

Abstract
Background
MicroRNAs (miRs) are small non‐coding RNAs that regulate gene (mRNA) expression. Although the pathological role of miRs have been studied in muscle wasting conditions such as myotonic and muscular dystrophy, their roles in cancer cachexia (CC) are still emerging.
Objectives
The objectives are (i) to profile human skeletal muscle expressed miRs; (ii) to identify differentially expressed (DE) miRs between cachectic and non‐cachectic cancer patients; (iii) to identify mRNA targets for the DE miRs to gain mechanistic insights; and (iv) to investigate if miRs show potential prognostic and predictive value.
Methods
Study subjects were classified based on the international consensus diagnostic criteria for CC. Forty‐two cancer patients were included, of which 22 were cachectic cases and 20 were non‐cachectic cancer controls. Total RNA isolated from muscle biopsies were subjected to next‐generation sequencing.
Results
A total of 777 miRs were profiled, and 82 miRs with read counts of ≥5 in 80% of samples were retained for analysis. We identified eight DE miRs (up‐regulated, fold change of ≥1.4 at P < 0.05). A total of 191 potential mRNA targets were identified for the DE miRs using previously described human skeletal muscle mRNA expression data (n = 90), and a majority of them were also confirmed in an independent mRNA transcriptome dataset. Ingenuity pathway analysis identified pathways related to myogenesis and inflammation. qRT‐PCR analysis of representative miRs showed similar direction of effect (P < 0.05), as observed in next‐generation sequencing. The identified miRs also showed prognostic and predictive value.
Conclusions
In all, we identified eight novel miRs associated with CC.
Keywords: Cancer cachexia, microRNAs, miRs, next‐generation sequencing, skeletal muscle, weight loss, muscle transcriptome, gene expression
Introduction
Cancer cachexia (CC) is a multifactorial syndrome characterized by severe depletion of skeletal muscle with or without fat loss. The pathophysiology includes systemic inflammation, reduced food intake and negative energy and protein balance.1 Patients with CC have poor treatment response, reduced survival and a severe compromise in their quality of life.2 CC is characterized by complex host–tumour interactions that remain to be fully elucidated. Tumour derived mediators lead to aberrant host tissue responses. Several proinflammatory and procachectic cytokines released from tumour cells contribute to systemic inflammation in the host and lead to metabolic alterations.3 Genes (mRNA) involved in the pathophysiology of CC have been fairly well studied.4, 5 However, the role of finer post‐transcriptional gene regulatory mechanisms and its implications on CC at the whole genome level has not been comprehensively explored.
MicroRNAs (miRs, 18‐25 nt) are a class of small non‐coding RNAs that are considered as global regulators of gene expression. They primarily bind to 3' untranslated region (3'UTR) of mRNA and cause either translational repression or mRNA degradation, depending on the degree of complementarity shared between the two molecules.6 Myo‐miRs, a suite of miRs highly enriched in skeletal muscle, are known to play a role in myogenesis.7 For example, miR‐1 and miR‐133 regulate skeletal muscle proliferation and differentiation.8 Similarly, diverse roles of miRs have been reported in physiological process such as adipogenesis, exercise and in general, health and disease states9, 10, 11 and have also been identified as promising biomarkers for many diseases.12, 13 Recent evidence has suggested that miR‐21 and miR‐378 promote muscle cell apoptosis and adipolysis, respectively, in CC.14, 15 While these studies have focused on animal models and human adipose tissues, it remains to be explored if miRs identified from human muscle associate with CC pathophysiology and can also act as potential biomarkers. Biomarkers for CC have previously been identified from preclinical models.16, 17 Nonetheless, many of these molecules are far from being universally accepted as these are yet to be validated in human subjects.
Whole genome miR profiling using next‐generation sequencing (NGS) are increasingly being utilized, as NGS offers better sensitivity and specificity compared with hybridization techniques.18 However, comprehensive profiling of miRs for CC using NGS platform has not yet been attempted. We hypothesized that deregulation of miRs contributes to the aetiology of CC. The study objectives were (i) to profile human skeletal muscle expressed miRs; (ii) to identify differentially expressed (DE) miRs between cachectic and non‐cachectic cancer patients; (iii) to identify mRNA targets for the DE miRs to gain mechanistic insights; and (iv) to explore the prognostic and predictive potential of miRs. We report eight novel miRs associated with CC pathophysiology. We have also identified gene targets (mRNA) for the miRs and potential regulation of canonical pathways by these miR‐mRNA pairs.
Methods
Recruitment of study subjects and acquisition of muscle samples
Skeletal muscle biopsies from patients who underwent laparotomy at the Foothills Medical Centre between 2006 and 2013 were obtained from the University of Calgary Hepatopancreaticobiliary/Gastrointestinal Tumour Bank. Patients were approached for consents in pre‐admission clinics (out‐patient) prior to surgery. All patients provided written informed consent for study participation, and the study was approved by the Conjoint Health Research Ethics Board at the University of Calgary (Ethics ID E‐17213). Biopsies of rectus abdominus were taken at the start of the surgery using sharp dissection, immediately frozen in liquid nitrogen to minimize ischemic shock post‐devitalization, and stored at −80°C until further use. Post‐biopsy molecular profiling of samples for NGS and collection of patient information was conducted under ethics protocol ETH‐21709, which was approved by Health Research Ethics Board of Alberta (HREBA)–Cancer Committee.
Study design and clinical annotation of cases
Weight loss (WL) information of patients (n = 42) was retrieved from medical charts. Computed tomography (CT) scans were available for majority of the samples (n = 35), and these were accessed for quantification of skeletal muscle and fat components. Patients were classified as cachectic based on the international consensus framework for CC; patients belonging to any one of the three diagnostic criteria were considered cachectic (more than 5% pre‐illness WL for a period of six months, body mass index (BMI) < 20 with any degree of WL >2% and lumbar skeletal sarcopenia, as defined by muscle index cut‐points using computed tomography).1 Non‐cachectic cancer patients had no WL over a period of 6 months compared with their pre‐illness weight. The exclusion criteria include as follows: (i) patients with no clinical chart or recorded WL information; (ii) below 18 years; and (iii) inability to give written informed consent. A subset of age matched patients in the bank meeting the aforementioned inclusion criteria and with available biopsies, and WL histories were accessed; of these 22 were cachectic cases (with mean WL of 11.8 ± 6.6%; hereafter referred to as cases) and 20 were non‐cachectic controls (hereafter referred to as controls). One patient had completed a course of chemotherapy (neo‐adjuvant) before surgery but had not received any chemotherapy 4 weeks prior to surgery. Remaining patients did not receive any chemotherapy before surgery.
Pancreatic and colorectal cancer (with liver metastasis) patients were considered for the present study from the collection of biopsies from the Hepatopancreaticobiliary/Gastrointestinal Tumour Bank to minimize the number of tumour types. Tumour stage is reported according to American Joint Committee on Cancer v7.
Computed tomography image quantification
Twenty cases and 15 controls had a single CT prior to surgery (70.75 ± 45.24 in days). The quantification protocol has been explained elsewhere in detail.19, 20 Briefly, the third lumbar vertebrae were used as a standard landmark to measure the cross‐sectional muscle area (cm2) and normalized to their stature (m2) to calculate the skeletal muscle index (SMI) (cm2/m2). Muscle attenuation (MA) (Hounsfield Units) was also captured for these patients. Sarcopenia status was assigned based on age and sex adjusted SMI values, as described earlier.21
RNA extraction and next‐generation sequencing profiling
Total RNA from muscle biopsies were isolated using Trizol (Sigma‐Aldrich, Oakville, ON, Canada) and purified using Qiagen's RNeasy Maxi kit (Mississauga, ON, Canada). Quantification of RNA was carried out using Nanodrop 1000 Spectrophotometer. All of the aforementioned protocols were carried out according to the manufacturer's instructions.
Services from PlantBiosis Ltd (Lethbridge, Alberta, Canada; http://www.plantbiosis.com/) were used from small RNA library preparation up to the generation of alignment files (.bam files), as described earlier.22 All the samples were sequenced in a single batch in a single lane to avoid batch effects. Sequencing of small RNA was carried out using Truseq Small RNA sequencing kit (Illumina), TruSeq SR Cluster Kit v5‐CS‐GA (Illumina) and TruSeq SBS Kit v5‐GA (Illumina), according to manufacturer's instructions. The samples were sequenced using MiSeq platform with 36 bases single‐end protocol. Base calling and demultiplexing was performed using MiSeq Reporter FastQ workflow. Cutadapt 1.4.1 (https://pypi.python.org/pypi/cutadapt/1.4.1) was used to trim the adapters. Only reads with a quality score of more than 30 on the Sanger scale were considered. Quality control for sequenced reads before and after adapter trimming was performed using FastQC v0.11.3 software. Burrows–Wheeler aligner version 0.6.1 was used to align trimmed reads to the reference genome23 (human genome 19 build), downloaded from Illumina iGenome website. Samtools version 0.1.18 was used to convert sequence aligned data file (.sam files) to its binary format (.bam format), which was used for downstream analyses. The raw files and normalized counts of the NGS data have been submitted to Gene Expression Omnibus (accession ID GSE75473).
Identification of differentially expressed microRNAs
Partek Genomics Suite 6.6 (PGS 6.6) was used to analyze the .bam files generated from NGS experiment. Raw data were filtered to include miRs with more than or equal to five read counts in at least 80% of the samples and were normalized using reads per kilobase per million (RPKM) method.24 DE miRs were identified with a fold change (FC) of >1.4 and P < 0.05 using one‐way analysis of variance. The DE miRs were also subjected to permutation test (n = 10 000), and miRs with permuted P‐values < 0.05 were considered for subsequent analysis.
qRT‐PCR validation of the identified microRNAs
From the eight DE miRs identified from NGS, representative miRs: hsa‐miR‐3184‐3p, hsa‐let‐7d‐3p and hsa‐miR‐1296‐5p were selected and validated using qRT‐PCR. Total RNA was reverse transcribed using TaqMan MicroRNA reverse transcription kit. RNU6B was used as the internal control. qPCR was performed using 7900HT fast real‐time PCR system (Applied Biosystems). The 20 μL reactions for each sample were run in triplicates according to manufacturer's protocol. The following thermal cycler conditions were used for reverse transcription: 30 min. at 16°C, 30 min. at 42°C and 5 min. at 95°C; and the following conditions were used for qPCR: 50°C for 2 min, 95°C for 20 s and 40 cycles of 95°C and 60°C for 1 s and 20 s, respectively. Data analysis was carried out using 2‐∆∆Ct method.25
Target predictions and putative functional annotation for mRNA targets of differentially expressed microRNAs
Detailed description of the in‐house muscle transcriptome gene (mRNA) expression data generated using Agilent platform is elaborated elsewhere26 and has been deposited in Gene Expression Omnibus repository (GSE41726). Initially, independent muscle biopsies (non‐matched data sets) were used for miR profiling (this study, n = 42) and mRNA profiling (n = 90, a previous study GSE41726 from the same lab). Briefly, using the same definitions as in the miR study, 29 cachectic cases (WL > 5%) and 61 weight stable (WS) controls were used to identify DE mRNAs. PGS 6.6 was used for differential expression analysis. Raw intensity files were quantile normalized, log 2 transformed and DE mRNAs were identified at ≥ 1.4 FC and P < 0.05 using one‐way analysis of variance. The direct binding of miR to 3'UTR of the mRNA is one of the most commonly recognized mechanisms in post‐transcriptional silencing of genes.27, 28 However, miRs may exert influence on target mRNAs in an indirect manner29, and hence, we considered all miR‐mRNA correlations. We interrogated the mRNA data showing inverse and similar directional effect of expression between miR and mRNA (i.e. miR up‐regulated and its corresponding mRNA down‐regulated and both miR and mRNA being up‐regulated/down‐regulated) to understand the biological role of identified miRs in CC.22 Putative gene targets for DE miRs were predicted in silico using TargetScan 7.0,30 based on the complementary binding of the seed region of miRs with 3'UTR of mRNAs. These predicted targets were then overlapped with the DE genes identified from in‐house muscle transcriptome dataset. Ingenuity pathway analysis (IPA) was used to identify pathways for the overlapped gene targets of eight DE miRs. We have further interrogated the mRNA targets (GSE85017) for the miRs in matched data sets, albeit of lower sample size (n = 42), compared with the unmatched mRNA data set (n = 90). Independent confirmation of findings from two sources, matched and unmatched data sets as an approach for functional validation of targets in a tissue‐specific context would strengthen the study findings.
Statistical analysis
Data are presented as mean ± standard deviation. Independent t‐test and χ² test were used for continuous and categorical variables, as appropriate. Sample size estimation was carried out using the following parameters: α = 0.05, β = 80% and a fold difference of 1.4 or more in miR expression. Nineteen samples per group were required to conduct the study (http://bioinformatics.mdanderson.org/MicroarraySampleSize/) to meet the statistical power and identify DE miRs with confidence.
Identification of microRNAs as prognostic and predictive factors
One of the objectives of this study was to identify miRs associated with overall survival (OS). OS was defined as the time period between the date of surgery and the date of death. The median follow‐up period was 1060 days (Range: 6 to 2041 days), from the date of muscle biopsy accrual till the last follow‐up date (March 2014). Of the 42 patients, 19 died, and 23 were alive at the time of completion of this study.
Prognostic value: we considered DE miRs as continuous variables and subjected the miRs to univariate cox proportional hazards regression model. For OS, composite risk score was constructed for each sample using the parameter estimates (co‐efficient) and the normalized counts obtained for the eight DE miRs. Receiver operating characteristics curve was used to determine the optimal cut‐point for the composite risk score. This was carried out to dichotomize the 42 samples into high‐risk and low‐risk groups. Hazard ratios (HR) along with 95% confidence intervals were reported for univariate and multivariate analyses. For multivariate analysis, the risk score was adjusted for age, BMI and tumour type, where appropriate. Log‐rank test was carried out to identify if any significant difference existed between the survival curves of two‐risk groups.
Predictive value: the association between the weight change (loss vs. stable) group and the miRNA score (composite risk score) was tested using binary logistic regression. Univariate and multivariate logistic regression models are reported. For the multivariate model, we adjusted for age and BMI as continuous variables and tumour type (colorectal vs. pancreas) as dichotomous variable. Odds ratio (OR) and the corresponding 95% confidence interval are reported. P‐value < 0.05 was used to define statistical significance. Two‐sided tests were used for the comparisons. All statistical analyses were conducted using spss v16.0 and sas (SAS Institute Inc., Cary, NC) v 9.3.
Results
Patient demographics and body composition analysis
Forty‐two patients who met the study criteria with information on age, gender, BMI, tumour type and physician‐documented WL information were selected. There was no significant difference between cases and controls in age, gender and tumour type. BMI was found to be significant between the groups (P = 0.01) (Table 1a). We investigated if the CT‐derived body composition measurements also are in concordance with the phenotypes of cachexia in the study cohort (Table 1b). We observed expected trends in SMI values between cases and controls, (P = 0.07 for SMI). MA was found to be reduced in cases compared with controls and were significant at P = 0.04.
Table 1a.
Patient demographics
| Characteristics | Cachectic cases (n = 22) | Non‐cachectic controls (n = 20) | P‐values |
|---|---|---|---|
| Weight loss, % mean | 11.8 ± 6.6 | — | |
| Age (mean, in years) [Range] | 64.9 ± 10.1 | 63.6 ± 7.9 | 0.6a |
| [40–83] | [45–76] | ||
| Tumour type (n) | 0.2b | ||
| Pancreatic | 12 | 7 | |
| Colorectal | 10 | 13 | |
| Tumour stage (n) | 0.6c | ||
| I | 2 | 1 | |
| II | 3 | 3 | |
| III | 2 | 0 | |
| IV | 15 | 16 | |
| Gender (n) | 0.7b | ||
| Male | 9 | 9 | |
| Female | 13 | 11 | |
| Body mass index (mean, in kg/m2) | 24.35 ± 2.5 | 27.02 ± 3.7 | 0.01a |
| [Range] | [19–29] | [20–40] |
Unpaired t‐test.
χ² test.
Fisher's exact test.
Data represented as mean ± standard deviation. Statistical analyses were carried out using spss v16. Independent t‐test was conducted for age and body mass index. χ² test was carried out for tumour type and gender. P < 0.05 was considered statistically significant.
Table 1b.
Body composition analysis for the study subjects
| Characteristics | Cachectic cases (n = 20) | Non‐cachectic controls (n = 15) | P‐values |
|---|---|---|---|
| Cross‐sectional skeletal muscle area (cm2)a | |||
| Male | 139.5 ± 15.4 | 158 ± 12.9 | 0.2 |
| Female | 96.2 ± 14.6 | 103.5 ± 14.1 | |
| Skeletal muscle index (cm2/m2)a | 0.07 | ||
| Male | 45.3 ± 5.9 | 49.1 ± 3.1 | |
| Female | 36.3 ± 5.6 | 41.52 ± 7.43a | |
| z‐score | −0.7 ± 0.7 | −0.08 ± 0.86 | 0.03 |
| Total adipose tissuea | 0.9 | ||
| Male | 226.8 ± 84.5 | 266.29 ± 77.2 | |
| Female | 328.8 ± 126.4 | 302.2 ± 122.7 | |
| Muscle attenuation (HU)a | 0.04 | ||
| Male | 34.14 ± 8.7 | 39.8 ± 6.9 | |
| Female | 29.46 ± 7.3 | 36.42 ± 8.4 |
Unpaired t‐test.
Body composition was calculated for subset of patients (n = 35) who had computed tomography prior to surgery. Statistical analysis was conducted between cases and controls. Muscle attenuation values were significant in the overall comparison between cachectic cases and non‐cachectic controls. z‐score is the difference expressed in standard deviation of patients' values from age and gender‐specific mean values.21
Technical variation, data processing and identification of differentially expressed microRNAs using next‐generation sequencing
Earlier reports suggested that the technical variance arising from the NGS platform is minimal.31 This has been validated in the current investigation as well. Technical replicates of four samples (two cases and two controls) were sequenced. Excellent concordance was observed (r > 0.99) between the replicates, suggesting that technical variance in the data is absent or minimal because of experimental procedures adopted ( Table S1).
A total of 7 926 299 and 9 155 808 reads were obtained from cases and controls (n = 42), respectively. A total of 97.12% (7 698 807) and 97.35% (8 913 914) of reads were retained after adapter trimming in cases and controls, respectively. A total of 87.78% (6 758 570) of reads in cases, and 87.64% (7 812 866) of reads in controls were aligned to the reference genome, of which 9.55% (646 069) of reads in cases and 8.14% (636 748) of reads in controls mapped to miRs (Table 2) and had read length distribution from 18 to 25 nucleotides (Figure 1).
Table 2.
Descriptive statistics for the data obtained from next‐generation sequencing
| Samples | Total reads | Reads retained after adapter trimming | Aligned reads | Reads mapped to miRNAs |
|---|---|---|---|---|
| Cachectic cases (n = 22) | 7 926 299 | 7 698 807 (97.12%) | 6 758 570 (87.78%) | 646 069 (9.55%) |
| Non‐cachectic controls (n = 20) | 9 155 808 | 8 913 914 (97.32%) | 7 812 866 (87.64%) | 636 748 (8.14%) |
Step‐wise filtering of next‐generation sequencing data is shown starting from total reads obtained from next‐generation sequencing for both cases and controls to reads mapped to miRNA. Furthermore, the reads mapped to miRNA were subjected to differential expression analysis.
Figure 1.
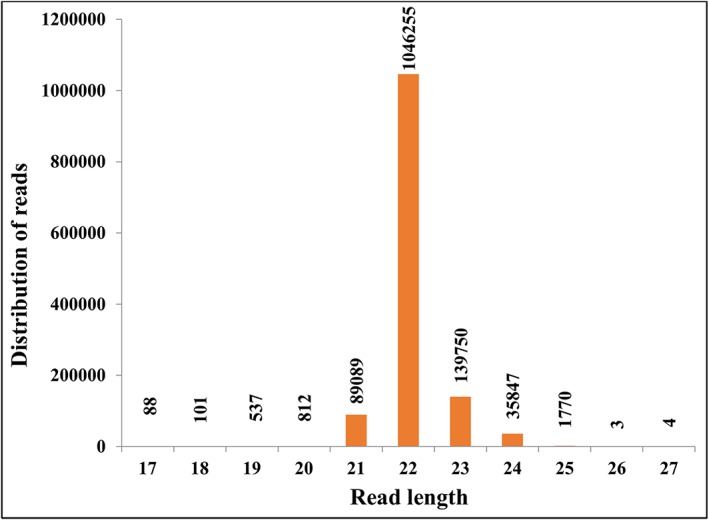
Length distribution of reads aligned to miRs: the aligned read length ranges from 17 to 27 nucleotides with the maximum distribution of reads captured between 18 and 25 nucleotides (reflecting miRNA read length).
A total of 777 miRs were profiled from the skeletal muscle tissues (out of a total of 2588 miRs annotated in miRbase v20, of which only a subset can be tissue specific). Eighty‐two miRs were retained after filtering and were subjected to DE analysis. A total of eight miRs were found to be up‐regulated with a FC of >1.4, P < 0.05 and with false discovery rate (FDR) ranging from 0.21 to 0.22. Permutation tests (n = 10 000) for the eight DE miRs were carried out and were also found to be significant with permuted P < 0.05. The eight up‐regulated DE miRs were hsa‐miR‐3184‐3p, hsa‐miR‐423‐5p, hsa‐let‐7d‐3p, hsa‐miR‐1296‐5p, hsa‐miR‐345‐5p, hsa‐miR‐532‐5p, hsa‐miR‐423‐3p and hsa‐miR‐199a‐3p (Table 3a). No down‐regulated miRs were identified in the current study under the stringent data filters applied and with the current sample size.
Table 3a.
Differentially expressed miRNAs
| miRNA | P‐value | FDR | Permutation P‐value | Fold change (Cachectic cases vs. non‐cachectic controls) | Direction of fold change |
|---|---|---|---|---|---|
| hsa‐let‐7d‐3p | 0.01 | 0.21 | 0.01 | 1.48 | Up |
| hsa‐miR‐345‐5p | 0.02 | 0.21 | 0.02 | 1.47 | Up |
| hsa‐miR‐423‐5p | 0.009 | 0.21 | 0.009 | 1.42 | Up |
| hsa‐miR‐532‐5p | 0.02 | 0.21 | 0.02 | 1.48 | Up |
| hsa‐miR‐1296‐5p | 0.03 | 0.22 | 0.03 | 1.44 | Up |
| hsa‐miR‐3184‐3p | 0.009 | 0.21 | 0.008 | 1.42 | Up |
| hsa‐miR‐423‐3p | 0.01 | 0.21 | 0.01 | 1.43 | Up |
| hsa‐miR‐199a‐3p | 0.03 | 0.22 | 0.01 | 2.01 | Up |
FDR, false discovery rate.
All eight differentially expressed miRNAs were up‐regulated in cachectic cases with a fold change of ≥1.4 at P < 0.05. Permutation test was carried out (n = 10 000) for these seven miRNAs to rule out observation by chance. The permuted P‐value was significant for all the eight differentially expressed miRNAs. FDR for all eight differentially expressed miRNAs were also represented.
Validation of select microRNAs using qRT‐PCR
Representative miRs such as hsa‐miR‐3184‐3p, hsa‐let‐7d‐3p and hsa‐miR‐1296‐5p were validated in an independent platform – qRT‐PCR. All the three miRs showed similar direction of expression as observed in NGS with significant P‐values (P < 0.05) (Figure 2).
Figure 2.
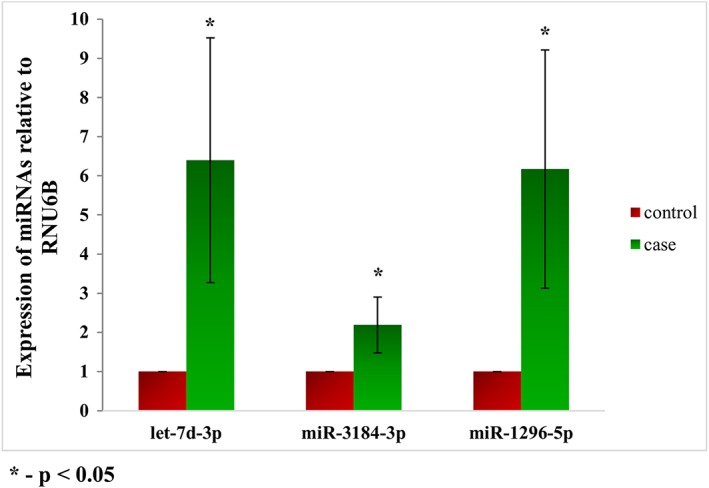
qRT‐PCR validation of miRs: representative miRNAs were validated using qRT‐PCR. Similar direction of effect was observed as seen in the next‐generation sequencing with statistical significance of P < 0.05.
Gene expression studies for identification of putative targets for differentially expressed microRNAs
TargetScan identified 20 988 mRNAs as potential targets for eight DE miRs. TargetScan in silico predicted targets included mRNAs from muscle and non‐muscle cell and tissue types. Therefore, to identify tissue‐specific gene (mRNA) targets for the DE miRs, an in‐house muscle transcriptome dataset was used.26 A total of 353 mRNAs with a FC of >1.4 and P < 0.05 were identified between cachectic cases (>5% WL) and non‐cachectic controls (WS), of which 127 were up‐regulated and 226 were down‐regulated (data not shown). Because we do not know a priori which among the 353 mRNAs serve as potential targets for the DE miRs in our study, we mapped the 353 mRNAs to the 25 348 mRNA targets predicted by TargetScan. In this analysis, we identified 191 mRNA targets as potentially regulated by the eight DE miRs (Table 3b). Up to 70% of the targets identified were down‐regulated, and is an expected consequence of the binding of miRs to 3'UTRs of mRNAs leading to gene silencing. To further confirm the direction of expression of these targets, an independent gene expression study for matched samples was conducted using Affymetrix Human Transcriptome Array 2.0 (GSE85017). We compared the direction of expression of the identified 191 mRNA targets in this independent dataset. We found 77% (147/191) of the mRNA targets expressed in the same direction, in these comparisons regardless of the P‐value and FC (data not shown). Further, the direction of expression of 10 representative mRNA targets chosen for discussion (see below) is indicated in Table S2. Although the P‐value and FC of these 10 mRNA targets were small in the independent dataset, perhaps due to the small sample size, we have confirmed the direction of expression in matching samples.
Table 3b.
Summary of target identification
| miRNA | Number of targets identified by target scan | Number of DE gene targets identified by in‐house datasets |
|---|---|---|
| hsa‐let‐7d‐3p | 479 | 4 |
| hsa‐miR‐345‐5p | 3515 | 34 |
| hsa‐miR‐423‐5p | 4437 | 41 |
| hsa‐miR‐532‐5p | 3227 | 32 |
| hsa‐miR‐1296‐5p | 2007 | 13 |
| hsa‐miR‐3184‐3p | 3531 | 28 |
| hsa‐miR‐423‐3p | 779 | 14 |
| hsa‐miR‐199a‐3p | 3013 | 25 |
Differentially expressed (DE) miRs were mapped to TargetScan 7.0. The identified gene targets from TargetScan 7.0 were overlapped to in‐house muscle transcriptome dataset to identify tissue‐specific gene targets for DE miRNA. The identified targets were interrogated for pathway analysis using ingenuity pathway analysis to understand the potential biological roles of the miRNAs in cancer cachexia.
Pathway analysis using ingenuity pathway analysis
The 191 mRNA targets identified for eight DE miRs were subjected to IPA to identify the corresponding pathways and understand their biological relevance (Table 4). The identified targets are known to play a role in adipogenesis, myogenesis (SULF1, BMPR1B and DLK1), inflammation and innate immune response (RPS6KA6) and also in signal transduction pathways (SFRP4 and DKK2) that may directly or indirectly contribute to the phenotype of CC (See Discussion for details).
Table 4.
List of significant pathways identified from ingenuity pathway analysis
| Ingenuity canonical pathways | Molecules | miRNA‐ID |
|---|---|---|
| Actin cytoskeleton signalling | FN1 | hsa‐miR‐199a‐3p |
| Adipogenesis pathway | DLK1 | hsa‐miR‐345‐5p, hsa‐miR‐423‐5p, hsa‐miR‐3184‐3p |
| BMP signalling pathway | GREM1 | hsa‐miR‐345‐5p, hsa‐miR‐3184‐3p, hsa‐miR‐199a‐3p |
| BMPR1B | hsa‐miR‐3184‐3p | |
| SULF 1 | hsa‐miR‐ 532‐5p | |
| Calcium signalling | CAMK2A | hsa‐hsa‐miR‐423‐3p |
| Cholesterol biosynthesis I | SQLE | hsa‐miR‐3184‐3p |
| CNTF signalling | RPS6KA6 | let‐7d‐3p, hsa‐miR‐1296, hsa‐miR‐199a‐3p, hsa‐miR‐532‐5p |
| Energy metabolism | NYP1R | hsa‐miR‐532‐5p |
| GDNF family ligand–receptor interactions | RET | hsa‐miR‐423‐5p, hsa‐miR‐3184‐3p |
| Glucocorticoid receptor signalling | PGR | let‐7d‐3p, hsa‐miR‐1296, hsa‐miR‐423‐5p |
| Glutamate receptor signalling | SLC1A7 | hsa‐miR‐345‐5p |
| IGF‐1 signalling | CYR61, NOV | hsa‐miR‐345‐5p |
| IL‐6 signalling | COL1A1 | hsa‐miR‐345‐5p, hsa‐miR‐423‐5p |
| IL‐8 signalling | EIF4EBP1 | hsa‐miR‐423‐5p, hsa‐miR‐199a‐3p |
| Insulin receptor signalling | EIF4EBP1 | hsa‐miR‐423‐5p |
| Integrin signalling | CAPN6 | let‐7d‐3p, hsa‐miR‐1296, hsa‐miR‐345‐5p, hsa‐miR‐423‐5p, hsa‐miR‐3184‐3p |
| Mitochondrial dysfunction | SOD2 | hsa‐miR‐345‐5p, hsa‐miR‐423‐5p, hsa‐miR‐3184‐3p, hsa‐miR‐199a‐3p |
| mTOR signalling | RPS6KA6 | let‐7d‐3p, hsa‐miR‐1296, hsa‐miR‐199a‐3p |
| EIF4EBP1 | hsa‐miR‐423‐5p, hsa‐miR‐199a‐3p | |
| NF‐κB signalling | BMPR1B | hsa‐miR‐3184‐3p |
| Oleate biosynthesis II | FADS2 | hsa‐miR‐423‐5p |
| Phospholipase C signalling | BLNK | hsa‐miR‐345‐5p |
| Regulation of cellular mechanics by calpain protease | CAPN6 | let‐7d‐3p, hsa‐miR‐1296, hsa‐miR‐345‐5p, hsa‐miR‐423‐5p, hsa‐miR‐3184‐3p |
| Serotonin receptor signalling | HTR2A | let‐7d‐3p, hsa‐miR‐1296, hsa‐miR‐3184‐3p, hsa‐miR‐199a‐3p |
| TGF‐β signalling | BMPR1B | hsa‐miR‐3184‐3p |
| Wnt/β‐catenin signalling | SFRP4 | let‐7d‐3p, hsa‐miR‐1296, hsa‐miR‐3184‐3p |
CNTF, ciliary neurotrophic factor; IGF‐1, insulin‐like growth factor gene 1; mTOR, mammalian target of rapamycin; TGF‐β, transforming growth factor beta.
All miRs selected for this analysis showed up‐regulation. Majority of the genes indicated in the table showed down‐regulation, as expected in the mRNA–miRNA correlations. Note that common molecules between pathways reflect in the redundancy of the genes due to multiple miRs regulating the same pathway.
microRNAs as potential independent prognostic factors
The eight DE miRs were considered as continuous variables and were subjected to univariate cox proportional hazards regression model. The parameter estimates thus obtained were used for constructing a risk score. A risk score cut‐point was estimated to dichotomize the patients into high‐risk and low‐risk groups. Cut‐point above 7.6 was considered as high risk and less than equal to 7.6 as low risk, following which the risk score was treated as categorical variable for univariate cox model. The risk score showed trend towards significance after adjusting for potential confounding factors (age, BMI and tumour type) in the multivariate analysis. The high‐risk group had a shorter OS when compared with the low‐risk group [HR: 2.32 (0.88–6.06), P = 0.08, Table 5a]. The log‐rank P‐value was significant in survival analysis between two‐risk groups (P = 0.001, Figure 3).
Table 5a.
Univariate and multivariate results for overall survival
| Parameter | Univariate analysis | Multivariate analysis | ||
|---|---|---|---|---|
| HR (95% CI) | P‐value | HR (95% CI) | P‐value | |
| Risk score | 3.49 (1.51–8.04) | 0.003 | 2.32 (0.88–6.06) | 0.08 |
| Age | 0.99 (0.94–1.04) | 0.81 | 0.95 (0.89–1.00) | 0.07 |
| BMI | 0.97 (0.86–1.10) | 0.67 | ||
| Tumour type | 0.21 (0.09–0.50) | 0.0005 | 0.22 (0.07–0.63) | 0.005 |
BMI, body mass index; CI, confidence interval; HR, hazards ratio.
Risk score and other clinical parameters were subjected to univariate cox proportional hazards model. In the multivariate analysis, risk score was marginally significant after adjusting for all potential confounders (age, BMI and tumour type).
Figure 3.
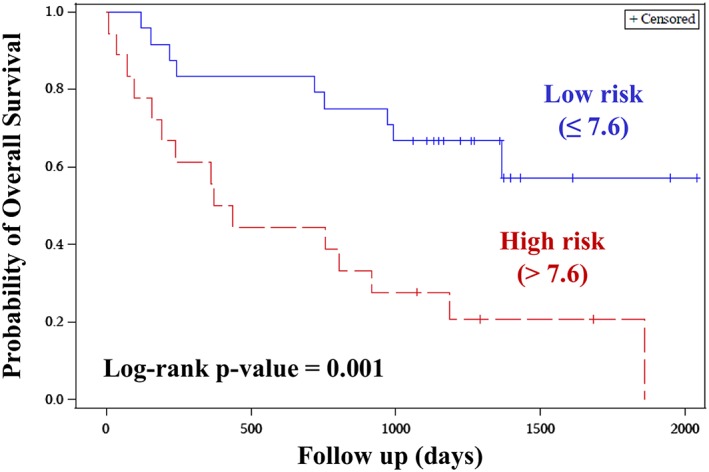
Kaplan–Meier plot for risk score: Kaplan–Meier plot was constructed to assess the survival function of high‐risk group vs. the low‐risk group, based on the risk score. The high‐risk group had a shorter OS when compared to the low‐risk group. The log rank P‐value was significant in survival analysis between the two‐risk groups.
microRNAs as potential independent predictive factors
The association between the dichotomous risk score obtained from OS analysis for eight miR signature and weight change (cases vs. controls) was analyzed using binary logistic regression. Results from this analysis indicated that the composite risk score is an independent predictor of weight change. The odds of WL were seven times higher in patients with a high‐risk score compared with the low‐risk score patients. When adjusted for age, BMI and tumour type, the odds were still higher, about 7.95 times higher (1.44–43.97 at 95% confidence interval, Table 5b). Area under the curve was found to be 0.84 after adjusting for potential confounding factors (age, BMI and tumour type). The wider confidence interval observed may be attributed to a relatively small sample size used in the study.
Table 5b.
Univariate and multivariate analysis for logistic regression
| Parameter | Univariate analysis | Multivariate analysis | ||
|---|---|---|---|---|
| OR (95% CI) | P‐value | OR (95% CI) | P‐value | |
| Risk score (high score vs. low score) | 7.00 (1.73–28.3) | 0.006 | 7.95 (1.44–44.0) | 0.01 |
| Age | 0.99 (0.92–1.06) | 0.8 | ||
| BMI | 0.72 (0.53–0.96) | 0.03 | 0.66 (0.45–0.96) | 0.03 |
| Tumour type (colorectal vs. pancreas) | 0.45 (0.13–1.56) | 0.21 | ||
BMI, body mass index; CI, confidence interval; OR, odds ratio.
The odds of weight loss was 7.95 times higher in patients with high‐risk score compared to low‐risk score after adjusting for potential confounders.
Discussion
We identified eight novel DE miRs from human skeletal muscle and also identified the potential contributions of miR‐mRNA correlations to the pathophysiology of CC. All eight DE miRs were up‐regulated, and none of the miRs reported here have been directly studied in the context of CC. This is the first study where NGS technology was used to comprehensively profile miRs on a genome‐wide scale for CC.
Forty‐two cancer patients were chosen and stratified as cases and controls for the study. BMI was less in cachectic cases when compared to controls (P = 0.01, Table 1a). In the current study, although cachectic cases have BMI in the overweight range, they have lost more than 10% of their weight in previous 6 months. In this population (Canada) considered for the study, WL regardless of BMI,32 as well as the combination of sarcopenia and WL, was shown to be associated with poor prognosis. Body composition analysis indicated that MA was reduced in cases compared with controls (P = 0.04). Previous studies suggest that cancer patients with reduced attenuation are associated with poor survival. In terms of attenuation values, we have observed a similar trend as reported in literature with large sample sizes.33 The reduced attenuation values in cases indicate that these muscles could have been infiltrated by fat.
Using in‐house muscle transcriptome dataset, we interrogated tissue (muscle) specific mRNA signatures to identify targets. The transcriptome dataset serves as a proxy for functional validation of eight DE miRs to gain mechanistic insights. A list of mRNA targets and inferred pathways for DE miRs is given in Table 4. However, further cell‐based assays would help confirm the specific targets and their regulation by their corresponding miRs. A brief discussion on eight DE miRs, their interacting partners (mRNAs) and their biological relevance to CC is described below. There is paucity of literature for the identified miRs associated with human CC, and hence, these comparisons are of high relevance.
-
i
We have identified Transferrin receptor (TFRC) as a putative target for let‐7d‐3p, in addition to three other targets. While let‐7d‐3p was up‐regulated in CC, its corresponding target, TFRC, was observed to be relatively down‐regulated in weight losing patients. Let‐7 members play a role in ageing; higher levels of let‐7 were observed in elderly subjects. They affect muscle cell proliferation by down‐regulating the target genes, eventually leading to reduced regenerative capacity of muscle.34 Transferrin, the ligand for TFRC, has been associated with myogenic differentiation, and down‐regulation of TFRC may impair myogenic differentiation. Furthermore, the expression of TFRC was observed to be down‐regulated in skeletal muscle and heart muscle in hereditary hemochromatosis and during muscle atrophy in conditions such as diabetes and uremia.35, 36 However, the role of TFRC in the context of CC remains to be elucidated.
-
ii
Gene targets identified for miR‐345‐5p include NOV, COL1A1 and CYR61. While miR‐345‐5p was up‐regulated in CC, its targets NOV and COL1A1 were down‐regulated in the muscle transcriptome dataset, whereas CYR61 was found to be up‐regulated. NOV and CYR61 are known to be involved in insulin‐like growth factor 1 gene signalling via the AKT and mammalian target of rapamycin pathway,37 impairment of which is related to suppression of protein synthesis leading to muscle atrophy. COL1A1 is an extracellular matrix protein, and its down‐regulation in animal models is reported to be associated with skeletal muscle atrophy. let‐7d‐3p and miR‐345‐5p regulate genes such as TFRC and COL1A1 that are known to be involved in skeletal muscle atrophy in diverse disease states including CC.36
-
iii
SQLE and FADS2 were the down‐regulated gene targets in muscle transcriptome dataset for miR‐423‐5p and miR‐3184‐3p; these miRs were up‐regulated and occur as clusters that are located within 10 kb distance.38 As they arise from the same clusters, their P‐value, FCs and direction of expression in the current study were found to be similar. Both these genes were found to be associated with lipid biosynthesis (Table 4) that is severely compromised in cachectic states. These genes contribute to sterol biosynthesis and fatty acid dehydrogenation. Additionally, miR‐423‐5p also regulates genes associated with energy homeostasis such as leptin. Perturbed levels of leptin in orexigenic and anorexigenic signals circuit and its impact on body weight regulation are fairly well understood. While increased leptin levels are observed in obesity, opposite effect is seen in CC.39 Leptin expression is low (but not lost) in mature skeletal muscle, but its expression is high in myocytes.40 Indeed, a similar trend was observed in our muscle transcriptome dataset, wherein leptin was down‐regulated in cases, relative to controls (data not shown). A similar argument may also be extended to miR‐423‐5p that is up‐regulated in CC (found in this study) and down‐regulated in morbid obese patients.41 miR‐423‐5p regulates DLK1 (down‐regulated in our study), which is known to be involved in skeletal muscle hypertrophy.42 However, the effect of this down‐regulation in CC needs to be further elucidated in future using model systems. miR‐423‐3p, which is derived from the same precursor miR as that of miR‐423‐5p, regulates CAMK2A (down‐regulated in this study), which is involved in calcium signalling. While CAMK2B is associated with inducing WL in CC,4 the role of CAMK2A remains to be elucidated.
miR‐3184‐3p, up‐regulated in the current study, is involved in Wnt/β‐catenin signalling, which plays a role in myogenic differentiation,43 and a defective signalling has been found to have an impact in muscle developmental defects.44 The targets of miR‐3184‐3p include BMPR1B and GREM1 that are up‐regulated and down‐regulated, respectively, in the muscle transcriptome dataset. They are involved in BMP signalling and transforming growth factor beta signalling, deregulation of which may contribute to CC.45
-
iv
Targets identified for miR‐532‐5p (up‐regulated) in this study include SULF1, RPS6KA6 and NPY1R. While SULF1 plays a role in regulating BMP signalling and is also required for somite development,46 NPY1R is involved in appetite regulation. The action of neuropeptideY (NPY) in energy metabolism is carried out through NPY1R and other NPY receptors,47 and their roles have been widely implicated in the pathogenesis of cachexia.48 RPSKA6 is involved in ciliary neurotrophic factor (CNTF) signalling (see below).
-
xxii
The targets identified for miR‐1296‐5p are HTR2A and RPS6KA6; both these genes were down‐regulated in muscle transcriptome dataset. HTR2A plays a role in serotonin signalling. Altered serotonin signalling has been demonstrated to have an impact on food intake in cachectic mouse models.49 Serotonin was also shown to be involved in myogenesis by promoting longitudinal growth of muscle fibres.50 RPS6KA6 (also regulated by miR‐532‐5p) is involved in CNTF signalling. CNTF is expressed in both central and peripheral nervous systems and has been shown to induce cachexia in model systems, when injected systemically.51
EIF4EBP1 (up‐regulated) was one of the targets identified for miR‐199a‐3p. EIF4EBP1 has been demonstrated to be involved in mammalian target of rapamycin signalling, which regulates muscle protein synthesis. This signalling pathway is shown to be impaired in cachectic states by the action of IL‐6.52 The other targets of miR‐199a‐3p are HTR2A and RPS6KA6.
Overall, based on these insights and inferences, the pleiotropic and redundant roles of miRs are observed against their binding partners regulating different pathways.
Biomarkers identified to‐date in CC, although promising, are not ready for translation to clinic. One of the foremost concerns is the availability of well‐annotated specimens for the discovery stages of the studies in sufficient numbers. Sample sizes used for gene expression studies are critical for confidence in the study findings. While it is important to have large number of samples, it is equally imperative to recognize the difficulties in obtaining biopsy material with detailed clinical annotations for CC phenotype. Despite these challenges, the current study had sample size of 42 for miR profiling and a sample size of 90 for mRNA profiling, well above the sample sizes used in earlier gene expression profiling experiments.4 Nevertheless, validating the identified biomarkers in independent datasets with larger sample size is warranted to confirm the study findings. Although the study has identified several targets and pathways that have potential implications in CC, as judged from the miR‐mRNA correlations, further characterization of the identified targets using model systems is needed to validate the overall biological relevance of the pathways. This would enable us to delineate the role of these identified miRs in CC and also gain functional insights.
Recent studies have highlighted the role of miRs as biomarkers from serum in myotonic dystrophy,53 and it remains to be seen if this could be extended to CC in future. In this proof of principle study, we have demonstrated potential utility of miRs as prognostic and predictive factors from muscle biopsies. However, muscle biopsy is invasive for routine analysis. Therefore, clinical application of the current findings awaits development of alternative methodologies and study designs to make it feasible in clinical setting. To understand the disease trajectory of CC, longitudinal studies can be conducted in plasma and serum as the same may not be possible with muscle biopsies. However, till such time, the challenges and logistics involved to carry out such investigations are addressed, and muscle biopsy remains the best option for CC biomarker and discovery studies. Identification and characterization of miRs that drive CC followed by functional testing in preclinical models may eventually lead to clinical trials to determine the efficacy of certain miRs for CC treatment.
Conclusions
We identified eight novel miRs that could potentially contribute to the aetiology of CC and as promising biomarkers. Continuing efforts in validating the miR signatures in independent cohorts and characterization of the identified miRs to understand its biological relevance to CC may eventually lead to the use of miRs as therapeutics.
Conflict of interest
The author(s) declare that they have no conflict of interest.
Supporting information
Table S1: Technical replicates
Table S2: Comparison of mRNA targets regulated by the differentially expressed miRs from two independent transcriptome datasets
Supporting info item
Supporting info item
Acknowledgements
The authors of this manuscript comply with the ethical guidelines for authorship and publishing in the Journal of Cachexia, Sarcopenia and Muscle: update 2015.54 Authors (S.D. and V.B.) thank Canadian Institute of Health Research (CIHR) for providing operating research grants. We would like to thank Lillian Cook and Jennifer Dufour for technical assistance.
Narasimhan, A. , Ghosh, S. , Stretch, C. , Greiner, R. , Bathe, O. F. , Baracos, V. , and Damaraju, S. (2017) Small RNAome profiling from human skeletal muscle: novel miRNAs and their targets associated with cancer cachexia. Journal of Cachexia, Sarcopenia and Muscle, 8: 405–416. doi: 10.1002/jcsm.12168.
References
- 1. Fearon K, Strasser F, Anker SD, Bosaeus I, Bruera E, Fainsinger RL, et al. Definition and classification of cancer cachexia: an international consensus. The Lancet Oncology 2011;12:489–95. [DOI] [PubMed] [Google Scholar]
- 2. Tan BH, Fearon KC. Cachexia: prevalence and impact in medicine. Current Opinion in Clinical Nutrition & Metabolic Care 2008;11:400–7. [DOI] [PubMed] [Google Scholar]
- 3. Argiles JM, Busquets S, Stemmler B, Lopez‐Soriano FJ. Cancer cachexia: understanding the molecular basis. Nature Reviews Cancer 2014;14:754–62. [DOI] [PubMed] [Google Scholar]
- 4. Stephens NA, Gallagher IJ, Rooyackers O, Skipworth RJ, Tan BH, Marstrand T, et al. Using transcriptomics to identify and validate novel biomarkers of human skeletal muscle cancer cachexia. Genome Medicine 2010;2:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Gallagher IJ, Stephens NA, MacDonald AJ, Skipworth RJ, Husi H, Greig CA, et al. Suppression of skeletal muscle turnover in cancer cachexia: evidence from the transcriptome in sequential human muscle biopsies. Clinical Cancer Research 2012;18:2817–27. [DOI] [PubMed] [Google Scholar]
- 6. Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 2004;116:281–97. [DOI] [PubMed] [Google Scholar]
- 7. Guller I, Russell AP. MicroRNAs in skeletal muscle: their role and regulation in development, disease and function. Journal of Physiology 2010;588:4075–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chen JF, Mandel EM, Thomson JM, Wu Q, Callis TE, Hammond SM, et al. The role of microRNA‐1 and microRNA‐133 in skeletal muscle proliferation and differentiation. Nature genetics 2006;38:228–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Sharma M, Juvvuna PK, Kukreti H, McFarlane C. Mega roles of microRNAs in regulation of skeletal muscle health and disease. Frontiers in Physiology. 2014;5:239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Chen L, Hou J, Ye L, Chen Y, Cui J, Tian W, et al. MicroRNA‐143 regulates adipogenesis by modulating the MAP2K5‐ERK5 signaling. Sci Rep. 2014;4:3819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ahn J, Lee H, Jung CH, Jeon TI, Ha TY. MicroRNA‐146b promotes adipogenesis by suppressing the SIRT1‐FOXO1 cascade. EMBO Molecular Medicine 2013;5:1602–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Romaine SP, Tomaszewski M, Condorelli G, Samani NJ. MicroRNAs in cardiovascular disease: an introduction for clinicians. Heart 2015;101:921–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Li X, Li Y, Zhao L, Zhang D, Yao X, Zhang H, et al. Circulating muscle‐specific miRNAs in Duchenne muscular dystrophy patients. Molecular TherapyNucleic Acids. 2014;3:e177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kulyte A, Lorente‐Cebrian S, Gao H, Mejhert N, Agustsson T, Arner P, et al. MicroRNA profiling links miR‐378 to enhanced adipocyte lipolysis in human cancer cachexia. American Journal of Physiology – Endocrinology & Metabolism 2014;306:E267–74. [DOI] [PubMed] [Google Scholar]
- 15. He WA, Calore F, Londhe P, Canella A, Guttridge DC, Croce CM. Microvesicles containing miRNAs promote muscle cell death in cancer cachexia via TLR7. Proceedings of the National Academy of Sciences of the United States of America 2014;111:4525–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bodine SC, Latres E, Baumhueter S, Lai VK, Nunez L, Clarke BA, et al. Identification of ubiquitin ligases required for skeletal muscle atrophy. Science 2001;294:1704–8. [DOI] [PubMed] [Google Scholar]
- 17. Gomes MD, Lecker SH, Jagoe RT, Navon A, Goldberg AL. Atrogin‐1, a muscle‐specific F‐box protein highly expressed during muscle atrophy. Proceedings of the National Academy of Sciences of the United States of America 2001;98:14440–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Tam S, de Borja R, Tsao MS, McPherson JD. Robust global microRNA expression profiling using next‐generation sequencing technologies. Laboratory Investigation 2014;94:350–8. [DOI] [PubMed] [Google Scholar]
- 19. Prado CM, Lieffers JR, McCargar LJ, Reiman T, Sawyer MB, Martin L, et al. Prevalence and clinical implications of sarcopenic obesity in patients with solid tumours of the respiratory and gastrointestinal tracts: a population‐based study. Lancet Oncology 2008;9:629–35. [DOI] [PubMed] [Google Scholar]
- 20. Martin L, Birdsell L, Macdonald N, Reiman T, Clandinin MT, McCargar LJ, et al. Cancer cachexia in the age of obesity: skeletal muscle depletion is a powerful prognostic factor, independent of body mass index. Journal of Clinical Oncology: Official Journal of the American Society of Clinical Oncology 2013;31:1539–47. [DOI] [PubMed] [Google Scholar]
- 21. Kazemi‐Bajestani SM, Mazurak VC, Baracos V. Computed tomography‐defined muscle and fat wasting are associated with cancer clinical outcomes. Seminars in Cell & Developmental Biology. 2016;54:2–10. [DOI] [PubMed] [Google Scholar]
- 22. Krishnan P, Ghosh S, Wang B, Li D, Narasimhan A, Berendt R, et al. Next generation sequencing profiling identifies miR‐574‐3p and miR‐660‐5p as potential novel prognostic markers for breast cancer. BMC Genomics 2015;16:735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Li H, Durbin R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 2009;25:1754–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B. Mapping and quantifying mammalian transcriptomes by RNA‐Seq. Nat Meth 2008;5:621–8. [DOI] [PubMed] [Google Scholar]
- 25. Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real‐time quantitative PCR and the 2(‐Delta Delta C(T)) Method. Methods 2001;25:402–8. [DOI] [PubMed] [Google Scholar]
- 26. Stretch C, Khan S, Asgarian N, Eisner R, Vaisipour S, Damaraju S, et al. Effects of sample size on differential gene expression, rank order and prediction accuracy of a gene signature. PLoS ONE [Electronic Resource] 2013;8:e65380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wightman B, Ha I, Ruvkun G. Posttranscriptional regulation of the heterochronic gene lin‐14 by lin‐4 mediates temporal pattern formation in C. Elegans. Cell n.d;75:855–62. [DOI] [PubMed] [Google Scholar]
- 28. Lee RC, Feinbaum RL, Ambros V. The C. elegans heterochronic gene lin‐4 encodes small RNAs with antisense complementarity to lin‐14. Cell n.d;75:843–54. [DOI] [PubMed] [Google Scholar]
- 29. Vasudevan S, Tong Y, Steitz JA. Switching from repression to activation: microRNAs can up‐regulate translation. Science 2007;318:1931–4. [DOI] [PubMed] [Google Scholar]
- 30. Agarwal V, Bell GW, Nam J‐W, Bartel DP. Predicting effective microRNA target sites in mammalian mRNAs. Elife. 2015;4, DOI: 10.7554/eLife.05005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Marioni JC, Mason CE, Mane SM, Stephens M, Gilad Y. RNA‐seq: an assessment of technical reproducibility and comparison with gene expression arrays. Genome Research 2008;18:1509–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Martin L, Senesse P, Gioulbasanis I, Antoun S, Bozzetti F, Deans C, et al. Diagnostic criteria for the classification of cancer‐associated weight loss. Journal of Clinical Oncology: Official journal of the American Society of Clinical Oncology 2015;33:90–9. [DOI] [PubMed] [Google Scholar]
- 33. Esfandiari N, Ghosh S, Prado CM, Martin L, Mazurak V, Baracos VE. Age, obesity, sarcopenia, and proximity to death explain reduced mean muscle attenuation in patients with advanced cancer. The Journal of Frailty & Aging 2014;3:3–8. [DOI] [PubMed] [Google Scholar]
- 34. Drummond MJ, McCarthy JJ, Sinha M, Spratt HM, Volpi E, Esser KA, et al. Aging and microRNA expression in human skeletal muscle: a microarray and bioinformatics analysis. Physiological Genomics 2011;43:595–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Rodriguez A, Hilvo M, Kytomaki L, Fleming R, Britton R, Bacon B, et al. Effects of iron loading on muscle: genome‐wide mRNA expression profiling in the mouse. BMC Genomics 2007;8:379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lecker SH, Jagoe RT, Gilbert A, Gomes M, Baracos V, Bailey J, et al. Multiple types of skeletal muscle atrophy involve a common program of changes in gene expression. The FASEB Journal 2004;18:39–51. [DOI] [PubMed] [Google Scholar]
- 37. Rommel C, Bodine SC, Clarke BA, Rossman R, Nunez L, Stitt TN, et al. Mediation of IGF‐1‐induced skeletal myotube hypertrophy by PI(3)K/Akt/mTOR and PI(3)K/Akt/GSK3 pathways. Nature Cell Biology 2001;3:1009–13. [DOI] [PubMed] [Google Scholar]
- 38. Zhang X, Azhar G, Wei JY. The expression of microRNA and microRNA clusters in the aging heart. PLoS ONE [Electronic Resource] 2012;7:e34688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Inui A, Meguid MM. Cachexia and obesity: two sides of one coin? Current Opinion in Clinical Nutrition & Metabolic Care 2003;6:395–9. [DOI] [PubMed] [Google Scholar]
- 40. Rigmor S, Vigdis A, Hege Thoresen G, Eili Tranheim K, Christian AD, Arild CR, et al. Leptin expression in human primary skeletal muscle cells is reduced during differentiation. Journal of Cellular Biochemistry 2005;96:89. [DOI] [PubMed] [Google Scholar]
- 41. Ortega FJ, Mercader JM, Catalan V, Moreno‐Navarrete JM, Pueyo N, Sabater M, et al. Targeting the circulating microRNA signature of obesity. Clinical Chemistry 2013;59:781–92. [DOI] [PubMed] [Google Scholar]
- 42. Waddell JN, Zhang P, Wen Y, Gupta SK, Yevtodiyenko A, Schmidt JV, et al. Dlk1 is necessary for proper skeletal muscle development and regeneration. PLoS ONE [Electronic Resource] 2010;5:e15055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Jones AE, Price FD, Le Grand F, Soleimani VD, Dick SA, Megeney LA, et al. Wnt/beta‐catenin controls follistatin signalling to regulate satellite cell myogenic potential. Skeletal Muscle. 2015;5:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Suzuki A, Scruggs A, Iwata J. The temporal specific role of WNT/β‐catenin signaling during myogenesis. Journal of Nature and Science 2015;1:e143. [PMC free article] [PubMed] [Google Scholar]
- 45. Zimmers TA, Davies MV, Koniaris LG, Haynes P, Esquela AF, Tomkinson KN, et al. Induction of cachexia in mice by systemically administered myostatin. Science 2002;296:1486–8. [DOI] [PubMed] [Google Scholar]
- 46. Meyers JR, Planamento J, Ebrom P, Krulewitz N, Wade E, Pownall ME. Sulf1 modulates BMP signaling and is required for somite morphogenesis and development of the horizontal myoseptum. Developmental Biology 2013;378:107–21. [DOI] [PubMed] [Google Scholar]
- 47. Sohn J‐W, Elmquist JK, Williams KW. Neuronal circuits that regulate feeding behavior and metabolism. Trends in Neurosciences 2013;36:504–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Inui A. Neuropeptide Y: a key molecule in anorexia and cachexia in wasting disorders? Molecular Medicine Today 1999;5:79–85. [DOI] [PubMed] [Google Scholar]
- 49. Dwarkasing JT, Boekschoten MV, Argilès JM, van Dijk M, Busquets S, Penna F, et al. Differences in food intake of tumour‐bearing cachectic mice are associated with hypothalamic serotonin signalling. J Cachexia Sarcopenia Muscle 2015;6:84–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Chandran S, Guo T, Tolliver T, Chen W, Murphy DL, McPherron AC. Effects of serotonin on skeletal muscle growth. BMC Proceedings. 2012;6, DOI: 10.1186/1753-6561-6-S3-O3 [Google Scholar]
- 51. Henderson JT, Seniuk NA, Richardson PM, Gauldie J, Roder JC. Systemic administration of ciliary neurotrophic factor induces cachexia in rodents. Journal of Clinical Investigation 1994;93:2632–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Durham WJ, Dillon EL, Sheffield‐Moore M. Inflammatory burden and amino acid metabolism in cancer cachexia. Current Opinion in Clinical Nutrition & Metabolic Care 2009;12:72–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Koutsoulidou A, Kyriakides TC, Papadimas GK, Christou Y, Kararizou E, Papanicolaou EZ, et al. Elevated muscle‐specific miRNAs in serum of myotonic dystrophy patients relate to muscle disease progress. PLoS ONE [Electronic Resource] 2015;10:e0125341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. von Haehling S, Morley JE, Coats AJS, Anker SD. Ethical guidelines for publishing in the Journal of Cachexia, Sarcopenia and Muscle: update 2015. J Cachexia Sarcopenia Muscle 2015;6:315–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1: Technical replicates
Table S2: Comparison of mRNA targets regulated by the differentially expressed miRs from two independent transcriptome datasets
Supporting info item
Supporting info item