

Abstract
Chemotherapy in patients with inoperable or advanced breast cancer inevitably results in low-dose exposure of tumor-cell subset and senescence. Metabolically active senescent cells secrete multiple tumor promoting factors making their elimination a therapeutic priority. Viscum album is one of the most widely used alternative anti-cancer medicines facilitating chemotherapy tolerance of breast cancer patients. The aim of this study was to model and investigate how Viscum album extracts execute additive anti-tumor activity with low-dose Dox using ER + MCF7 breast cancer cells. We report that cotreatment of MCF7 with Viscum album and Dox abrogates G2/M cycle arrest replacing senescence with intrinsic apoptotic program. Mechanistically, this switch was associated with down-regulation of p21, p53/p73 as well as Erk1/2 and p38 activation. Our findings, therefore, identify a novel mechanistic axis of additive antitumor activity of Viscum album and low dose-Dox. In conclusion, ER + breast cancer patients may benefit from addition of Viscum album to low-dose Dox chemotherapy due to suppression of cancer cell senescence and induction of apoptosis.
Introduction
Despite significant progress made in early diagnosis and treatment, breast cancer remains the most frequent cancer amongst women and the second most common cancer type in the world1, 2.
Hormone receptor positive breast cancers represent two thirds of all breast cancers diagnosed today3, 4. Despite effective targeted treatment strategies4 the use of chemotherapeutics is indicated for patients with primary inoperable and advanced ER/PR positive breast cancers unresponsive to first line therapy4. Doxorubicin (Dox), a naturally occurring anthracycline antibiotic5 has long been used as a chemotherapeutic component in breast cancer patient treatment6–8.
Antitumor activity of Dox has been assigned to induction of DNA damage and ROS production9. The amount of genotoxic stress and total ROS production dictates possible outcomes such as cell death or G1 and/or G2 cell cycle arrest and senescence10–12. Senescence induced by chemotherapeutics (therapy induced senescence, TIS) has been well studied in vitro and, more recently, detected in breast tumors of patients undergoing pre-operative neoadjuvant chemotherapy13. Though TIS has been long considered a desirable therapeutic outcome and a promising strategy in overcoming therapy resistance13–15, a growing body of work has indicated that TIS cells may alter response to chemotherapy16, escape cell cycle arrest17, 18 and promote tumor growth19 (reviewed in refs20,21). Most of these detrimental effects have been attributed to both autocrine and paracrine activity of senescent cell secretome designated as Senescence Associated Secretory Phenotype or SASP.
SASP components contributing to relapse and aggressive cancer occurrence22 include: interleukins 6 and 8 (IL-6, IL-8)23; amphiregulin (AREG) and growth-related oncogene (GRO) α24, 25, VEGF26, 27 or matrix metalloproteinases (MMPs)25, 28, 29. SASP has recently been linked to immune surveillance of damaged normal and tumor cells30–32. During acute normal and tumor tissue injury, one of the key SASP functions is to attract immune cells facilitating clearance of damaged senescent cells33, 34. However, under conditions of persistent tissue injury, damaged normal and tumor cells undergo immunoediting escaping immune surveillance35, an effect recently linked to SASP secretome36.
Therefore, compelling evidence indicates that non-cell autonomous action of SASP secretome could drive cancer relapse making eradication of therapy induced senescent cells a priority for researchers today. Complementary Alternative Medicines have long been used in oncotherapy both as therapeutic efficacy enhancers whilst facilitating tolerance of its side effects37–44. Phytochemical preparations of mistletoe, including aqueous extracts, are among the most frequently prescribed complementary and alternative therapies for cancer in Europe45. Despite well documented research and clinical studies supporting beneficial effects of Mistletoe as a complementary cancer medicine37–44, the most challenging obstacle towards its definitive inclusion in oncotherapy is a lack of preparation with standardized anti-tumor activity. While Viscum Album Extracts (VAE) exhibit potent tumor toxicity where several isolated extract compounds, such as Mistletoe Lectins (MLs), have been demonstrated to have strong apoptosis-inducing effects46–48. ML-induced apoptosis is primarily triggered by PI3K/Akt-, MAPK-, TLR-signalling resulting in the activation of caspases49–51. Its cytotoxic and anti-metastatic effect has been demonstrated in different solid tumours and leukaemia cell lines in vitro and in vivo52–55. MLs also display cytotoxic effects on multidrug-resistant cancer cells (e.g. MDR + colon cancer cells56 and enhance cytotoxicity of anticancer drugs57, 58.
Furthermore, VAEs seem to interfere with tumour angiogenesis59, 60. Injected into tumour-bearing animals, VAEs display growth-inhibiting and tumor reducing effects61, 62. Iscador Qu 5 mg Spezial represents Mistletoe aqueous extract suitable for subcutaneous injections with standardized dry substance, viscotoxin and mistletoe lectin content63, 64. Antitumor-activity of Mistletoe extracts has been shown to vary depending on both harvest season and host plant species65 allowing for designations such as Qu, M or P identifying Mistletoe hosts as Quercus (oak), Malus (Apple) or Pinus (Pine) respectively.
We have previously shown that Viscum album/aqueous extract (VAE) increases anti-leukemic effectiveness of doxorubicin by preventing G2/M arrest and inducing apoptosis66. The aim of this study was to investigate whether Viscum album preparation (Iscador Qu) potentiates Dox toxicity at sub-therapeutic concentrations in MCF7 ER + breast cancer cells as well as its mechanism of action.
Results
Isc Qu treatment of MCF7 cells abrogates low-dose Dox induced G2/M arrest
Baring in mind that low-dose chemotherapy induces cell cycle arrest and senescence, we wanted to ask the question whether Mistletoe extract (Isc Qu) anti-tumor activity targets this cell population.
In a series of 48 and 72 h treatments of MCF7 cells with increasing Isc Qu and Dox concentrations (Fig. S1A left and right tables, respectively), we have observed the strongest synergistic cytotoxicity after 72 h treatment with 50 nM Dox and 85 ug/mL Isc Qu (Fig. S1A). Furthermore, 72 h treatment of MCF7 cells with 50 nM Dox had the lowest impact on cell viability (Fig. 1A, top left) while potently inducing G2/M arrest (Fig. 1B). MTT assay67, 68 results after 72 h treatment of MCF7 cells with a range of Isc Qu concentrations revealed that IC50 value for Iscador Qu was 43.4 µg/mL (Fig. 1A, top right). Interestingly, 72 h treatment of MRC5 non-tumorigenic fibroblast cell line with Isc Qu did not exhibit marked toxicity even in the concentration range well above MCF7 IC50 value (300; 100; 33.3; 11.1 and 3.7 µg/mL) (Fig. 1A, bottom). Furthermore, all concentrations used for MRC5 control cell treatment seem to fall well below the IC50 mark (Fig. 1A bottom).
Figure 1.
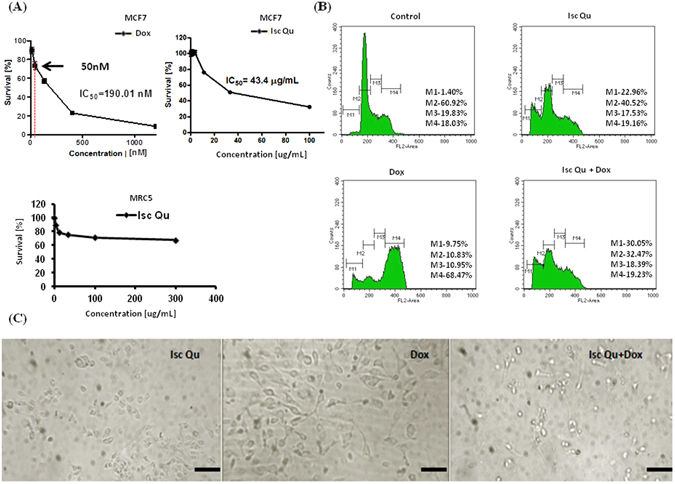
Isc Qu treatment of MCF7 cells abrogates low-dose Dox induced G2/M arrest. (A) MCF7 survival plots upon 72 h monotreatment with: Dox (top left), Isc Qu (top right). MRC5 survival plot upon 72 h monotreatment with Isc Qu (bottom). (B) Changes in cell cycle phase distribution of MCF7 cells upon 72 h treatment with Isc Qu (85 µg/mL), Dox (50 nM) or combination Isc Qu (85 µg/mL) + Dox (50 nM) are shown. After treatment, cells were stained with propidium iodide and analyzed by flow cytometry for cell cycle phase distribution changes. M1- apoptotic cells with DNA content corresponding to sub-G1 fraction; M2- cells with DNA content corresponding to G0/G1 phases; M3- cells with DNA content corresponding to S phase; M4- cells with DNA content corresponding to G2/M phases. (C) Light microscope photographs of morphological changes of MCF7 cells induced by Dox. Magnification, 10X.
Thus, during 72 h cotreatment experiments with 50 nM Dox, Iscador Qu concentration of 85 µg/mL induced the strongest synergistic antitumor activity (Fig. S1A, right table). In addition, Isc Qu concentration of 85 µg/mL had no significant impact on MRC5 fibroblast viability (Fig. 1A, bottom) and was therefore selected as a minimal, most effective and well tolerated concentration.
MCF7 mono-treatment with 50 nM Dox resulted in an minor increase in subG1 population and cell death (Fig. 1B, bottom left and Fig. 1C, right) and massive expansion of G2/M arrested population from 18, 03% in untreated (Fig. 1B, top left) to 68, 47% in Dox treated group (Fig. 1B bottom left). Interestingly, cotreatment with 85 ug/mL Isc Qu abrogated Dox induced G2/M cell cycle block (Fig. 1B, bottom right) accompanied by concomitant increase of dying sub-G1 population (Fig. 1B, bottom right). Collectively, our data confirm tumor-selective Isc Qu toxicity and reveal that additive antitumor activity of Isc Qu with low-dose Dox occurs by preventing G2/M arrest and triggering cell death.
G2/M arrest override during MCF7 cotreatment with Isc Qu and Dox activates intrinsic cell death pathway
To gain insight into events underlying Isc Qu mediated switch from senescence to cell death of Dox treated MCF7 cells, we asked what type of cell death was prevalent under cotreatment conditions. Compared to Dox treated controls, 72 hour cotreatment of MCF7 cells with 85 µg/mL of Isc Qu and 50 nM Dox led to strong increase in Annexin V labeled early apoptotic cell population (Fig. 2A), as well as late apoptotic cells detectable by double Annexin V and 7AAD staining (Fig. 2A). Consistent with increased early and late apoptotic marker positivity (Fig. 2A), cotreatment with Isc Qu and Dox increased the detachment of MCF7 cells (Fig. 1C, right) with nearly 100% trypan blue positivity detected among cells in suspension (not shown). Cytotoxic drugs have been shown to activate intrinsic/mitochondrial apoptotic pathway69 reflected by measurable changes in mitochondrial membrane potential (∆ψm)70, 71, release of cytochrome C, induction and/or activation of proapoptotic factor Bax and effector caspase372–75. FACS Rh123 fluorochrome incorporation assay76 revealed a dramatic loss of mitochondrial membrane potential (Δψm) in MCF7 cells upon 72 hour cotreatment with Isc Qu (Fig. 2B) indicating initiation of mitochondrial apoptotic pathway program. Moreover, FACS and qPCR analysis revealed a prominent increase of Bax/Bcl-2 ratio during cotreatment with Isc Qu compared to monotreatment conditions (Fig. 2C) confirming the activity of the apoptotic program. Thus, synergistic anti-tumor activity observed during MCF7 cotreatment with sub-apoptotic concentrations of Dox and Isc Qu is achieved by activation of intrinsic apoptotic program.
Figure 2.
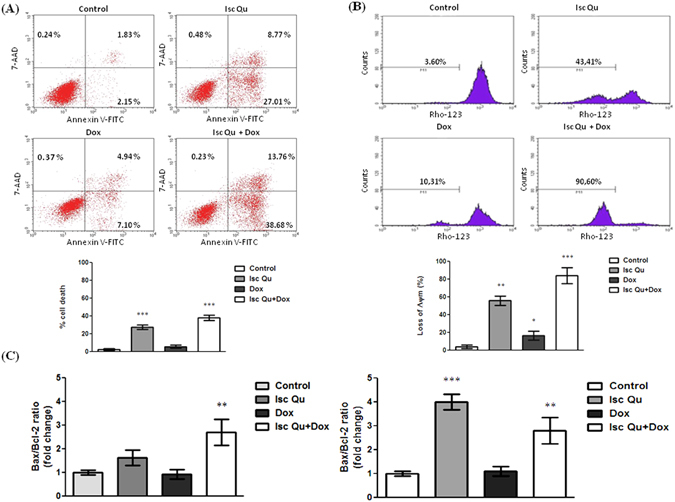
Cotreatment of MCF7 with Isc Qu and Dox activates intrinsic cell death pathway. (A) Proportion of early apoptotic and late apoptotic/necrotic cells was measured by the bivariate Annexin V/7AAD flow cytometry after 72 h treatment of MCF7 cells with Isc Qu (85 µg/mL), Dox (50 nM) or combination Isc Qu (85 µg/mL) + Dox (50 nM) (top). Bar graph showing results presented as the mean ± SEM of three independent experiments (bottom). (B) Dissipation of mitochondrial membrane potential was assessed by flow cytometry using rhodamine 123 staining (top). Bar graph showing results presented as the mean ± SEM of three independent experiments (bottom). (C) Bax/Bcl-2 ratio levels. Analysis of Bcl-2 and Bax protein(left) and mRNA expression (right). Detected mRNA levels were normalized to GAPDH. Results are presented as the mean ± SEM of three independent experiments. Asterisks denote statistical significance compared to control cells (*p < 0.05; **p < 0.01; ***p < 0.001). Representative dot plots from three independent experiments performed in triplicate are shown.
Isc Qu cotreatment prevents low-dose Dox induced senescence and SASP in MCF7 cells
Next, we sought to address the mechanistic basis underlining Isc Qu mediated switch from Dox induced MCF7 cell senescence to apoptosis. Sub-apoptotic doses of chemotherapeutics have been shown to induce cell-cycle arrest and senescence both in vitro and in breast cancer patients13, 77–80. Considering that Dox induced G2/M accumulation of MCF7 cells was lost upon co treatment with Isc Qu (Fig. 1B), we next asked whether Isc Qu prevents accumulation of senescent tumor cells.
Compared to Dox treated controls, 72 hour cotreatment of MCF7 cells with 85 µg/mL of Iscador Qu and 50 nM Dox dramatically reduced appearance of senescent cells characterized by flattened morphology with enlarged nuclei (Fig. 1C) and SA-ß-Gal-positivity (Fig. S1B). Cancer cell response to genotoxic stress is mediated by p53/p21 and p16INK4a/pRB tumor suppressive pathways81–87. Both pathways are complex; each having multiple upstream and downstream regulators and modifying side branches88, 89. Moreover, these pathways were shown to cross-regulate each other90–92. Using flow cytometry, immunofluorescence staining and western blot, we next measured expression levels of both proteins under treatment conditions described. A significant increase in both p21/WAF1 and p53 protein was observed 72 hours after MCF7 Dox treatment (Fig. 3A–C). qPCR data revealed that gene expression of both p21 and tp53 has increased under all treatment conditions (Fig. S1C and S1D) but resulted in protein stabilization and accumulation only upon Dox treatment (Fig. 3A–C). These results show that unlike during Dox treatment, Isc Qu induced p21 promoter activity is independent of p53 likely due to absence of efficient DDR pathway activation.
Figure 3.
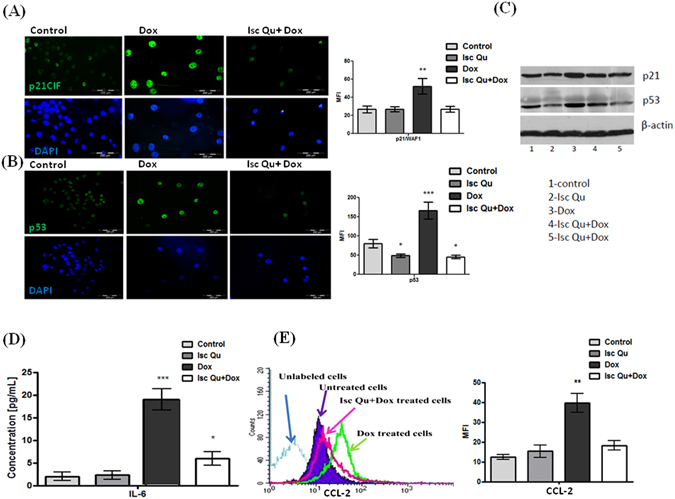
Isc Qu cotreatment prevents low-dose Dox induced senescence and SASP in MCF7 cells. Confluent MCF7 cells were treated with Isc Qu (85 µg/mL) and Dox (50 nM) alone or in combination for 72 hours and were subjected to immunofluorescence, FACS analyses and western blot. Immunofluorescent labeling of (A) p21 and (B) p53 protein expression, as well as (C) FACS analysis and immunobloting. Cell nuclei are labeled with DAPI (blue). (D) IL-6 protein levels in the supernatant of cultured MCF7 cells upon 72 h treatment with Isc Qu (85 µg/mL) and Dox (50 nM) alone or in combination, were measured using an ELISA. IL-6 activity was converted to pg according to the manufacturer’s instructions (Affymetrix, eBiosciences, CA, USA). (E) Protein expression of CCL-2 in treated MCF7 cells was determined by flow cytometry (left) and presented as MFI- the mean fluorescence intensity (right). Results are presented as the mean ± SEM of three independent experiments. Asterisks denote statistical significance compared to control cells (***p < 0.001). Representative photos and histogram for at least three independent experiments performed in triplicate are shown.
SASP represents a specific program triggered by genotoxic stress in cancer cells25. SASP components may differ depending on the cell type, tissue type or genomic stressor93. Though efforts are still ongoing to identify SASP markers unique for specific cell context, IL-6 and IL-8 are among most prominently detected SASP cytokines that seem to be directly controlled by persistent DNA-damage signaling94. Thus, we essayed expression of IL-6 by enzyme linked immunosorbent assay (ELISA). 72 h Doxorubicin treatment of MCF7 cells resulted in a ten-fold induction of IL-6 secretion (Fig. 3D) and increased STAT3 activity (Fig. S1E) supporting previously described role for IL-6/STAT3 signaling in senescence establishment and SASP maintenance95, 96. Conversely, cotreatment with Isc Qu reduced IL-6 secretion (Fig. 3D) and STAT3 activation (Fig. S1E).
Though IL-6 levels detected upon Isc Qu and Dox cotreatment were higher compared to controls (Fig. 3D), STAT3 activation seems to be efficiently blocked by Isc Qu and remained significantly lower compared to untreated controls (Fig. S1E). Viscum album extracts have been reported to increase tumor infiltration and immune system activity97. Among SASP secretome components with potent adverse activity, CCL-2 has been recently described as an essential mediator of immune surveillance suppression allowing for tumor progression and metastases13, 98, 99. To investigate whether G2 arrested senescent cells produce CCl-2 and if this production is affected by Isc Qu cotreatment, we have assayed for CCL2 expression under described treatment conditions using FACS. Whereas 72 h Dox treatment of MCF7 cells potently induced CCL-2 protein and mRNA expression, addition of Isc Qu strongly abrogated these effects (Fig. 3E). Collectively, our results show that addition of Isc Qu to low-dose Dox efficiently blocks senescence from G2/M cell cycle arrested cells and SASP.
Isc Qu induced G2/M senescence switch to apoptosis is accompanied by p38 and Erk1/2 inhibition
One of the well studied mechanisms of Dox toxicity in tumor cells is induction of ROS100. While mitochondria can generate damaging ROS101, mitochondrial defects were demonstrated to promote cellular senescence102–104. Finally, Mistletoe extracts have been reported to have a potent antioxidant activity105–109.Therefore, we asked whether alterations of ROS levels and corresponding signaling pathways could be responsible for the switch from senescence to apoptosis observed during IscQu cotreatment with low-dose Dox.
Data presented in Fig. 4A reveal that all treatment conditions resulted in substantial increase of mitochondrial ROS (mROS) production with maximal levels detected in cells undergoing Dox monotreatment. Interestingly, total ROS levels did not mimic alterations observed in the mitochondrial compartment though maximum total ROS production was also detected upon Dox monotreatmet (Fig. 4A and B). These results suggest that Isc Qu likely acts by suppressing ROS generation from other cellular sources such as increased signaling, peroxisome, hyperactivation of oxidases, cyclooxygenases, lipoxigenases etc.110.
Figure 4.
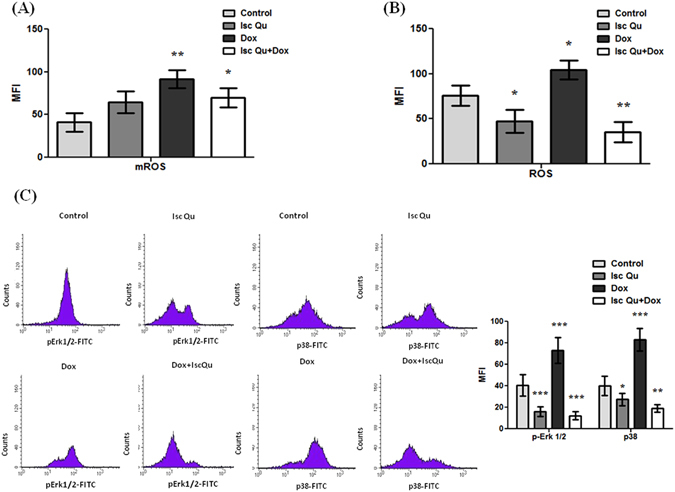
Isc Qu induced G2/M senescence switch to apoptosis is accompanied by p38 and Erk1/2 inhibition. (A) After 72 hour treatment with Isc Qu (85 µg/mL) and Dox (50 nM) alone or in combination, MCF7 cells where stained with dyhydrorhodamine 123 for 20 min in order to measurelevels of mitochondrial ROS, or (B) incubated 30 min with DCFH-DA to measure total ROS levels. (C) Phosphorylated pErk1/2 and p38 protein levels were assessed by flow cytometry. MFI-mean fluorescence intensity. Results are presented as the mean ± SEM of three independent experiments. Asterisks denote statistical significance compared to control cells (*p < 0.05; **p < 0.01; ***p < 0.001). Representative histograms for at least three independent experiments performed in triplicate are shown.
By increasing production of distinct ROS species111, 112 Doxorubicin was shown to activate Erk1/2 and p38, two MAP kinases described to have a central role in establishment of senescence113, 114, SASP induction115 and survival upon oxidative injury116, 117. To investigate whether Isc Qu mediates a switch from senescence to apoptosis via inhibition of Erk1/2 and p38 MAP kinases, we examined the status of Erk1/2 and p38 by flow cytometry. Increased levels of both activated Erk1/2 and p-38 could be readily observed upon 72 h treatment with Dox (Fig. 4C) in line with the peak ROS production (Fig. 4A and B). Isc Qu addition almost completely abolished this effect reducing both total ROS and activated p38 well below Isc Qu mono-treatment or control levels (Fig. 4B and C, respectively). Conclusively, these data suggest that Isc Qu blocks Dox induced ROS production, consequent activation of Erk1/2 and p38 kinases preventing senescence establishment and SASP.
Discussion
Tumor suppressor function of cellular senescence has been identified in the context of the irreversible cell cycle exit118 as well as autocrine and paracrine SASP mediated effects: cell cycle arrest reinforcement119–122 propagation of senescent phenotype on neighboring damaged cells123 and supporting tissue regeneration124–126. Moreover, SASP can elicit immune surveillance and clearance of both tumor127–129 and neighboring damaged cells124.
Paradoxically, more recent studies have demonstrated that SASP can promote tumorigenesis by supporting proliferation and invasion of surrounding tumor cells21, 23, 130, promoting angiogenesis131 or creating immunosuppressive environment allowing for tumor cell escape from immune system recognition and clearance98, 99.
Thus, beneficial and/or detrimental aspects of SASP appear to be critically dependent on the senescence trigger and specific SASP secretome production132.
Our study was designed to identify whether and how Viscum album extracts could potentiate anti-tumor activity of sub-apoptotic Doxorubicin concentrations in MCF7 breast cancer cells. In this study we report for the first time that VAE potently inhibits chemotherapy induced senescence and triggers apoptosis. We have also confirmed previous reports describing selective anti-tumor toxicity of VAE/Isc Qu66. We show that 72 h MCF7 cell treatment with sub-apoptotic Dox concentrations induces G2/M cell cycle arrest, senescence and SASP which were blocked by cotreatment with Isc Qu. The switch from G2/M senescence entry into apoptosis seems to be regulated by preventing Dox mediated ROS accumulation and Erk1/2 p38 activation. Our data suggest that G2/M cell cycle exit and senescence are regulated by both p53-p21 and p38 activated INK4A/Rb pathway probably owing to their extensive crosstalk. A trigger stimulus for Dox induced G2/M senescence seems to be moderate ROS production leading to sustained p38 activation114, 133. Persistent p38 activity was shown to induce SASP96, 116 largely via facilitating NF-kB activation and its recruitment to inflammatory gene promoters such as IL-6 and CCL-2134. Previous work has shown that IL-6 plays a key role in oncogene induced senescence establishment25 and inflammatory secretome maintenance135. Thus, Increased ROS production during Dox treatment would likely trigger p38 activity136 establishing a positive feedback loop in IL-6 secretion and secretome maintenance. We report that additive antitumor activity of low-dose Dox (50 nM) with Iscador Qu triggers apoptosis while reducing ROS, p53 and p21 levels. Our results mimic the findings of Delebinski and colleagues in lauekaemia cells137 where mistletoe lectins where shown to suppress p53 and induce TRAIL expression and apoptosis in a dose dependent manner. It is possible that, as previously shown138–140, low-dose-Dox treatment sensitizes MCF7 cells to Mistletoe lectin-induced TRAIL expression and apoptosis while ROS quenching by the extract attenuates p38, Erk1/2, p53 and p21 activation. Considering that we haven’t analyzed temporal profiles of ROS production as well as Erk1/2 and p38 activity, it is not clear whether putative cross-talk with TRAIL signaling and resulting apoptotic shift takes place at the onset of p38 activation or establishment of the positive feedback loop mediated by inflammatory signaling. To our knowledge, this is the first report showing that Viscum Album extract could prevent chemotherapy induced senescence by redirecting cells into apoptosis. Further studies elucidating the exact timing and molecular mechanism of this “switch” are of special interest both for the therapeutic purposes and prevention of ageing-related diseases. In conclusion, our data indicate that Isc Qu can sensitize MCF7 cells to apoptotic cell death at senescent concentration of the Dox by down-regulation of ROS, p21/WAF1, and p53 and by suppressing SASP profile through prevention of activation of Erk1/2, p38 and STAT3. Therapeutic interventions with Isc Qu applied as CAM, whether by dampening the SASP profile or by preventing the therapeutic senescence through induction of apoptotic cell death, should be much more effective. Whether Isc Qu is effective in targeting endogenously senescent tumor or non-tumorigenic cells remains to be addressed.
Materials and Methods
Drug
Doxorubicin hydrochloride (Sigma-Aldrich, St. Louis, USA) was diluted in sterile water. For the purpose of the study, we used Iscador Qu Spezial (Iscador AG, Switzerland) in 1 mL ampules of 5 mg. This preparation is fermented aqueous extracts of mistletoe plants growing on oak trees. As confirmed with the manufacturer, Iscador Qu Spezial batches used in our experiments are standardized on the basis of their contents of mistletoe lectins 391 ± 18.3 ng/mL and viscotoxins 13 ± 3 µg/mL.
Cell line
All cell lines used in this study were obtained from the American Type Culture Collection (Rockville, MD) unless specified otherwise. Human breast adenocarcinoma cells (MCF-7) and a single human normal cell line (MRC-5) were maintained as monolayer culture in a nutrient medium, MRC-5 cells in the Roswell Park Memorial Institute (RPMI) 1640 medium, while MCF7 cells in the DMEM medium. Powdered RPMI 1640 medium, and DMEM modified medium, were purchased from Sigma Chemicals Co, USA. Nutrient medium RPMI 1640 was prepared in sterile deionized water, supplemented with penicillin (192 U/mL), streptomycin (200 µg/mL), 4-(2-hydroxyethyl) piperazine-1-ethanesulfonic acid (HEPES) (25 mM), L-glutamine (3 mM) and 10% of heat-inactivated fetal calf serum (FCS) (pH 7.2). Modified DMEM nutrient medium was prepared in sterile deionized water, supplemented with penicillin (192 U/mL), streptomycin (200 µg/mL) and 10% of heat-inactivated FCS. Cells were grown at 37 °C in 5% CO2 and humidified air atmosphere, by twice weekly subculture.
Cytotoxicity assay
Cytotoxic activity of Isc Qu and Dox on MCF7 and MRC5 cells was assessed using the MTT assay67, 68. After treatment in 96-well plates, 20 µL of MTT solution (3-(4, 5-dimethylthiazol-2-yl)-2, 5-dyphenyl tetrazolium bromide) (Sigma-Aldrich, St. Louis, USA) was added to each well. Samples were incubated for 4 h, followed by the addition of 100 μL of 10% SDS and incubated at 37 °C. Absorbance at 570 nm was measured the next day.
Cell survival (%) was calculated as an absorbance (A570 nm) ratio between treated and control cells multiplied by 100. IC50 was defined as the concentration of the agent that inhibited cell survival by 50% compared to the vehicle control.
Flow-cytometric analysis of cell cycle phase distribution
Briefly, 2 × 105 cells/Petri dish (dimensions 60 × 15 mm, NUNC) were treated with investigated drugs as indicated. After collection, cells were fixed with ethanol and stained with propidium iodide (PI) (Sigma-Aldrich, St. Louis, USA)141. Cell cycle phase distribution was analyzed by FACS Calibur Becton Dickinson flow cytometer using Cell Quest computer software (Becton Dickinson, Heidelberg, Germany)
Apoptotic assay
Apoptotic rates were assessed with flow cytometry using the Annexin V–fluorescein isothiocyanate/and 7AAD (BD Pharmingen, San Diego, CA, USA). Samples were prepared according to manufacturer’s instructions. Flow cytometry analysis was performed using a FACS-Calibur cytometer using Cell Quest computer software (Becton Dickinson, Heidelberg, Germany).
Quantification of mitochondrial transmembrane potential
Mitochondrial transmembrane potential (Δψm) was measured using a cationic fluorochrome Rodamine 123 (Rh123) (Sigma-Aldrich, St. Louis, USA) as described by Yan et al.76. Briefly, 1 × 106 cells resuspended in 200 μL of phosphate buffered saline were stained with Rh123 (2.5 μg/mL) for 30 min at 37 °C. After washing, samples were analyzed by flow cytometry using Cell Quest software (Becton Dickinson, Heidelberg, Germany).
Intracellular staining
Cells stained for FACS analysis were treated as described above. For intracellular staining the following antibodies were used: mouse anti-p21WAF1, clone CP74 (5 µg/mL, Merck Millipore, Darmstadt, Germany), mouse anti-Bax, clone 3/Bax (1:100, BD Pharmingen, San Diego, CA, USA), FITC-conjugated monoclonal Bcl-2 antibody, clone Bcl-2/100 (1:100, BD Pharmingen, San Diego, CA, USA), mouse anti-human Phospho-p44/42 MAPK (Erk1/2) (Thr202/Tyr204) antibody (1:200, Cell Signaling, Danvers, MA, USA), mouse anti-human phospho-p38 MAPK (Thr180/Tyr182) antibody, clone 28B10 (1:800, Cell Signaling, Danvers, MA, USA), mouse anti-p53 antibody, clone DO7 (dilution 1:100, Dako, Glostrup, Denmark) and mouse anti-p73, clone ER-13 (5 µg/mL, Merck Millipore, Darmstadt, Germany).
Briefly, cells (5 × 105 cells/flask 25 cm2) were allowed to adhere for 24 h in standard conditions, and then treated as described in Results. After the stimulation period, cells were fixed the cells immediately by adding pre-warmed Cytofix Buffer for 10 to 12 minutes at 37 °C, and washed twice with PBS containing 1% BSA. After permeabilization of the cells using of Perm Buffer for 20 minutes at room temperature and washing, cells were incubated with antibodies at room temperature for 60 minutes protected from light, and washed twice with PBS containing 1% BSA. After appropriate incubation, cells were washed three times with PBS containing 1% BSA and incubated with the corresponding FITC-coupled secondary antibodies (dilution 1:100, BD Pharmingen, San Diego, CA, USA). Samples were analyzed on a FACS-Calibur cytometer using Cell Quest software (Becton Dickinson, Heidelberg, Germany).
Immunofluorescence Labeling
Cells (5 × 104 cells/cover slip) were allowed to adhere for 24 h in standard conditions, and then treated as described in Results. Immunofluorescent labeling was performed as previously described142. Briefly, cells were fixed in 4% paraformaldehyde, permeabilized in 0.1% Triton-X100 (Sigma Aldrich), and then immunolabeled with primary antibodies: mouse anti-p21WAF1, clone CP74 (5 µg/mL, Merck Millipore, Darmstadt, Germany), mouse anti-p53, clone DO7 (dilution 1:100, Dako, Glostrup, Denmark). Samples incubated in 1% BSA in PBS served as negative control. After appropriate incubation (indicated in Results), cells were washed three times with PBS and incubated with the corresponding FITC-coupled secondary antibodies (dilution 1:100, from Pharmingen) and 1 µg/mL of DAPI (Sigma-Aldrich, St. Louis, USA) for nucleus labeling for 1 h at room temperature. Mounted cells were analyzed using an epifluorescent microscope (Olympus, Japan).
Analysis of gene expression by Real-time PCR
Total RNA was isolated using TRI REAGENT® BD kit (Sigma-Aldrich, St. Louis, USA). cDNA synthesis from total RNA using random primers and MultiScribe™ Reverse Transcriptase from High-Capacity cDNA Reverse Transcription kit (Applied Biosystems, CA, USA). All target transcripts were detected using TaqMan® Gene Expression Assays Applied Biosystems, CA, USA. All reactions were performed in duplicate and the data represent mean ± SEM of three independent experiments. Results were analyzed using the classical delta-delta-Ct method. GraphPad Prism 5.04 (GraphPad Software, CA, USA) was used to compare means by two way analysis of variance (ANOVA) and Bonferroni method to adjust the p-value for multiple comparisons. Differences were considered significant if p < 0.05. P-values for each analysis are indicated in Fig. legends.
Senescence associated β-galactosidase staining
Cells (2 × 105 per well) were seeded into 6-well plates in DMEM culture medium and after 24 hours treated with investigated drugs for 72 hours. SA-β-gal staining was performed using Senescence β-galactosidase staining kit (Cell Signaling) according to the manufactures’ instructions. Briefly, cells were fixed for 15 min in fixative solution, washed and incubated at 37 °C with X-gal (1 mg/mL), dissolved in a solution containing 40 mM citric acid pH 6.5, 5 mM potassium ferrocyanide, 5 mM potassium ferricyanide, 150 mM NaCl and 2 mM MgCl2. After 24 h incubation, photographs were taken using a standard light microscope (Olympus, Japan).
Enzyme-linked immunosorbent assay
MCF7 cells (5 × 103 per well) were plated in 96-well plates and allowed to adhere for 24 h in standard conditions. Upon adherence, cells were treated with investigated drugs for 72 hours and the culture supernatants were collected. Supernatant concentrations of IL-6, TNF-α and TGF-β were measured using ELISA according to manufacturer’s protocols (Affymetrix, eBiosciences, CA, USA).
Measurement of mitochondrial reactive oxygen species
MCF7 mitochondrial reactive oxygen species (ROS) levels were measured using a ROS sensitive fluorescent probe, 1, 2, 3 dihydrorhodamine (DHR). Fluorescent marker dihydrorhodamine 123 was purchased from Molecular Probes, prepared as a 5 mM stock solution in dimethyl sulfoxide, and used at 1 μM. Briefly, cultured MCF7 cells were treated with Isc Qu, Dox and their combination with untreated cells maintained as controls. Following 72 h incubation, cells were harvested, washed twice, resuspended in 1 μM DHR and incubated at 37 °C for 20 min in the dark. Levels of intracellular ROS were examined with flow cytometry (FACS Calibur, BD Biosciences, USA). Excitation wavelength was 485 nm with peak emission measured at 530 nm. Data acquisition and analyses were carried out using Cell Quest software (BD Biosciences, USA).
Measurement of total intracellular reactive oxygen species
Generation of reactive oxygen species (ROS) in MCF7 cells was measured using a ROS sensitive fluorescent probe, 2,7-dichlorodihydrofluorescein diacetate (DCFH-DA). This probe can be oxidized to 2′,7′-dichlorofluorescein (DCF) by ROS and exhibits increased green fluorescence intensity. Briefly, cultured MCF7 cells were treated with Isc Qu, Dox and their combination with untreated cells maintained as controls. Following 72 h incubation, cells were harvested, washed twice, re-suspended in 10 mM DCFH-DA and incubated at 37 °C for 30 min in the dark. Levels of intracellular ROS were examined using flow cytometry (FACS Calibur, BD Biosciences, USA). Excitation wavelength used in measurements was 485 nm with peak emission measured at 530 nm. Data acquisition and analyses were carried out using Cell Quest software (BD Biosciences, USA).
Western blot analysis
Following 72 h treatment, protein samples extracted from MCF7 cells were used for immunoblot assay. Cells were washed twice with ice-cold PBS, and lysed in Cell Lysis Buffer (20 mM Tris pH 7.5, 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1% Triton X-100, 2.5 mM sodium pyrophosphate, 1 mM b-glycerolphosphate, 1 mM Na3PO4, 1 mg/mL Leupeptin, 1 mM PMSF). Cell lysates were centrifuged then normalized by the Lowry assay; proteins in lysates were separated by sodium dodecyl sulfate (SDS)- polyacrylamaide gel electrophoresis (PAGE). Proteins were transferred to nitrocellulose membranes and membranes were blocked with TBST containing 5% non-fat milk. Following overnight incubation of membranes with primary antibodies at 4 °C, membranes were washed with TBST containing 0.1% Tween 20, and incubated with goat antimouse peroxidase- conjugated secondary antibodies for 60 min at room temperature. Blots were probed with an enhanced chemiluminescence (ECL) substrate (Pierce, Thermo scientific) and exposed to Hyperfilm ECL to visualize the immunoreactive bands.
Statistical analysis
Data are presented as mean standard deviation (SEM) of three independent experiments. Comparisons between two groups were performed using Student’s t-test. p-value 0.05 was considered to be significant.
Electronic supplementary material
Acknowledgements
This work was supported by a grant from Ministry of Education and Science of the Republic of Serbia (Grant number III 41026).
Author Contributions
T.S.-R., J.F.S., and N.T.-M. performed all experiments. K.K., M.C., A.K.-R. and N.K. participated in the design of the experiments, revised critically the manuscript and contributed to the discussion. D.G. and M.J. revised critically the manuscript and added important points to the discussion. T.Z. and T.S.-R. designed the study, analyzed and interpreted data and wrote the manuscript. All authors approved the final draft of this manuscript.
Competing Interests
The authors declare that they have no competing interests.
Footnotes
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Change history
10/26/2018
A correction to this article has been published and is linked from the HTML and PDF versions of this paper. The error has been fixed in the paper.
Electronic supplementary material
Supplementary information accompanies this paper at doi:10.1038/s41598-017-03898-0
References
- 1.Ferlay J, et al. Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int. J. Cancer. 2010;127:2893–2917. doi: 10.1002/ijc.25516. [DOI] [PubMed] [Google Scholar]
- 2.Gonzalez-Angulo AM, Morales-Vasquez F, Hortobagyi GN. Overview of resistance to systemic therapy in patients with breast cancer. Adv. Exp. Med. Biol. 2007;608:1–22. doi: 10.1007/978-0-387-74039-3_1. [DOI] [PubMed] [Google Scholar]
- 3.Uy GB, et al. Breast cancer hormone receptor assay results of core needle biopsy and modified radical mastectomy specimens from the same patients. Clin Breast Cancer. 2010;10:154–159. doi: 10.3816/CBC.2010.n.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Reinert T, Barrios CH. Optimal management of hormone receptor positive metastatic breast cancer in 2016. Ther Adv Med Oncol. 2015;7:304–320. doi: 10.1177/1758834015608993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Minotti G, Menna P, Salvatorelli E, Cairo G, Gianni L. Anthracyclines: molecular advances and pharmacologic developments in antitumor activity and cardiotoxicity. Pharmacol. Rev. 2004;56:185–229. doi: 10.1124/pr.56.2.6. [DOI] [PubMed] [Google Scholar]
- 6.Sinha BK, Mimnaugh EG, Rajagopalan S, Myers CE. Adriamycin activation and oxygen free radical formation in human breast tumor cells: protective role of glutathione peroxidase in adriamycin resistance. Cancer Res. 1989;49:3844–3848. [PubMed] [Google Scholar]
- 7.Tassone P, et al. BRCA1 expression modulates chemosensitivity of BRCA1-defective HCC1937 human breast cancer cells. Br. J. Cancer. 2003;88:1285–1291. doi: 10.1038/sj.bjc.6600859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Weiss RB. The anthracyclines: will we ever find a better doxorubicin? Semin Oncol. 1992;19:670–686. [PubMed] [Google Scholar]
- 9.Gewirtz DA. A critical evaluation of the mechanisms of action proposed for the antitumor effects of the anthracycline antibiotics adriamycin and daunorubicin. Biochem Pharmacol. 1999;57:727–741. doi: 10.1016/S0006-2952(98)00307-4. [DOI] [PubMed] [Google Scholar]
- 10.Lu T, Finkel T. Free radicals and senescence. Exp Cell Res. 2008;314:1918–22. doi: 10.1016/j.yexcr.2008.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.D’Adda di Fagagna F. Living on a break: Cellular senescence as a DNA-damage response. Nat Rev Cancer. 2008;8:512–522. doi: 10.1038/nrc2440. [DOI] [PubMed] [Google Scholar]
- 12.Lord CJ, Ashworth A. The DNA damage response and cancer therapy. Nature. 2012;481:287–294. doi: 10.1038/nature10760. [DOI] [PubMed] [Google Scholar]
- 13.te Poele RH, Okorokov AL, Jardine L, Cummings J, Joel SP. DNA damage is able to induce senescence in tumor cells in vitro and in vivo. Cancer Res. 2002;62:1876–1883. [PubMed] [Google Scholar]
- 14.Braig M, et al. Oncogene-induced senescence as an initial barrier in lymphoma development. Nature. 2005;436:660–665. doi: 10.1038/nature03841. [DOI] [PubMed] [Google Scholar]
- 15.Schmitt CA, et al. A senescence program controlled by p53 and p16INK4a contributes to the outcome of cancer therapy. Cell. 2002;109:335–346. doi: 10.1016/S0092-8674(02)00734-1. [DOI] [PubMed] [Google Scholar]
- 16.Jackson JG, et al. p53-mediated senescence impairs the apoptotic response to chemotherapy andclinical outcome in breast cancer. Cancer Cell. 2012;21:793–806. doi: 10.1016/j.ccr.2012.04.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang Qin, Wu Peter C., Dong David Z., Ivanova Iana, Chu Elizabeth, Zeliadt Steven, Vesselle Hubert, Wu Daniel Y. Polyploidy road to therapy-induced cellular senescence and escape. International Journal of Cancer. 2012;132(7):1505–1515. doi: 10.1002/ijc.27810. [DOI] [PubMed] [Google Scholar]
- 18.Wang Q, et al. Survivin and escaping in therapy induced cellular senescence. Int J Cancer. 2011;128:1546–58. doi: 10.1002/ijc.25482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Henry J, et al. Acquired stem cell properties in therapy-induced senescence of lymphomas and acute leukemias in vitro and in vivo. Blood. 2013;122:4193. [Google Scholar]
- 20.Campisi J. Aging, cellular senescence, and cancer. Annu Rev Physiol. 2013;75:685–705. doi: 10.1146/annurev-physiol-030212-183653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rodier F, Campisi J. Four faces of cellular senescence. J Cell Biol. 2011;192:547–556. doi: 10.1083/jcb.201009094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cahu J, Bustany S, Sola B. Senescence-associated secretory phenotype favors the emergence of cancer stem-like cells. Cell Death Dis. 2012;3:e446. doi: 10.1038/cddis.2012.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Coppé JP, et al. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol. 2008;6:2853–68. doi: 10.1371/journal.pbio.0060301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bavik C, et al. The gene expression program of prostate fibroblast senescence modulates neoplastic epithelial cell proliferation through paracrine mechanisms. Cancer Res. 2006;66:794–802. doi: 10.1158/0008-5472.CAN-05-1716. [DOI] [PubMed] [Google Scholar]
- 25.Coppe JP, Desprez PY, Krtolica A, Campisi J. The senescence-associated secretory phenotype: the dark side of tumor suppression. Annu. Rev. Pathol. 2010;5:99–118. doi: 10.1146/annurev-pathol-121808-102144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Coppé JP, Kauser K, Campisi J, Beauséjour CM. Secretion of vascular endothelial growth factor by primary human fibroblasts at senescence. J Biol Chem. 2006;281:29568–29574. doi: 10.1074/jbc.M603307200. [DOI] [PubMed] [Google Scholar]
- 27.Ksiazek K, et al. Vulnerability to oxidative stress and different patterns of senescence in human peritoneal mesothelial cell strains. Am J Physiol Regul Integr Comp Physiol. 2009;296:R374–382. doi: 10.1152/ajpregu.90451.2008. [DOI] [PubMed] [Google Scholar]
- 28.Millis AJ, McCue HM, Kumar S, Baglioni C. Metalloproteinase and TIMP-1 gene expression during replicative senescence. Exp Gerontol. 1992;27:425–8. doi: 10.1016/0531-5565(92)90076-C. [DOI] [PubMed] [Google Scholar]
- 29.Kang MK, et al. Senescence-associated genes in normal human oral keratinocytes. Exp Cell Res. 2003;287:272–81. doi: 10.1016/S0014-4827(03)00061-2. [DOI] [PubMed] [Google Scholar]
- 30.Yevsa T, Kang TW, Zender L. Immune surveillance of pre-cancerous senescent hepatocytes limits hepatocellular carcinoma development. Oncoimmunology. 2012;1:398–399. doi: 10.4161/onci.19128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hoenicke L, Zender L. Immune surveillance of senescent cells–biological significance in cancer- and non-cancer pathologies. Carcinogenesis. 2012;33:1123–1126. doi: 10.1093/carcin/bgs124. [DOI] [PubMed] [Google Scholar]
- 32.Sagiv A, Krizhanovsky V. Immunosurveillance of senescent cells: the bright side of the senescence program. Biogerontology. 2013;14:617–28. doi: 10.1007/s10522-013-9473-0. [DOI] [PubMed] [Google Scholar]
- 33.Muñoz-Espín D, Serrano M. Cellular senescence: from physiology to pathology. Nat Rev Mol Cell Biol. 2014;15:482–96. doi: 10.1038/nrm3823. [DOI] [PubMed] [Google Scholar]
- 34.Pérez-Mancera PA, Young AR, Narita M. Inside and out: the activities of senescence in cancer. Nat Rev Cancer. 2014;14:547–58. doi: 10.1038/nrc3773. [DOI] [PubMed] [Google Scholar]
- 35.Dunn GP, Bruce AT, Ikeda H, Old LJ, Schreiber RD. Cancer immunoediting: from immunosurveillance to tumor escape. Nat Immunol. 2002;3:991–8. doi: 10.1038/ni1102-991. [DOI] [PubMed] [Google Scholar]
- 36.Kim R, Emi M, Tanabe K. Cancer immunoediting from immune surveillance to immune escape. Immunology. 2007;121:1–14. doi: 10.1111/j.1365-2567.2007.02587.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Augustin M, Bock PR, Hanisch J, Karasmann M, Schneider B. Safety and efficacy of the long-term adjuvant treatment of primary intermediate- to highrisk malignant melanoma (UICC/AJCC stage II and III) with a standardized fermented European mistletoe (Viscum album L.) extract. Results from a multicenter, comparative, epidemiological cohort study in Germany and Switzerland. Arzneimittelforschung. 2005;55:38–49. doi: 10.1055/s-0031-1296823. [DOI] [PubMed] [Google Scholar]
- 38.Bussing A, et al. Prevention of surgery-induced suppression of granulocyte function by intravenous application of a fermented extract from Viscum album L. in breast cancer patients. Anticancer Res. 2005;25:4753–4757. [PubMed] [Google Scholar]
- 39.Kienle GS, Kiene H. Review article: Influence of Viscum album L (European mistletoe) extracts on quality of life in cancer patients: a systematic review of controlled clinical studies. Integr Cancer Ther. 2010;9:142–157. doi: 10.1177/1534735410369673. [DOI] [PubMed] [Google Scholar]
- 40.Tröger W, et al. Quality of life and neutropenia in patients with early stage breast cancer: a randomized pilot study comparing additional treatment with mistletoe extract to chemotherapy alone. Breast Cancer (Auckl). 2009;3:35–45. doi: 10.4137/bcbcr.s2905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tröger W, et al. Viscum album [L.] extract therapy in patients with locally advanced or metastatic pancreatic cancer: a randomised clinical trial on overall survival. Eur J Cancer. 2013;49:3788–3797. doi: 10.1016/j.ejca.2013.06.043. [DOI] [PubMed] [Google Scholar]
- 42.Tröger W, Zdrale Z, Tišma N, Matijašević M. Additional Therapy with a Mistletoe Product during Adjuvant Chemotherapy of Breast Cancer Patients Improves Quality of Life: An Open Randomized Clinical Pilot Trial. Evid Based Complement Alternat Med. 2014;2014:430518. doi: 10.1155/2014/430518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tröger W, et al. Quality of life of patients with advanced pancreatic cancer during treatment with mistletoe. Dtsch Arztebl Int. 2014;111:493–502. doi: 10.3238/arztebl.2014.0493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dicato, M. A. Side effects of medical cancer therapy. In Prevention and Treatment. (ed. Springer-Verlag) (London 2012).
- 45.Kienle GS, et al. Mistletoe in cancer - a systematic review on controlled clinical trials. Eur J Med Res. 2003;8:109–19. [PubMed] [Google Scholar]
- 46.Eggenschwiler J, et al. Mistletoe lectin is not the only cytotoxic component in fermented preparations of Viscum album from white fir (Abies pectinata) BMC Complement. Altern. Med. 2007;7:14. doi: 10.1186/1472-6882-7-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Büssing A, Schietzel M. Apoptosis-inducing properties of Viscum album L. extracts from different host trees, correlate with their content of toxic mistletoe lectins. Anticancer. Res. 1999;19:23–28. [PubMed] [Google Scholar]
- 48.Elsässer-Beile U, Lusebrink S, Grussenmeyer U, Wetterauer U, Schultze-Seemann W. Comparison of the effects of various clinically applied mistletoe preparations on peripheral blood leukocytes. Arzneimittelforschung. 1998;48:1185–1189. [PubMed] [Google Scholar]
- 49.Bussing A, et al. Expression of mitochondrial Apo2.7 molecules and caspase-3 activation in human lymphocytes treated with the ribosome-inhibiting mistletoe lectins and the cell membrane permeabilizing viscotoxins. Cytometry. 1999;37:133–139. doi: 10.1002/(SICI)1097-0320(19991001)37:2<133::AID-CYTO6>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- 50.Bussing A, et al. Induction of apoptosis in human lymphocytes treated with Viscum album L. is mediated by the mistletoe lectins. Cancer Lett. 1996;99:59–72. doi: 10.1016/0304-3835(95)04038-2. [DOI] [PubMed] [Google Scholar]
- 51.Bantel H, Engels IH, Voelter W, Schulze-Osthoff K, Wesselborg S. Mistletoe lectin activates caspase-8/FLICE independently of death receptor signaling and enhances anticancer drug-induced apoptosis. Cancer Res. 1999;59:2083–90. [PubMed] [Google Scholar]
- 52.Duong Van Huyen JP, et al. Interleukin-12 is associated with the in vivo anti-tumor effect of mistletoe extracts in B16 mouse melanoma. Cancer Lett. 2006;243:32–37. doi: 10.1016/j.canlet.2005.11.016. [DOI] [PubMed] [Google Scholar]
- 53.Klingbeil MF, et al. Cytotoxic effects of mistletoe (Viscum album L.) in head and neck squamous cell carcinoma cell lines. Oncology reports. 2013;30:2316–2322. doi: 10.3892/or.2013.2732. [DOI] [PubMed] [Google Scholar]
- 54.Park YK, Do YR, Jang BC. Apoptosis of K562 leukemia cells by Abnobaviscum F(R), a European mistletoe extract. Oncology reports. 2012;28:2227–2232. doi: 10.3892/or.2012.2026. [DOI] [PubMed] [Google Scholar]
- 55.Seifert G, et al. Molecular mechanisms of mistletoe plant extract-induced apoptosis in acute lymphoblastic leukemia in vivo and in vitro. Cancer Lett. 2008;264:218–28. doi: 10.1016/j.canlet.2008.01.036. [DOI] [PubMed] [Google Scholar]
- 56.Valentiner U, Pfüller U, Baum C, Schumacher U. The cytotoxic effect of mistletoe lectins I, II and III on sensitive and multidrug resistant human colon cancer cell lines in vitro. Toxicology. 2002;171:187–199. doi: 10.1016/S0300-483X(01)00581-9. [DOI] [PubMed] [Google Scholar]
- 57.Siegle I, Fritz P, McClellan M, Gutzeit S, Murdter TE. Combined cytotoxic action of Viscum album agglutinin-1 and anticancer agents against human A549 lung cancer cells. Anticancer. Res. 2001;21:2687–2691. [PubMed] [Google Scholar]
- 58.Bantel H, Engels IH, Voelter W, Schulze-Osthoff K, Wesselborg S. Mistletoe lectin activates caspase-8/FLICE independently of death receptor signaling and enhances anticancer drug induced apoptosis. Cance.r Research. 1999;59:2083–2090. [PubMed] [Google Scholar]
- 59.Park W-B, et al. Inhibition of tumor growth and metastasis by Korean mistletoe lectin is associated with apoptosis and antiangiogenesis. Cancer. Biother. Radiopharm. 2001;16:439–447. doi: 10.1089/108497801753354348. [DOI] [PubMed] [Google Scholar]
- 60.Van Huyen JP, et al. Induction of apoptosis of endothelial cells by Viscum album: a role for anti-tumoral properties of mistletoe lectins. Mol. Med. 2002;8:600–606. doi: 10.1007/BF03402170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kienle, G. S. & Kiene, H. Die Mistel in der Onkologie: Fakten und konzeptionelle Grundlagen. Stuttgart, Germany: Schattauer Verlag (2003).
- 62.Büssing A, ed. Mistletoe. The Genus Viscum. Amsterdam, the Netherlands: Hardwood Academic (2000).
- 63.Gorter RW, van Wely M, Stoss M, Wollina U. Subcutaneous infiltrates induced by injection of mistletoe extracts (Iscador) Am J Ther. 1998;5:181–7. doi: 10.1097/00045391-199805000-00009. [DOI] [PubMed] [Google Scholar]
- 64.Urech Konrad, Schaller Gerhard, Jäggy Christoph. Viscotoxins, Mistletoe Lectins and their Isoforms in Mistletoe (Viscum album L.) Extracts Iscador. Arzneimittelforschung. 2011;56(06):428–434. doi: 10.1055/s-0031-1296808. [DOI] [PubMed] [Google Scholar]
- 65.Marvibaigi M, Supriyanto E, Amini N, Abdul Majid FA, Jaganathan SK. Preclinical and clinical effects of mistletoe against breast cancer. Biomed Res Int. 2014;2014:785479. doi: 10.1155/2014/785479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Srdic-Rajic T, et al. Sensitization of K562 Leukemia Cells to Doxorubicin by the Viscum album Extract. Phytother Res. 2016;30:485–495. doi: 10.1002/ptr.5554. [DOI] [PubMed] [Google Scholar]
- 67.Mosmann T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Methods. 1983;65:55–63. doi: 10.1016/0022-1759(83)90303-4. [DOI] [PubMed] [Google Scholar]
- 68.Ohno M, Abe T. Rapid colorimetric assay for the quantification of leukemia inhibitory factor (LIF) and interleukin-6 (IL-6) J Immunol Methods. 1991;145:199–203. doi: 10.1016/0022-1759(91)90327-C. [DOI] [PubMed] [Google Scholar]
- 69.Fulda S, Debatin KM. Apoptosis signaling in tumor therapy. Ann N Y Acad Sci. 2004;1028:150–156. doi: 10.1196/annals.1322.016. [DOI] [PubMed] [Google Scholar]
- 70.Decaudin D, Marzo I, Brenner C, Kroemer G. Mitochondria in chemotherapy-induced apoptosis: a prospective novel target of cancer therapy (review) Int J Oncol. 1998;12:141–52. [PubMed] [Google Scholar]
- 71.Green DR, Kroemer G. The pathophysiology of mitochondrial cell death. Science. 2004;305:626–629. doi: 10.1126/science.1099320. [DOI] [PubMed] [Google Scholar]
- 72.Marzo I, et al. Bax and adenine nucleotide translocator cooperate in the mitochondrial control of apoptosis. Science. 1998;281:2027–2031. doi: 10.1126/science.281.5385.2027. [DOI] [PubMed] [Google Scholar]
- 73.Petit PX, et al. Implication of mitochondria in apoptosis. Mol Cell Biochem. 1997;174:185–188. doi: 10.1023/A:1006848205880. [DOI] [PubMed] [Google Scholar]
- 74.Saleh A, Srinivasula SM, Acharya S, Fishel R, Alnemri ES. Cytochrome c and dATP-mediated oligomerization of Apaf-1 is a prerequisite for procaspase-9 activation. J Biol Chem. 1999;274:17941–17945. doi: 10.1074/jbc.274.25.17941. [DOI] [PubMed] [Google Scholar]
- 75.Shimizu S, Narita M, Tsujimoto Y. Bcl-2 family proteins regulate the release of apoptogenic cytochrome c by the mitochondrial channel VDAC. Nature. 1999;399:483–487. doi: 10.1038/20959. [DOI] [PubMed] [Google Scholar]
- 76.Yan M, Zhu P, Liu HM, Zhang HT, Liu L. Ethanol induced mitochondria injury and permeability transition pore opening: role of mitochondria in alcoholic liver disease. World J Gastroenterol. 2007;13:2352–2356. doi: 10.3748/wjg.v13.i16.2352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Collado M, Serrano M. Senescence in tumours: evidence from mice and humans. Nat Rev Cancer. 2010;10:51–57. doi: 10.1038/nrc2772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Jackson JG, Pereira-Smith OM. Primary and compensatory roles for RB family members at cell cycle gene promoters that are deacetylated and downregulated in doxorubicin-induced senescence of breast cancer cells. Mol Cell Biol. 2006;26:2501–2510. doi: 10.1128/MCB.26.7.2501-2510.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Elmore LW, et al. Adriamycin-induced senescence in breast tumor cells involves functional p53 and telomere dysfunction. J Biol Chem. 2002;277:35509–35515. doi: 10.1074/jbc.M205477200. [DOI] [PubMed] [Google Scholar]
- 80.Chang BD, et al. A senescence-like phenotype distinguishes tumor cells that undergo terminal proliferation arrest after exposure to anticancer agents. Cancer Res. 1999;59:3761–3767. [PubMed] [Google Scholar]
- 81.Di Micco R, et al. Oncogene-induced senescence is a DNA damage response triggered by DNA hyper-replication. Nature. 2006;444:638–642. doi: 10.1038/nature05327. [DOI] [PubMed] [Google Scholar]
- 82.Mallette FA, Gaumont-Leclerc MF, Ferbeyre G. The DNA damage signaling pathway is a critical mediator of oncogene-induced senescence. Genes Dev. 2007;21:43–48. doi: 10.1101/gad.1487307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Bunz F, et al. Requirement for p53 and p21 to sustain G2 arrest after DNA damage. Science. 1998;282:1497–501. doi: 10.1126/science.282.5393.1497. [DOI] [PubMed] [Google Scholar]
- 84.Baus F, Gire V, Fisher D, Piette J, Dulić V. Permanent cell cycle exit in G2 phase after DNA damage in normal human fibroblasts. EMBO J. 2003;22:3992–4002. doi: 10.1093/emboj/cdg387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Campisi J, d’Adda di Fagagna F. Cellular senescence: when bad things happen to good cells. Nat. Rev. Mol. Cell Biol. 2007;8:729–740. doi: 10.1038/nrm2233. [DOI] [PubMed] [Google Scholar]
- 86.Adams PD. Healing and hurting: molecular mechanisms, functions and pathologies of cellular senescence. Mol. Cell. 2009;36:2–14. doi: 10.1016/j.molcel.2009.09.021. [DOI] [PubMed] [Google Scholar]
- 87.Collins CJ, Sedivy JM. Involvement of the INK4a/Arf gene locus in senescence. Aging Cell. 2003;2:145–50. doi: 10.1046/j.1474-9728.2003.00048.x. [DOI] [PubMed] [Google Scholar]
- 88.Levine AJ, Oren M. The first 30 years of p53: growing ever more complex. Nat. Rev. Cancer. 2009;9:749–58. doi: 10.1038/nrc2723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Chau BN, Wang JY. Coordinated regulation of life and death by RB. Nat. Rev. Cancer. 2003;3:130–38. doi: 10.1038/nrc993. [DOI] [PubMed] [Google Scholar]
- 90.Takeuchi S, et al. Intrinsic cooperation between p16INK4a and p21Waf1/Cip1 in the onset of cellular senescence and tumor suppression in vivo. Cancer Res. 2010;70:9381–90. doi: 10.1158/0008-5472.CAN-10-0801. [DOI] [PubMed] [Google Scholar]
- 91.Zhang J, Pickering CR, Holst CR, Gauthier ML, Tlsty TD. p16INK4a modulates p53 in primary human mammary epithelial cells. Cancer Res. 2006;66:10325–31. doi: 10.1158/0008-5472.CAN-06-1594. [DOI] [PubMed] [Google Scholar]
- 92.Yamakoshi K, et al. Real-time in vivo imaging of p16Ink4a reveals cross talk with p53. J. Cell Biol. 2009;186:393–407. doi: 10.1083/jcb.200904105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Özcan S, et al. Unbiased analysis of senescence associated secretory phenotype (SASP) to identify common components following different genotoxic stresses. Aging (Albany NY). 2016;8:1316–1329. doi: 10.18632/aging.100971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Rodier F, et al. Persistent DNA damage signalling triggers senescence-associated inflammatory cytokine secretion. Nat Cell Biol. 2009;11:973–979. doi: 10.1038/ncb1909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Kojima H, Inoue T, Kunimoto H, Nakajima K. IL-6-STAT3 signaling and premature senescence. JAKSTAT. 2013;2:e25763. doi: 10.4161/jkst.25763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Kuilman T, et al. Oncogene-induced senescence relayed by an interleukin-dependent inflammatory network. Cell. 2008;133:1019–1031. doi: 10.1016/j.cell.2008.03.039. [DOI] [PubMed] [Google Scholar]
- 97.Strüh CM, et al. Triterpenoids amplify anti-tumoral effects of mistletoe extracts on murine B16.f10 melanoma in vivo. PLoS One. 2013;8:e62168. doi: 10.1371/journal.pone.0062168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Ruhland MK, et al. Stromal senescence establishes an immunosuppressive microenvironment that drives tumorigenesis. Nat Commun. 2016;7:11762. doi: 10.1038/ncomms11762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Eggert T, et al. Distinct Functions of Senescence-Associated Immune Responses in Liver Tumor Surveillance and Tumor Progression. Cancer Cell. 2016;30:533–547. doi: 10.1016/j.ccell.2016.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Doroshow JH, Davies KJ. Redox cycling of anthracyclines by cardiac mitochondria. II. Formation of superoxide anion, hydrogen peroxide, and hydroxyl radical. J Biol Chem. 1986;261:3068–3074. [PubMed] [Google Scholar]
- 101.Quinlan CL, Perevoschikova IV, Goncalves RL, Hey-Mogensen M, Brand MD. The determination and analysis of site-specific rates of mitochondrial reactive oxygen species production. Methods Enzymol. 2013;526:189–217. doi: 10.1016/B978-0-12-405883-5.00012-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Passos JF, et al. Mitochondrial dysfunction accounts for the stochastic heterogeneity in telomere-dependent senescence. PLoS Biol. 2007;5:e110. doi: 10.1371/journal.pbio.0050110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Moiseeva O, Bourdeau V, Roux A, Deschênes-Simard X, Ferbeyre G. Mitochondrial dysfunction contributes to oncogene-induced senescence. Mol Cell Biol. 2009;29:4495–4507. doi: 10.1128/MCB.01868-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Velarde MC, Flynn JM, Day NU, Melov S, Campisi J. Mitochondrial oxidative stress caused by Sod2 deficiency promotes cellular senescence and aging phenotypes in the skin. Aging (Albany NY). 2012;4:3–12. doi: 10.18632/aging.100423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Onay-Uçar E, Karagöz A, Arda N. Antioxidant activity of Viscum album ssp. album. Fitoterapia. 2006;77:556–60. doi: 10.1016/j.fitote.2006.08.001. [DOI] [PubMed] [Google Scholar]
- 106.Leu YL, Hwang TL, Chung YM, Hong PY. The inhibition of superoxide anion generation in human neutrophils by Viscum coloratum. Chem Pharm Bull (Tokyo). 2006;54:1063–106. doi: 10.1248/cpb.54.1063. [DOI] [PubMed] [Google Scholar]
- 107.Yao H, et al. Antioxidative flavanone glycosides from the branches and leaves of Viscum coloratum. Chem Pharm Bull (Tokyo). 2006;54:133–5. doi: 10.1248/cpb.54.133. [DOI] [PubMed] [Google Scholar]
- 108.Vicas S, Prokisch J, Rugina D, Socaciu C. Hydrophilic and Lipophilic Antioxidant Activities of Mistletoe (Viscum album) as determined by FRAP method. Notulae Botanicae Cluj-Napoca. 2009;37:112–116. [Google Scholar]
- 109.Choudhary MI, et al. Characterization and antiglycation activity of phenolic constituents from Viscum album (European Mistletoe) Chem Pharm Bull (Tokyo). 2010;58:980–982. doi: 10.1248/cpb.58.980. [DOI] [PubMed] [Google Scholar]
- 110.Storz P. Reactive oxygen species in tumor progression. Front Biosci. 2005;10:1881–1896. doi: 10.2741/1667. [DOI] [PubMed] [Google Scholar]
- 111.Doroshow JH, Davies KJ. Redox cycling of anthracyclines by cardiac mitochondria. II. Formation of superoxide anion, hydrogen peroxide, and hydroxyl radical. J Biol Chem. 1986;261:3068–3074. [PubMed] [Google Scholar]
- 112.Davies KJ, Doroshow JH. Redox cycling of anthracyclines by cardiac mitochondria. I. Anthracycline radical formation by NADH dehydrogenase. J Biol Chem. 1986;261:3060–3067. [PubMed] [Google Scholar]
- 113.Deschênes-Simard X, et al. Tumor suppressor activity of the ERK/MAPK pathway by promoting selective protein degradation. Genes Dev. 2013;27:900–915. doi: 10.1101/gad.203984.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Ostman A, Frijhoff J, Sandin A, Böhmer FD. Regulation of protein tyrosine phosphatases by reversible oxidation. J Biochem. 2011;150:345–56. doi: 10.1093/jb/mvr104. [DOI] [PubMed] [Google Scholar]
- 115.Schieven GL. The p38alpha kinase plays a central role in inflammation. Curr Top Med Chem. 2009;9:1038–1048. doi: 10.2174/156802609789630974. [DOI] [PubMed] [Google Scholar]
- 116.Guyton KZ, Liu Y, Gorospe M, Xu Q, Holbrook NJ. Activation of mitogen-activated protein kinase by H2O2. Role in cell survival following oxidant injury. J Biol Chem. 1996;271:4138–4142. doi: 10.1074/jbc.271.8.4138. [DOI] [PubMed] [Google Scholar]
- 117.Chiarugi P. PTPs versus PTKs: the redox side of the coin. Free Radic Res. 2005;39:353–364. doi: 10.1080/10715760400027987. [DOI] [PubMed] [Google Scholar]
- 118.Salama R, Sadaie M, Hoare M, Narita M. Cellular senescence and its effector programs. Genes Dev. 2014;28:99–114. doi: 10.1101/gad.235184.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Acosta JC, et al. Chemokine signaling via the CXCR2 receptor reinforces senescence. Cell. 2008;133:1006–1018. doi: 10.1016/j.cell.2008.03.038. [DOI] [PubMed] [Google Scholar]
- 120.Kuilman T, et al. Oncogene induced senescence relayed by an interleukin-dependent inflammatory network. Cell. 2008;133:1019–1031. doi: 10.1016/j.cell.2008.03.039. [DOI] [PubMed] [Google Scholar]
- 121.Wajapeyee N, Serra RW, Zhu X, Mahalingam M, Green MR. Oncogenic BRAF induces senescence and apoptosis through pathways mediated by the secreted protein IGFBP7. Cell. 2008;132:363–374. doi: 10.1016/j.cell.2007.12.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Kortlever RM, Higgins PJ, Bernards R. Plasminogen activator inhibitor-1 is a critical downstream target of p53 in the induction of replicative senescence. Nat Cell Biol. 2006;8:877–884. doi: 10.1038/ncb1448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Lujambio A, Villanueva A. The usual SASPects of liver cancer. Aging (Albany NY). 2015;7:348–349. doi: 10.18632/aging.100758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Krizhanovsky V, et al. Implications of cellular senescence in tissue damage response, tumor suppression, and stem cell biology. Cold Spring Harb Symp Quant Biol. 2008;73:513–522. doi: 10.1101/sqb.2008.73.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Jun JI, Lau LF. Cellular senescence controls fibrosis in wound healing. Aging (Albany NY). 2010;2:627–631. doi: 10.18632/aging.100201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Demaria M, et al. An essential role for senescent cells in optimal wound healing through secretion of PDGF-AA. Dev Cell. 2014;31:722–733. doi: 10.1016/j.devcel.2014.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Ventura A, et al. Restoration of p53 function leads to tumour regression in vivo. Nature. 2007;445:661–665. doi: 10.1038/nature05541. [DOI] [PubMed] [Google Scholar]
- 128.Xue W, et al. Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature. 2007;445:656–660. doi: 10.1038/nature05529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Iannello A, Thompson TW, Ardolino M, Lowe SW, Raulet DH. p53-dependent chemokine production by senescent tumor cells supports NKG2D-dependent tumor elimination by natural killer cells. J Exp Med. 2013;210:2057–2069. doi: 10.1084/jem.20130783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Laberge RM, Awad P, Campisi J, Desprez PY. Epithelial-mesenchymal transition induced by senescent fibroblasts. Cancer Microenviron. 2012;5:39–44. doi: 10.1007/s12307-011-0069-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Kelly J, Ali Khan A, Yin J, Ferguson TA, Apte RS. Senescence regulates macrophage activation and angiogenic fate at sites of tissue injury in mice. J Clin Invest. 2007;117:3421–3426. doi: 10.1172/JCI32430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Toso A, Di Mitri D, Alimonti A. Enhancing chemotherapy efficacy by reprogramming the senescence-associated secretory phenotype of prostate tumors: A way to reactivate the antitumor immunity. Oncoimmunology. 2015;4:e994380. doi: 10.4161/2162402X.2014.994380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Keyse SM. Dual-specificity MAP kinase phosphatases (MKPs) and cancer. Cancer Metastasis Rev. 2008;27:253–261. doi: 10.1007/s10555-008-9123-1. [DOI] [PubMed] [Google Scholar]
- 134.Saccani S, Pantano S, Natoli G. p38-Dependent marking of inflammatory genes for increased NF-kappa B recruitment. Nat Immunol. 2002;3:69–75. doi: 10.1038/ni748. [DOI] [PubMed] [Google Scholar]
- 135.Kuilman T, et al. Oncogene-induced senescence relayed by an interleukin-dependent inflammatory network. Cell. 2008;133:1019–1031. doi: 10.1016/j.cell.2008.03.039. [DOI] [PubMed] [Google Scholar]
- 136.Matsuoka T, Hardy C, Tavassoli M. Characterization of membrane homing receptors in two cloned murine hemopoietic progenitor cell lines. J Clin Invest. 1989;83:904–911. doi: 10.1172/JCI113975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Delebinski CI, et al. A Natural Combination Extract of Viscum album L. Containing Both Triterpene Acids and Lectins Is Highly Effective against AML In Vivo. PLoS One. 2015;10:e0133892. doi: 10.1371/journal.pone.0133892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Dumitru CA, Carpinteiro A, Trarbach T, Hengge UR, Gulbins E. Doxorubicin enhances TRAIL-induced cell death via ceramide-enriched membrane platforms. Apoptosis. 2007;12:1533–1541. doi: 10.1007/s10495-007-0081-9. [DOI] [PubMed] [Google Scholar]
- 139.Kelly MM, Hoel BD, Voelkel-Johnson C. Doxorubicin pretreatment sensitizes prostate cancer cell lines to TRAIL induced apoptosis which correlates with the loss of c-FLIP expression. Cancer. Biol. Ther. 2002;1:520–527. doi: 10.4161/cbt.1.5.169. [DOI] [PubMed] [Google Scholar]
- 140.Kang J, Bu J, Hao Y, Chen F. Subtoxic concentration of doxorubicin enhances TRAIL-induced apoptosis in human prostate cancer cell line LNCaP. Prostate. Cancer. Prostatic. Dis. 2005;8:274–279. doi: 10.1038/sj.pcan.4500798. [DOI] [PubMed] [Google Scholar]
- 141.Ormerod, M. G. Analysis of DNA-General Methods. Flow Cytometry, a Practical Approach (ed. Ormerod M.G.) 119–125 (Oxford University Press, New York, 1994).
- 142.Kocic J, et al. Interleukin-17 modulates myoblast cell migration by inhibiting urokinase type plasminogen activator expression through p38 mitogen-activated protein kinase. Int. J. Biochem. Cell Biol. 2012;45:464–475. doi: 10.1016/j.biocel.2012.11.010. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.