

Abstract
Hydrogen-deuterium exchange (HDX) coupled with mass spectrometry (HDXMS) is a rapid and effective method for localizing and determining protein stability and dynamics. Localization is routinely limited to a peptide resolution of 5 to 20 amino acid residues. HDXMS data can contain information beyond that needed for defining protein stability at single amide resolution. Here we present a method for extracting this information from an HDX dataset to generate a HDXMS protein stability fingerprint. High resolution (HR)-HDXMS was applied to the analysis of a model protein of a spectrin tandem repeat that exemplified an intuitive stability profile based on the linkage of two triple helical repeats connected by a helical linker. The fingerprint recapitulated expected stability maximums and minimums with interesting structural features that corroborate proposed mechanisms of spectrin flexibility and elasticity. HR-HDXMS provides the unprecedented ability to accurately assess protein stability at the resolution of a single amino acid. The determination of HDX stability fingerprints may be broadly applicable in many applications for understanding protein structure and function as well as protein ligand interactions.
Introduction
Rapid characterization of protein conformation, dynamics, and mapping of protein-ligand interface sites is increasingly being performed by employing hydrogen-deuterium exchange mass spectrometry (HDXMS)1. This methodology is advantageous in terms of sensitivity, speed and capability of analyzing large proteins and protein-ligand complexes2. HDX is pH- and temperature-dependent3, 4 and involves the reversible exchange of backbone amide hydrogens in a protein with deuterium (D) from the solvent. HDX rates of proteins are determined by labeling of backbone amides with deuterium oxide (D2O) and are measured by liquid chromatography mass spectrometry (LCMS). In order for HDX to occur, the amide hydrogen must physically contact the D from the solvent through a local or global unfolding mechanism that may necessitate disruption of any hydrogen bonding.
The relationship of HDX and protein structure dynamics has been well-characterized5, 6. It is an invaluable method for interrogating protein structure fluctuations and representing localized dynamics6. The HDX rate of protein amides is greatly impacted by their solvent accessibility in the context of the protein structure and may vary as many as 9 orders of magnitude on the time scale7–10. This wide range of HDX rates represents enormous differences in the protein stability spectrum and might also be expressed as the free energy of exchange (ΔGex)8. Consequently, the stability fingerprint of a protein structure may then also be defined as the ΔGex versus the number of amino acids.
The ultimate advantages of HDXMS in many areas of life sciences10–15 has led to a rapid expansion of this technology1 and fostered the development of software packages for high-throughput analysis16, 17. The HDXMS workflow has been thoroughly described in numerous applications17–21. Briefly, proteins or their complexes are incubated in deuterated buffer and after defined time points the reaction is quenched by acidic quench buffer acids (pH 2.7). Subsequently, samples are digested proteolytically to generate peptide fragments17. Localization and quantification of D-incorporation into each peptide is performed by LCMS with resulting data analyzed using specialized software18. Ultimately, the HDXMS data are then either projected as the percentage of D-incorporation (%D) onto the amino acid sequence (ribbon heat maps) or onto a protein structure (3D heat maps)19, 22. At present, HDXMS analysis is mainly restricted to peptide resolution governed by the length of the obtained peptides. Analysis of HDXMS data at single amino acid resolution would lead to a much greater insight and understanding of protein structure, dynamics and function. However, software tools enabling this task have yet to be (further) developed.
In this study, we present a software tool enabling HDXMS analysis at highest resolution (HR-HDXMS) by determining single amide HDX rates from acquired HDXMS centroid data. HR-HDXMS utilizes D-incorporation from multiple overlapping peptide fragments and multiple D-labeling time points to determine single amide HDX rates in proteins. These can be collectively presented as the HDX stability fingerprint of a given protein. To demonstrate the feasibility of our approach, we used the structurally and dynamically well-characterized α-spectrin variant as a model protein23. Our software improved the spatial resolution of protein dynamics by providing a superior visualization of the spatial distribution of the stability profile of the structure. Our application demonstrates the utility of HR-HDXMS and HDX stability profiles for evaluating and understanding protein structure and function.
Results
Conventional HDXMS analysis of spectrin
For the development of HR-HDXMS we chose a structurally well-characterized variant of α-spectrin from chicken brain containing repeats 16 and 17 (further called R1617)23. R1617 is composed of two triple-helical bundles joined by a 5 amino acid linker with α-helical features23 (Fig. 1A). In order to generate a sufficient amount of overlapping peptides covering the complete amino acid sequence of spectrin, we employed a combination of the proteases pepsin and protease type XIII from Aspergillus saitoi (FPXIII) during LCMS analysis. We obtained approximately 200 peptides ranging from 4 to 41 amino acids and covering the entire amino acid sequence of spectrin (Fig. S1 and Table S1). Each peptide was manually reviewed for the correct assignment of charge state and the quality of the spectrum. The combined use of pepsin and FPXIII rather than each alone produced many small peptides with high overlap (Fig. S1). Moreover, this approach significantly increased the redundancy in coverage per amino acid in most regions greater than 2-fold and in some regions even greater than 5-fold. (Fig. S1). Furthermore, we were able to obtain many peptides with charge states ranging from +1 to +3. Multiple charge states are beneficial to estimate the degree of D-incorporation more accurately because the mass of the peptide should be the same at each charge state.
Figure 1.
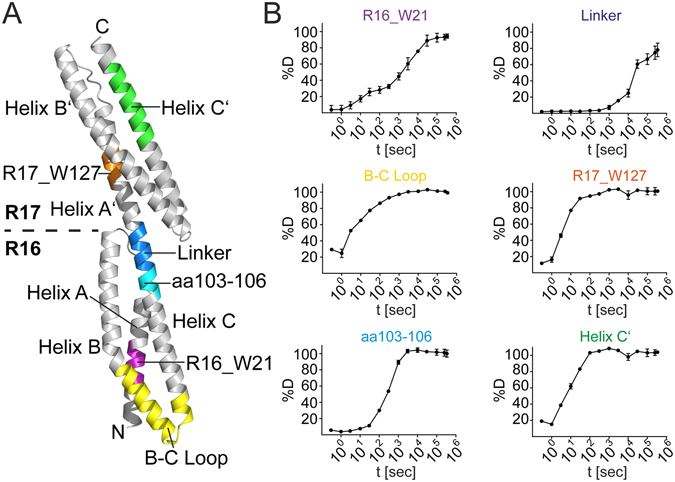
Conformational dynamics of R1617. (A) The crystal structure of repeats 16 & 17 of α-spectrin (PDB: 1CUN23) is shown in cartoon representation. Regions shown in (B) are colored and indicated. (B) Deuterium uptake of regions of R1617: R16_W21 (aa 15–21), B-C Loop (aa 61–84), aa 103–106, Linker (aa 108–114), R17_W127 (aa 125–128) and Helix C′ (aa 200–213).
The percentage of D-incorporation over time for each peptide depends on the localization of the peptide within the protein structure. The use of 13 time points for the measurement of %D provided complete and accurate information about which regions of R1617 were less or more accessible to HDX and how fast the exchange occurred (Fig. 1A and B). We classified different regions of R1617 based on their velocity of HDX by determining the time required for 50% D-incorporation to occur on a peptide (Table S1). With this approach, peptides with 50% D-incorporation <1000 seconds were classified as fast exchanging (135 peptides), those with 50% D-incorporation between 1000–3000 seconds were intermediate exchanging (36 peptides), and peptides that reached 50% D-incorporation above 3000 seconds as slow exchanging (31 peptides). Examples for the fast exchange were the B-C Loop (<3 seconds), Helix C′ (<10 seconds) and a peptide in repeat 17 spanning W127 (R17_W127) residing in helix A′ (<10 seconds, Fig. 1B). Also, amino acids 103–106 preceding the linker region of spectrin incorporate over 50%D after just 300 seconds, while the slow exchanging linker (i.e. amino acids 108–114) requires 100-times longer to reach similar deuterium incorporation (Fig. 1B). A peptide (R16_W21) in repeat 16 that includes W21 residing in Helix A of R1617 displays an intermediate D-incorporation. However, two distinct kinetics of deuterium uptake were apparent from the time course with a rather fast and slow exchanging species being overlaid (Fig. 1B). Our data confirm that R1617 is a highly dynamic molecule. Nevertheless, with the conventional way of analyzing HDXMS data subtle differences within the dynamic behavior of spectrin cannot be identified or be attributed to single amino acid residues.
Theoretical concept of HR-HDXMS
To improve the resolution in visualizing HDXMS data, we aimed to develop a computational algorithm to determine the hydrogen-deuterium exchange rates and the thermodynamic stability of single amides from an HDXMS dataset of a protein (Fig. 2). Central to this approach is the generation of a peptide library containing multiple overlapping peptides that are produced by combined proteolysis of the protein using pepsin and FPXIII followed by the determination of subfragments between all peptides. In this a subfragment X is defined as the smallest common overlapping sequence between two given peptides (Fig. 2A). Subsequently, the amount of deuterium incorporated on each subfragment X is computationally determined by a linear least squares minimization of a global error (GE) function (Fig. 2B and Fig. S2). Deuterium shifts for each subfragment are then determined from the difference of D-incorporation of the subfragment between each successive labeling time point. Next, the HDX rates (k ex) for each subfragment are estimated by fitting the exponent in equation 1 using the D-shifts of the calculated subfragments (Fig. 2B and Fig. S3). Consequently, k ex for each X represents an average of the exchange rates from all amides within X. The exchange rates from the subfragments are used to seed initial starting rates for the non-linear least squares optimization function that solves for the k ex of all amides (Fig. 2B and Fig. S3). The so-obtained exchange rates k ex of each amide can then be converted into the free energy of hydrogen-deuterium exchange (ΔGex) according to the Gibbs free energy equation allowing for a precise understanding of protein dynamics on the single amino acid level (Fig. 2C and D). Taken together, this procedure should enable the determination of k ex values of single amides from HDXMS datasets containing a sufficiently large number of overlapping peptides with redundant sequence coverage.
Figure 2.
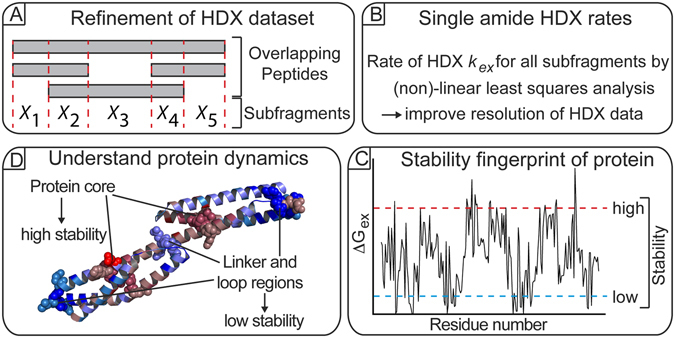
Workflow of HR-HDXMS. (A) Amino acid subfragments are determined from overlapping peptides generated by proteolytic digest of the protein. (B) The rate of hydrogen-deuterium exchange (k ex) of each subfragment is calculated by linear and non-linear least squares analysis. (C) The free energy value ΔGex derived from k ex is used to generate a stability fingerprint of the protein. (D) The protein fingerprint helps in understanding protein dynamics and allows predictions about residues that might be essential for protein stability and/or function.
Subfragmentation improves the spatial resolution of HDXMS
To learn more about subtle changes within the dynamics of R1617 that might not be visible by conventional HDXMS evaluation, we applied our HR-HDXMS software to the HDXMS dataset. Subfragments were determined from overlapping peptides followed by determination of the deuterium incorporation and the rate of hydrogen-deuterium exchange k ex of each subfragment by linear and non-linear least squares analysis (see above). These steps drastically improved the spatial resolution of the dynamics of R1617 (Fig. S4). In those areas of R1617 where many overlapping peptide fragments were available, deuterium uptake could even be determined for single amino acids (compare to Figs S1 and S4).
To visually inspect the improvements in resolution, we projected the results of ‘conventional’ HDXMS (see above) and HR-HDXMS data analysis on the crystal structure of R1617 (Fig. 3). Helix C′ could now be subdivided into four segments with different dynamic properties rather than two seen with the conventional approach (from the N- to C-terminus of helix C′: >90, 50, 20 and >90% D) (Fig. 3). Another example was found for the dynamics of the B-C Loop. While the conventional approach displayed intermediate-high dynamics (70% D), the HR-HDXMS heat map was further subdivided into three categories of dynamics including regions of very high (>90% D) over intermediate (50% D) to low (20% D) dynamics from the N- to the C-terminus of the B-C Loop (Fig. 3). Moreover, within the linker region the number of segments exhibiting different dynamics could be increased approximately two-fold allowing for a more detailed understanding (see below). Taken together, our approach of generating a subfragment map clearly refines analysis of HDXMS as exemplified by R1617 (Fig. 3).
Figure 3.
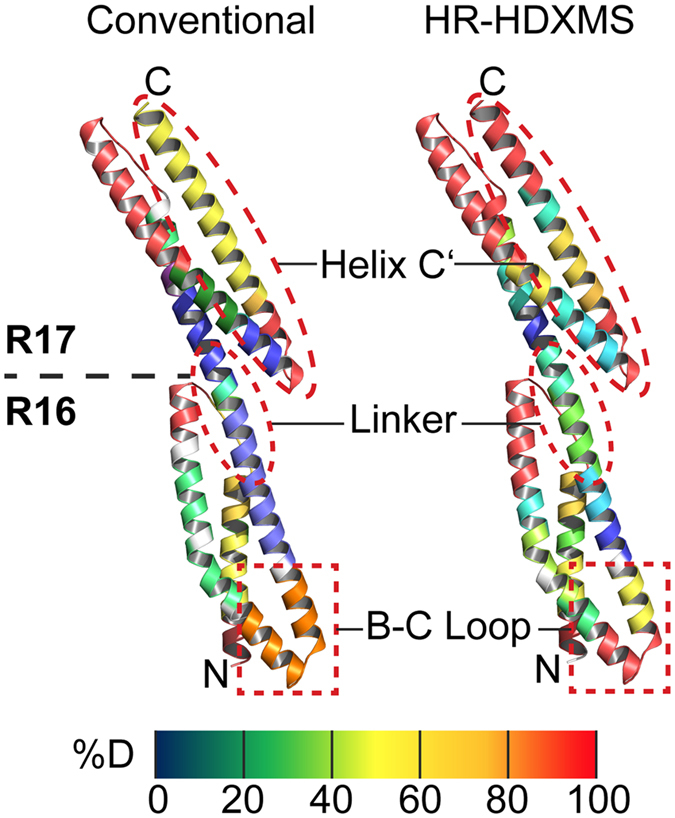
Improved resolution of R1617 conformational dynamics by HR-HDXMS. Deuterium uptake of R1617 mapped onto its crystal structure (PDB: 1CUN23). Data analysis by HR-HDXMS (right) provides a superior spatial resolution of deuterium uptake compared to ‘conventional’ HDX data analysis (left).
HR-HDXMS allows stability fingerprints at single amino acid residue resolution
Following our theoretical considerations (see above), we wanted to take our analysis further, eventually to determine stability fingerprints at the single amide level. Therefore, the exchange rates from each subfragment were used to seed initial starting rates for the nonlinear least squares function solving for the exchange rate of each amide (k ex) (equation 7). The k ex was converted into the free energy of exchange (ΔGex) according to the Gibbs Free Energy equation (ΔGexchange,i = −RT ln(k ex,i/kint,i)) where k ex,i and k int,i are the experimental and intrinsic (random coil) exchange rates, respectively, at an amide i as determined from the intrinsic rates of random coil model peptides24, 25. These ΔGex for each amide were plotted versus the position of the amino acid residue (Fig. 4A). The ΔGex values range from 0 kcal/mol (i.e. prolines lacking the amide hydrogen) to 14 kcal/mol. Generally, lower and higher ΔGex values correlate to faster and slower HDX rates of the amide, respectively6, 7, 26. Therefore, lower ΔGex values represent amides of amino acid residues that are more dynamic and, vice versa, higher ΔGex represent amides of residues that are less dynamic.
Figure 4.
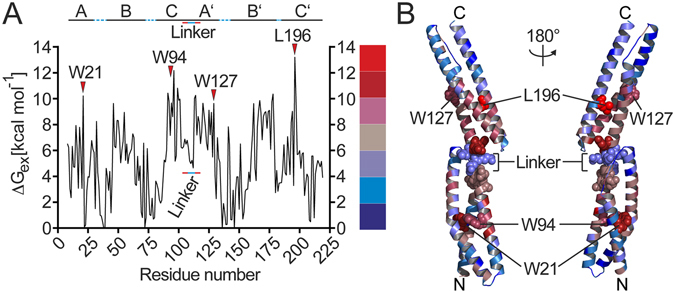
Stability fingerprint of R1617 determined by HR-HDXMS. (A) Plot of the free energy value ΔGex for each amino acid residue of R1617. The linker region between repeats 16 and 17 and amino acids adjacent to the linker are indicated by solid blue and red lines, respectively. Amino acid residues exhibiting high stability are indicated. (B) The crystal structure of R1617 (PDB: 1CUN23) colored according to the ΔGex values shown in (A).
The HDX stability fingerprint of R1617 as determined by our HR-HDXMS approach displays a jagged profile with an observable pattern of stability with a large window of variation in amide dynamics (Fig. 4A). Generally, higher ΔGex values are observed around the centers of the α-helices and lower ΔGex values are observed in the loop regions connecting the α-helices. Noteworthy, the amides of four amino acids (i.e. amino acids 109–112) constituting the linker that connects the repeats 16 and 17 exhibit a local minimum in ΔGex while residues preceding and following the linker showed much higher ΔGex values (Fig. 4A). The functional consequences of this keen discrimination between the linker region and the C- and N-terminal α-helices of the spectrin repeats 16 and 17 are discussed below.
To challenge the validity of our approach, we examined the accuracy upon which the HDX stability fingerprint could correctly identify amino acids or locations on the structure of R1617 that have previously been shown to confer stability or impose flexibility23, 27, 28. For this, the ΔGex values were projected onto the crystal structure of R1617 resulting in a superior spatial resolution of dynamics when compared to our previous subfragment analysis (Fig. 4B, and compare with Fig. 3). Our analysis revealed the amides of three tryptophan residues (i.e. W21, W94 within repeat 16 and W127 in repeat 17) and leucine at position 196 to exhibit very high ΔGex values. These residues localize in the center of the triple helical bundles forming repeat 16 and 17 (ref. 23). Notably, W21 and W94 have already been shown to significantly confer stability of the triple helical core27, 28, consolidating the validity of our HR-HDXMS approach to determine a stability fingerprint.
Taken together, these results clearly demonstrate that our HR-HDXMS approach allows unraveling the contribution of single amino acids to the overall dynamics of protein structures exemplified by spectrin.
HR-HDXMS agrees with single amide HDX rates obtained by NMR
Traditionally, single amide HDX rates have been determined by nuclear magnetic resonance (NMR)29, 30. Therefore, we extended the validation of our HR-HDXMS method against data obtained by NMR. To do so, we performed HR-HDXMS on a dataset obtained for Staphylococcus nuclease (SNase) comprising 341 peptides and 10 sampled time points31. In the initial determination of deuterium incorporation by linear least squares analysis (equation 1), a large number of residues exhibited a high residual error (Fig. S5A). To circumvent this problem that might be caused by poor data quality or incorrect peptide assignment, we eliminated peptides with an error ≥2 deuterium. After exclusion of those peptides, the residual error for each peptide from the linear optimization (equation 2) could be reduced below 1 deuterium for the remaining 206 filtered fragments (Fig. S5B). Although approximately 40% of peptides are lost during this procedure reducing the obtainable resolution, the quality of deuterium incorporation assignment for the remaining peptides is significantly raised.
For comparison of the stability fingerprints obtained by HR-HDXMS and NMR, the analogous NMR exchange rate data of SNase served as a gold standard9, 26. At first, the k ex rates obtained by NMR were used to determine the stability fingerprint of SNase by the HR-HDXMS algorithm. The ΔGex values obtained by global error minimization applying HR-HDXMS are almost identical to the ΔGex values obtained from the experimental data (Fig. S6A). Also, the stability fingerprint of SNase obtained by determination of k ex values from HDXMS dataset resulted in a good overlay with the NMR-derived stability fingerprint (Fig. S6B). These results further demonstrate the feasibility of HR-HDXMS even when suboptimal HDX data are available (see above) and show that the accuracy of the fingerprint relies on the quality of the seeding rates in the nonlinear solver.
Discussion
HR-HDXMS as tool to refine HDX data to single amide resolution
In this work, we present HR-HDXMS as suitable tool for refining HDXMS data to single amide resolution. Therefore, HR-HDXMS opens a new route to delineate the individual contribution of single amino acids to the overall dynamics of proteins and their macromolecular assemblies. To this end, suitability of HR-HDXMS was tested with repeats 16 & 17 of chicken brain α-spectrin (R1617), an enormously well characterized protein in terms of structure23, 32, biophysical characteristics27, 33, and postulated mechanisms of function. As exemplified, HR-HDXMS was able to recapitulate all known features of spectrin structure and dynamics, but much more provided further insights into the dynamic features of spectrin (see below). However, whether these are of functional relevance remains to be further investigated.
The HR-HDXMS approach involves a simple two-phase numerical technique. A linear least squares fit is performed for the initial estimation of amide exchange rates from overlapping peptide fragments followed by a non-linear least squares fit solving for individual amide exchange rates within each peptide. We generated an HDXMS dataset of repeats 16 & 17 of chicken brain α- spectrin, which provided 2600 overlapping constraints that were a product of 200 peptides and 13 time points of deuterium labeling for performing the linear and non-linear fits. Our study showed that successful application of HR-HDXMS critically relies on: the generation of multiple overlapping peptide fragments, general data quality, correction for D-loss into the solvent by incorporating residue-specific back-exchange rates in the non-linear fit, and imposing appropriate boundaries for HDX rates during non-linear optimization to serve as constraints for the fit. We believe that the generation of multiple overlapping peptides (allowing the subfragment assignment) is most critical for the success of HR-HDXMS. This issue can be addressed by the use of the two proteases pepsin and FPXIII. Overall, HR-HDXMS seems to offer a promising way to analyze HDXMS datasets at single amide resolution. However, further studies are required to challenge the suitability of our approach especially with respect to larger proteins and macromolecular assemblies where the amount of overlapping peptides and redundant sequence coverage becomes a greater challenge.
Advantages of HR-HDXMS over other methods and alternatives
Traditionally, single amide HDX rates have been determined by nuclear magnetic resonance (NMR)29, 30. While NMR is powerful to accurately measure single amide HDX rates, it is usually limited in protein size (<30 kDa molecular weight). A great advantage of HDXMS is its ability to analyze proteins and macromolecular assemblies extending molecular weights over 100 kDa. Moreover, our study shows that HR-HDXMS is able to recapitulate single amide HDX rates obtained by NMR as exemplified by SNase.
Mass spectrometry is also capable of determining the D-incorporation at a single amide through stepwise peptide fragmentation employing electron capture dissociation (ECD) and electron transfer dissociation (ETD)34–37. Collision-induced dissociation (CID) fragmentation approaches have commonly been used to improve coverage and sublocalization of D-labeling. However, this approach typically suffers from deuterium scrambling38 among adjacent amides making reliable sublocalization challenging. HR-HDXMS alleviates the drawbacks of CID, because it relies on the total D-incorporation on the peptide irrespective of any scrambling occurring during CID fragmentation.
Fragmentation methods such as ECD39 and ETD36, 40 also allow for determination of single amide D-incorporation by inducing stepwise fragmentation. However, the conventional use of these methods to determine D-incorporation at the single amide level is limited. ETD requires peptides with higher charge states to work effectively41. Therefore, not all peptides will be amenable to ETD fragmentation.
Other mathematical and analytical approaches have recently been developed for determining single amide rates and/or improving the resolution in HDXMS datasets31, 42–46. These approaches include a mathematical model for predicting the assignment of HDX rate classes to amino acid segments from overlapping peptides46, a statistical approach to quantify the difference of HDX between two states42, residue averaging of the mid-point values of HDX43, and utilization of the shape of the isotopic envelope to determine the distribution of deuterium on the amides of the peptide by the software HDSite31. HDSite demonstrates the feasibility of determining exchange rates at single amide resolution, but it functions optimally only with high-resolution data providing enough information to produce a well-defined isotopic envelope. Implementation of HDSite involves a separate iteration for each peptide to determine the amide D-occupancy in the peptide and a separate fit to an exponential to determine the HDX rate for each residue based on the previously determined amount of D on the amide at each time point. In contrast to HDSite, HR-HDXMS performs a single global linear optimization iteration to simultaneously determine the D level on all subfragments from overlapping peptides and then a single global non-linear iteration using a non-linear optimizer to simultaneously determine all HDX rates for all amides. One drawback of the HR-HDXMS software is that it assumes all HDX to be governed by EX2 kinetics, and in its present version does not take EX1 kinetics into account.
Taken together, both HDSite and HR-HDXMS depend on a sufficiently high number of overlapping fragments, number of time points, length of the sequence, and number of replicates. These issues represent the most challenging limitation of both approaches. Nevertheless, our validation of HR-HDXMS demonstrated its robustness to correctly calculate the stability profile of a protein.
Lessons learned on the dynamics of spectrin by HR-HDXMS
Our HR-HDXMS approach improved the spatial resolution of the dynamics of the α-spectrin tandem repeat (R1617) to the single amide level. Based on the crystal structure of R1617, two models of elasticity and flexibility were proposed23. The first model described the flexibility imposed by the linker23, 47 through helix bending at the linker region. The second one suggested high flexibilities along the α-helices and their connecting loops due to their conformational rearrangement18. Interestingly, our HR-HDXMS analysis supports both models and extends them further. We found substantial stability gradients at the center of the α-helices that gradually decrease towards the helical ends (Fig. 4B). Probably, gradients of stability near the helical ends may facilitate the structural transition from helical propensity into loop geometry. With respect to the linker, conventional analysis of our HDXMS supports the highly dynamic behavior of the linker as already inferred from the crystal structure (Fig. 3). However, the refinement in spatial resolution provided by our HR-HDXMS approach revealed that residues 104–106 preceding the linker exhibited higher ΔGex than the linker residues 108–112 that exhibited a local minimum in ΔGex. Thereby, we could identify specific amino acids that possibly confer less or more stability within the linker region. Therefore, our HR-HDXMS analysis of the spectrin tandem repeat R1617 supports a model combining the ones previously postulated and suggests a much more complex scenario in which all the structural elements of spectrin contribute to the overall dynamics required for its functionality. Moreover, our analysis clearly shows that the triple helical bundles of repeats 16 and 17 require structural stabilization that is provided through intramolecular contacts by a number of central tryptophane residues27, 48, 49.
Taken together, HR-HDXMS might represent a powerful tool to analyze protein dynamics at single amide resolution.
Materials and Methods
Protein preparation
The cDNA of repeats 16 and 17 from chicken brain α-spectrin was cloned and expressed in Escherichia coli BL21 (DE3) (Novagen) and purified as previously described23.
Sample preparation and fragmentation optimization
Conditions for optimal protein digestion were determined by adding 30 μl of quench solution with increasing concentrations of Guanidine Hydrochloride (GuHCl) (0/0.8/1.6/3.2/6.4 M GuHCl in 0.8% (v/v) formic acid) to 20 μl of sample (final concentration 0.5% (v/v) formic acid, 0/0.5/1/2/4 M GuHCl) containing 10–15 μg of protein in Tris-buffered saline (TBS: 50 mM Tris-HCl, pH 7.0 and 150 mM NaCl). Samples were then frozen on dry ice within one minute after addition of quench solution and stored at −80 °C until LCMS analysis. Sequest (Thermo Finnigan Inc) was used for peptide identification and peptides were verified using the “DXMS Explorer” software (Sierra Analytics, LLC, Modesto, CA)19.
Deuterium labeling of proteins
Exchange-deuterated samples of spectrin were prepared by the addition of 15 μl of D2O, containing 50 mM Tris, pD 2.7 and 150 mM NaCl to 5 μl of protein stock (3 mg/ml in TBS pH 7.0) and incubated at room temperature 25 °C for 3, 10, 30, 100, 300, 103, 3 × 103, 104, 2.5 × 105 and 3.4 × 105 seconds, with the reaction quenched by addition of 30 μl quench solution (0.8% (v/v) formic acid, 0.8 M GuHCl) at 0 °C, and samples immediately placed on dry ice and frozen at −80 °C until further processed (as described above). Time points of 3 and 10 seconds were incubated on ice to simulate 0.3 and 1 seconds deuteration time as there would be a 10-fold decrease in the exchange rate for every 25 °C decrease in temperature51. Data on the deuterated sample sets were acquired in a single automated 8 hour run, and subsequent data reduction was performed on the data reduction software (see above). Corrections for loss of D-labelling by individual fragments during HDXMS analysis (after “quench”) were made by determination of loss of deuterium from reference samples that had been fully deuterated by overnight incubation under denaturing conditions, as previously described52–55.
LCMS analysis
Frozen quenched samples were placed in a Spectraphysics AS3000 autosampler kept at −45 °C with dry ice powder, rapidly thawn within 30 seconds followed by immediate injection into LC sample loop pre-cooled at 0 °C. LCMS analysis was performed at 0 °C and pH 2.3 as previously described56, 57. Online proteolysis involved the sample passing through an initial immobilized pepsin (30 mg/ml) column (66 μl column bed volume) that was coupled to a 20AL resin from PerSeptive Biosystems in 0.05% (w/v) trifluoroacetic acid (TFA) followed by passage through a column (66 μl column bed volume) containing immobilized protease type XIII from Aspergillus saitoi (20 mg/ml)58 at 250 μl/min flow rate. Reversed-phase separation of digested peptides utilized a C18 column, eluted by a linear acetonitrile gradient (5 to 45% B in 30 minutes; 50 μl/min; solvent A, 0.05% TFA; solvent B, 80% acetonitrile, 20% water, 0.01% TFA), with data acquisition using a Thermo Finnigan Classic LCQ electrospray ion trap type mass spectrometer or an electrospray Micromass Q-TOF mass spectrometer operated with a capillary temperature at 200 °C.
HDXMS analysis
Mass spectrometry data was analyzed using DXMS data reduction software (Sierra Analytics, Version 1.01 (beta))19. Deuterated peptides were identified in DXMS and pasted into an excel macro that determines the D-incorporation of each peptide fragment at each timepoint. Protein fragmentation map, ribbon heat maps, and D-incorporation labeling curves were generated as previously described56. All structure figures were generated with Pymol (Delano Scientific).
Linear least squares analysis
Given m overlapping fragments spanning residues a → b, a subfragment x can be defined as the overlapping and non-overlapping region(s) between fragments (Fig. S2). Given a number of subfragments N within a given fragment F, the total amount of experimentally measured deuterium incorporated on fragment n at each timepoint t can be represented as the sum of the deuterium incorporated on each subfragment plus a residual error as depicted in equation 1, where j is the defined subfragment, N is the total number of subfragments spanning fragment n, and p is the number of prolines that are subtracted from the number of exchangeable amides as they do not contain amide hydrogens.
| 1 |
The application of the linear program based on approximation of initial exchange rates of subfragments is illustrated in Fig. S2. The three peptide fragments F 1, F 2, F 3 spanning the first 29 amino acids of spectrin define subfragments (x 1, …, x 5). The amount of D on each subfragment at each timepoint is determined by linear least square optimization to minimize the sum of the residual error over all fragments (M), and all time points (T) as defined by the global error function in equation 2.
| 2 |
Non-Linear Least Squares Analysis
The collective exchange rate k ex of the amides within each subfragment is determined by simultaneously fitting the D-incorporation data to the first-order reaction kinetics for D-incorporation of an amide in equation 3, where Di,t is the amount of D incorporated on residue i at time t.
| 3 |
The rate constant is dependent on pH, temperature, protein sequence, and protein conformation. In a completely unstructured polypeptide chain, all peptide amide hydrogens are freely accessible to water and exchange at their maximal possible rate, with a half-life of exchange of approximately one second at 0 °C and pH 7.0 (ref. 25). Exact off-exchange rates for amide hydrogens in a fully unstructured protein sequence can be reliably calculated from knowledge of the temperature, pH and primary amino acid sequence involved24, 25. The precise rate of HDX of a particular amide in random coil can vary more than 30-fold from the average rates for all amides in a peptide under such conditions, with the precise rate depending upon the identity of the two amino acids flanking the particular amide bond. Because the deuterated samples are subjected to quench conditions during the LC process, a correction for back-exchange must be made to account for the loss of label from the time the sample is injected to the time it is eluted, also referred to as the lag time . We can model the back-exchange on subfragment x as the fraction of remaining deuterium after a lag time of an amide at quench conditions through:
| 4 |
where is the calculated back-exchange rate24, 25 of amino acid under quench conditions at lag time .
Optimizing on-exchange rates by incorporating back-exchange rates
The amount of D on a single amide i, in a peptide fragment corrected for back-exchange can be described as the product of the amount of D-incorporated on the amide and the back-exchange according to:
| 5 |
It follows that the experimental D-accumulation of a given peptide fragment F, including back-exchange corrections is the sum of the individual exchange rates of each amide plus a residual error defined by equation 6.
| 6 |
Here, is summed over all amide position I, m < i < l, where m and l are the first and last residues of fragment n, respectively. The non-linear least squares algorithm solves for the rate constants k ex that minimize the squared difference between the fitted and experimental deuteration level of all fragments. This concept is illustrated in Fig. S3. The objective function aims at minimizing the global error for all fragments () at all time points ():
| 7 |
The success of HR-HDXMS relies on the extent of overlapping fragments and the number of sampled on-exchange time points. Upper and lower boundaries are set as constraints for the fit using the intrinsic back-exchange rates determined by Molday et al.24 from spreadsheet calculations obtained at http://hx2.med.upenn.edu/download.html). The exchange rate of all prolines was set to 0 during the non-linear fit. Validation of HR-HDXMS was performed by determination of the HDX stability profile of SNase from HDXMS data and comparison with the NMR-derived stability profile. Acquisition of NMR data was performed as previously described9.
HR-HDXMS was written in MatLab utilizing the optimization toolbox implementing the linear and non-linear functions. The MatLab algorithm is available under http://www.hr-hdxms.us.
Electronic supplementary material
Acknowledgements
This work is dedicated to the memory of Dr. Virgil L. Woods. We thank David Wemmer, Pat Jennings, David Baker, Russell Doolittle, Philip Bourne, Halbert White, and Karin Maxson for support and guidance. We thank Pavel Pevzner, Neil Jones, and Bryant Forsgren for their guidance in the design and development of the precursor of the HR-HDXMS stability profile algorithm, Eleftheria Mavridou for assistance in preparation of the figures and manuscript, Spiro Pantazatos, Ruby I. MacDonald and Michel Alain Cuendet for their valuable comments and review of the manuscript. This work was supported by NIH grant CA099835 (VLW) and by grants from the University of California BioStar and LSI programs, grants S97-90, S99-44, L98-30 (VLW). The LOEWE excellence program of the state of Hessen (Germany) supported this work (GB). We gratefully acknowledge funding from the Hellenic American Foundation (DP).
Author Contributions
S.B. performed NMR and HDXMS experiments on SNase. J.S. performed the fitting for the NMR data and its analysis. D.P. designed, performed and analyzed HDX experiments and wrote the MatLab code. C.G. assisted in the development of the MatLab code and performed computational validation studies with the NMR and HDXMS data. V.L.W. conceived, co-designed and supported experiments and conducted data interpretation. T.J.W. provided insight in manuscript preparation and supported the study. G.B., W.S. and D.P. prepared the manuscript.
Competing Interests
The authors declare that they have no competing interests.
Footnotes
Virgil L. Woods is deceased.
Electronic supplementary material
Supplementary information accompanies this paper at doi:10.1038/s41598-017-03922-3
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Pirrone GF, Iacob RE, Engen JR. Applications of hydrogen/deuterium exchange MS from 2012 to 2014. Anal Chem. 2015;87:99–118. doi: 10.1021/ac5040242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wales TE, Engen JR. Hydrogen exchange mass spectrometry for the analysis of protein dynamics. Mass Spectrometry Reviews. 2006;25:158–170. doi: 10.1002/mas.20064. [DOI] [PubMed] [Google Scholar]
- 3.Linderstrom-Lang K. The pH-dependence of the deuterium exchange of insulin. Biochim Biophys Acta. 1955;18:308. doi: 10.1016/0006-3002(55)90084-6. [DOI] [PubMed] [Google Scholar]
- 4.Zhang Z, Smith DL. Determination of amide hydrogen exchange by mass spectrometry: a new tool for protein structure elucidation. Protein Sci. 1993;2:522–31. doi: 10.1002/pro.5560020404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Berger A, Linderstrom-Lang K. Deuterium exchange of poly-DL-alanine in aqueous solution. Arch Biochem Biophys. 1957;69:106–18. doi: 10.1016/0003-9861(57)90478-2. [DOI] [PubMed] [Google Scholar]
- 6.Englander SW, Mayne L, Bai Y, Sosnick TR. Hydrogen exchange: the modern legacy of Linderstrom-Lang. Protein Science. 1997;6:1101–9. doi: 10.1002/pro.5560060517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Maity H, Lim WK, Rumbley JN, Englander SW. Protein hydrogen exchange mechanism: local fluctuations. Protein Sci. 2003;12:153–60. doi: 10.1110/ps.0225803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Milne JS, Mayne L, Roder H, Wand AJ, Englander SW. Determinants of protein hydrogen exchange studied in equine cytochrome c. Protein Sci. 1998;7:739–45. doi: 10.1002/pro.5560070323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Skinner JJ, Lim WK, Bedard S, Black BE, Englander SW. Protein hydrogen exchange: testing current models. Protein Sci. 2012;21:987–95. doi: 10.1002/pro.2082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Houde D, Berkowitz SA, Engen JR. The utility of hydrogen/deuterium exchange mass spectrometry in biopharmaceutical comparability studies. J Pharm Sci. 2011;100:2071–86. doi: 10.1002/jps.22432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wei H, et al. Hydrogen/deuterium exchange mass spectrometry for probing higher order structure of protein therapeutics: methodology and applications. Drug Discov Today. 2014;19:95–102. doi: 10.1016/j.drudis.2013.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Barclay LA, et al. Inhibition of Pro-apoptotic BAX by a noncanonical interaction mechanism. Mol Cell. 2015;57:873–86. doi: 10.1016/j.molcel.2015.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Melnyk RA, et al. Structural determinants for the binding of anthrax lethal factor to oligomeric protective antigen. J Biol Chem. 2006;281:1630–5. doi: 10.1074/jbc.M511164200. [DOI] [PubMed] [Google Scholar]
- 14.Hamuro Y, et al. Hydrogen/deuterium-exchange (H/D-Ex) of PPARgamma LBD in the presence of various modulators. Protein Sci. 2006;15:1883–92. doi: 10.1110/ps.062103006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ramachandran G, Udgaonkar JB. Difference in fibril core stability between two tau four-repeat domain proteins: a hydrogen-deuterium exchange coupled to mass spectrometry study. Biochemistry. 2013;52:8787–9. doi: 10.1021/bi4014352. [DOI] [PubMed] [Google Scholar]
- 16.Hamuro Y, Weber PC, Griffin PR. High-throughput analysis of protein structure by hydrogen/deuterium exchange mass spectrometry. Methods Biochem Anal. 2005;45:131–57. [PubMed] [Google Scholar]
- 17.Gallagher ES, Hudgens JW. Mapping Protein-Ligand Interactions with Proteolytic Fragmentation, Hydrogen/Deuterium Exchange-Mass Spectrometry. Methods Enzymol. 2016;566:357–404. doi: 10.1016/bs.mie.2015.08.010. [DOI] [PubMed] [Google Scholar]
- 18.Mayne L, et al. Many overlapping peptides for protein hydrogen exchange experiments by the fragment separation-mass spectrometry method. J Am Soc Mass Spectrom. 2011;22:1898–905. doi: 10.1007/s13361-011-0235-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Garcia RA, Pantazatos D, Villarreal FJ. Hydrogen/deuterium exchange mass spectrometry for investigating protein-ligand interactions. Assay Drug Dev Technol. 2004;2:81–91. doi: 10.1089/154065804322966342. [DOI] [PubMed] [Google Scholar]
- 20.Steinchen W, et al. Catalytic mechanism and allosteric regulation of an oligomeric (p)ppGpp synthetase by an alarmone. Proc Natl Acad Sci USA. 2015;112:13348–53. doi: 10.1073/pnas.1505271112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schuhmacher JS, et al. MinD-like ATPase FlhG effects location and number of bacterial flagella during C-ring assembly. Proc Natl Acad Sci USA. 2015;112:3092–7. doi: 10.1073/pnas.1419388112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hamuro Y, et al. Rapid analysis of protein structure and dynamics by hydrogen/deuterium exchange mass spectrometry. J Biomol Tech. 2003;14:171–82. [PMC free article] [PubMed] [Google Scholar]
- 23.Grum VL, Li D, MacDonald RI, Mondragon A. Structures of two repeats of spectrin suggest models of flexibility. Cell. 1999;98:523–35. doi: 10.1016/S0092-8674(00)81980-7. [DOI] [PubMed] [Google Scholar]
- 24.Molday RS, Englander SW. Primary structure effects on peptide group hydrogen exchange. Biochemistry. 1972;11:150. doi: 10.1021/bi00752a003. [DOI] [PubMed] [Google Scholar]
- 25.Bai Y, Milne JS, Mayne L, Englander SW. Primary structure effects on peptide group hydrogen exchange. Proteins. 1993;17:75–86. doi: 10.1002/prot.340170110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Skinner JJ, Lim WK, Bedard S, Black BE, Englander SW. Protein dynamics viewed by hydrogen exchange. Protein Sci. 2012;21:996–1005. doi: 10.1002/pro.2081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pantazatos DP, MacDonald RI. Site-directed mutagenesis of either the highly conserved Trp-22 or the moderately conserved Trp-95 to a large, hydrophobic residue reduces the thermodynamic stability of a spectrin repeating unit. J Biol Chem. 1997;272:21052–9. doi: 10.1074/jbc.272.34.21052. [DOI] [PubMed] [Google Scholar]
- 28.MacDonald RI, Musacchio A, Holmgren RA, Saraste M. Invariant tryptophan at a shielded site promotes folding of the conformational unit of spectrin. Proc Natl Acad Sci USA. 1994;91:1299–303. doi: 10.1073/pnas.91.4.1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cortajarena AL, Mochrie SG, Regan L. Mapping the energy landscape of repeat proteins using NMR-detected hydrogen exchange. J Mol Biol. 2008;379:617–26. doi: 10.1016/j.jmb.2008.02.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Henry GD, Weiner JH, Sykes BD. Backbone dynamics of a model membrane protein: measurement of individual amide hydrogen-exchange rates in detergent-solubilized M13 coat protein using 13C NMR hydrogen/deuterium isotope shifts. Biochemistry. 1987;26:3626–34. doi: 10.1021/bi00386a056. [DOI] [PubMed] [Google Scholar]
- 31.Kan ZY, Walters BT, Mayne L, Englander SW. Protein hydrogen exchange at residue resolution by proteolytic fragmentation mass spectrometry analysis. Proc Natl Acad Sci USA. 2013;110:16438–43. doi: 10.1073/pnas.1315532110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kusunoki H, Minasov G, Macdonald RI, Mondragon A. Independent movement, dimerization and stability of tandem repeats of chicken brain alpha-spectrin. J Mol Biol. 2004;344:495–511. doi: 10.1016/j.jmb.2004.09.019. [DOI] [PubMed] [Google Scholar]
- 33.Li D, et al. Structural and Thermodynamic Studies of Cloned Fragments of Spectrin. Cell Mol Biol Lett. 2001;6:218. [PubMed] [Google Scholar]
- 34.Rand KD, Pringle SD, Morris M, Engen JR, Brown JM. ETD in a traveling wave ion guide at tuned Z-spray ion source conditions allows for site-specific hydrogen/deuterium exchange measurements. J Am Soc Mass Spectrom. 2011;22:1784–93. doi: 10.1007/s13361-011-0196-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rand KD, Zehl M, Jensen ON, Jorgensen TJ. Protein hydrogen exchange measured at single-residue resolution by electron transfer dissociation mass spectrometry. Anal Chem. 2009;81:5577–84. doi: 10.1021/ac9008447. [DOI] [PubMed] [Google Scholar]
- 36.Zehl M, Rand KD, Jensen ON, Jorgensen TJ. Electron transfer dissociation facilitates the measurement of deuterium incorporation into selectively labeled peptides with single residue resolution. J Am Chem Soc. 2008;130:17453–9. doi: 10.1021/ja805573h. [DOI] [PubMed] [Google Scholar]
- 37.Konermann L, Pan J, Liu YH. Hydrogen exchange mass spectrometry for studying protein structure and dynamics. Chem Soc Rev. 2011;40:1224–34. doi: 10.1039/C0CS00113A. [DOI] [PubMed] [Google Scholar]
- 38.Ferguson PL, Konermann L. Nonuniform isotope patterns produced by collision-induced dissociation of homogeneously labeled ubiquitin: implications for spatially resolved hydrogen/deuterium exchange ESI-MS studies. Anal Chem. 2008;80:4078–86. doi: 10.1021/ac8001963. [DOI] [PubMed] [Google Scholar]
- 39.Abzalimov RR, Kaplan DA, Easterling ML, Kaltashov IA. Protein conformations can be probed in top-down HDX MS experiments utilizing electron transfer dissociation of protein ions without hydrogen scrambling. J Am Soc Mass Spectrom. 2009;20:1514–7. doi: 10.1016/j.jasms.2009.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rand KD, et al. Gas-phase hydrogen/deuterium exchange in a traveling wave ion guide for the examination of protein conformations. Anal Chem. 2009;81:10019–28. doi: 10.1021/ac901897x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Abzalimov RR, Kaltashov IA. Controlling hydrogen scrambling in multiply charged protein ions during collisional activation: implications for top-down hydrogen/deuterium exchange MS utilizing collisional activation in the gas phase. Anal Chem. 2010;82:942–50. doi: 10.1021/ac9021874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Saltzberg, D. J. et al. A Residue Resolved Bayesian Approach to Quantitative Interpretation of Hydrogen Deuterium Exchange from Mass Spectrometry: Application to Characterizing Protein-Ligand Interactions. J Phys Chem B (2016). [DOI] [PMC free article] [PubMed]
- 43.Keppel TR, Weis DD. Mapping residual structure in intrinsically disordered proteins at residue resolution using millisecond hydrogen/deuterium exchange and residue averaging. J Am Soc Mass Spectrom. 2015;26:547–54. doi: 10.1007/s13361-014-1033-6. [DOI] [PubMed] [Google Scholar]
- 44.Hu W, et al. Stepwise protein folding at near amino acid resolution by hydrogen exchange and mass spectrometry. Proc Natl Acad Sci USA. 2013;110:7684–9. doi: 10.1073/pnas.1305887110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fajer PG, Bou-Assaf GM, Marshall AG. Improved sequence resolution by global analysis of overlapped peptides in hydrogen/deuterium exchange mass spectrometry. J Am Soc Mass Spectrom. 2012;23:1202–8. doi: 10.1007/s13361-012-0373-3. [DOI] [PubMed] [Google Scholar]
- 46.Althaus E, et al. Computing H/D-exchange rates of single residues from data of proteolytic fragments. BMC Bioinformatics. 2010;11:424. doi: 10.1186/1471-2105-11-424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kusunoki H, MacDonald RI, Mondragon A. Structural insights into the stability and flexibility of unusual erythroid spectrin repeats. Structure. 2004;12:645–56. doi: 10.1016/j.str.2004.02.022. [DOI] [PubMed] [Google Scholar]
- 48.MacDonald RI, Cummings JA. Stabilities of folding of clustered, two-repeat fragments of spectrin reveal a potential hinge in the human erythroid spectrin tetramer. Proc Natl Acad Sci USA. 2004;101:1502–7. doi: 10.1073/pnas.0308059100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yan Y, et al. Crystal structure of the repetitive segments of spectrin. Science. 1993;262:2027–30. doi: 10.1126/science.8266097. [DOI] [PubMed] [Google Scholar]
- 50.Krezel A, Bal W. A formula for correlating pKa values determined in D2O and H2O. J Inorg Biochem. 2004;98:161–6. doi: 10.1016/j.jinorgbio.2003.10.001. [DOI] [PubMed] [Google Scholar]
- 51.Englander JJ, Calhoun DB, Englander SW. Measurement and calibration of peptide group hydrogen-deuterium exchange by ultraviolet spectrophotometry. Anal Biochem. 1979;92:517–24. doi: 10.1016/0003-2697(79)90693-6. [DOI] [PubMed] [Google Scholar]
- 52.Woods-Jr. VL, Hamuro YH. Resolution, High- Throughput Amide Deuterium Exchange- Mass Spectrometry (DXMS) Determination of Protein Binding Site Structure and Dynamics: Utility in Pharmaceutical Design. Journal of Cellular Biochemistry. 2001;37:89–98. doi: 10.1002/jcb.10069. [DOI] [PubMed] [Google Scholar]
- 53.Woods-Jr., V. L. Methods for identifying hot- spot residues of binding proteins and small compounds that bind to the same. in US Patent # 6,599,707. (USA, 2003).
- 54.Hamuro Y, et al. Dynamics of cAPK Type IIb Activation Revealed by Enhanced Amide H/2H Exchange Mass Spectrometry (DXMS) J. Mol. Biol. 2003;327:1065–1076. doi: 10.1016/S0022-2836(03)00234-1. [DOI] [PubMed] [Google Scholar]
- 55.Englander JJ, et al. Protein structure change studied by hydrogen-deuterium exchange, functional labeling, and mass spectrometry. Proc. Nat. Acad. Sci. 2003;100:7057–7062. doi: 10.1073/pnas.1232301100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pantazatos D, Gessner CR, Woods VL, Jr., Gilbert GE. Changes in the Factor VIII C2 domain upon membrane binding determined by hydrogen-deuterium exchange MS. Biochem J. 2014;461:443–51. doi: 10.1042/BJ20140121. [DOI] [PubMed] [Google Scholar]
- 57.Garcia RA, et al. Molecular interactions between matrilysin and the matrix metalloproteinase inhibitor doxycycline investigated by deuterium exchange mass spectrometry. Mol Pharmacol. 2005;67:1128–36. doi: 10.1124/mol.104.006346. [DOI] [PubMed] [Google Scholar]
- 58.Cravello L, Lascoux D, Forest E. Use of different proteases working in acidic conditions to improve sequence coverage and resolution in hydrogen/deuterium exchange of large proteins. Rapid Commun Mass Spectrom. 2003;17:2387–93. doi: 10.1002/rcm.1207. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.