of RNA replication. Proc Natl Acad Sci U S A. 2007;104:9463–9468. doi: 10.1073/pnas.0700518104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrer-Orta C, de la Higuera I, Caridi F, Sanchez-Aparicio MT, Moreno E, Perales C, Singh K, Sarafianos SG, Sobrino F, Domingo E, Verdaguer N. Multifunctionality of a picornavirus polymerase domain: nuclear localization signal and nucleotide recognition. J Virol. 2015;89:6848–6859. doi: 10.1128/JVI.03283-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrer-Orta C, Sierra M, Agudo R, de la Higuera I, Arias A, Perez-Luque R, Escarmis C, Domingo E, Verdaguer N. Structure of foot-and-mouth disease virus mutant polymerases with reduced sensitivity to ribavirin. J Virol. 2010;84:6188–6199. doi: 10.1128/JVI.02420-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzgerald KD, Semler BL. Bridging IRES elements in mRNAs to the eukaryotic translation apparatus. Biochim Biophys Acta. 2009;1789:518–528. doi: 10.1016/j.bbagrm.2009.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gnadig NF, Beaucourt S, Campagnola G, Borderia AV, Sanz-Ramos M, Gong P, Blanc H, Peersen OB, Vignuzzi M. Coxsackievirus B3 mutator strains are attenuated in vivo. Proc Natl Acad Sci U S A. 2012;109:E2294–2303. doi: 10.1073/pnas.1204022109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gohara DW, Crotty S, Arnold JJ, Yoder JD, Andino R, Cameron CE. Poliovirus RNA-dependent RNA polymerase (3Dpol): structural, biochemical, and biological analysis of conserved structural motifs A and B. J Biol Chem. 2000;275:25523–25532. doi: 10.1074/jbc.M002671200. [DOI] [PubMed] [Google Scholar]
- Gohara DW, Ha CS, Kumar S, Ghosh B, Arnold JJ, Wisniewski TJ, Cameron CE. Production of “authentic” poliovirus RNA-dependent RNA polymerase (3D(pol)) by ubiquitin-protease-mediated cleavage in Escherichia coli. Protein Expr Purif. 1999;17:128–138. doi: 10.1006/prep.1999.1100. [DOI] [PubMed] [Google Scholar]
- Gong P. A structural view of the RNA-dependent RNA polymerases from the Flavivirus genus. Virus Research IN THE SAME ISSUE OF VIRUS RESEARCH. 2017 doi: 10.1016/j.virusres.2017.01.020. [DOI] [PubMed] [Google Scholar]
- Gong P, Kortus MG, Nix JC, Davis RE, Peersen OB. Structures of coxsackievirus, rhinovirus, and poliovirus polymerase elongation complexes solved by engineering RNA mediated crystal contacts. PLoS One. 2013;8:e60272. doi: 10.1371/journal.pone.0060272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong P, Peersen OB. Structural basis for active site closure by the poliovirus RNA-dependent RNA polymerase. Proc Natl Acad Sci U S A. 2010;107:22505–22510. doi: 10.1073/pnas.1007626107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorbalenya A, Lauber C. Origin and Evolution of the Picornaviridae Proteome. In: Ehrenfeld E, Domingo A, Roos RP, editors. The Picornaviruses. ASM Press; Washington, DC: 2010. pp. 253–270. [Google Scholar]
- Gruez A, Selisko B, Roberts M, Bricogne G, Bussetta C, Jabafi I, Coutard B, De Palma AM, Neyts J, Canard B. The crystal structure of coxsackievirus B3 RNA-dependent RNA polymerase in complex with its protein primer VPg confirms the existence of a second VPg binding site on Picornaviridae polymerases. J Virol. 2008;82:9577–9590. doi: 10.1128/JVI.00631-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen JL, Long AM, Schultz SC. Structure of the RNA-dependent RNA polymerase of poliovirus. Structure. 1997;5:1109–1122. doi: 10.1016/s0969-2126(97)00261-x. [DOI] [PubMed] [Google Scholar]
- Harris KS, Reddigari SR, Nicklin MJ, Hammerle T, Wimmer E. Purification and characterization of poliovirus polypeptide 3CD, a proteinase and a precursor for RNA polymerase. J Virol. 1992;66:7481–7489. doi: 10.1128/jvi.66.12.7481-7489.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hobdey SE, Kempf BJ, Steil BP, Barton DJ, Peersen OB. Poliovirus polymerase residue 5 plays a critical role in elongation complex stability. J Virol. 2010;84:8072–8084. doi: 10.1128/JVI.02147-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hope DA, Diamond SE, Kirkegaard K. Genetic dissection of interaction between poliovirus 3D polymerase and viral protein 3AB. J Virol. 1997;71:9490–9498. doi: 10.1128/jvi.71.12.9490-9498.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin Z, Chuang Y-CC, Kao CC, Deval J. Structure(s), Function(s), and Inhibition of the RNA-Dependent RNA Polymerase of Noroviruses. Virus Res IN THE SAME ISSUE OF VIRUS RESEARCH. 2017 doi: 10.1016/j.virusres.2016.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin Z, Leveque V, Ma H, Johnson KA, Klumpp K. Assembly, purification and pre-steady-state kinetic analysis of an active RNA-dependent RNA polymerase elongation complex. The Journal of biological chemistry. 2012;287:10674–10683. doi: 10.1074/jbc.M111.325530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karr JP, Peersen OB. ATP Is an Allosteric Inhibitor of Coxsackievirus B3 Polymerase. Biochemistry. 2016;55:3995–4002. doi: 10.1021/acs.biochem.6b00467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley LA, Mezulis S, Yates CM, Wass MN, Sternberg MJ. The Phyre2 web portal for protein modeling, prediction and analysis. Nature protocols. 2015;10:845–858. doi: 10.1038/nprot.2015.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kempf BJ, Kelly MM, Springer CL, Peersen OB, Barton DJ. Structural features of a picornavirus polymerase involved in the polyadenylation of viral RNA. J Virol. 2013;87:5629–5644. doi: 10.1128/JVI.02590-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kempf BJ, Peersen OB, Barton DJ. Poliovirus Polymerase Leu420 Facilitates RNA Recombination and Ribavirin Resistance. J Virol. 2016;90:8410–8421. doi: 10.1128/JVI.00078-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korboukh VK, Lee CA, Acevedo A, Vignuzzi M, Xiao Y, Arnold JJ, Hemperly S, Graci JD, August A, Andino R, Cameron CE. RNA virus population diversity, an optimum for maximal fitness and virulence. J Biol Chem. 2014;289:29531–29544. doi: 10.1074/jbc.M114.592303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kortus MG, Kempf BJ, Haworth KG, Barton DJ, Peersen OB. A template RNA entry channel in the fingers domain of the poliovirus polymerase. Journal of molecular biology. 2012;417:263–278. doi: 10.1016/j.jmb.2012.01.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauring AS, Andino R. Quasispecies theory and the behavior of RNA viruses. PLoS pathogens. 2010;6:e1001005. doi: 10.1371/journal.ppat.1001005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauring AS, Frydman J, Andino R. The role of mutational robustness in RNA virus evolution. Nature reviews. Microbiology. 2013;11:327–336. doi: 10.1038/nrmicro3003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauring AS, Jones JO, Andino R. Rationalizing the development of live attenuated virus vaccines. Nature biotechnology. 2010;28:573–579. doi: 10.1038/nbt.1635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesburg CA, Cable MB, Ferrari E, Hong Z, Mannarino AF, Weber PC. Crystal structure of the RNA-dependent RNA polymerase from hepatitis C virus reveals a fully encircled active site. Nat Struct Biol. 1999;6:937–943. doi: 10.1038/13305. [DOI] [PubMed] [Google Scholar]
- Liu X, Yang X, Lee CA, Moustafa IM, Smidansky ED, Lum D, Arnold JJ, Cameron CE, Boehr DD. Vaccine-derived mutation in motif D of poliovirus RNA-dependent RNA polymerase lowers nucleotide incorporation fidelity. J Biol Chem. 2013;288:32753–32765. doi: 10.1074/jbc.M113.484428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love RA, Maegley KA, Yu X, Ferre RA, Lingardo LK, Diehl W, Parge HE, Dragovich PS, Fuhrman SA. The crystal structure of the RNA-dependent RNA polymerase from human rhinovirus: a dual function target for common cold antiviral therapy. Structure. 2004;12:1533–1544. doi: 10.1016/j.str.2004.05.024. [DOI] [PubMed] [Google Scholar]
- Lowry K, Woodman A, Cook J, Evans DJ. Recombination in enteroviruses is a biphasic replicative process involving the generation of greater-than genome length ‘imprecise’ intermediates. PLoS pathogens. 2014;10:e1004191. doi: 10.1371/journal.ppat.1004191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu G, Gong P. Crystal Structure of the Full-Length Japanese Encephalitis Virus NS5 Reveals a Conserved Methyltransferase-Polymerase Interface. PLoS pathogens. 2013;9:e1003549. doi: 10.1371/journal.ppat.1003549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcotte LL, Wass AB, Gohara DW, Pathak HB, Arnold JJ, Filman DJ, Cameron CE, Hogle JM. Crystal structure of poliovirus 3CD protein: virally encoded protease and precursor to the RNA-dependent RNA polymerase. J Virol. 2007;81:3583–3596. doi: 10.1128/JVI.02306-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald S, Block A, Beaucourt S, Moratorio G, Vignuzzi M, Peersen OB. Design of a Genetically Stable High Fidelity Coxsackievirus B3 Polymerase That Attenuates Virus Growth in Vivo. J Biol Chem. 2016;291:13999–14011. doi: 10.1074/jbc.M116.726596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McIntyre CL, Savolainen-Kopra C, Hovi T, Simmonds P. Recombination in the evolution of human rhinovirus genomes. Arch Virol. 2013;158:1497–1515. doi: 10.1007/s00705-013-1634-6. [DOI] [PubMed] [Google Scholar]
- Mestas SP, Sholders AJ, Peersen OB. A fluorescence polarization-based screening assay for nucleic acid polymerase elongation activity. Analytical biochemistry. 2007;365:194–200. doi: 10.1016/j.ab.2007.03.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosley RT, Edwards TE, Murakami E, Lam AM, Grice RL, Du J, Sofia MJ, Furman PA, Otto MJ. Structure of HCV Polymerase in Complex with Primer-Template RNA. Journal of virology. 2012;86:6503–6511. doi: 10.1128/JVI.00386-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moustafa IM, Korboukh VK, Arnold JJ, Smidansky ED, Marcotte LL, Gohara DW, Yang X, Sanchez-Farran MA, Filman D, Maranas JK, Boehr DD, Hogle JM, Colina CM, Cameron CE. Structural dynamics as a contributor to error-prone replication by an RNA-dependent RNA polymerase. J Biol Chem. 2014;289:36229–36248. doi: 10.1074/jbc.M114.616193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moustafa IM, Shen H, Morton B, Colina CM, Cameron CE. Molecular dynamics simulations of viral RNA polymerases link conserved and correlated motions of functional elements to fidelity. Journal of molecular biology. 2011;410:159–181. doi: 10.1016/j.jmb.2011.04.078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng KK, Cherney MM, Vazquez AL, Machin A, Alonso JM, Parra F, James MN. Crystal structures of active and inactive conformations of a caliciviral RNA-dependent RNA polymerase. J Biol Chem. 2002;277:1381–1387. doi: 10.1074/jbc.M109261200. [DOI] [PubMed] [Google Scholar]
- O’Farrell D, Trowbridge R, Rowlands D, Jager J. Substrate complexes of hepatitis C virus RNA polymerase (HC-J4): structural evidence for nucleotide import and de-novo initiation. J Mol Biol. 2003;326:1025–1035. doi: 10.1016/s0022-2836(02)01439-0. [DOI] [PubMed] [Google Scholar]
- Palmenberg AC, Neubauer D, Skern T. Genome Organization and Encoded Proteins. In: Ehrenfeld E, Domingo A, Roos RP, editors. The Picornaviruses. ASM Press; Washington, DC: 2010. pp. 3–17. [Google Scholar]
- Paul AV, Rieder E, Kim DW, van Boom JH, Wimmer E. Identification of an RNA hairpin in poliovirus RNA that serves as the primary template in the in vitro uridylylation of VPg. J Virol. 2000;74:10359–10370. doi: 10.1128/jvi.74.22.10359-10370.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul AV, Yin J, Mugavero J, Rieder E, Liu Y, Wimmer E. A “slide-back” mechanism for the initiation of protein-primed RNA synthesis by the RNA polymerase of poliovirus. J Biol Chem. 2003;278:43951–43960. doi: 10.1074/jbc.M307441200. [DOI] [PubMed] [Google Scholar]
- Pfeiffer JK, Kirkegaard K. A single mutation in poliovirus RNA-dependent RNA polymerase confers resistance to mutagenic nucleotide analogs via increased fidelity. Proc Natl Acad Sci U S A. 2003;100:7289–7294. doi: 10.1073/pnas.1232294100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeiffer JK, Kirkegaard K. Increased fidelity reduces poliovirus fitness and virulence under selective pressure in mice. PLoS pathogens. 2005;1:e11. doi: 10.1371/journal.ppat.0010011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeiffer JK, Kirkegaard K. Bottleneck-mediated quasispecies restriction during spread of an RNA virus from inoculation site to brain. Proc Natl Acad Sci U S A. 2006;103:5520–5525. doi: 10.1073/pnas.0600834103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadeghipour S, Bek EJ, McMinn PC. Ribavirin-resistant mutants of human enterovirus 71 express a high replication fidelity phenotype during growth in cell culture. J Virol. 2013;87:1759–1769. doi: 10.1128/JVI.02139-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez-Aparicio MT, Rosas MF, Sobrino F. Characterization of a nuclear localization signal in the foot-and-mouth disease virus polymerase. Virology. 2013;444:203–210. doi: 10.1016/j.virol.2013.06.011. [DOI] [PubMed] [Google Scholar]
- Schein CH, Oezguen N, van der Heden van Noort GJ, Filippov DV, Paul A, Kumar E, Braun W. NMR solution structure of poliovirus uridylyated peptide linked to the genome (VPgpU) Peptides. 2010;31:1441–1448. doi: 10.1016/j.peptides.2010.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schein CH, Oezguen N, Volk DE, Garimella R, Paul A, Braun W. NMR structure of the viral peptide linked to the genome (VPg) of poliovirus. Peptides. 2006;27:1676–1684. doi: 10.1016/j.peptides.2006.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schein CH, Ye M, Paul AV, Oberste MS, Chapman N, van der Heden van Noort GJ, Filippov DV, Choi KH. Sequence specificity for uridylylation of the viral peptide linked to the genome (VPg) of enteroviruses. Virology. 2015;484:80–85. doi: 10.1016/j.virol.2015.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen H, Sun H, Li G. What Is the Role of Motif D in the Nucleotide Incorporation Catalyzed by the RNA-dependent RNA Polymerase from Poliovirus? PLoS computational biology. 2012;8:e1002851. doi: 10.1371/journal.pcbi.1002851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sholders AJ, Peersen OB. Distinct conformations of a putative translocation element in poliovirus polymerase. J Mol Biol. 2014;426:1407–1419. doi: 10.1016/j.jmb.2013.12.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shu B, Gong P. Structural basis of viral RNA-dependent RNA polymerase catalysis and translocation. Proc Natl Acad Sci U S A. 2016;113:E4005–4014. doi: 10.1073/pnas.1602591113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sierra M, Airaksinen A, Gonzalez-Lopez C, Agudo R, Arias A, Domingo E. Foot-and-mouth disease virus mutant with decreased sensitivity to ribavirin: implications for error catastrophe. J Virol. 2007;81:2012–2024. doi: 10.1128/JVI.01606-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song Y, Liu Y, Ward CB, Mueller S, Futcher B, Skiena S, Paul AV, Wimmer E. Identification of two functionally redundant RNA elements in the coding sequence of poliovirus using computer-generated design. Proc Natl Acad Sci U S A. 2012;109:14301–14307. doi: 10.1073/pnas.1211484109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steil BP, Barton DJ. Poliovirus cis-acting replication element-dependent VPg Uridylylation lowers the Km of the initiating nucleoside triphosphate for viral RNA replication. J Virol. 2008;82:9400–9408. doi: 10.1128/JVI.00427-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steitz TA. A mechanism for all polymerases. Nature. 1998;391:231–232. doi: 10.1038/34542. [DOI] [PubMed] [Google Scholar]
- Sun Y, Wang Y, Shan C, Chen C, Xu P, Song M, Zhou H, Yang C, Xu W, Shi PY, Zhang B, Lou Z. Enterovirus 71 VPg uridylation uses a two-molecular mechanism of 3D polymerase. J Virol. 2012;86:13662–13671. doi: 10.1128/JVI.01712-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svensen N, Peersen OB, Jaffrey SR. Peptide Synthesis on a Next-Generation DNA Sequencing Platform. Chembiochem : a European journal of chemical biology. 2016;17:1628–1635. doi: 10.1002/cbic.201600298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theobald DL, Steindel PA. Optimal simultaneous superpositioning of multiple structures with missing data. Bioinformatics (Oxford, England) 2012;28:1972–1979. doi: 10.1093/bioinformatics/bts243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theobald DL, Wuttke DS. Accurate structural correlations from maximum likelihood superpositions. PLoS computational biology. 2008;4:e43. doi: 10.1371/journal.pcbi.0040043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson AA, Albertini RA, Peersen OB. Stabilization of Poliovirus Polymerase by NTP Binding and Fingers-Thumb Interactions. J Mol Biol. 2007;366:1459–1474. doi: 10.1016/j.jmb.2006.11.070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson AA, Peersen OB. Structural basis for proteolysis-dependent activation of the poliovirus RNA-dependent RNA polymerase. Embo J. 2004;23:3462–3471. doi: 10.1038/sj.emboj.7600357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vignuzzi M, Stone JK, Arnold JJ, Cameron CE, Andino R. Quasispecies diversity determines pathogenesis through cooperative interactions in a viral population. Nature. 2006;439:344–348. doi: 10.1038/nature04388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vignuzzi M, Wendt E, Andino R. Engineering attenuated virus vaccines by controlling replication fidelity. Nat Med. 2008;14:154–161. doi: 10.1038/nm1726. [DOI] [PubMed] [Google Scholar]
- Vives-Adrian L, Lujan C, Oliva B, van der Linden L, Selisko B, Coutard B, Canard B, van Kuppeveld FJM, Ferrer-Orta C, Verdaguer N. The Crystal Structure of a Cardiovirus RNA-Dependent RNA Polymerase Reveals an Unusual Conformation of the Polymerase Active Site. Journal of Virology. 2014;88:5595–5607. doi: 10.1128/JVI.03502-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weeks SA, Lee CA, Zhao Y, Smidansky ED, August A, Arnold JJ, Cameron CE. A Polymerase mechanism-based strategy for viral attenuation and vaccine development. The Journal of biological chemistry. 2012;287:31618–31622. doi: 10.1074/jbc.C112.401471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao Y, Rouzine IM, Bianco S, Acevedo A, Goldstein EF, Farkov M, Brodsky L, Andino R. RNA Recombination Enhances Adaptability and Is Required for Virus Spread and Virulence. Cell host & microbe. 2016;19:493–503. doi: 10.1016/j.chom.2016.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X, Smidansky ED, Maksimchuk KR, Lum D, Welch JL, Arnold JJ, Cameron CE, Boehr DD. Motif D of viral RNA-dependent RNA polymerases determines efficiency and fidelity of nucleotide addition. Structure. 2012;20:1519–1527. doi: 10.1016/j.str.2012.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X, Welch JL, Arnold JJ, Boehr DD. Long-range interaction networks in the function and fidelity of poliovirus RNA-dependent RNA polymerase studied by nuclear magnetic resonance. Biochemistry. 2010;49:9361–9371. doi: 10.1021/bi100833r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Rijnbrand R, McKnight KL, Wimmer E, Paul A, Martin A, Lemon SM. Sequence requirements for viral RNA replication and VPg uridylylation directed by the internal cis-acting replication element (cre) of human rhinovirus type 14. J Virol. 2002;76:7485–7494. doi: 10.1128/JVI.76.15.7485-7494.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin J, Paul AV, Wimmer E, Rieder E. Functional dissection of a poliovirus cis-acting replication element [PV-cre(2C)]: analysis of single- and dual-cre viral genomes and proteins that bind specifically to PV-cre RNA. J Virol. 2003;77:5152–5166. doi: 10.1128/JVI.77.9.5152-5166.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamyatkin DF, Parra F, Alonso JM, Harki DA, Peterson BR, Grochulski P, Ng KK. Structural insights into mechanisms of catalysis and inhibition in Norwalk virus polymerase. The Journal of biological chemistry. 2008;283:7705–7712. doi: 10.1074/jbc.M709563200. [DOI] [PubMed] [Google Scholar]