

Abstract
The characteristics of intestinal microbial communities may be affected by changes in the pathophysiology of patients with end-stage liver disease. Here, we focused on the characteristics of intestinal fecal microbial communities in post-liver transplantation (LT) patients in comparison with those in the same individuals pre-LT and in healthy individuals. The fecal microbial communities were analyzed via MiSeq-PE250 sequencing of the V4 region of 16S ribosomal RNA and were then compared between groups. We found that the gut microbiota of patients with severe liver disease who were awaiting LT was significantly different from that of healthy controls, as represented by the first principal component (p = 0.0066). Additionally, the second principal component represented a significant difference in the gut microbiota of patients between pre-LT and post-LT surgery (p = 0.03125). After LT, there was a significant decrease in the abundance of certain microbial species, such as Actinobacillus, Escherichia, and Shigella, and a significant increase in the abundance of other microbial species, such as Micromonosporaceae, Desulfobacterales, the Sarcina genus of Eubacteriaceae, and Akkermansia. Based on KEGG profiles, 15 functional modules were enriched and 21 functional modules were less represented in the post-LT samples compared with the pre-LT samples. Our study demonstrates that fecal microbial communities were significantly altered by LT.
Introduction
With the development of sequencing techniques, accumulating research on the connection between the human gut microbiota and disease has been conducted. This research includes studies on the association of gut microbiota alterations with liver diseases, such as obesity-related liver disease1, non-alcoholic fatty liver disease2, 3, autoimmune liver disease4, and liver cirrhosis5, 6. Liver cirrhosis is characterized by the pathological changes of end-stage liver diseases, and its clinical manifestations include a series of complications associated with portal hypertension, such as hepatic encephalopathy, spontaneous bacterial peritonitis, portal hypertensive gastropathy, and bleeding from esophageal varices. Many studies have reported that these clinical manifestations are associated with the intestinal microbial community7–10. Liver transplantation (LT) resolves the associated problems in patients with liver failure or portal hypertension; following LT, the liver function of patients gradually returns to normal and portal hypertension-related gastrointestinal diseases can be alleviated11.
Currently, no study has reported on the differences in the intestinal microbiota of patients who received LT, as measured using 16S ribosomal RNA (rRNA) sequencing technology. Lu et al.12 analyzed the variation in the microbiome during the perioperative period in LT patients by performing denaturing gradient gel electrophoresis (DGGE), and their results showed that the diversity in predominant intestinal microbes decreased substantially in patients who experienced infection during the perioperative period.
Using MiSeq-PE250 sequencing technology combined with bioinformatics analysis, we studied the characteristics of the intestinal microbiota from patients with end-stage liver disease before and after they received LT. The purpose of this study was to analyze the differences in the intestinal microflora of patients between pre-LT and post-LT to investigate the effect of LT therapy and subsequent immunosuppressive treatment on the intestinal microflora, thereby providing experimental data further revealing the mechanism by which liver diseases alter the gut microbiota.
Methods
Study population
This study included 33 samples from 24 participants, nine of whom initially had an end-stage liver disease (1 sample each pre- and post-LT, for a total of 18 samples) and 15 of whom were healthy controls. All details of the entire study design and procedures involved were established in accordance with the Declaration of Helsinki. Written informed consent forms were signed before the time of sample collection. This protocols were approved by the Ethics Committee of Tianjin First Central Hospital. Of the nine LT patients, four had liver failure because of decompensated cirrhosis, and five had hepatocellular carcinoma (HCC) (the characteristics of the patients are presented in Supplementary Tables 1, 2 and 3). All patients were undergoing primary non-bypass orthotopic LT for the first time, and the method of biliary anastomosis was end-to-end anastomosis. Furthermore, all nine patients were male and 49.4 ± 13.9 years old. Before LT, the patients provided fecal samples and underwent blood tests. Each patient was re-examined 3 months after LT, at which time the patients provided a second fecal sample. The 15 healthy controls were individuals who visited the physical examination center of the hospital during the study period and who had normal blood test results. Among the 15 healthy controls, 11 were male and 4 were female, aged 48 ± 4.19 years (see Supplementary Table 4). There was no significant difference (see Supplementary Tables 5 and 6). None of the individuals in this study used antimicrobial agents in the month prior to enrollment or had any evidence of systemic bacterial, viral, or fungal infection. All post-LT patients received immunosuppression therapy with tacrolimus and methylprednisolone. In the first 5 to 7 days after surgery, the patients received antibiotic treatment with cephalosporin to prevent infection. No vascular complications or infections occurred through the time of patient discharge.
Sampling and DNA extraction
Fecal samples were frozen at −80 °C immediately upon collection and then stored for later use. At the beginning of the experiment, 180–200 mg of each sample was weighed out and transferred to a 2-ml centrifuge tube, which was then placed on ice. DNA was extracted from the samples using the repeated bead beating plus column (RBB+C) method. Briefly, ASL buffer (1.4 ml) was added to each sample, followed by the addition of 0.4 g of sterile zirconia beads (0.3 g of 0.1-mm beads and 0.1 g of 0.5-mm beads). The samples were subjected to bead beating (3 min, maximum speed) using a Mini-BeadBeater-16 cell disruptor (BioSpec Products, Bartlesville, OK, USA) prior to an initial incubation for lysis via heating at 70 °C for 15 min, with thorough shaking every 5 min. Subsequent steps of the DNA extraction protocol followed the instructions from the manufacturer of the QIAamp Stool Mini Kit (Qiagen, Germany) for bacterial DNA extraction.
16S rRNA gene amplicon sequencing using a MiSeq sequencer
For sequencing, isolated fecal DNA was used as a template for amplification, and the V4 region of 16S rRNA was amplified by performing PCR assays using the universal bacterial primer set 515F (5′-GTGCCAGCMGCCGCGGTAA-3′) and 806R (5′-GGACTACHVGGGTWTCTAAT-3′).
The PCR reaction mixture (30 μl) contained 2× High-Fidelity PCR Master Mix, 3 µl of primer (2 μM), and 10 μl of gDNA, and the reaction steps were as follows: 98 °C for 1 min, followed by 35 cycles of 98 °C for 10 s, 50 °C for 30 s, and 72 °C for 30 s, with a final elongation at 72 °C for 5 min. The reactions were conducted in a Veriti 96-Well Thermal Cycler (Biometra, Germany). The resulting PCR products were purified via separation on a 2% agarose gel, followed by DNA isolation using the QIAquick Gel Extraction Kit (Qiagen, Santa Clarita, CA, USA) according to the manufacturer’s instructions. The purified DNA was quantified using a Qubit® 2.0 Quantitation Starter Kit (Invitrogen, USA) according to the manufacturer’s instructions. The partial 16S rRNA genes were sequenced using an Illumina MiSeq sequencer.
Bioinformatics analysis
Data processing
Paired end reads were overlapped using FLASH-1.2.1013 with the parameters ‘-m 150 -M 250 -x 0.15 -O’; no mismatches in primers or barcodes were allowed, and no ambiguous bases (N) in whole overlapped reads were allowed. When assigning reads to samples, the barcodes had to be consistent with the reference barcode sequences. Finally, we removed chimeras using Uchime14.
Binning of operational taxonomic units (OTUs)
Usearch15 was used to bin reads into OTUs with a 0.97 identity cut-off. Samples with more sequenced reads had more observed OTUs (rho = 0.78; p = 2.533 × 10−07, Spearman’s rank correlation analysis) when all reads were binned into OTUs. Thus, we randomly chose reads with the same number (10,000) and then identified representative OTU reads. Finally, we mapped all randomly chosen reads against representative OTU reads to obtain the OTU composition of all samples.
Diversity and richness calculation
Shannon, simpson, and invsimpson indices were calculated using the ‘vegan’ package in R16 with a normalized OTU matrix. The observed species, Chao1, and ICE indices were calculated using the ‘fossil’ package in R17 with a non-normalized OTU matrix.
Beta-diversity calculation
The Hellinger distance was calculated with the ‘topicmodels’ package in R18. The JSD distance was calculated with a custom R script provided by the European Molecular Biology Laboratory enterotyping tutorial (http://enterotype.embl.de/enterotypes.html). Spearman’s rank correlation distance was calculated using the following command in R: ‘D < −1 - cor.test (X, method = “spearman”)’, where X is the OTU composition of all samples. All three distances were calculated based on the OTU composition.
To calculate the unifrac distance, we constructed an abundance profile of unique reads from all randomly chosen reads (singletons were removed). We used MUSCLE v3.8.3119 to generate multiple alignments with the default parameters and used FastTree version 2.1.3 SSE320 to construct a phylogenetic tree with a generalized time-reversible model. Finally, we used the ‘phyloseq’ package in R21 to calculate the unifrac distance with the following command: ‘Weighted_Unifrac_distance < -UniFrac (x1, weighted = T, normalized = T, fast = T)’, where ‘x1’ is a combination of the phylogenetic tree and the abundance of unique reads (the ‘weighted = F’ parameter was used to calculate the unweighted unifrac distance).
KEGG orthology and pathway profiles
KEGG orthology profiles were constructed using the piCrust version 1.0.0 pipeline22. First, our custom representative OTU reads were aligned against the greenGene_Database_16S_rRNA Fasta reference database (2013 version), and the abundances of representative OTU reads from the same 16S rRNA reference were then summed. The reference profile normalized by the 16S rRNA copy number was used to predict the KEGG orthology profile, and HUMAnN software version 0.9923 was subsequently used to determine the abundance of KEGG pathways and modules.
Statistical analysis
The Wilcoxon rank sum test based on the null hypothesis was performed to evaluate alpha diversity, principal coordinates, and KEGG module profiles between the different cohorts. A paired test was used to compare samples from the same individual (pre-LT and post-LT cohorts) with the following command in R: ‘wilcox.test (a, b, paired = T, alternative = “less”)’, where a and b are numeric vectors of the data for the pre-LT and post-LT samples, respectively. LEfSe24 was used to compare the bacterial populations between different cohorts. When using LEfSe to perform a Wilcoxon rank sum test between the pre-LT and post-LT samples, we made a replace correction using the p-values generated from a paired test.
Enterotype clustering
We used the ‘NbClust’ package in R25 to identify the number of clusters into enterotypes based on the OTU composition. We then used an exhaustive combination of two clustering methods (k-means method and complete cluster linkage method) and five different beta-diversities (weighted unifrac distance, unweighted unfired distance, JSD distance, Spearman’s rank correlation distance, and Euclidean distance), with indices integrated in NbClust to confirm the optimal number of clusters. Finally, the number of clusters was identified according to a major rule.
Results
DNA sequencing results
All 33 samples were pooled into one sequencing library, and a total of 1,907,569 raw reads were sequenced. After quality control and chimera removal, 1,281,639 reads remained for downstream analysis (Supplementary Table 7). To avoid bias caused by sequencing unevenness, we randomly downsized the sequences of all samples at the same level with a minimum cut-off number of 10,000.
Randomly chosen reads were binned into OTUs with a 0.97 identity cut-off, and 505 OTUs were considered to be representative OTUs. We then mapped all clean reads (1,281,639 reads before downsizing) against these representative reads, and 90.341% of reads were able to be re-binned. No significant differences in microbial richness (observed species, Chao1, and ICE indices) or diversity (shannon, simpson, and invsimpson indices) were found between any of the three cohorts (pre-LT patients, post-LT patients, and healthy controls) (p < 0.05) (Table 1).
Table 1.
PCoA based on OTU composition.
| P value | pre_control | pre_post | control_post |
|---|---|---|---|
| PC1 | 0.00661413 | 0.375 | 0.36840933 |
| PC2 | 0.05555165 | 0.03125 | 0.07771601 |
| PC3 | 0.83659349 | 0.29688 | 0.26659396 |
Changes to gut microbiota composition in recipients of LT surgery
Samples pre_7 and post_1 were considered to be outliers and were excluded from our analysis. We also excluded the corresponding samples from the same two individuals: post_7 and pre_1 (for details, see Supplementary Figs 1 and 2). After exclusion of these samples, 29 (7 pre-LT, 7 post-LT, and 15 control samples) remained for downstream analysis. The results of a principal component analysis (PCoA; weighted unifrac distance) based on OTU composition showed that the gut microbiota of patients with severe liver disease who were awaiting LT surgery (pre-LT) were significantly different from that of control patients, which corresponded to the first principal component (PC1, p = 0.0066). Additionally, a second principal component (PC2) corresponded to a significant difference in the gut microbiota of the same patients between pre-LT and post-LT (p = 0.03125). However, there was no significant difference in the top three principal components between the post-LT patients and the controls (Fig. 1a and Table 1). We observed a reduction in the unweighted unifrac distance between the post-LT and control groups compared with that between the pre-LT and control groups (Fig. 1b, p = 0.03823), indicating that the gut microbiota composition of LT recipients became more similar to that of healthy controls after the LT surgery.
Figure 1.
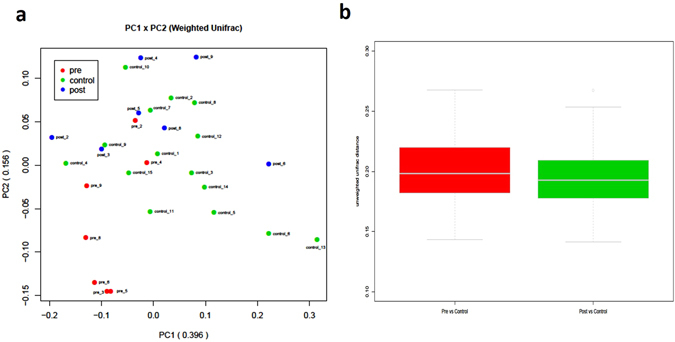
The results of a PCoA based on OTU composition are shown as graphs of the (a) LT-filtered weighted unifrac distance and (b) the unifrac distance from the pre-LT and post-LT samples to the control samples.
Previous studies have demonstrated a clear stratification of human gut microbiota compositions, i.e., enterotypes, in populations that is neither nation- nor continent-specific26. Thus, we aimed to determine whether enterotype is associated with the changes in the gut microbiota observed in a patient after LT surgery. All samples could be clustered into three enterotypes according to a major rule (Fig. 2a and b, weighted unifrac distance, k-means clustering method). We found that the enterotype distribution was not associated with LT surgery (Fig. 2c, Fisher’s exact test of the pre-LT and post-LT groups). Based on the relative abundance of Bacteroides, Prevotella, and Faecalibacterium in the gut microbial population, we clustered the samples into three enterotypes (Fig. 2d). While the enterotype of three recipients (cases 3, 8, and 6) changed following LT, the gut microbiotas of two of these individuals (3 and 8) had completely recovered (shifted from E1 to E2 with significantly reduced beta-diversity). For case 6, although the weighted unifrac distance from the healthy control group was not significantly changed, the sample after LT surgery (post_6) was binned into E3, which otherwise consisted solely of healthy controls.
Figure 2.
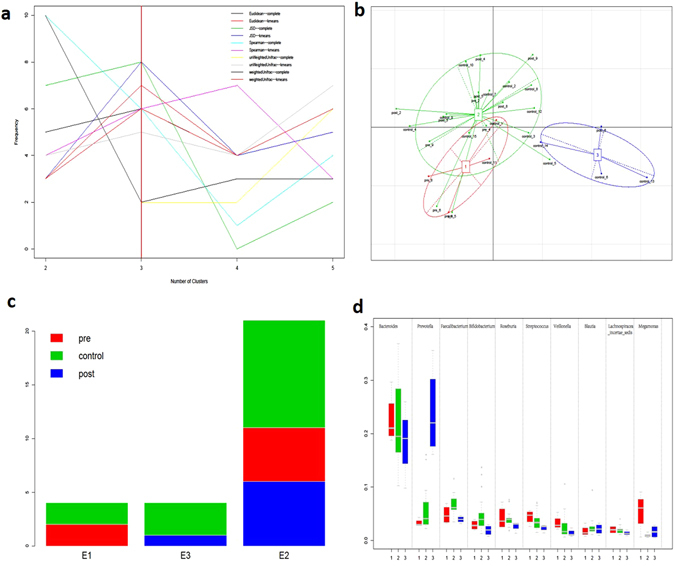
The enterotypes of samples from the pre-LT, post-LT, and healthy control groups were analyzed. (a) The number of enterotype clusters. (b) The enterotypes of all samples. (c) The distribution of enterotypes. (d) The genus distribution of all enterotypes.
Of the four recipients without an enterotype change after receiving LT (cases 2, 4, 5, and 9), the gut microbiota of two cases (2 and 4) significantly skewed from that of the healthy controls after receiving LT surgery (beta-diversity was significantly increased). Furthermore, these two samples (pre_2 and pre_4) were homogeneously mixed with the control samples (Fig. 1a), indicating that LT surgery might have slightly disturbed the gut microbiota in these two cases. A follow-up trial showed that biliary complications (biliary anastomotic stricture) occurred in these two samples, but our data are insufficient to confirm an association between the gut microbiota and biliary complications. The gut microbiota of case 5 (post_5) had also recovered, even though no enterotype change was observed (Table 2).
Table 2.
Changes to gut microbiota composition following LT surgery.
| Case | Enterotype changea | P valueb | More similar |
|---|---|---|---|
| 2 | E2-E2 | 1.72E-05 | Prec |
| 4 | E2-E2 | 0.033748 | Pre |
| 3 | E1-E2 | 0.011748 | Postd |
| 5 | E2-E2 | 4.49E-05 | Post |
| 8 | E1-E2 | 0.000132 | Post |
| 6 | E2-E3 | 0.564903 | Nonee |
| 9 | E2-E2 | 0.10084 | None |
aEnterotype change represents the enterotype of individual samples taken before (pre, left) and after (post, right) LT.
bBased on a unilateral Wilcoxon rank sum test of the weighted unifrac distances between the pre-LT or post-LT sample and samples from the 15 healthy controls.
cPre indicates that the weighted unifrac distance from the pre-LT sample to the healthy control samples is shorter than that from the post-LT sample to the healthy controls samples.
dPost indicates that the weighted unifrac distance from the post-LT sample to the healthy control samples is shorter than that from the pre-LT sample to the healthy control samples.
eNone indicates that there is no significant difference (p > 0.05) in the weighted unifrac distances to the samples from the 15 healthy controls between the pre-LT and post-LT samples.
Differential species abundance
We used a LEfSe analysis to detect differential abundances of species between the pre-LT, post-LT, and control samples. Because the pre-LT and post-LT samples belonged to the same individuals, we performed a correction for unilateral inspection, using the results of a Wilcoxon rank sum test in our LEfSe analysis. We then identified differential distributions of species using the default cut-offs (LDA score > 2.0; p value < 0.05).
We found that the relative abundances of Actinobacillus, Escherichia, Shigella, Anaerolineaceae, Fusobacteriales, Clostridium (sensu stricto), Fusobacteriaceae, Aeromonas, and Clostridium cluster XVIII, among others, were significantly decreased but that the relative abundances of Micromonosporaceae, Desulfobacterales, Eubacteriaceae, Sarcina, Akkermansia, Chitinophagaceae, and Coriobacteriaceae were significantly increased in post-LT samples compared with pre-LT samples (Figs 3 and 4). Notably, there were no differences in the relative abundances of these species between the post-LT samples and the control samples (Fig. 5; low-abundance species are not shown). Although not all of the cases showed an enterotype change following the LT surgery, the relative abundance of Akkermansia changed after LT surgery in all individuals (mean fold-change: 2.92, range: 1.67–20.49; Fig. 6). Additionally, the relative abundances of Clostridium cluster XIVa, Corynebacterium, and Blautia were significantly increased following LT surgery, and their relative abundances in the post-LT samples were greater than those in the control samples.
Figure 3.
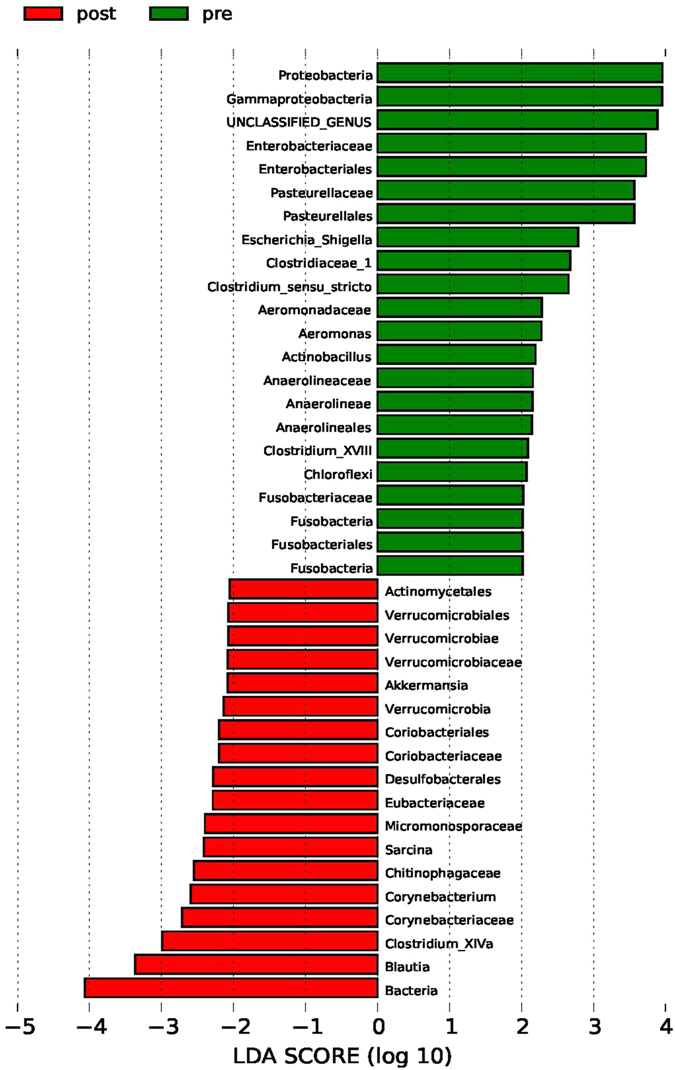
LEfSe analysis of differential species abundance between the pre-LT and post-LT groups was performed. The LDA score is used as the abscissa, and higher scores indicate a greater difference. The red and green horizontal bars represent the species enrichment in the post-LT and pre-LT groups, respectively.
Figure 4.
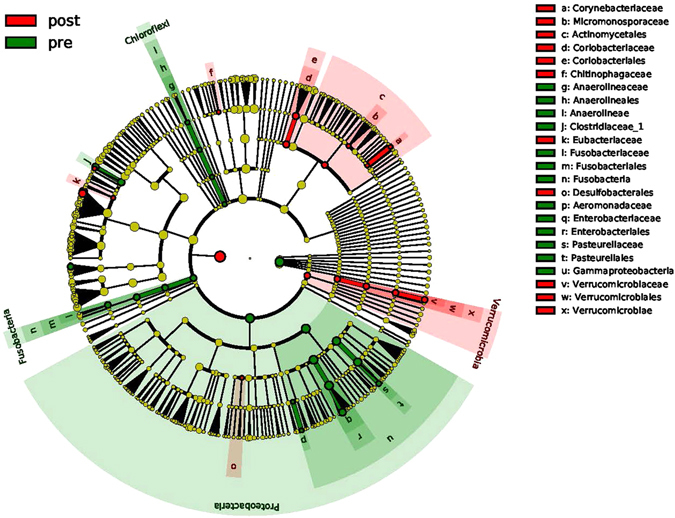
A cladogram plot of differential species abundance between the pre-LT and post-LT groups is shown. The red and green areas represent the species enrichment in the post-LT and pre-LT groups, respectively.
Figure 5.
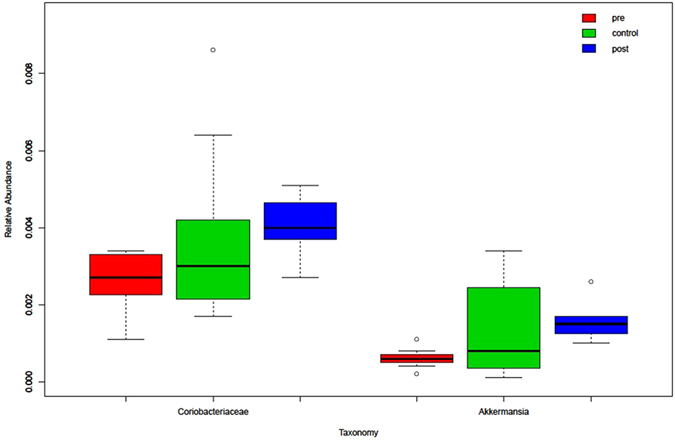
The distributions of Coriobacteriaceae and Akkermansia in all samples from the pre-LT, post-LT, and healthy control groups are shown.
Figure 6.
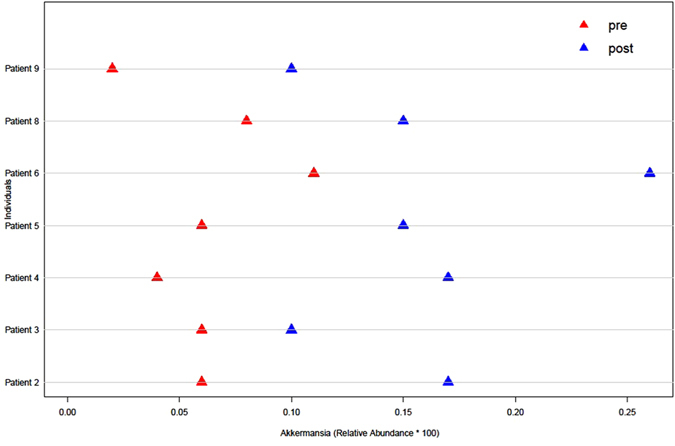
The abundance of Akkermansia in samples from patients collected before and after LT surgery is shown.
Differences in metabolic pathways between groups
We transformed the composition of the OTU sequence into KEGG orthology to analyze the differences in the metabolic pathways represented in the gut microbiota between pre-LT and post-LT samples. We generated the KEGG profiles and then compared the components of functional genomics in KEGG pathways, yielding 118 KEGG modules. Of these, 15 functional modules were enriched in the post-LT samples and 21 functional modules were less represented in the post-LT samples compared with the pre-LT samples (Tables 3 and 4).
Table 3.
Modules enriched in post-LT samples compared with pre-LT samples.
| Enriched post-LT | P | Functional description |
|---|---|---|
| M00233 | 0.0234375 | Glutamate_transport_system |
| M00051 | 0.0234375 | Uridine_monophosphate_biosynthesis,_glutamine_(+_PRPP)_=>_UMP |
| M00144 | 0.0234375 | Complex_I_(NADH_dehydrogenase),_NADH_dehydrogenase_I |
| M00232 | 0.0234375 | General_L-amino_acid_transport_system |
| M00088 | 0.0234375 | Ketone_body_biosynthesis,_acetyl-CoA_=>_acetoacetate/3-hydroxybutyrate/acetone |
| M00342 | 0.029529115 | Bacterial_proteasome |
| M00176 | 0.029529115 | Sulfur_reduction,_sulfate_=>_H2S |
| M00116 | 0.029586034 | Menaquinone_biosynthesis,_chorismate_=>_menaquinone |
| M00206 | 0.029586034 | Cellobiose_transport_system |
| M00036 | 0.029586034 | Leucine_degradation,_leucine_=>_acetoacetate_+_acetyl-CoA |
| M00200 | 0.029586034 | Sorbitol/mannitol_transport_system |
| M00156 | 0.029586034 | Complex_IV_(Cytochrome_c_oxidase),_cytochrome_c_oxidase,_cbb3-type |
| M00174 | 0.029586034 | Methane_oxidation,_methylotroph,_methane_=>_CO2 |
| M00277 | 0.0390625 | PTS_system,_N-acetylgalactosamine-specific_II_component |
| M00033 | 0.046746242 | Ectoine_biosynthesis |
Table 4.
Modules enriched in pre-LT samples compared with post-LT samples.
| Enriched pre-LT | P | Functional description |
|---|---|---|
| M00017 | 0.0078125 | Methionine_biosynthesis,_aspartate_=>_homoserine_=>_methionine |
| M00229 | 0.0078125 | Arginine_transport_system |
| M00019 | 0.0078125 | Leucine_biosynthesis,_pyruvate_=>_2-oxoisovalerate_=>_leucine |
| M00324 | 0.0078125 | Dipeptide_transport_system |
| M00150 | 0.0078125 | Complex_II_(succinate_dehydrogenase_/_fumarate_reductase),_fumarate_reductase |
| M00121 | 0.0078125 | Heme_biosynthesis,_glutamate_=>_protoheme/siroheme |
| M00064 | 0.0078125 | ADP-L-glycero-D-manno-heptose_biosynthesis |
| M00025 | 0.0156250 | Tyrosine_biosynthesis,_chorismate_=>_tyrosine |
| M00260 | 0.0234375 | DNA_polymerase_III_complex,_bacteria |
| M00335 | 0.0234375 | Sec_(secretion)_system |
| M00136 | 0.0234375 | GABA_biosynthesis,_prokaryotes,_putrescine_=>_GABA |
| M00230 | 0.0234375 | Glutamate/aspartate_transport_system |
| M00060 | 0.0234375 | Lipopolysaccharide_biosynthesis,_KDO2-lipid_A |
| M00276 | 0.0390625 | PTS_system,_mannose-specific_II_component |
| M00336 | 0.0390625 | Twin-arginine_translocation_(Tat)_system |
| M00008 | 0.0390625 | Entner-Doudoroff_pathway,_glucose-6P_=>_glyceraldehyde-3P_+_pyruvate |
| M00117 | 0.0390625 | Ubiquinone_biosynthesis,_prokaryotes,_chorismate_=>_ubiquinone |
| M00226 | 0.0390625 | Histidine_transport_system |
| M00225 | 0.0390625 | Lysine/arginine/ornithine_transport_system |
| M00300 | 0.0390625 | Putrescine_transport_system |
| M00012 | 0.0390625 | Glyoxylate_cycle |
Discussion
The microbes in our bodies are estimated to collectively account for up to 100 trillion cells and may encode 100-fold more unique genes than our own genome27–29. Not surprisingly, the intestinal microflora play key roles in the maintenance of a healthy state, including influencing the absorption of food and the synthesis and digestion of polysaccharides, vitamins, and other composite materials30–32. The human intestine and liver are closely related and interact with each other through bile, hormones, inflammatory mediators, and products of digestion and absorption. Notably, the amount, type, and composition of intestinal microflora can have both direct and indirect effects on the physiological function of the liver and can impact the occurrence and development of liver diseases2, 4, 33.
In patients with end-stage liver disease, liver cells show degeneration, necrosis, and other pathological changes that damage the normal liver structure, ultimately leading to cirrhosis and portal hypertension. Increased resistance to portal blood flow is the primary factor in the pathophysiology of portal hypertension, and it mainly develops via morphological changes that occur in chronic liver diseases. Another factor contributing to the exacerbation of portal hypertension is significantly increased portal blood flow, which is caused by arteriolar splanchnic vasodilation and hyperkinetic circulation34. Due to portal hypertension, portal hypertensive gastropathy and bowel disease can appear. In addition, intestinal mucosal damage causes an increase in mucosal permeability and a decrease in defense functions, resulting in bacterial translocation and changes to substance metabolism35.
Grąt et al. investigated LT candidates and revealed that pre-LT dysbiosis was significantly correlated with Enterococcus 36. Several other studies have also revealed that Enterobacteriaceae and Enterococcus were more abundant in LT candidates with liver cirrhosis than in healthy individuals. Furthermore, potentially pathogenic Enterobacteriaceae have been described as major determinants of gut dysbiosis in cirrhosis patients and have been identified as harmful components of the intestinal microbiota37–39. However, current knowledge on the gut microbiota and its alterations in relation to post-LT outcomes including early infections, acute cellular rejection and long-term complications are insufficient due to the limited number of human trials and small sample sizes40.
The results of our PCoA based on OTU composition showed that the gut microbiota of patients with severe liver disease who were awaiting LT surgery was significantly different from that of control subjects; however, there was no significant difference in the gut microbiota between post-LT and control samples. We also found significantly decreased relative abundances of certain species following LT, including Actinobacillus, Escherichia, Shigella (belonging to Enterobacteriaceae), and Fusobacteriales. However, the relative abundances of other species, such as Akkermansia and Corynebacterium, were significantly increased after LT. Notably, the abundance of Akkermansia was higher in all post-LT samples.
Several recently published reports in human and animal settings indicated that the abundance of Escherichia species may be related to the presence of HCC41. Our study comparing HCC and non-HCC samples showed no significant differences in the overall OTU composition of the gut microbiota between HCC and non-HCC samples. In particular, Escherichia is not enriched in HCC samples, and this finding is not consistent with some other studies. We propose that cirrhosis might be the major symptom of HCC patients with liver cirrhosis. A previous study showed that Escherichia is enriched in the gut microbiota of liver cirrhosis patients, compared with healthy controls; thus, we are unsure whether Escherichia is solely associated with liver carcinoma. However, the abundance of Escherichia was significantly decreased after LT surgery (the fold-change in abundance was not significantly different between HCC and non-HCC samples), and this decrease might be partially attributable to LT in patients with liver cirrhosis prior to the LT surgery. Future studies will need an expanded sample size, as the sample size of the current study was small.
Akkermansia plays a role in intestinal protection; it can consolidate epithelial cells to strengthen the intestinal barrier. This species has the capacity not only to degrade mucins but also to stimulate mucin synthesis, which is illustrative of an autocatalytic process42. Akkermansia spp. appear to be important for a healthy mucus layer in the human gut with respect to mucus production and thickness. The mucus layer is continuously reshaped and refreshed, and in this way, a healthy environment for the underlying epithelial cells is created43.
Some Corynebacterium spp., such as Corynebacterium glutamicum, which can utilize a variety of carbohydrates and organic acids as its sole source of carbon and energy, can also be of benefit to their hosts44. Corynebacterium glutamicum can produce amino acids including L-glutamic acid, L-threonine, and L-lysine45. L-glutamic acid is involved in protein and sugar metabolism, promotes oxidation processes, and can combine with ammonia in the body to form non-toxic L-glutamine, thereby decreasing blood ammonia levels.
We generated KEGG profiles and compared the representations of KEGG functional pathways. In the post-LT samples, 15 functional modules were enriched and 21 function modules were less represented compared with the pre-LT samples. For example, M00116 is a member of a metabolic pathway for the synthesis of vitamin K, which is a major cofactor that can help convert prothrombin into thrombin through a series of enzymatic reactions. Importantly, intestinal bacterial compounds are a main source of material for vitamin K production. Previous reports indicated that the synthesis of vitamin K increases and blood clotting function improves in patients after LT46, 47. M00232/M00233 is an amino acid transporter in a metabolic system. The increased amino acid metabolism in patients after LT indicates that the level of intestinal nutrients is increased and the digestive and absorptive functions of the intestinal tract are improved in these patients48, 49.
Liver function gradually improves and portal venous pressure is relieved in recipients after LT. Our study indicates that fecal microbial communities were significantly altered following LT and that the gut microbiota composition of LT recipients was more similar to that of healthy controls after the LT surgery. The current study is limited by its small sample size, which prevents subgroup analysis. Therefore, further research should be conducted.
Electronic supplementary material
Acknowledgements
We thank Mr. Ang Li, Mr. Zu-Jiang Yu, Mr. Zhi-Gang Ren, Mr. Ran-Ran Sun (the First Affiliated Hospital of Zhengzhou University) and Mrs Xu-Qun Lin (Shanghai Shannon-seqnote Biotech Company) for bioinformatics analysis and Mrs Yuan Zhang for DNA extraction (College of Medicine, NanKai University). This study was supported by the National Natural Science Foundation of China (Grant No. 81570586) and the Ascent Plan of the Beijing Municipal Administration of Hospitals (Code: DFL20150101).
Author Contributions
Li-Ying Sun was involved in data acquisition, experiment operation and manuscript writing. Yun-Sheng Yang designed and supervised the study. Wei Qu was involved in DNA extraction and sample collection. Zhi-Jun Zhu, Lin Wei, Zhi-Gui Zeng, Ying Liu, Jian-Rui Zhang and Xiao-Ye Sun were involved in patient recruitment, sample collection and sample transportation. Zhi-Sheng Ye was involved in statistical analysis.
Competing Interests
The authors declare that they have no competing interests.
Footnotes
Electronic supplementary material
Supplementary information accompanies this paper at doi:10.1038/s41598-017-03476-4
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Turnbaugh PJ, et al. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature. 2006;444:1027–1031. doi: 10.1038/nature05414. [DOI] [PubMed] [Google Scholar]
- 2.Abu-Shanab A, Quigley EM. The role of the gut microbiota in nonalcoholic fatty liver disease. Nat Rev Gastroenterol Hepatol. 2010;7:691–701. doi: 10.1038/nrgastro.2010.172. [DOI] [PubMed] [Google Scholar]
- 3.Aron-Wisnewsky J, Gaborit B, Dutour A, Clement K. Gut microbiota and non-alcoholic fatty liver disease: new insights. Clin Microbiol Infect. 2013;19:338–348. doi: 10.1111/1469-0691.12140. [DOI] [PubMed] [Google Scholar]
- 4.Terjung B, Spengler U. Atypical p-ANCA in PSC and AIH: a hint toward a “leaky gut”? Clin Rev Allergy Immunol. 2009;36:40–51. doi: 10.1007/s12016-008-8088-8. [DOI] [PubMed] [Google Scholar]
- 5.Chen Y, et al. Characterization of fecal microbial communities in patients with liver cirrhosis. Hepatology. 2011;54:562–572. doi: 10.1002/hep.24423. [DOI] [PubMed] [Google Scholar]
- 6.Zaman A, Kareem R, Mahmood R, Hameed K, Khan EM. Frequency of microbial spectrum of spontaneous bacterial peritonitis in established cirrhosis liver. J Ayub Med Coll Abbottabad. 2011;23:15–17. [PubMed] [Google Scholar]
- 7.Basile AS, Jones EA. Ammonia and GABA-ergic neurotransmission: interrelated factors in the pathogenesis of hepatic encephalopathy. Hepatology. 1997;25:1303–5. doi: 10.1002/hep.510250636. [DOI] [PubMed] [Google Scholar]
- 8.Guarner C, Soriano G. Spontaneous bacterial peritonitis. Semin Liver Dis. 1997;17:203–217. doi: 10.1055/s-2007-1007198. [DOI] [PubMed] [Google Scholar]
- 9.Campillo B, et al. Intestinal permeability in liver cirrhosis: relationship with severe septic complications. Eur J Gastroenterol Hepatol. 1999;11:755–759. doi: 10.1097/00042737-199907000-00013. [DOI] [PubMed] [Google Scholar]
- 10.Husova L, et al. Bacterial infection and acute bleeding from upper gastrointestinal tract in patients with liver cirrhosis. Hepatogastroenterology. 2005;52:1488–1490. [PubMed] [Google Scholar]
- 11.Hillert C, Fischer L, Broering DC, Rogiers X. Liver transplantation in patients with liver cirrhosis and esophageal bleeding. Langenbecks Arch Surg. 2003;388:150–154. doi: 10.1007/s00423-003-0378-2. [DOI] [PubMed] [Google Scholar]
- 12.Lu H, et al. Assessment of microbiome variation during the perioperative period in liver transplant patients: a retrospective analysis. Microb Ecol. 2013;65:781–791. doi: 10.1007/s00248-013-0211-6. [DOI] [PubMed] [Google Scholar]
- 13.Magoč T, Salzberg SL. FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics. 2011;27:2957–2963. doi: 10.1093/bioinformatics/btr507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Edgar RC, Haas BJ, Clemente JC, Quince C, Knight R. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics. 2011;27:2194–2200. doi: 10.1093/bioinformatics/btr381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Edgar RC. UPARSE: highly accurate OTU sequences from microbial amplicon reads. Nat Methods. 2013;10:996–998. doi: 10.1038/nmeth.2604. [DOI] [PubMed] [Google Scholar]
- 16.Jari O, et al. vegan: Community Ecology Package. R package version. 2016;2:3–5. [Google Scholar]
- 17.Vavrek Matthew, J. Fossil: palaeoecological and palaeogeographical analysis tools. Palaeontologia Electronica 14.1.1T (2011).
- 18.Bettina G, Kurt H. topicmodels: An R Package for Fitting Topic Models. Journal of Statistical Software. 2011;40:1–30. [Google Scholar]
- 19.Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–7. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Price MN, Dehal PS, Arkin AP. FastTree 2–approximately maximum-likelihood trees for large alignments. PLoS One. 2010;5:e9490. doi: 10.1371/journal.pone.0009490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.McMurdie PJ, Holmes S. phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PLoS One. 2013;8:e61217. doi: 10.1371/journal.pone.0061217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Langille MG, et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat Biotechnol. 2013;31:814–21. doi: 10.1038/nbt.2676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Abubucker S, et al. Metabolic reconstruction for metagenomic data and its application to the human microbiome. PLoS Comput Biol. 2012;8:e1002358. doi: 10.1371/journal.pcbi.1002358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Segata N, et al. Metagenomic biomarker discovery and explanation. Genome Biol. 2011;12:R60. doi: 10.1186/gb-2011-12-6-r60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Charrad M, et al. NbClust: An R Package for Determining the Relevant Number of Clusters in a Data Set. Journal of Statistical Software. 2014;61:1–36. doi: 10.18637/jss.v061.i06. [DOI] [Google Scholar]
- 26.Arumugam M, et al. Enterotypes of the human gut microbiome. Nature. 2011;473:174–80. doi: 10.1038/nature09944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ley RE, Peterson DA, Gordon JI. Ecological and evolutionary forces shaping microbial diversity in the human intestine. Cell. 2006;124:837–848. doi: 10.1016/j.cell.2006.02.017. [DOI] [PubMed] [Google Scholar]
- 28.Gill SR, et al. Metagenomic analysis of the human distal gut microbiome. Science. 2006;312:1355–1359. doi: 10.1126/science.1124234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Arumugam M, et al. A human gut microbial gene catalogue established by metagenomics sequencing. Nature. 2010;464:U59–U70. doi: 10.1038/nature08821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Clemente JC, Ursell LK, Parfrey LW, Knight R. The impact of the gut microbiota on human health: an integrative view. Cell. 2012;148:1258–1270. doi: 10.1016/j.cell.2012.01.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Marques TM, et al. Programming infant gut microbiota: influence of dietary and environmental factors. Curr Opin Biotechnol. 2010;21:149–156. doi: 10.1016/j.copbio.2010.03.020. [DOI] [PubMed] [Google Scholar]
- 32.Backhed F. Programming of host metabolism by the gut microbiota. Ann Nutr Metab. 2011;58:44–52. doi: 10.1159/000328042. [DOI] [PubMed] [Google Scholar]
- 33.Szabo G, Bala S, Petrasek J, Gattu A. Gut-liver axis and sensing microbes. Dig Dis. 2010;28:737–744. doi: 10.1159/000324281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bosch J, Garcia-Pagan JC. Complications of cirrhosis. I. Portal hypertension. J Hepatol. 2000;32(Suppl 1):141–156. doi: 10.1016/S0168-8278(00)80422-5. [DOI] [PubMed] [Google Scholar]
- 35.Teltschik Z, et al. Intestinal bacterial translocation in rats with cirrhosis is related to compromised Paneth cell antimicrobial host defense. Hepatology. 2012;55:1154–1163. doi: 10.1002/hep.24789. [DOI] [PubMed] [Google Scholar]
- 36.Grąt M, et al. The relevance of intestinal dysbiosis in liver transplant candidates. Transpl Infect Dis. 2015;17:174–84. doi: 10.1111/tid.12352. [DOI] [PubMed] [Google Scholar]
- 37.Wu ZW, et al. Changes of gut bacteria and immune parameters in liver transplant recipients. Hepatobiliary Pancreat Dis Int. 2012;11:40–50. doi: 10.1016/S1499-3872(11)60124-0. [DOI] [PubMed] [Google Scholar]
- 38.Lu H, et al. Assessment of microbiome variation during the perioperative period in liver transplant patients: a retrospective analysis. Microb Ecol. 2013;5:781–91. doi: 10.1007/s00248-013-0211-6. [DOI] [PubMed] [Google Scholar]
- 39.Wei X, et al. Abnormal fecal microbiota community and functions in patients with hepatitis B liver cirrhosis as revealed by a metagenomic approach. BMC Gastroenterol. 2013;13:175. doi: 10.1186/1471-230X-13-175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Doycheva I, Leise MD, Watt KD. The Intestinal Microbiome and the Liver Transplant Recipient: What We Know and What We Need to Know. Transplantation. 2016;100:61–8. doi: 10.1097/TP.0000000000001008. [DOI] [PubMed] [Google Scholar]
- 41.Tao X, Wang N, Qin W. Gut Microbiota and Hepatocellular Carcinoma. Gastrointest Tumors. 2015;2:33–40. doi: 10.1159/000380895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Derrien, M., Belzer, C. & de Vos, W. M. Akkermansia muciniphila and its role in regulating host functions Microb Pathog pii: S0882-4010(15)30178-9 (2016). [DOI] [PubMed]
- 43.Belzer C, de Vos WM. Microbes inside–from diversity to function: the case of Akkermansia. ISME J. 2012;6:1449–58. doi: 10.1038/ismej.2012.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Uhde A, et al. Glucosamine as carbon source for amino acid-producing Corynebacterium glutamicum. Appl Microbiol Biotechnol. 2013;97:1679–87. doi: 10.1007/s00253-012-4313-8. [DOI] [PubMed] [Google Scholar]
- 45.Hermann T. Industrial production of amino acids by Coryneform bacteria. Journal of Biotechnology. 2003;104:155–172. doi: 10.1016/S0168-1656(03)00149-4. [DOI] [PubMed] [Google Scholar]
- 46.Widhalm JR, van Oostende C, Furt F, Basset GJ. A dedicated thioesterase of the Hotdog-fold family is required for the biosynthesis of the naphthoquinone ring of vitamin K1. Proc Natl Acad Sci USA. 2009;106:5599–603. doi: 10.1073/pnas.0900738106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.van Tegelen LJ, Moreno PR, Croes AF, Verpoorte R, Wullems GJ. Purification and cDNA cloning of isochorismate synthase from elicited cell cultures of Catharanthus roseus. Plant Physiol. 1999;119:705–12. doi: 10.1104/pp.119.2.705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pernil R, Picossi S, Mariscal V, Herrero A, Flores E. ABC-type amino acid uptake transporters Bgt and N-II of Anabaena sp. strain PCC 7120 share an ATPase subunit and are expressed in vegetative cells and heterocysts. Mol Microbiol. 2008;67:1067–80. doi: 10.1111/j.1365-2958.2008.06107.x. [DOI] [PubMed] [Google Scholar]
- 49.Kronemeyer W, Peekhaus N, Krämer R, Sahm H, Eggeling L. Structure of the gluABCD cluster encoding the glutamate uptake system of Corynebacterium glutamicum. J Bacteriol. 1995;177:1152–8. doi: 10.1128/jb.177.5.1152-1158.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.