

Abstract
Background
Exercise train (ET) stimulates muscle response in pathological conditions, including aging. The molecular mechanisms by which exercise improves impaired adiponectin/adiponectin receptor 1 (AdipoR1)‐related muscle actions associated with aging are poorly understood. Here we observed that in a senescence‐accelerated mouse prone 10 (SAMP10) model, long‐term ET modulated muscle‐regenerative actions.
Methods
25‐week‐old male SAMP10 mice were randomly assigned to the control and the ET (45 min/time, 3/week) groups for 4 months. Mice that were maintained in a sedentary condition served controls.
Results
ET ameliorated aging‐related muscle changes in microstructure, mitochondria, and performance. The amounts of proteins or mRNAs for p‐AMPKα, p‐Akt, p‐ERK1/2, p‐mTOR, Bcl‐XL, p‐FoxO3, peroxisome proliferators‐activated receptor‐γ coactivator, adiponectin receptor1 (adpoR1), and cytochrome c oxidase‐IV, and the numbers of CD34+/integrin‐α7 + muscle stem cells (MuSCs) and proliferating cells in the muscles and bone‐marrow were enhanced by ET, whereas the levels of p‐GSK‐3α and gp91phox proteins and apoptotic cells were reduced by ET. The ET also resulted in increased levels of plasma adiponectin and the numbers of bone‐marrow (BM)‐derived circulating CD34+/integrin‐α7 + MuSCs and their functions. Integrin‐α7 + MuSCs of exercised mice had improved changes of those beneficial molecules. These ET‐mediated aged muscle benefits were diminished by adiponectin and AdipoR1 blocking as well as AMPK inhibition. Finally, recombinant mouse adiponectin enhanced AMPK and mTOR phosphorylations in BM‐derived integrin‐α7 + cells.
Conclusions
These findings suggest that ET can improve aging‐related impairments of BM‐derived MuSC regenerative capacity and muscle metabolic alterations via an AMPK‐dependent mechanism that is mediated by an adiponectin/AdipoR1 axis in SAMP10 mice.
Keywords: Aging, Exercise, Muscle stem cell, Sarcopenia, SAMP10, Adiponectin
Introduction
In a wide range of mammals, age‐associated muscle weakness and wasting (often called sarcopenia) is characterized by an accumulation of adipose tissue and decreases in the number and size of muscle fibres, slower contraction speed, a shift in the composition of fibre types, and changes in various metabolic parameters.1, 2, 3, 4, 5, 6 Many attempts (including nutritional, hormonal, and pharmacological approaches) to prevent muscle atrophy have been proposed that are directed at reversing myofiber loss or promoting myofiber hypertrophy, and these approaches are largely designed to target mitochondria and metabolic mechanisms in aging‐related sarcopenia and frailty.7, 8, 9, 10 Despite the advances that have been achieved, no potential pharmacological approaches are currently in clinical use that mitigate or reverse the decline in muscle strength in aged humans,11, 12, 13 which constitutes a costly and ever‐increasing healthcare problem.12
Regular exercise training (ET) has been extensively studied because of its beneficial effects in restoring organ and tissue physical functions, thus preventing age‐related diseases such as muscle atrophy and peripheral arterial disease, possibly by activating cellular metabolism and regeneration.9, 14, 15, 16 ET‐induced changes in systematic and local tissue vascular growth hormones and cytokines have been shown to regulate vascular actions in response to ischemic stress.14, 17, 18 Consistent with these vascular protective actions, a numbers of clinical and experimental studies showed that ET also helped prevent muscle wasting in aged humans and animals.19, 20, 21 Over the last few years, comprehensive reviews highlighted the actions of pleiotropic adiponectin in various tissues and cells in response to stresses.22, 23 Aging and metabolic disorders were observed to result in a decrease in plasma adipose tissue‐derived adiponectin protein levels and tissue adiponectin receptor 1 (AdipoR1) levels.20, 24, 25 It was also demonstrated that decreased levels of adiponectin and AdipoR1 in obesity may have causal roles in mitochondrial dysfunction and insulin resistance in the skeletal muscles of diabetes.7 Moreover, long‐term ET alone or combined with diet or the use of an anti‐oxidant drug was shown to alter the plasma or/and adipose tissue adiponectin levels in animals and humans.20, 26 A single recent study provided circumstantial evidence that adiponectin promotes angiogenesis via a phosphoinositide 3 kinase/Akt‐hypoxia‐inducible factor‐1α pathway.27 However, regarding the ET‐related changes of aged animals, the link between the adiponectin/AdipoR1 axis and muscle regenerative capacity remains uncertain.
It has become clear that the function and the number of bone‐marrow (BM)‐derived stems cells are modified by pathological conditions such as aging and metabolic disorders.28, 29, 30, 31 The ability of therapeutic strategies to improve the regenerative capacity of muscle stem cells (MuSCs) located in muscle fibres is likely to contribute to muscle repair under the changed systemic and local microenvironments associated with aging.29, 32 As is the case with vascular endothelial progenitor cells (EPCs), a limited number of experimental and clinical studies have reported that bone marrow MuSCs contributed to muscle homeostasis and regeneration in humans and animals that underwent an exercise intervention.33, 34
Here, we report that long‐term ET improved the declines in muscle regeneration and performance via an AMP‐activated protein kinase (AMPK)‐dependent mechanism that is mediated by the adiponectin/AdipoR1 axis in a senescence‐accelerated mouse prone 10 (SAMP10) model.
Materials and methods
Animal care and use
All mouse experiments were performed with the approval of the Institutional Animal Care and Use Committee at Nagoya University and were in accordance with the U.S. National Institutes of Health (NIH) Guide for the Care and Use of Laboratory Animals. For all experiments described herein, we used male SAMP10 mice (SAMP10/TaSlc, Japan SLC, Hamamatsu, Japan) that were 8 weeks old when we obtained them. Young (8‐week‐old). The animals were housed in a room with controlled temperature (22 ± 2°C) and a 12‐h light–dark cycle, with ad libitum access to food and water.
Exercise and evaluation of endurance
We used a motorized rodent treadmill (S‐CON MINI‐Z: Tokyo Engineering) to determine the endurance capacity for running as shown in the exercise programme (Supporting Information, Figure S1). In the preliminary training sessions, 24‐week‐old mice were made to run on the treadmill at an inclination of 0° for 2 weeks (3×/week) as follows: warm‐up (5 min), 7 m/min; exercise (35 min), 17 m/min; cool‐down (5 min), 7 m/min.
For the measurement of endurance capacity, when the mice were aged 25 weeks, their initial running times for ET were determined according to the following programme at an inclination of 5° for 10 weeks (3×/week): warm‐up (5 min), 10 m/min; exercise (35 min), 18 m/min; cool‐down (5 min), 10 m/min. When these mice reached the age of 37 weeks, the initial running times to exhaustion were determined according to the following programme at an inclination of 5° for 4 weeks for the measurement of endurance capacity: warm‐up (5 min), 10 m/min; exercise (35 min), 20 m/min; cool‐down (5 min), 10 m/min.
To reduce the inherent variation in running capacity among the mice, we eliminated the mice whose running time was 25% longer or shorter than the average from the study. The average running times and body weights were matched among the groups. At 25 weeks of age, the mice were divided into a non‐ET group and an ET group, and they began an exercise programme on a treadmill 3×/week. At 4, 8, 12, and 16 wks after the start of the experiment, the endurance capacity of each mouse for running was measured as described.20
During the experimental period, a shock‐bar electric‐pressure was kept at 20 V, and we also counted the drop‐out times (as called the touching times to the shock‐bar herein, expressed as times/30 min). A mouse was deemed to be fatigued when it was no longer able to continue to run on the treadmill.
Grip strength
We also studied grip strength by using a small‐animal grip strength meter (Columbus Co., Largo, FL). When the forelimbs of a mouse whose tail was pulled horizontally by an examiner's hand were no longer able to grasp the strength meter, the indicated force was deemed the maximum grip strength. Grip strength was measured every 2 weeks and averaged over five measurements for each mouse. On the last day of the ET experiment, all of the mice were euthanized by an overdose of sodium pentobarbital, and the respective isolated muscles (soleus and gastrocnemius) were subjected to biochemical and morphological analyses.
Adiponectin/AdipoR1 blocking and AMPK inhibitor experiments
For the neutralizing antibody experiment, mice that had undergone the ET programme were injected subcutaneously with either control IgG (Cont group, ab37355, Abcam) or rabbit polyclonal antibody (pAb) against adiponectin (pAb‐Adip group; 450 µg/kg/d, ab3455, Cambridge, UK) 1×/week for 8 weeks, and they underwent endurance and drop‐up tests (1×/month); For the AdipoR1 inhibition experiment, mice that had undergone the ET programme were injected subcutaneously with either control mouse IgG (Cont group, ab37355) or rabbit pAb against adipoR1 (pAb‐AdipoR1 group; 450 µg/kg/day, ab70362, Abcam) 1×/week for 8 weeks, and they underwent endurance and drop‐up tests (1×/month); For a specific AMPK inhibition experiment, mice that had undergone the ET programme were injected subcutaneously with either dimethyl sulfoxide (DMSO group, Sigma‐Aldrich) or AMPK inhibitor compound C (Com‐C group; 10 mg/kg, 2 times/week, EMD Chemicals Inc, Gibbstown, NJ) 1×/week for 8 weeks, and they underwent endurance and drop‐up tests (1×/month). They were eventually sacrificed for biochemical and morphological analyses.
Body weight and other metabolic parameters
The body weights of all mice were measured weekly. The amounts of food intake, urine, and excrement were measured for each mouse with a metabolic and diuresis cage (TECNIPLAST, Tokyo) every 2 or 3 days, and we then calculated the average of each week's measurements.
Morphometry and immunohistochemistry analyses
We prepared cross‐cryosections (4 µm thick) and stained them with haematoxylin and eosin (H&E). The area per muscle fibre was measured in three randomly chosen microscopic fields from six different sections in each tissue block and averaged for each animal. For the immunohistochemistry, corresponding sections on separate slides were treated with mouse monoclonal antibodies (mAbs) against fast myosin light chain (MLC; 1:100, Sigma‐Aldrich, St. Louis, MO), proliferating cell nuclear antigen (PCNA; 1:SAMP10, ab29, Abcam, Cambridge, MA), rabbit pAb against slow myosin heavy chain (MHL; 1:100, ab11083, Abcam), and rat mAb against macrophages (Mac 3; 1:40, Clone M3/84, BD Pharmingen, San Diego, CA). The sections were then visualized with an ABC substrate kit (SK‐4400, Vector Laboratories, Burlingame, CA) in accord with the manufacturer's instructions.
Double immunofluorescence was performed using rabbit pAb to laminin‐5 (BS‐6713R, Bioss Antibodies, Woburn, MA), mouse mAb to desmin (1:100; Clone 33, Dako, Carpinteria, CA), goat pAb to integrin‐α7 (1:100; sc‐27706, Santa Cruz Biotechnology, Santa Cruz, CA), and rabbit mAb to CD34 (1: 100; ab110643, Abcam). The sections were visualized using Zenon rabbit and mouse IgG labelling kits (1:200; Molecular Probes, Eugene, OR) according to the manufacturer's instructions. The slides of the tissues (muscles and femurs) and the cells were mounted in glycerol‐based Vectashield medium (Vector Laboratories) containing the nucleus stain 4',6‐diamidino‐2‐phenylindole. For negative controls, the primary antibodies were replaced with nonimmune immunoglobulin G or zenon‐labelled rabbit or mouse IgG.
The ELISA and the biochemical analyses
Using 40‐week‐old mice, we determined the contents of plasma adiponectin, tumour necrosis factor‐alpha (TNF‐α), basic‐fibroblast growth factor (bFGF, 3139‐FB), and vascular endothelial growth factor (VEGF, MMV00) in the above‐described experimental groups by using a commercially available enzyme‐linked immunosorbent assay (ELISA) kit (R&D Systems, Minneapolis, MN) according to the manufacturer's instructions. Plasma glucose, triglyceride (TG), low‐density lipoprotein (LDL), high‐density lipoprotein (HDL), blood urea nitrogen (BUN), and creatinine were examined by the Nagoya University Clinical Research Laboratory.
Western blot analysis
Protein was extracted with the use of a RIPA lysis buffer, and the proteins were then Western‐blotted against antibodies for phospho‐mammalian target of rapamycinser2448 (p‐mTORser2448, #2971), total mTOR (#4517), phospho‐Aktser473 (p‐Aktser473, #4060), total Akt (#2967), p‐AMP‐activated protein kinaseαthr172 (p‐AMPKαthr172, #2535), total AMPKα, p‐extracellular signal‐regulated kinase1/2thr202/tyr204 (p‐Erk1/2thr202/tyr204, #4377), total Erk1/2 (#9107), phospho‐glycogene synthesis kinase‐3βser9 (p‐GSK‐3βser9, #9336), total GSK‐3β (#5676), phospho‐FoxO3aser294 (p‐FoxO3aser294, #5538), FoxO3 (#2497), Bcl‐2 (#2764), Bcl‐XL, (#2764, Cell Signaling Technology, Beverly, MA), gp91phox (clone 53, BD Transduction Laboratories, Lexington, KY), glyceraldehyde 3‐phosphate dehydrogenase (GAPDH, sc‐20357, Santa Cruz Biotechnology), and β‐actin (AC‐15, Sigma‐Aldrich; both used later as loading controls) (1:1000 for each antibody).
Quantitative real‐time gene expression assay
RNA was harvested from tissue with an RNeasy Fibrous Tissue Mini‐Kit (Qiagen, Hilden, Germany) in accord with the manufacturer's instructions. The mRNA was reverse‐transcribed to cDNA with an RNA PCR Core kit (Applied Biosystems, Foster City, CA). Quantitative gene expression was studied using the ABI 7300 real‐time polymerase chain reaction (PCR) system with Universal PCR Master Mix (Applied Biosystems).35 All experiments were performed in triplicate. The sequences of primers for adiponectin, AdipoR1, atrogin‐1, cytochrome c oxidase (COX)‐III, COX‐IV, glucose transporter‐4 (GLUT‐4), peroxisome proliferator‐activated receptor‐γ coactivator‐1α (PGC‐1α), PGC‐1β, and GAPDH genes are shown in Supporting Information, Table S1. The transcription of targeted genes was normalized to GAPDH.36
Electron microscopic analysis of myocardial mitochondria
Electron microscopy was performed as described.37 Muscle samples were cut into approx. 1 mm3 pieces and fixed first for 24 h with 2% glutaraldehyde in 0.16 M phosphate‐buffered saline (PBS; pH 7.2) and then for 1 h with 1% osmium tetroxide. The fixed tissues were dehydrated in a graded series of ethanol solutions before exposure to propylene oxide and embedding in Epon. The sections were cut at a thickness of 60–70 nm, stained with uranyl acetate and lead citrate, and observed with a transmission electron microscope (JEM‐1400, JEOL, Tokyo) operating at 100 kV. The quantitation of mitochondrial number and size was performed at a magnification of 15 000× by counting the corresponding number of pixels with the use of Adobe Photoshop CS5 software. A total of 60–80 mitochondrial cross‐sectional areas from six sections were measured for each mouse, and histograms were generated separately for each experimental group.
MuSC mobilization assay
At 8 and 16 weeks post‐ET, BM and peripheral blood (PB) samples were obtained from the two experimental groups (ET and Cont), and erythrocytes were lysed with ammonium chloride and separated into pellets. The cells were washed with PBS and sorted by flow cytometry using fluorescein isothiocyanate (FITC)‐labelled CD34 (Clone RAM34, eBioscience, San Diego, CA) and phycoerythrin‐labelled integrin‐α7 (3C12, Medical & Biological Laboratories Co., Ltd, Nagoya, Japan) as described.38
BM‐derived integrin α7+ MuSC isolation
BM‐derived cells were obtained from the ET and Cont groups of SAMP10 mice (n = 5 per group). Following the isolation of lineage− mononuclear cells, BM‐derived integrin‐α7 + cells were isolated by using integrin‐α7 MicroBeads and MACS (Miltenyli Biotec, Bergisch Gladbach, Germany) according to the manufacturer's instructions. Integrin‐α7 + BM cells were >90% positive for integrin‐α7 +.39 These cells expressed MuSC surface markers such as CD34 and integrin α7. After being cultured on fibronectin‐coated dishes (Wako, Osaka, Japan) in Dulbecco's Modified Eagle Medium (DMEM) and 2% foetal bovine serum for 7 days, the CD34+ cells were collected and used for the cellular assay.
Cell proliferation assay
In vitro MuSC proliferation was assessed with the Cell Titer 96AQ Assay kit (Promega, Madison, WI).40 Cells were plated on collagen‐coated 96‐well plates at 5000 cells in 100 μL of 0.3% bovine serum albumin (BSA)/DMEM‐2 per well and incubated in DMEM with insulin growth factor‐1 (IGF‐1) (50 ng/mL, 791‐MG, R&D Systems) for 48 h. Then, 20 μL of a mixture of tetrazolium compound and phenazine methosulfate was added (for an MTS assay), and the absorbance was determined at 492 nm. Experiments were performed five separate times for each group in triplicate.
Adiponection treatment assay
BM‐derived integrin‐α7 +cells were seeded on 6‐well plates and then cultured with DMEM containing 10% foetal bovine serum for 1 day. After starvation for overnight, the cells were treated with mouse recombinant adiponectin (Cat. 5095‐AC, R&D Systems) in serum‐free DMEM at indicated concentrations and time points (45 min for phosphorylation experiments; 24 h for protein expression experiments), and the levels of targeted proteins were analyzed by Western blot.
Cell migration and invasion assays
The cell migration and invasion assays were performed on Transwell (Costar®, Sigma‐Aldrich) 24‐well tissue culture plates as described.41 The cells that invaded the outer side of the membrane were stained and counted in 6–8 randomly chosen fields of the duplicate chambers at a magnification of 200× for each sample.
TUNEL staining
After the attachment of BM‐derived CD34+cells (cells density at 2 × 104 cells/mL) to coverslips precoated with gelatin, the cells were cultured in serum‐free DMEM containing 250 μM H2O2 for 24 h and then subjected to terminal deoxynucleotidyl transferase‐mediated dUTP nick end labelling (TUNEL, Cat. 11684795910) staining as described.39 The apoptotic cells in the muscles were also evaluated by TUNEL staining.
Immunocytofluorescence
After the attachment of BM‐derived CD34+cells (cells density at 2 × 104 cells/mL) to coverslips precoated with gelatin, the cells were fixed for 10 min with 8% paraformaldehyde and washed with 1% glycerol. Following blocking with 0.1% BSA/PBS, the cells were characterized using immunofluorescence.41
Statistical analysis
The data are expressed as means ± standard error of the mean (SEM). Student's t‐tests (for comparisons between two groups) or one‐way analysis of variance (ANOVA) (for comparisons of three or more groups) followed by Tukey post hoc tests were used for the statistical analyses. The nonparametric Kruskal–Wallis test (Tukey‐type multiple comparison) was used for the ANOVA of the gene expression data. The endurance and metabolic parameters were subjected to a two‐way repeated‐measures ANOVA and Bonferroni post hoc tests. The comparative incidence of drop‐out times and grip strength was evaluated by the χ2 test. SPSS software version 17.0 (SPSS, Chicago, IL) was used. P‐values <0.05 were considered significant.
Results
SAMP10 mice underwent an exercise programme from 24 weeks of age. We evaluated the changes in metabolic parameters, plasma lipid profiles, and other parameters at the indicated time points. There were no significant differences in food intake, drinking water, urine volume, excrement weight, or body weight between the control and ET groups during the experimental period (Supporting Information, Figures S2, S3A). Likewise, at 4 months after the start of the intervention, with the exception of HDL, there were no significant differences in plasma total cholesterol (T‐Cho), HDL, TG, glucose, BUN, or creatinine between the two groups (Table 1).
Table 1.
Levels of lipid and targeted plasma growth factors and cytokines and genes in the exercise training (ET) and Non‐ET groups at 40 weeks
| Parameter | Non‐ET | ET |
|---|---|---|
| T‐cho (mg/dL) | 68.9 ± 5.5 | 60.2 ± 4.0 |
| LDL (mg/dL) | 3.48 ± 1.1 | 4.4 ± 1.2 |
| HDL (mg/dL) | 7.9 ± 1.0 | 15.1 ± 2.5* |
| Triglyceride | 48.3 ± 10.2 | 36.1 ± 5.8 |
| Glucose (mg/dL) | 115.0 ± 8.8 | 116.0 ± 8.4 |
| BUN (mg/dL) | 27.4 ± 1.5 | 28.1 ± 1.8 |
| Cre (mg/dL) | 0.06 ± 0.01 | 0.08 ± 0.01 |
| TNF‐α (pg/mL) | 95.6 ± 9.6 | 108.7 ± 8.4 |
| Adiponectin (ng/mL) | 1052 ± 22 | 7518 ± 641** |
| VEGF (pg/mL) | 30.8 ± 3.5 | 37.4 ± 3.6 |
| bFGF (pg/mL) | 73.6 ± 4.1 | 82.1 ± 1.6 |
| RT‐PCR | ||
| Soleus muscle | ||
| Atrogin‐1 | 31.2 ± 5.07 | 3.98 ± 0.05** |
| COX4 | 0.70 ± 0.25 | 2.57 ± 1.09 |
| COX3 | 0.03 ± 0.01 | ‐1.61 ± 0.35** |
| GLUT4 | 0.65 ± 0.12 | 8.45 ± 2.49* |
| PGC‐1α | 1.05 ± 0.18 | 2.53 ± 0.39* |
| PGC‐1β | 0.09 ± 0.03 | 1.15 ± 0.15** |
| AdipoR1 | 0.61 ± 0.12 | 1.43 ± 0.24** |
| Gastrocnemius | ||
| Atrogin‐1 | 4409.83 ± 0.15.23 | 244.39 ± 15.63** |
| COX4 | 2.74 ± 0.55 | 3.06 ± 0.96 |
| COX3 | 0.19 ± 0.07 | 0.74 ± 0.48 |
| GLUT4 | 0.54 ± 0.22 | 3.21 ± 1.20* |
| PGC‐1α | 0.05 ± 0.02 | 1.12 ± 0.42* |
| PGC‐1β | 0.07 ± 0.04 | 1.15 ± 0.40* |
| AdipoR1 | 0.79 ± 0.31 | 3.01 ± 0.83* |
Values are mean ± SEM. T‐cho, total cholesterol; LDL, low‐density lipoprotein; HDL, high‐density lipoprotein; TG, triglyceride; BUN, blood urea nitrogen; Cre, creatinine, TNF‐α, tumour necrosis‐factor‐α; VEGF, vascular endothelial growth factor; RT‐PCR, real‐time polymerase reaction chain; AdipoR1, adiponectin receptor R1: MuRF‐1, muscle RING‐finger protein‐1, COX‐III, cytochrome c oxidase; COX‐IV, cytochrome c oxidase‐IV, GLUT‐4, glucose transporter‐4; PGC‐1α, peroxisome proliferator‐activated receptor‐γ coactivator‐1α; GAPDH, glyceraldehyde 3‐phosphate dehydrogenase.
P < 0.05,
P < 0.01 vs. control group.
The ET ameliorated the muscle loss and dysfunction in SAMP10 mice
SAMP10 strains can be useful for studying muscular aging.19 We examined the impact of long‐term ET on age‐associated muscle morphological changes and dysfunction in SAMP10 mice at 40 weeks after ET, and the results demonstrated that ET significantly improved the impaired grip strength and running endurance capacity (Supporting Information, Figure S3B,C). The ET group had significantly higher soleus muscle and gastrocnemius mass values compared with the control group (Supporting Information, Figure S3D,E). Likewise, the ET resulted in increased myofiber size and slow MHC positive myofiber rate (Supporting Information, Figure S4).
In addition, as the deterioration of cell proliferation and the increase in apoptosis seemed to be tightly associated with age‐related muscle loss, we tested the effects of ET on cell proliferation. At 4 months post‐ET, we observed that that ET activated cell proliferation in the soleus and gastrocnemius muscles (Figure 1A–1C). In addition, smaller numbers of apoptotic cells were observed in both muscles of the ET‐group mice compared with the controls (Figure 1E–1G). Thus, an ET‐mediated amelioration of morphological changes and restoration of the balance between muscle apoptosis and proliferation may account for the prevention of age‐related muscle dysfunction and wasting.
Figure 1.
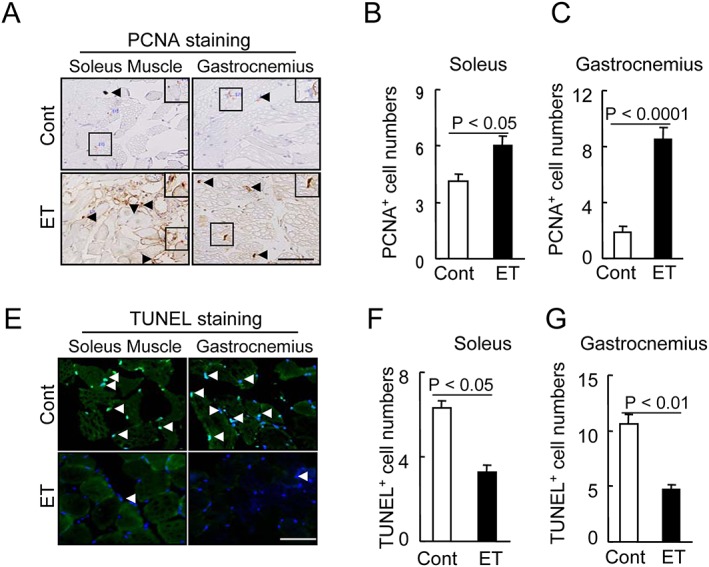
Effects of exercise training (ET) on muscle proliferation and apoptosis in 40‐week‐old mice. (A) Representative PCNA immunostaining with the mouse mAb used to assess the content of proliferated cells. (B,C) Quantitative data for PCNA‐positive cells. (E) Representative TUNEL staining used to assess the content of apoptotic cells. (F,G) Quantitative data for TUNEL‐positive cells. Data are mean ± SEM (n = 12–15). Arrowheads: related positive‐staining cells. Scale bar: 50 µm.
Adiponectin/AdipoR1 signalling activation is involved ET‐mediated muscle benefits
A close association between decreased levels of adipocytes‐derived adiponectin and muscle mitochondrial dysfunction has been characterized in pathological conditions, including aging and diabetes.9 As shown in Table 1, we observed that the ET mice had 6.3‐fold increased levels of plasma adiponectin protein compared with the non‐ET SAMP10 mice. The ET resulted in an increase in AdipoR1 gene in the soleus muscle and gastrocnemius (Table 1).
To further examine the consequences of ET‐induced molecular changes, we analyzed the extractions of the muscles to investigate the role of the adiponectin/AdipoR1 signalling pathway. The results indicated that the ET enhanced the phosphorylation of AMPKα protein, a key player in adiponectin/AdipoR1 signalling, in the soleus and gastrocnemius (Figure 2A–2C). The ET was also able to induce the phosphorylation of Akt, Erk1/2, and mTOR. Likewise, the ET resulted in decreased levels of p‐GSK and gp91phox proteins and increased levels of Bcl‐XL in both the soleus and gastrocnemius muscles (Figure 2A–2D). Thus, the ET‐mediated muscle cell protective response by adiponectin/AdipoR1 signalling activation and anti‐oxidative stress may be responsible for the preventive effects on age‐related atrophy. On the other hand, atrogin‐1 has been identified as a likely targes of drug therapy for muscle atrophy.42 Our present quantitative real‐time PCR data demonstrated that the ET suppressed the atrogin‐1 gene expression in both the soleus and gastrocnemius muscles (Table 1). The ET also resulted in an increase in the levels of p‐FoxO‐3a protein (Figure 2A–2D), leading us to speculate that ET‐mediated FoxO3a phosphorylation and atrogin‐1 inactivation likely contribute to the mitigation of intracellular protein degradation in the muscle loss process.
Figure 2.
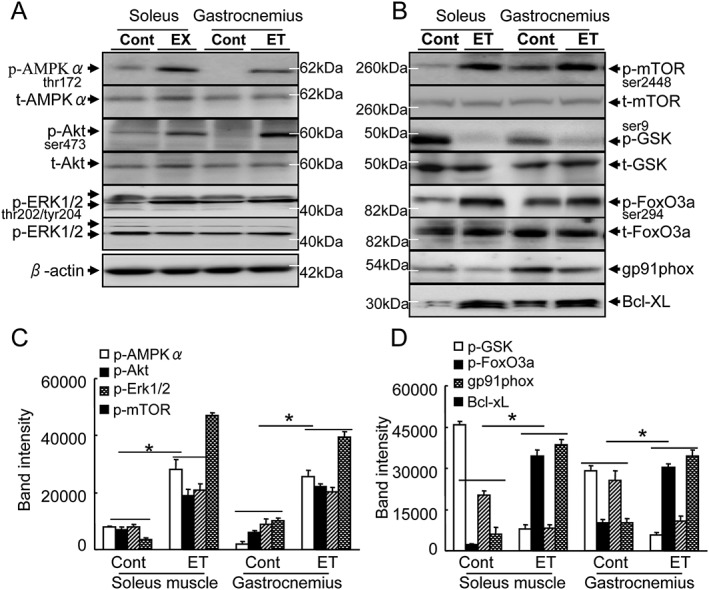
The effects of exercise training (ET) on targeted protein expression and phosphorylation in 40‐week‐old mice. (A,B) Representative immunoblots show the levels of the targeted protein expression or phosphorylation in the soleus and gastrocnemius. (C,D) Quantitative analysis of Western blots for the levels of p‐AMPKα, p‐Akt, p‐Erk1/2, p‐mTOR (C), p‐GSK, p‐FoxO3a, gp91phox, and Bcl‐XL proteins (D) in the two groups. The expression level of each targeted protein was normalized with a Western blot antibody to β‐actin. Data are mean ± SEM (n = 5). *P < 0.01 vs. control group (Cont.)
The ET improved muscle mitochondrial biogenesis and dysfunction
As seen in Supporting Information, Figure S5A, the electron microscopy showed that severe degeneration in the cristae of mitochondria had occurred in both the soleus and gastrocnemius muscles of the SAMP10 mice. The numbers of damaged mitochondria, the percentage of damaged mitochondria and the numbers of lipid droplets were significantly lower in the ET group compared with the control group (Supporting Information, Figure S5B–D). The quantitative PCR also revealed that the levels of the cytochrome c oxidase subunit 5B (COX5B, called respiratory complex IV) gene, a key enzyme of the respiratory chain, were higher in the exercised soleus and gastrocnemius muscles of SAMP10 mice (Table 1). The exercised muscles also had increased levels of PGC‐1α gene expression. Adiponectin has been shown to be involved in PGC‐1α signalling pathway activation in cardiomyopathy.43 Moreover, Iwabu and colleagues demonstrated that the adiponectin/AdipoR1 axis can modulate mitochondrial biogenesis via transcriptional coactivator PGC‐1α activation in muscles.9 Here, the ET resulted in increased levels of plasma adipose tissue‐derived adiponectin (Table 1), suggesting that the improvement of physical performance might be due to the PGC‐1α activation‐mediated mitochondrial biogenesis and energy metabolism by the up‐regulation of adiponectin in response to the long‐term ET. However, there were significant differences in VEGF, bFGF, and TNF‐α between the ET and control mice (Table 1).
The ET stimulated MuSC mobilization and improved the intrinsic function of MuSCs
To visualize the regenerative process, we performed double immunofluorescence for desmin (an intermediate filament protein that in the control mice was expressed at a lower level) and for laminin 5, a basement membrane protein that was disorganized in immature muscle fibres of the soleus and gastrocnemius muscles of the control mice. As shown by Figure 3A and 3B, at 4 months post‐ET, the ET muscles showed strong desmin and laminin 5 expressions and ordered organization. Likewise, the numbers of CD34+/integrin‐α7 + MuSCs were much higher in the muscles of the ET mice compared with those of the control mice (Figure 3C–3E). Moreover, the flow cytometry revealed that the ET resulted in increased numbers of CD34+/integrin α7 + MuSCs in the BM and the PB (Figure 4A and 4B). BM‐derived stem cells isolated with integrin‐α7 + microbeads expressed MuSC surface markers such as CD34 and integrin‐α7 (Figure 4C). A Western blot assay revealed that the ET resulted in increased levels of p‐Akt and Bcl‐2 protein and decreased levels of gp91phox in the integrin‐α7+ MuSCs of the SAMP10 mice (Figure 5A and 5B). We investigated whether ET would influence the BM‐derived MuSC dysfunction associated with aging. The results indicated that the ET enhanced the MuSC migration and proliferation and mitigated MuSC apoptosis (Figure 5C–5E). Taken together, these findings suggest that the improvement of BM‐derived MuSC mobilization/homing and dysfunction by ET could represent a common cellular mechanism in the protection of muscle tissue against aging stress.
Figure 3.
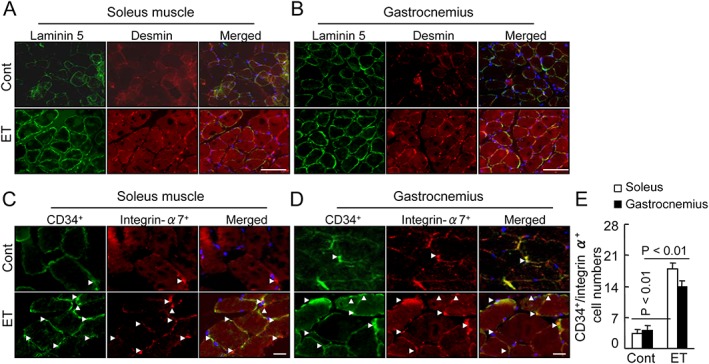
Exercise training (ET) ameliorated the laminin and the desmin expressions in the soleus and gastrocnemius at 4 months post‐ET. (A,B) Fluorescence staining of muscles with laminin 5 rabbit pAb (green) and desmin mAb (red). (C,D) Representative images for muscle stem cells identified by double immunofluorescent staining with rabbit mAb against CD34 and goat pAb against integrin‐α7. (E) Quantitative data for CD34 and integrin‐α7 double positive cells. White arrowheads indicate CD34+/integrin‐α7 + cells. Data are mean ± SEM (n = 12). Scale bars: 50 µm.
Figure 4.
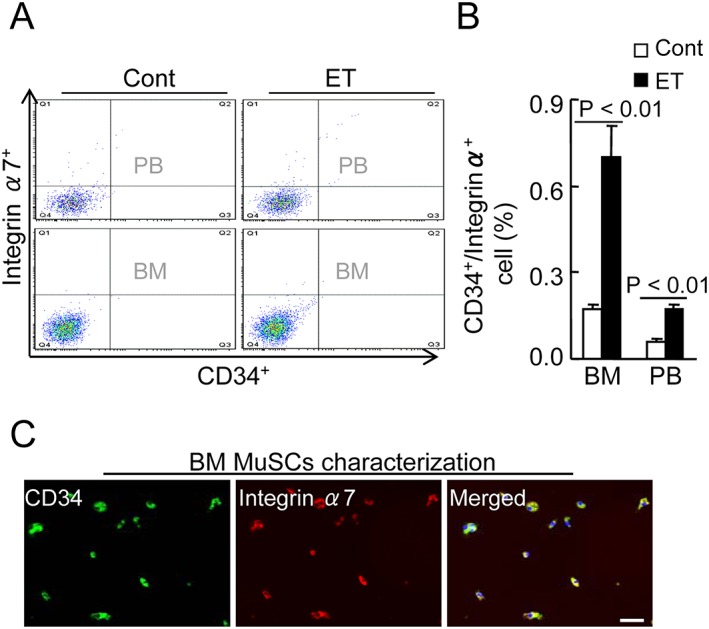
Effects of exercise training (ET) on muscle stem cells (MuSC) production and mobilization in 40‐week‐old mice. (A,B) CD34+/integrin‐α7 + MuSCs were measured by flow cytometry in BM and PB of both experimental group mice. Representative images and quantitative data for CD34+/integrin α7 + cell plots (A) and their percentages (B) in BM and PB of both groups (total 2 × 104 cells). Data are mean ± SEM (n = 6). (C) BM‐derived integrin‐α7 cells were isolated using CD34 MicroBeads and cultured DMEM containing muscle growth factors for 7 days. Double labelling of MuSCs for green (CD34) and red (integrin‐α7), showing yellow (co‐expression merged).
Figure 5.
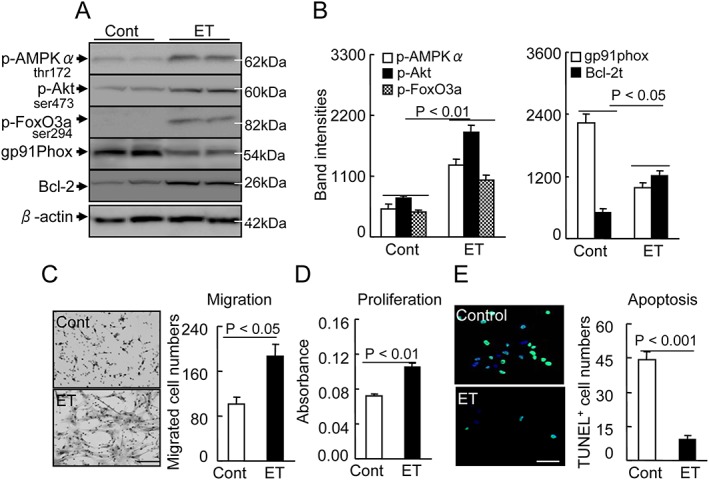
Effects of exercise training (ET) on targeted protein expression and phosphorylation in 40‐week‐old mice. (A,B) Representative images and quantitative data show the levels of p‐AMPKα, p‐Akt, p‐FoxO3a, gp91phox, and Bcl‐2 proteins in the integrin‐α7 + BM stem cells of both groups. (C) To evaluate the migration capacity of muscle stem cells (MuSCs) from non‐ET vs. ET mice, we subjected isolated integrin‐α7 + MuSCs to a Boyden chamber assay. Representative images and combined quantitative data show migrated cells. (D) The BM‐derived integrin‐α7 + stem cell proliferation capacity was analyzed by an MTT cell proliferation assay expressed as absorbance. (E) To evaluate the cell apoptosis, we cultured isolated integrin‐α7+ stem cells in the presence of H2O2 (500 μM) for 24 h, and then the MuSCs were treated with TUNEL staining agents for the apoptosis count. Representative imags and combined quantitative data show TUNEL+ cells in both experimental groups. Data are means ± SEM (n = 8–12). Scale bars: 50 µm.
Adiponectin blocking diminished the ET‐mediated muscle benefits in SAMP10 mice
To further explore the molecular mechanisms underlying the ET‐mediated prevention of aging‐related muscle damage, we analyzed SAMP10 mice treated with control IgG and neutralizing rabbit pAb against adiponectin (pAb‐Adip) for 2 months. Although there was no difference in body weight between the two groups, adiponectin blocking decreased the endurance capacity and enhanced the drop‐out times at the indicated time points (Figure 6A–6C). The muscle mass and the myofiber size were significantly lower in both muscles of the pAb‐Adip mice that underwent ET (Figure 6D–6F and 6H). Interestingly, we observed that that adiponectin depletion suppressed cell proliferation in the soleus and gastrocnemius muscles (Supporting Information, Figure S6). In contrast, greater numbers of apoptotic cells were observed in both muscles of the pAb‐Adip mice compared with the controls (Figure 6G and 6I). In addition, the pAb‐Adip mice had decreased levels of the p‐AMPKα, p‐Akt, p‐mTOR, and Bcl‐XL proteins (Supporting Information, Figure S7), indicating that adiponectin blocking diminished the ET‐mediated prevention of muscle morphological changes and dysfunction via the inactivation of AMPK/p‐Akt‐mTOR signalling. However, there were no significant differences in the plasma T‐Cho, HDL, TG, glucose, BUN, or creatinine levels between the two groups (Supporting Information, Table S2). With the exception of white blood cells and the percentages of neutrophils, there were also no significant differences in PB parameters between the two groups (Supporting Information, Table S2).
Figure 6.
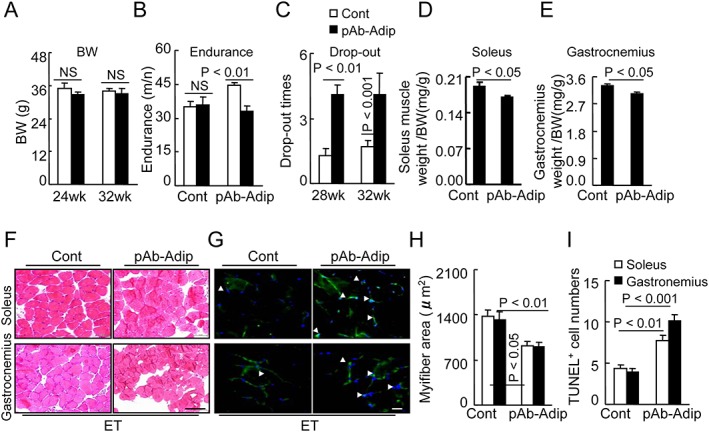
Adiponectin depletion diminished exercise training (ET)‐mediated muscle benefits. (A–C) Body weight (BW), endurance, and drop‐out were recorded in the Cont and pAb‐Adip groups at the indicated time points. (D,E) The ratios of soleus muscle to BW and ratios of gastrocnemius to BW were calculated in the two groups. Data are means ± SEM (n = 8–9). (F,H) Representative haematoxylin and eosin images and quantitative data showing the myofiber size in both muscles of Cont and pAb‐Adip mice. (G,I) Representative TUNEL images and quantitative data showing the numbers of apoptotic cells in both muscles of the two groups. White arrowheads indicate TUNEL‐positive cells. Data are mean ± SEM (n = 8–9). Scale bars: 50 µm.
Adiponectin blocking disturbed the ET‐mediated mitochondrial biogenic actions
We examined whether adiponectin blocking would influence the ET‐mediated mitochondrial morphological changes and function. The results indicated that adiponectin depletion enhanced the damage to mitochondria and the lipid droplet accumulation in the SAMP10 mice that underwent ET (Supporting Information, Figure S8). As seen in Supporting Information, Table S3, the adiponectin depletion decreased the levels of COX4 and PGC‐1α genes compared with the control mice. Collectively, these findings suggest that these antimusculoprotective actions of adiponectin blocking are mediated, at least in part, through the inactivation of an adiponectin‐dependent PGC‐1α signalling pathway in the muscles of SAMP10 mice.
Adiponectin blocking impaired the MuSC mobilization and function in the ET mice
To further explore the molecular mechanisms of adiponectin blocking‐mediated inhibitory effects on MuSC regeneration capacity, we characterized MuSCs from the soleus and gastrocnemius of 32‐week‐old mice by double immunofluorescent staining. The numbers of CD34+/c‐Kit+ MuSCs were lower in both muscles of the pAb‐Adip mice compared with the ET‐alone mice (Figure 7A–7C). Immunofluorescence showed that the adiponectin depletion reduced the positive staining for the desmin and laminin 5 proteins in the soleus and gastrocnemius muscles of the ET‐alone mice (Figure 7D and 7E). The flow cytometry revealed that the adiponectin depletion also resulted in decreased numbers of CD34+/integrin α7 + MuSCs in the BM and the PB (Figure 8A and 8B). The pAb‐Adip mice had decreased levels of p‐AMPKα, p‐mTOR, and Bcl‐2 proteins in the BM‐derived integrin‐α7+ stem cells (Figure 8C). H&E staining shows the BM structures of the femurs of both groups (Figure 8D). As anticipated, adiponectin depletion reduced the numbers of PCNA+ and PCNA+/integrin‐α7+ stem cells in the BM (Figure 8E–8G). Recombinant mouse adiponectin enhanced the levels of p‐AMPKα, p‐mTOR, and Bcl‐2 proteins the BM‐derived integrin‐α7+ stem cells (Supporting Information, Figure S9). Thus, these findings suggest that adiponectin blocking may impair MuSC mobilization and function in ET mice.
Figure 7.
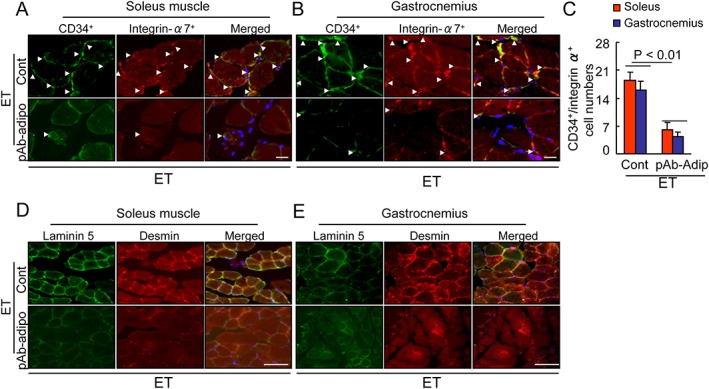
Effects of adiponectin depletion on muscle stem cell regeneration in the soleus muscles and gastrocnemius at 2 months post‐ET. (A–C) Representative images and quantitative data for muscle stem cell regeneration identified by double immunofluorescent staining with rabbit mAb against CD34 (green) and goat pAb against integrin‐α7 (red). Arrowheads: related positive‐staining cells. Data are mean ± SEM (n = 8–9). (D,E) Fluorescence staining of muscles with laminin 5 rabbit pAb (green) and desmin mAb (red). Scale bar: 50 µm.
Figure 8.
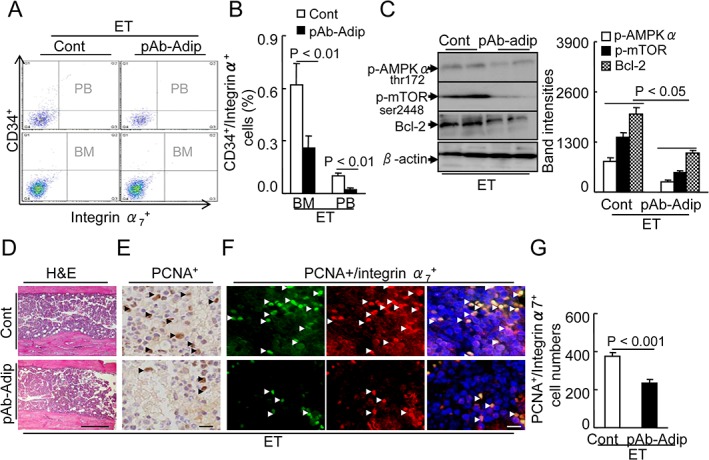
Effects of adiponectin depletion on muscle stem cells production and mobilization. (A,B) At 2 months post‐ET, CD34+/integrin‐α7 + was measured by flow cytometry in the BM and PB of the control and ET mice. Representative images and quantitative data show CD34+/integrin‐α7 + cell plots (A) and their percentages (B) in the BM and PB of both groups (total 2 × 104 cells). (C) Representative images and combined quantitative data show the levels of p‐AMPKα, p‐mTOR, and Bcl‐2 proteins in the integrin‐α7 + BM stem cells of both groups. (D) Representative images of the H&E staining of the femur BM structure. E: Representative images for PCNA+ proliferating cells. (F,G) Representative images and quantitative data for integrin‐α7 +/PCNA+ proliferating muscle stem cells of both groups. Arrowheads: positive cells. Data are mean ± SEM (n = 6–8). Scale bars: 50 µm.
AdipoR1 inhibition abrogated the ET‐mediated muscle benefits
To obtain further evidence of adiponectin/AdipoR1‐mediated aged muscle beneficial effects in SAMP10 mice received long‐term exercise, we conducted AdipoR1 inhibition experiments. AdipoR1 inhibition abrogated grip strength, endurance capacity, and muscle mass of ET mice (Supporting Information, Figure S10A–D). The numbers of CD34+/c‐Kit+ MuSCs were lower in both muscles of the AdipoR1‐I mice compared with the control mice (Supporting Information, Figure S10E). The flow cytometry revealed that the adipoR1 inhibition also resulted in decreased numbers of CD34+/integrin α7 + MuSCs in the BM and the PB (Supporting Information, Figure S10F). Interestingly, we observed that that AdipoR blocking suppressed cell proliferation in the soleus and gastrocnemius muscles (Supporting Information, Figure S10G). Likewise, the AdipoR1‐I mice had decreased levels of the p‐AMPKα, p‐mTOR, and Bcl‐XL proteins in the muscles (Supporting Infomation, Figure 10H). Therefore, these results indicate that AdipoR1 blocking abrogated the ET‐mediated prevention of muscle morphological changes and dysfunction and improvements of MuSC mobilization and dysfunction in SAMP 10 mice.
Pharmacological inhibition of AMPK abrogated the ET‐mediated muscle benefits
Finally, we examined whether pharmacological inhibition of AMPK would influence the ET‐mediated muscle changes and function. The results indicated that AMPK inhibition with compound C decreased the endurance capacity and muscle mass and enhanced the drop‐out times (Supporting Information, Figure S11A–E). As anticipated, the numbers of apoptotic cells were higher in both muscles of the AMPK‐I mice compared with the control mice (Supporting Information, Figure S10F), indicating that AMPK inhibition appears to abrogate ET‐mediated muscle actions in SMAP10 mice.
Discussion
Regular long‐term ET‐induced systemic and individual tissue cell responses represent the leading cause of various benefits.44, 45, 46, 47, 48, 49 Exploring the potential mechanisms of ET‐related modulations of the features of age‐associated muscle diseases will contribute to therapeutic strategies to combat sarcopenia and frailty. The significant findings of our present work are that the mice that underwent ET as early as at 24 weeks of age showed improved physical performance in contrast to aging‐associated muscle wasting. Here, at the molecular and cellular levels, the exercise enhanced not only the circulating adiponectin levels and muscle AdipoR1 gene expression but also the AMPK/PGC‐1α‐related mitochondrial biogenesis and Akt/mTOR‐mediated protein synthesis and proliferation. Simultaneously, the ET retarded the Akt/p‐FoxO3‐related atrogin‐1 activation and Nox2‐related oxidative stress production and restored the anti‐apoptotic (Bcl‐1 or/and Bcl‐XL) molecule expression. Adiponectin blocking diminished these beneficial intracellular actions and the muscle dysfunction improvement in the ET mice. Pharmacological interventions targeted towards AdipoR1 and AMPK also abrogated ET‐related muscle beneficial actions in SAMP10 mice. Moreover, recombinant mouse adiponectin resulted in increased levels of p‐AMPKα, p‐mTOR, and Bcl‐2 in BM‐derived integrin‐α7+ cells. The ET stimulated BM‐derived CD34+/integrin‐α7+ MuSC protection and mobilization and homing into injured musculature and proliferation, and it retarded the mitochondria damage and cell apoptosis via an AMPK‐dependent mechanism that is mediated by the adiponectin/AdipoR1 axis in SAMP10 mice. The mechanisms underlying the improvement of aging‐related muscle regeneration and dysfunction in a SAMP10 mouse model are schematically represented in Supporting Information, Figure S9.
It is well established that regular ET is beneficial against various types of cardiovascular and muscle diseases.9, 14 The data presented here show that exercise improved the grip strength and endurance of SAMP10 mice. The ET also enhanced the desmin expression and slowed the HMC‐positive myofiber rate in the soleus and gastrocnemius muscles of the mice. The ET improved the aging‐related unbalanced cell proliferation and apoptosis in both muscles. The exercised mice had increased levels of p‐AMPKα, p‐Akt, p‐ERK1/2, p‐mTOR, and p‐FoxO3 proteins and AdipoR1 gene and decreased levels of astrogin‐1 gene. Taken together with the results of our adiponectin and AdipoR1 blocking experiments, these findings indicate that the benefits of exercise are likely to improve muscle integrity via an adiponectin/AdipoR1‐related restoration of the balance of protein synthesis and proteolysis.
It has been clear that the function and the numbers of BM‐derived stem cells are modified by pathological conditions such as age‐associated disease and by therapeutic exercise intervention.14, 39 In the present study, the ET caused an increase in the numbers of CD34+/integrin‐α7 + MuSCs in the bone marrow and PB. The muscles of the ET mice showed strong desmin and laminin 5 expressions and ordered organization. The ET also enhanced the numbers of CD34+/integrin‐α7+ MuSCs in the soleus and the gastrocnemius. In addition, the migration and proliferative responses of BM‐derived integrin‐α7+ stem cells to IGF‐1 were higher in the ET mice than in the control mice. Likewise, the ET reduced the cell apoptosis after exposure to H2O2. Moreover, the ET resulted in increased levels of AdipoR1 gene and p‐AMPKα, p‐Akt, p‐FoxO3a, and Bcl‐2 proteins and decreased levels of gp91phox in the BM‐derived integrin‐α7+ MuSCs. The ELISA revealed that the plasma levels of adipose tissue‐derived adiponectin were increased in the ET mice. In addition, recombinant mouse adiponectin resulted in increased levels of p‐AMPKα, p‐mTOR, and Bcl‐2 in BM‐derived integrin‐α7+ cells. Adiponectin protects EPCs against apoptosis and therefore could modulate EPCs' ability to induce the repair of vascular damage.50 Another study demonstrated that adiponectin deficiency resulted in decreased BM‐derived EPC‐like cell survival, decreased proliferation, and decreased differentiation ability in mice.51 Thus, the ability of ET to improve MuSCs' intrinsic function and mobilization has a salutary effect on the bone marrow and the muscles even under conditions of aging‐related oxidative stress, by the activation of an adiponectin/AdipoR1‐AMPK signalling pathway, thereby promoting muscle regeneration. This notion was further supported by our finding that the pharmacological interventions targeted towards adiponectin and AdipoR1 diminished the ET‐induced MuSC regenerative actions including production, mobilization, and homing into the injured musculature via AMPK activation in SAMP10 mice.
Aging and metabolic disorders caused decreases in the levels of circulating adiponectin proteins and tissue adiponectin receptor1 (AdipoR1).20, 24, 25 The ability of ET to increase adiponectin levels is likely contribute to the prevention of muscle loss under experimental conditions. In agreement with a previous report that exercise reduced endothelial cell apoptosis in ischemic muscles at advanced ages,39 our present findings demonstrated that cell apoptosis in the soleus and gastrocnemius were reduced by ET, and these effects were reduced by adiponectin depletion. Similar to the results of a clinical study,52 our present findings show that exercise significantly increased the circulating adiponectin levels in SAMP10 mice. ET reduced the muscle p‐GSK and gp91phox levels and enhanced the Bcl‐XL levels. Moreover, adiponectin and AdipoR1 blocking reduced the muscle Bcl‐XL level. As seen in AMPK inhibition experiments, compound C resulted in decreased grip strength, endurance capacity, and muscle mass and increased the numbers of TUNEL+ cells in both soleus muscle and gastrocnemius in ET mice. Adiponectin/adipoR1 has been reported to protect against cardiac cell and muscle apoptosis in vivo and in vitro.43, 53 Thus, exercise appears to prevent muscle loss via adiponectin/AdipoR1‐AMPK‐mediated anti‐apoptotic and anti‐oxidative actions.
In addition, the effect of exercise on PGC‐1α activity is likely to be of great clinical and experimental significance when the expression of the transcriptional coactivator is reduced.54 Accumulating evidence indicates that PGC‐1α expression in skeletal muscle declines with age and with diabetic disease.19, 54 Our present findings show that ET retarded age‐associated mitochondrial damage and lipid droplet accumulation. It was recently reported that exercise reversed obesity‐related decreased levels of adiponectin and adipoR1 and mitochondrial dysfunction in the muscle.9 Our present observations demonstrate that adiponectin blocking suppressed exercise‐induced AMPKα phosphorylation and PGC‐1α and COX4 expressions and accelerated the mitochondrial morphological changes. Because PGC‐1α overexpression rescues mitochondrial injury, we propose that the enhancement of the adiponectin/adipoR1‐depedent AMPK‐PGC‐1α activation‐mediated mitochondrial biogenic capacity is likely attributable, at least in part, to contribute to the ET‐mediated muscle‐protecting actions.
There were several study limitations. First, we did not design to calculate the amounts of subcutaneous and inguinal fat mass in any way in the experimental settings. Second, this study was not designed to compare the difference between SAMR1 and SAMP10.
Summary
Exercise stimulates the muscles' responses in pathological conditions, including sarcopenia. The molecular mechanisms by which exercise improves the muscle loss and dysfunction associated with aging are poorly understood. The present study's results indicate that aging‐associated muscle wasting in SAMP10 mice can be ameliorated by ET via the improvement of adiponection‐AdipoR1‐dependent AMPK/Akt‐mTOR signalling‐mediated protein catabolic and anabolic responses, oxidative stress‐related cell apoptosis, and adiponectin/PGC‐1α activation‐related biogenesis. The ability of ET to restore the ‘young’ muscle response can be recommended as a powerful strategy to prevent age‐associated declines in muscle regeneration and function by recruiting and improving the delivery of MuSCs to the damaged muscle tissues. This might also allow the therapeutic use of nonpharmacological interventions, such as voluntary exercising, to complement pharmacological or cell therapies.
Author contribution statement
A.I.: Major contributor to the collection and assembly of data; manuscript writing; morphological analysis; ELISA and laboratory analysis; real‐time PCR analysis; collection and assembly of samples and data, data analysis and interpretation; X.W.C.: Major contributor to the collection and assembly of data; manuscript writing; Western blotting and electron microscopy; financial support; conception, design, and editing of the manuscript; H.J., G.Z., Y.L., G.Y., and X.L.: Collection and assembly of samples and data, data analysis, and interpretation; R.K. and L.P.: ELISA and laboratory analysis; L.H. and H.W.: Bone‐marrow transplantation and in vitro MuSCs analysis; Z.H., T.S., and K.I.: Western blotting, morphological analysis, and electron microscope analysis; K.S., Y.B., and T.K.: Reviewed/edited the manuscript; M.K.: Financial support; conception, design, and editing of the manuscript.
Conflicts of interest
The authors declare no conflicts of interest.
Supporting information
Table S1. Primer sequences used in the quantitative real‐time polymerase chain reaction (PCR)
Table S2. Levels of plasma lipid, targeted growth factors, cytokines and others in two experimental groups at indicated time points
Table S3. Levels of targeted genes in two experimental groups at 32 wks
Figure S1. Schematic representation of the exercise training (ET) program.
Figure S2. Effects of ET on metabolic parameters. A–D: Food intake, drinking water, urine volume and excrement weight were recorded in the control (Cont) and exercise training (ET) groups at the indicated time points. Data are mean ± SEM (n = 6).
Figure S3. The effects of ET on muscle function and mass at 4 mos post‐ET. A–C: Body weight (BW), grip strength/BW and endurance were recorded in the control (Cont) and exercise training (ET) groups. D,E: The ratios of soleus muscle to BW and ratios of gastrocnemius to BW were calculated at 40 wks of age in both groups. Data are mean ± SEM (n = 10–12). NS: not significant.
Figure S4. The effects of ET on the myofiber size and the slow MHC rate at 4 mos post‐ET. A: Representative H&E staining of soleus and gastrocnemius of Cont and ET mice. B: Quantitative data showing the myofiber size in both muscles. C: Representative MHC staining images used to assess the content of MHC+ myofibers in the soleus and gastrocnemius of both groups. D: Quantitative data showing the ratios of the MHC+ myofibers to total myofibers in both muscles. Data are mean ± SEM (n = 10–12). Scale bars: 50 µm.
Figure S5. ET ameliorated the mitochondria damage and lipid droplet accumulation in the soleus and gastrocnemius at 4 mos post‐ET. A: Representative electron microscopy 7 images show a relatively preserved mitochondrial configuration as well as a small amount of lipid droplets. B–D: Quantitative data of damaged mitochondrial numbers, percentage of damaged mitochondria. and lipid droplet numbers. Data are mean ± SEM (n = 4–6). Scale bar: 500 nm.
Figure S6. Administration of neutralizing pAb against adiponectin (pAb‐Adip) suppressed muscle proliferation in 32‐wk‐old mice. A: Representative PCNA immunostaining with the mouse mAb used to assess the content of proliferated cells in both groups. B: Quantitative data for PCNA‐positive cells. Data are mean ± SEM (n = 8–9). Arrowheads: related positive‐staining cells. Scale bar: 50 µm.
Figure S7. Effects of adiponectin depletion on intracellular signal molecule changes in the muscles at 2 mos of treatment. A,B: Representative Western blot images and quantitative data for the changes of p‐AMPKα, p‐Akt, p‐mTOR, and Bcl‐xL proteins in the muscle of both groups. Data are mean ± SEM (n = 3).
Figure S8. Adiponectin blocking diminished the ET‐mediated amelioration of mitochondria damage and lipid droplet accumulation in the soleus muscles and gastrocnemius at 2 mos of treatment. Representative electron microscopy shows a relatively preserved mitochondrial configuration as well as a small amount of lipid droplets.
Figure S9. Recombinant mouse adiponectin enhanced the levels of of p‐AMPKα, 8 p‐mTOR, and Bcl‐2 proteins the BM‐derived integrin‐α7+ stem cells. Representative Western blot images and quantitative data for the changes of p‐AMPKα, p‐mTOR, and Bcl‐2 proteins in a dose‐dependent manner. Data are mean ± SEM (n = 3).
Figure S10. AdipoR1 inhibition abrogated beneficial consequences of exercise on the muscle of SAMP10 mice (24‐wk‐old). A,B: Grip strength/BW and endurance were recorded in exercised mice treated mouse control IgG (Cont group; 450 µg/kg, one/week) or AdipoR1 pAb (AdipoR1‐I group; 450 µg/kg, one/week), respectively, for 8 weeks. C,D: The ratios of soleus muscle to BW and ratios of gastrocnemius to BW were calculated at 32 wks of age in both groups. E: Qauntitative data from double immunofluorescence show the numbers of CD34+/integrin‐α7 + in soleus muscles and gastrocnemius. F: At 2 mos post‐ET, CD34+/integrin‐α7 + was measured by flow cytometry in the BM and PB of both experimental groups. G: Representative PCNA immunostaining and combined quantitative data show the content of proliferated cells in the soleus muscles and gastrocnemius of the two experimental groups. H: Representative Western blot images and quantitative data show the changes of p‐AMPKα, p‐mTOR, and Bcl‐XL proteins in the muscle of both groups. Data are mean ± SEM (n = 3 for Western blots, n = 5‐6 for others).
Figure S11. AMPK inhibition abrogated beneficial consequences of exercise on aged muscle (24‐wk‐old). A–C: BW, endurance, and drop‐out were recorded in exercised mice treated with DMSO (Cont group; 50 µl/day, twice/week) or AMPK inhibitor compound C (AMPK‐I group; 10 mg/kg/50 µl, twice/week), respectively, for 8 weeks. D,E: The ratios 9 of soleus muscle to BW and ratios of gastrocnemius to BW were calculated at 32 wks of age in both groups. F: Representative TUNEL images and quantitative data showing the numbers of apoptotic cells in both muscles of the two groups. White arrowheads indicate TUNEL‐positive cells. Data are mean ± SEM (n = 5–6). Scar bar, 50 µm.
Figure S12. Proposed mechanism of ET‐mediated alleviation of muscle regeneration and dysfunction in a SAMP10 mouse model. MuSCs, muscle stem cells; AMPK, AMP‐activated protein kinase; PGC‐1α, peroxisome proliferator‐activated receptor‐γ coactivator‐1α; mTOR, mammalian target of rapamycin.
Supporting info item
Acknowledgements
This work was supported in part by grants from the Ministry of Education, Culture, Sports, Science, and Technology of Japan (nos. 23390208, 22390143, 15H04801, and 15H04802); from the Japan Takeda Science Foundation (no. 26‐007596); and from the Novartis Aging and Geriatrics Research Foundation (no. 25‐7778). We thank M. Hasegawa and K. Suzuki for the technical assistance. The authors certify that they comply with the ethical guidelines for publishing in the Journal of Cachexia, Sarcopenia and Muscle: update 2015.55
Inoue, A. , Cheng, X. W. , Huang, Z. , Hu, L. , Kikuchi, R. , Jiang, H. , Piao, L. , Sasaki, T. , Itakura, K. , Wu, H. , Zhao, G. , Lei, Y. , Yang, G. , Zhu, E. , Li, X. , Sato, K. , Koike, T. , and Kuzuya, M. (2017) Exercise restores muscle stem cell mobilization, regenerative capacity and muscle metabolic alterations via adiponectin/AdipoR1 activation in SAMP10 mice. Journal of Cachexia, Sarcopenia and Muscle, 8: 370–385. doi: 10.1002/jcsm.12166.
References
- 1. Lexell J. Human aging, muscle mass, and fiber type composition. J Gerontol A Biol Sci Med Sci 1995;50Spec No:11–16. [DOI] [PubMed] [Google Scholar]
- 2. Alnaqeeb MA, Goldspink G. Changes in fibre type, number and diameter in developing and ageing skeletal muscle. J Anat 1987;153:31–45. [PMC free article] [PubMed] [Google Scholar]
- 3. Ansved T, Larsson L. Effects of ageing on enzyme‐histochemical, morphometrical and contractile properties of the soleus muscle in the rat. J Neurol Sci 1989;93:105–124. [DOI] [PubMed] [Google Scholar]
- 4. Sheard PW, Anderson RD. Age‐related loss of muscle fibres is highly variable amongst mouse skeletal muscles. Biogerontology 2012;13:157–167. [DOI] [PubMed] [Google Scholar]
- 5. Schiaffino S, Dyar KA, Ciciliot S, Blaauw B, Sandri M. Mechanisms regulating skeletal muscle growth and atrophy. FEBS J 2013;280:4294–4314. [DOI] [PubMed] [Google Scholar]
- 6. Sanchez AM, Candau RB, Bernardi H. FoxO transcription factors: their roles in the maintenance of skeletal muscle homeostasis. Cell Mol Life Sci 2014;71:1657–1671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Sandri M, Sandri C, Gilbert A, Skurk C, Calabria E, Picard A, et al. Foxo transcription factors induce the atrophy‐related ubiquitin ligase atrogin‐1 and cause skeletal muscle atrophy. Cell 2004;117:399–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Greenlund LJ, Nair KS. Sarcopenia–consequences, mechanisms, and potential therapies. Mech Ageing Dev 2003;124:287–299. [DOI] [PubMed] [Google Scholar]
- 9. Iwabu M, Yamauchi T, Okada‐Iwabu M, Sato K, Nakagawa T, Funata M, et al. Adiponectin and AdipoR1 regulate PGC‐1alpha and mitochondria by Ca(2+) and AMPK/SIRT1. Nature 2010;464:1313–1319. [DOI] [PubMed] [Google Scholar]
- 10. Mounier R, Theret M, Lantier L, Foretz M, Viollet B. Expanding roles for AMPK in skeletal muscle plasticity. Trends Endocrinol Metab 2015;26:275–286. [DOI] [PubMed] [Google Scholar]
- 11. Brotto M, Abreu EL. Sarcopenia: pharmacology of today and tomorrow. J Pharmacol Exp Ther 2012;343:540–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Brass EP, Sietsema KE. Considerations in the development of drugs to treat sarcopenia. J Am Geriatr Soc 2011;59:530–535. [DOI] [PubMed] [Google Scholar]
- 13. Flack KD, Davy KP, Hulver MW, Winett RA, Frisard MI, Davy BM. Aging, resistance training, and diabetes prevention. J Aging Res 2010. 127315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Cheng XW, Kuzuya M, Kim W, Song H, Hu L, Inoue A, et al. Exercise training stimulates ischemia‐induced neovascularization via phosphatidylinositol 3‐kinase/Akt‐dependent hypoxia‐induced factor‐1 alpha reactivation in mice of advanced age. Circulation 2010;122:707–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lee S, Kwak HB. Effects of interventions on adiponectin and adiponectin receptors. J Exerc Rehabil 2014;10:60–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Macaluso F, Myburgh KH. Current evidence that exercise can increase the number of adult stem cells. J Muscle Res Cell Motil 2012;33:187–198. [DOI] [PubMed] [Google Scholar]
- 17. Jensen L, Bangsbo J, Hellsten Y. Effect of high intensity training on capillarization and presence of angiogenic factors in human skeletal muscle. J Physiol 2004;557:571–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ryan NA, Zwetsloot KA, Westerkamp LM, Hickner RC, Pofahl WE, Gavin TP. Lower skeletal muscle capillarization and VEGF expression in aged vs. young men. J Appl Physiol (1985) 2006;100:178–185. [DOI] [PubMed] [Google Scholar]
- 19. Haramizu S, Ota N, Hase T, Murase T. Aging‐associated changes in physical performance and energy metabolism in the senescence‐accelerated mouse. J Gerontol A Biol Sci Med Sci 2011;66:646–655. [DOI] [PubMed] [Google Scholar]
- 20. Murase T, Haramizu S, Ota N, Hase T. Suppression of the aging‐associated decline in physical performance by a combination of resveratrol intake and habitual exercise in senescence‐accelerated mice. Biogerontology 2009;10:423–434. [DOI] [PubMed] [Google Scholar]
- 21. Salanova M, Gambara G, Moriggi M, Vasso M, Ungethuem U, Belavy DL, et al. Vibration mechanosignals superimposed to resistive exercise result in baseline skeletal muscle transcriptome profiles following chronic disuse in bed rest. Sci Rep 2015;5. doi:10.1038/srep17027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Yamauchi T, Kadowaki T. Adiponectin receptor as a key player in healthy longevity and obesity‐related diseases. Cell Metab 2013;17:185–196. [DOI] [PubMed] [Google Scholar]
- 23. Ntaios G, Gatselis NK, Makaritsis K, Dalekos GN. Adipokines as mediators of endothelial function and atherosclerosis. Atherosclerosis 2013;227:216–221. [DOI] [PubMed] [Google Scholar]
- 24. Njajou OT, Kanaya AM, Holvoet P, Connelly S, Strotmeyer ES, Harris TB, et al. Association between oxidized LDL, obesity and type 2 diabetes in a population‐based cohort, the health, aging and body composition study. Diabetes Metab Res Rev 2009;25:733–739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Gatselis NK, Ntaios G, Makaritsis K, Dalekos GN. Adiponectin: a key playmaker adipocytokine in non‐alcoholic fatty liver disease. Clin Exp Med 2014;14:121–131. [DOI] [PubMed] [Google Scholar]
- 26. Lakhdar N, Denguezli M, Zaouali M, Zbidi A, Tabka Z, Bouassida A. Six months training alone or combined with diet alters HOMA‐AD, HOMA‐IR and plasma and adipose tissue adiponectin in obese women. Neuro Endocrinol Lett 2014;35:373–379. [PubMed] [Google Scholar]
- 27. Lee HP, Lin CY, Shih JS, Fong YC, Wang SW, Li TM, et al. Adiponectin promotes VEGF‐A‐dependent angiogenesis in human chondrosarcoma through PI3K, Akt, mTOR, and HIF‐alpha pathway. Oncotarget 2015;6:36746–36761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Cheng XW, Kuzuya M, Nakamura K, Maeda K, Tsuzuki M, Kim W, et al. Mechanisms underlying the impairment of ischemia‐induced neovascularization in matrix metalloproteinase 2‐deficient mice. Circ Res 2007;100:904–913. [DOI] [PubMed] [Google Scholar]
- 29. Sacco A, Doyonnas R, Kraft P, Vitorovic S, Blau HM. Self‐renewal and expansion of single transplanted muscle stem cells. Nature 2008;456:502–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Bernet JD, Doles JD, Hall JK, Kelly Tanaka K, Carter TA, Olwin BB. p38 MAPK signaling underlies a cell‐autonomous loss of stem cell self‐renewal in skeletal muscle of aged mice. Nat Med 2014;20:265–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Tierney MT, Aydogdu T, Sala D, Malecova B, Gatto S, Puri PL, et al. STAT3 signaling controls satellite cell expansion and skeletal muscle repair. Nat Med 2014;20:1182–1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Brack AS, Rando TA. Tissue‐specific stem cells: lessons from the skeletal muscle satellite cell. Cell Stem Cell 2012;10:504–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Jackson JR, Kirby TJ, Fry CS, Cooper RL, McCarthy JJ, Peterson CA, et al. Reduced voluntary running performance is associated with impaired coordination as a result of muscle satellite cell depletion in adult mice. Skelet Muscle 2015;5. doi:10.1186/s13395‐015‐0065‐3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Joanisse S, Gillen JB, Bellamy LM, McKay BR, Tarnopolsky MA, Gibala MJ, et al. Evidence for the contribution of muscle stem cells to nonhypertrophic skeletal muscle remodeling in humans. FASEB J 2013;27:4596–4605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Jiang H, Cheng XW, Shi GP, Hu L, Inoue A, Yamamura Y, et al. Cathepsin K‐mediated Notch1 activation contributes to neovascularization in response to hypoxia. Nat Commun 2014;5. doi:10.1038/ncomms4838. [DOI] [PubMed] [Google Scholar]
- 36. Li X, Cheng XW, Hu L, Wu H, Guo P, Hao CN, et al. Cathepsin S activity controls ischemia‐induced neovascularization in mice. Int J Cardiol 2015;183:198–208. [DOI] [PubMed] [Google Scholar]
- 37. Cheng XW, Kuzuya M, Sasaki T, Inoue A, Hu L, Song H, et al. Inhibition of mineralocorticoid receptor is a renoprotective effect of the 3‐hydroxy‐3‐methylglutaryl‐coenzyme A reductase inhibitor pitavastatin. J Hypertens 2011;29:542–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Cosgrove BD, Gilbert PM, Porpiglia E, Mourkioti F, Lee SP, Corbel SY, et al. Rejuvenation of the muscle stem cell population restores strength to injured aged muscles. Nat Med 2014;20:255–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Di Q, Cheng Z, Kim W, Liu Z, Song H, Li X, et al. Impaired cross‐activation of beta3 integrin and VEGFR‐2 on endothelial progenitor cells with aging decreases angiogenesis in response to hypoxia. Int J Cardiol 2013;168:2167–2176. [DOI] [PubMed] [Google Scholar]
- 40. Hu L, Cheng XW, Song H, Inoue A, Jiang H, Li X, et al. Cathepsin K activity controls injury‐related vascular repair in mice. Hypertension 2014;63:607–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Cheng XW, Song H, Sasaki T, Hu L, Inoue A, Bando YK, et al. Angiotensin type 1 receptor blocker reduces intimal neovascularization and plaque growth in apolipoprotein E‐deficient mice. Hypertension 2011;57:981–989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kimura K, Cheng XW, Inoue A, Hu L, Koike T, Kuzuya M. beta‐Hydroxy‐beta‐methylbutyrate facilitates PI3K/Akt‐dependent mammalian target of rapamycin and FoxO1/3a phosphorylations and alleviates tumor necrosis factor alpha/interferon gamma‐induced MuRF‐1 expression in C2C12 cells. Nutr Res 2014;34:368–374. [DOI] [PubMed] [Google Scholar]
- 43. Lin H, Lian WS, Chen HH, Lai PF, Cheng CF. Adiponectin ameliorates iron‐overload cardiomyopathy through the PPARalpha‐PGC‐1‐dependent signaling pathway. Mol Pharmacol 2013;84:275–285. [DOI] [PubMed] [Google Scholar]
- 44. Safdar A, Little JP, Stokl AJ, Hettinga BP, Akhtar M, Tarnopolsky MA. Exercise increases mitochondrial PGC‐1alpha content and promotes nuclear‐mitochondrial cross‐talk to coordinate mitochondrial biogenesis. J Biol Chem 2011;286:10605–10617. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 45. Claudino MA, Franco‐Penteado CF, Priviero FB, Camargo EA, Teixeira SA, Muscara MN, et al. Upregulation of gp91phox subunit of NAD(P)H oxidase contributes to erectile dysfunction caused by long‐term nitric oxide inhibition in rats: reversion by regular physical training. Urology 2010;75:961–967. [DOI] [PubMed] [Google Scholar]
- 46. Song W, Kwak HB, Lawler JM. Exercise training attenuates age‐induced changes in apoptotic signaling in rat skeletal muscle. Antioxid Redox Signal 2006;8:517–528. [DOI] [PubMed] [Google Scholar]
- 47. Emmons R, Niemiro GM, Owolabi O, De Lisio M. Acute exercise mobilizes hematopoietic stem and progenitor cells and alters the mesenchymal stromal cell secretome. J Appl Physiol (1985) 2016;120:624–632. [DOI] [PubMed] [Google Scholar]
- 48. Pasini E, Le Douairon LS, Flati V, Assanelli D, Corsetti G, Speca S, et al. Effects of treadmill exercise and training frequency on anabolic signaling pathways in the skeletal muscle of aged rats. Exp Gerontol 2012;47:23–28. [DOI] [PubMed] [Google Scholar]
- 49. Ziaaldini MM, Koltai E, Csende Z, Goto S, Boldogh I, Taylor AW, et al. Exercise training increases anabolic and attenuates catabolic and apoptotic processes in aged skeletal muscle of male rats. Exp Gerontol 2015;67:9–14. [DOI] [PubMed] [Google Scholar]
- 50. Lavoie V, Kernaleguen AE, Charron G, Farhat N, Cossette M, Mamarbachi AM, et al. Functional effects of adiponectin on endothelial progenitor cells. Obesity (Silver Spring) 2011;19:722–728. [DOI] [PubMed] [Google Scholar]
- 51. Eren P, Camus S, Matrone G, Ebrahimian TG, Francois D, Tedgui A, et al. Adiponectinemia controls pro‐angiogenic cell therapy. Stem Cells 2009;27:2712–2721. [DOI] [PubMed] [Google Scholar]
- 52. Hayashino Y, Jackson JL, Hirata T, Fukumori N, Nakamura F, Fukuhara S, et al. Effects of exercise on C‐reactive protein, inflammatory cytokine and adipokine in patients with type 2 diabetes: a meta‐analysis of randomized controlled trials. Metabolism 2014;63:431–440. [DOI] [PubMed] [Google Scholar]
- 53. Khan MP, Singh AK, Joharapurkar AA, Yadav M, Shree S, Kumar H, et al. Pathophysiological mechanism of bone loss in type 2 diabetes involves inverse regulation of osteoblast function by PPARgamma coactivator‐1alpha and skeletal muscle atrogenes: adiponectin receptor 1 as a potential target for reversing diabetes‐induced osteopenia. Diabetes 2015;64:2609–2623. [DOI] [PubMed] [Google Scholar]
- 54. Harkin A. Muscling in on depression. N Engl J Med 2014;371:2333–2334. [DOI] [PubMed] [Google Scholar]
- 55. von Haehling S, Morley JE, Coats AJS, Anker SD. Ethical guidelines for publishing in the Journal of Cachexia, Sarcopenia and Muscle: update 2015. J Cachexia Sarcopenia Muscle 2015;6:315–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Primer sequences used in the quantitative real‐time polymerase chain reaction (PCR)
Table S2. Levels of plasma lipid, targeted growth factors, cytokines and others in two experimental groups at indicated time points
Table S3. Levels of targeted genes in two experimental groups at 32 wks
Figure S1. Schematic representation of the exercise training (ET) program.
Figure S2. Effects of ET on metabolic parameters. A–D: Food intake, drinking water, urine volume and excrement weight were recorded in the control (Cont) and exercise training (ET) groups at the indicated time points. Data are mean ± SEM (n = 6).
Figure S3. The effects of ET on muscle function and mass at 4 mos post‐ET. A–C: Body weight (BW), grip strength/BW and endurance were recorded in the control (Cont) and exercise training (ET) groups. D,E: The ratios of soleus muscle to BW and ratios of gastrocnemius to BW were calculated at 40 wks of age in both groups. Data are mean ± SEM (n = 10–12). NS: not significant.
Figure S4. The effects of ET on the myofiber size and the slow MHC rate at 4 mos post‐ET. A: Representative H&E staining of soleus and gastrocnemius of Cont and ET mice. B: Quantitative data showing the myofiber size in both muscles. C: Representative MHC staining images used to assess the content of MHC+ myofibers in the soleus and gastrocnemius of both groups. D: Quantitative data showing the ratios of the MHC+ myofibers to total myofibers in both muscles. Data are mean ± SEM (n = 10–12). Scale bars: 50 µm.
Figure S5. ET ameliorated the mitochondria damage and lipid droplet accumulation in the soleus and gastrocnemius at 4 mos post‐ET. A: Representative electron microscopy 7 images show a relatively preserved mitochondrial configuration as well as a small amount of lipid droplets. B–D: Quantitative data of damaged mitochondrial numbers, percentage of damaged mitochondria. and lipid droplet numbers. Data are mean ± SEM (n = 4–6). Scale bar: 500 nm.
Figure S6. Administration of neutralizing pAb against adiponectin (pAb‐Adip) suppressed muscle proliferation in 32‐wk‐old mice. A: Representative PCNA immunostaining with the mouse mAb used to assess the content of proliferated cells in both groups. B: Quantitative data for PCNA‐positive cells. Data are mean ± SEM (n = 8–9). Arrowheads: related positive‐staining cells. Scale bar: 50 µm.
Figure S7. Effects of adiponectin depletion on intracellular signal molecule changes in the muscles at 2 mos of treatment. A,B: Representative Western blot images and quantitative data for the changes of p‐AMPKα, p‐Akt, p‐mTOR, and Bcl‐xL proteins in the muscle of both groups. Data are mean ± SEM (n = 3).
Figure S8. Adiponectin blocking diminished the ET‐mediated amelioration of mitochondria damage and lipid droplet accumulation in the soleus muscles and gastrocnemius at 2 mos of treatment. Representative electron microscopy shows a relatively preserved mitochondrial configuration as well as a small amount of lipid droplets.
Figure S9. Recombinant mouse adiponectin enhanced the levels of of p‐AMPKα, 8 p‐mTOR, and Bcl‐2 proteins the BM‐derived integrin‐α7+ stem cells. Representative Western blot images and quantitative data for the changes of p‐AMPKα, p‐mTOR, and Bcl‐2 proteins in a dose‐dependent manner. Data are mean ± SEM (n = 3).
Figure S10. AdipoR1 inhibition abrogated beneficial consequences of exercise on the muscle of SAMP10 mice (24‐wk‐old). A,B: Grip strength/BW and endurance were recorded in exercised mice treated mouse control IgG (Cont group; 450 µg/kg, one/week) or AdipoR1 pAb (AdipoR1‐I group; 450 µg/kg, one/week), respectively, for 8 weeks. C,D: The ratios of soleus muscle to BW and ratios of gastrocnemius to BW were calculated at 32 wks of age in both groups. E: Qauntitative data from double immunofluorescence show the numbers of CD34+/integrin‐α7 + in soleus muscles and gastrocnemius. F: At 2 mos post‐ET, CD34+/integrin‐α7 + was measured by flow cytometry in the BM and PB of both experimental groups. G: Representative PCNA immunostaining and combined quantitative data show the content of proliferated cells in the soleus muscles and gastrocnemius of the two experimental groups. H: Representative Western blot images and quantitative data show the changes of p‐AMPKα, p‐mTOR, and Bcl‐XL proteins in the muscle of both groups. Data are mean ± SEM (n = 3 for Western blots, n = 5‐6 for others).
Figure S11. AMPK inhibition abrogated beneficial consequences of exercise on aged muscle (24‐wk‐old). A–C: BW, endurance, and drop‐out were recorded in exercised mice treated with DMSO (Cont group; 50 µl/day, twice/week) or AMPK inhibitor compound C (AMPK‐I group; 10 mg/kg/50 µl, twice/week), respectively, for 8 weeks. D,E: The ratios 9 of soleus muscle to BW and ratios of gastrocnemius to BW were calculated at 32 wks of age in both groups. F: Representative TUNEL images and quantitative data showing the numbers of apoptotic cells in both muscles of the two groups. White arrowheads indicate TUNEL‐positive cells. Data are mean ± SEM (n = 5–6). Scar bar, 50 µm.
Figure S12. Proposed mechanism of ET‐mediated alleviation of muscle regeneration and dysfunction in a SAMP10 mouse model. MuSCs, muscle stem cells; AMPK, AMP‐activated protein kinase; PGC‐1α, peroxisome proliferator‐activated receptor‐γ coactivator‐1α; mTOR, mammalian target of rapamycin.
Supporting info item