

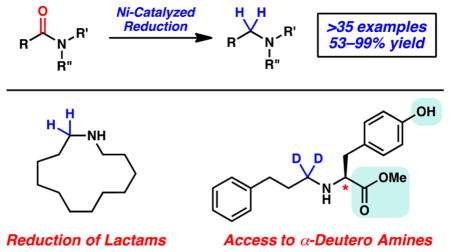
Abstract
The nickel-catalyzed reduction of secondary and tertiary amides to give amine products is reported. The transformation is tolerant of extensive variation with respect to the amide substrate, proceeds in the presence of esters and epimerizable stereocenters, and can be used to achieve the reduction of lactams. Moreover, this methodology provides a simple tactic for accessing medicinally relevant α-deuterated amines.
Graphical abstract
The ability to manipulate amides using nickel catalysis represents a rapidly growing area of research.1–4 Interest in this field has been driven in part by the stability of amides and their consequential promise for applications in multistep synthesis.5,6 The high abundance, low cost, and minimal CO2 footprint associated with nickel also render these transformations highly attractive.7 Over the past two years alone, several nickel-catalyzed reactions of amides have been developed (Figure 1), including esterification,1a,f transamidation,1c Suzuki–Miyaura couplings,1b Negishi couplings,1d,2c,d and decarbonylative processes.2a,b
Figure 1.
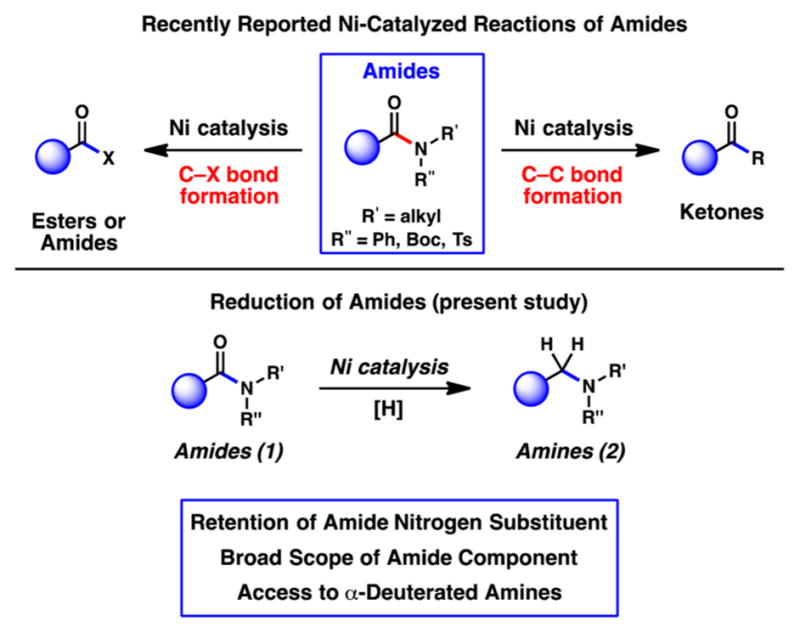
Nickel-catalyzed reactions of amides (prior studies) and nickel-catalyzed reduction of amides (present study).
With the aim of expanding the scope of base-metal-catalyzed reactions of amides, we became interested in the nickel-catalyzed reduction of amides (Figure 1, 1 → 2). Amide reduction is a powerful synthetic tool and has been the topic of recent investigations8 with notable breakthroughs including contributions by Beller (Fe or Ru, 2° or 3° amides; Zn, 3° amides; Cu, 2° amides),8a,b,e,g Nagashima (Pt or Fe, 3° amides),8c,k and Adolfsson (Mo, 3° amides).8d Beller’s Fe-catalyzed amide reduction8a is the most general of those methodologies that utilize nonprecious metal catalysis and provides a particularly impressive means to reduce tertiary amides.9 Surprisingly, however, only scattered reports of nickel-catalyzed amide reduction have been reported over the past century, with no general methodologies having been disclosed. Most known examples of nickel-catalyzed amide reduction require the use of Raney nickel, high temperatures, and high pressures of H2 gas.10,11 Only one exception exists, which is the double reduction of α-ketoanilides to the corresponding α-hydroxylamines.12 A general catalytic platform to achieve the Ni-catalyzed reduction of amides has remained elusive.
Herein, we report a simple catalyst system that enables the reduction of both secondary and tertiary amide substrates. The transformation is tolerant of extensive variation with respect to the amide C- and N-substituents. Furthermore, the nickel-catalyzed amide reduction can be performed in the presence of esters and epimerizable stereocenters. Finally, the methodology provides a simple tactic for the synthesis of α-deuterated amines, which are important motifs in medicinal chemistry.13
To initiate our studies, we examined the nickel-catalyzed reduction of several classes of amide substrates (Table 1).14 N-Methyl-N-phenylbenzamide (3), the substrate used in our initial amide-coupling studies,1a was tested under a variety of reaction conditions.15 Ultimately, we found that 3 underwent efficient reduction to amine 4 in the presence of PhSiH316 and a catalytic amount of the air-stable Ni(II) precatalyst NiCl2(dme)17 (entry 1). An external ligand was not required. With this exciting result in hand, we directed our attention to assessing other amide substrate classes, especially those that did not undergo coupling in our prior studies.1 Amide 5, a substrate bearing two aliphatic nitrogen substituents, was reduced to give amine 6 in 92% yield (entry 2). Moreover, the use of secondary amide 7 as a substrate furnished amine 8 in excellent yield (entry 3). In the final example shown, we evaluated amide 9, a substrate derived from an aliphatic carboxylic acid (entry 4). This reduction delivered amine 10 in 93% yield. Given that most reported nickel-catalyzed reactions of amides require the use of benzamide-type substrates and activating groups on the nitrogen,18 the examples shown in entries 2–4 were especially encouraging.19
Table 1.
Evaluation of Amide Substratesa
| |||
|---|---|---|---|
| entry | amide | amine | yieldb |
| 1 |
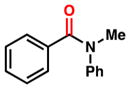 3 |
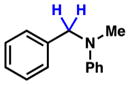 4 |
86% |
| 2 |
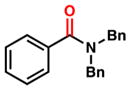 5 |
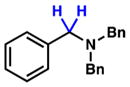 6 |
92% |
| 3 |
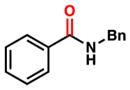 7 |
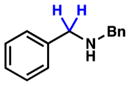 8 |
95% |
| 4 |
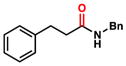 9 |
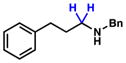 10 |
93% |
Conditions unless otherwise stated: NiCl2(dme) (10 mol %), substrate (1.0 equiv, 0.2 mmol), PhSiH3 (2.0 equiv), and toluene (1.0 M) at 115 °C for 24 h in a sealed vial.
Yields determined by 1H NMR analysis using hexamethylbenzene as an internal standard.
Having identified conditions to effect the nickel-catalyzed reduction of secondary and tertiary amides, the scope of this methodology was evaluated.20 We first tested substituent effects with regard to the carbon-component of the amide carbonyl using secondary amides derived from benzylamine (Figure 2). Reduction of the parent benzamide 7 gave 8 in 94% isolated yield. Electron-donating groups on the aromatic ring were tolerated, as exemplified by the formation of amines 11–13 in high yield. From the latter two cases, it should be emphasized that a tertiary amine and a phenol, respectively, did not hinder catalytic reduction. As suggested by products 14 and 15, the electron-withdrawing groups – CF3 and – F also withstood the reaction conditions. Several heteroaromatic substrates were also examined. To our delight, a free indole and a pyrazole were tolerated, as shown by the formation of amines 16 and 17 in 75 and 67% yield, respectively. In addition to nitrogen-based heterocycles, sulfur- and oxygen- containing heterocycles could be utilized to give thiophene 18 and furan 19, both in synthetically useful yields.
Figure 2.

Reduction of secondary amides with varying C-substituents. (a) Conditions unless otherwise stated: NiCl2(dme) (10 mol %), substrate (1.0 equiv, 0.2 mmol), PhSiH3 (2.0 equiv), and toluene (1.0 M) at 115 °C for 24 h in a sealed vial. Yields shown reflect the average of two isolation experiments. (b) PhSiH3 (4.0 equiv) was used. (c) Yields determined by 1H NMR analysis using hexamethylbenzene as an internal standard.
As noted previously, one of the key limitations of most nickel-catalyzed reactions of amides is the necessity to employ benzamide-type substrates. Thus, we were eager to probe a range of amides derived from aliphatic carboxylic acid substrates (Figure 2). A secondary amide derived from decanoic acid underwent reduction to give product 20 in 75% isolated yield. α-Branched substrates could also be reduced in excellent yields, as shown by products 21 and 22. It is noteworthy that significant steric bulk did not hinder the transformation. Olefin-containing compounds were also probed under our reaction conditions. Although not all tests were successful, product 23 was obtained in high yield, without competitive reduction of the alkene. Finally, heterocyclic amides derived from aliphatic carboxylic acids were found to undergo smooth reduction, as seen by the formation of 24–27. The tolerance of this methodology to oxygen- and nitrogen-containing heterocycles, including free NHs and carbamates, bodes well for future synthetic applications.
As shown in Figure 3, the scope of the methodology is broad with respect to the amide nitrogen substituents. Secondary amides beyond N-Bn,H-amides can be utilized as shown by the formation of 28 and 29. The latter example also suggests that large groups on the amide nitrogen are tolerated. A series of tertiary amides were evaluated, which led to the smooth formation of reduced products 30–36. These results collectively demonstrate that acyclic, cyclic, branched, and heterocyclic fragments can be utilized in this methodology.
Figure 3.
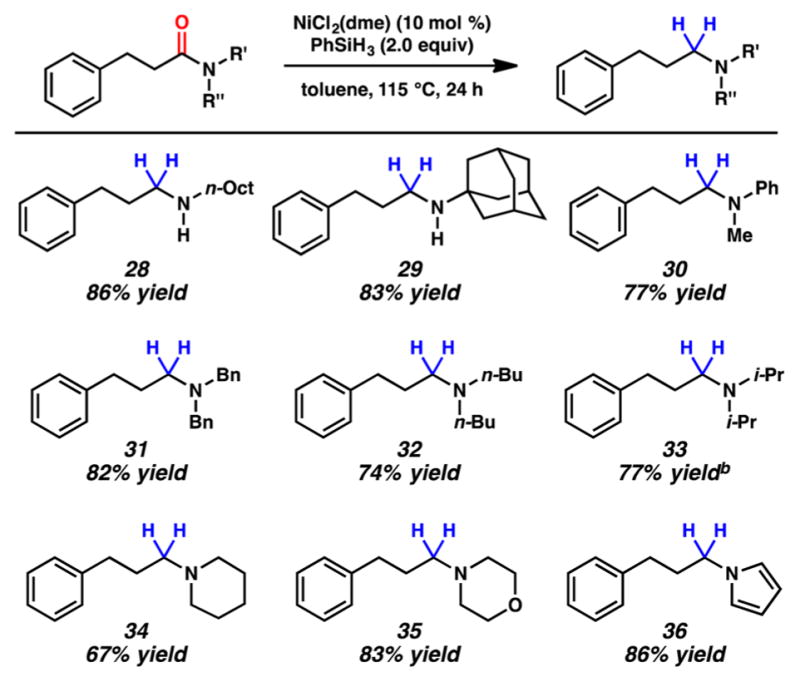
Reduction of aliphatic substrates. (a) Conditions unless otherwise stated: NiCl2(dme) (10 mol %), substrate (1.0 equiv, 0.2 mmol), PhSiH3 (2.0 equiv), and toluene (1.0 M) at 115 °C for 24 h in a sealed vial. Yields shown reflect the average of two isolation experiments. (b) Yield determined by 1H NMR analysis using hexamethylbenzene as an internal standard.
Having demonstrated the broad scope of this methodology for the reduction of acyclic amides, we sought to test its applicability to the reduction of lactam substrates. Lactam reduction is most often performed using LiAlH4, Red-Al, or two-step protocols.21 As shown in Figure 4, we found that a 4-membered lactam underwent reduction to give azetidine 37 in 64% yield. Additionally, 6- and 7-membered ring substrates could be reduced to give piperidine (38) and azepane (39), respectively. Finally, reduction of a 13-membered ring substrate delivered macrocyclic amine 40 in quantitative yield. Thus, our nickel-based approach offers a simple and relatively mild alternative to conventional lactam reduction methods and can be used to access cyclic amines of varying ring sizes.
Figure 4.
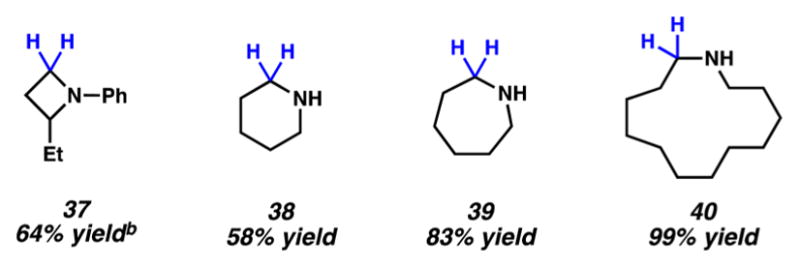
Cyclic amines prepared by reduction of the corresponding lactams. (a) Conditions unless otherwise stated: NiCl2(dme) (10 mol %), substrate (1.0 equiv, 0.2 mmol), PhSiH3 (2.0 equiv), and toluene (1.0 M) at 115 °C for 24 h in a sealed vial. Yield determined by 1H NMR analysis using hexamethylbenzene as an internal standard due to product volatility. (b) Yield shown reflects the average of two isolation experiments.
As a final test of this methodology, a variety of optically enriched amino acid derivatives were reduced using PhSiD3 to give the corresponding α-deuteroamines (Figure 5).22 α-Deuteroamines are less prone to undergo metabolism compared to their nondeutero counterparts and are therefore sought after in drug discovery.13,23 We found that a valine-derived amide bearing a methyl ester could be reduced to give deuteroamine 41. Similarly, reduction of a tyrosine derivative proceeded in the presence of the free alcohol to furnish 42. In both cases, the ester remained intact and no epimerization was observed. The same features were noted upon reduction of a proline-derived substrate, which gave deuterated aminoester 43. In the last two examples, we examined the reduction of two prolinol derivatives, one bearing a free alcohol and the other bearing an acetoxy group. In both cases, reduction proceeded smoothly to give the corresponding amines 44 and 45, respectively. Thus, these results not only underscore the utility of our methodology for the preparation of α-deuteroamines but also re-emphasize the relatively mild nature of the reaction conditions.
Figure 5.
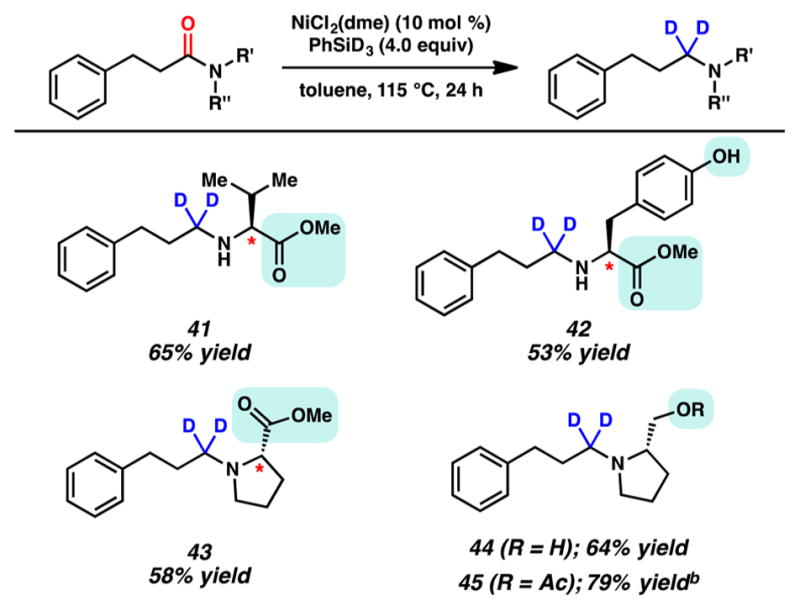
Reduction of amino acid derivatives for the synthesis of α-deuteroamines. (a) Conditions unless otherwise stated: NiCl2(dme) (10 mol %), substrate (1.0 equiv, 0.1 mmol), PhSiD3 (4.0 equiv), and toluene (1.0 M) at 115 °C for 24 h in a sealed vial. Yields shown reflect the average of two isolation experiments. (b) PhSiD3 (2.0 equiv) was used.
We have developed a facile means to achieve the reduction of secondary and tertiary amides using nonprecious metal catalysis. The transformation is tolerant of extensive variation with respect to the amide substrate, proceeds in the presence of esters and epimerizable stereocenters, and can be used to reduce lactams. Moreover, by employing a deuterated silane as the reducing agent, this methodology provides a simple tactic for the synthesis of medicinally relevant α-deuterated amines. Our discovery complements the most general solutions available to the problem of amide reduction known to date.
Supplementary Material
Acknowledgments
We are grateful for financial support from the NIH-NIGMS (R01-GM117016), the NSF (DGE-1144087 for B.J.S.), the Fulbright Research Fellowship (M.H.), the Oskar Jeger Scholarship Program (M.K.J.), and the University of California, Los Angeles. These studies were supported by shared instrumentation grants from the NSF (CHE-1048804) and the NIH NCRR (S10RR025631).
Footnotes
Notes
The authors declare no competing financial interest.
NOTE ADDED AFTER ASAP PUBLICATION
References 8n,o,p were added March 29, 2017.
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.orglett.7b00683.
Experimental details and compound characterization data (PDF)
References
- 1.(a) Hie L, Fine Nathel NF, Shah TK, Baker EL, Hong X, Yang Y-F, Liu P, Houk KN, Garg NK. Nature. 2015;524:79. doi: 10.1038/nature14615. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Weires NA, Baker EL, Garg NK. Nat Chem. 2016;8:75. doi: 10.1038/nchem.2388. [DOI] [PubMed] [Google Scholar]; (c) Baker EL, Yamano MM, Zhou Y, Anthony SM, Garg NK. Nat Commun. 2016;7:11554. doi: 10.1038/ncomms11554. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Simmons BJ, Weires NA, Dander JE, Garg NK. ACS Catal. 2016;6:3176. doi: 10.1021/acscatal.6b00793. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Dander JE, Weires NA, Garg NK. Org Lett. 2016;18:3934. doi: 10.1021/acs.orglett.6b01758. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Hie L, Baker EL, Anthony SM, Desrosiers JN, Senanayake C, Garg NK. Angew Chem, Int Ed. 2016;55:15129. doi: 10.1002/anie.201607856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.For nickel-catalyzed amide C–N bond activation studies from other laboratories, see: Hu J, Zhao Y, Zhang Y, Shi Z. Angew Chem, Int Ed. 2016;55:8718. doi: 10.1002/anie.201603068.Shi S, Meng G, Szostak M. Angew Chem, Int Ed. 2016;55:6959. doi: 10.1002/anie.201601914.Shi S, Szostak M. Chem - Eur J. 2016;22:10420. doi: 10.1002/chem.201602202.Shi S, Szostak M. Org Lett. 2016;18:5872. doi: 10.1021/acs.orglett.6b02952.Liu L, Chen P, Sun Y, Wu Y, Chen S, Zhu J, Zhao Y. J Org Chem. 2016;81:11686. doi: 10.1021/acs.joc.6b02093.Dey A, Sasmal S, Seth K, Lahiri GK, Maiti D. ACS Catal. 2017;7:433.
- 3.For palladium-catalyzed amide C–N bond activation, see: Meng G, Szostak M. Org Lett. 2015;17:4364. doi: 10.1021/acs.orglett.5b02209.Meng G, Szostak M. Org Biomol Chem. 2016;14:5690. doi: 10.1039/c6ob00084c.Liu C, Meng G, Liu Y, Liu R, Lalancette R, Szostak R, Szostak M. Org Lett. 2016;18:4194. doi: 10.1021/acs.orglett.6b01836.Meng G, Szostak M. Angew Chem, Int Ed. 2015;54:14518. doi: 10.1002/anie.201507776.Meng G, Shi S, Szostak M. ACS Catal. 2016;6:7335.Liu C, Meng G, Szostak M. J Org Chem. 2016;81:12023. doi: 10.1021/acs.joc.6b02294.Li X, Zou G. Chem Commun. 2015;51:5089. doi: 10.1039/c5cc00430f.Li X, Zou G. J Organomet Chem. 2015;794:136.Yada A, Okajima S, Murakami M. J Am Chem Soc. 2015;137:8708. doi: 10.1021/jacs.5b05308.Lei P, Meng G, Szostak M. ACS Catal. 2017;7:1960.Cui M, Wu H, Jian J, Wang H, Liu C, Daniel S, Zeng Z. Chem Commun. 2016;52:12076. doi: 10.1039/c6cc06428k.Liu C, Liu Y, Liu R, Lalancette R, Szostak R, Szostak M. Org Lett. 2017;19:1434. doi: 10.1021/acs.orglett.7b00373.
- 4.(a) Meng G, Shi S, Szostak M. Synlett. 2016;27:2530. [Google Scholar]; (b) Dander JE, Garg NK. ACS Catal. 2017;7:1413. doi: 10.1021/acscatal.6b03277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Amides are considered to be ideally suited for use as synthons in chemical synthesis. For select recent examples involving amide directing groups in transition-metal-catalyzed methodologies, see: Topczewski JJ, Cabrera PJ, Saper NI, Sanford MS. Nature. 2016;531:220. doi: 10.1038/nature16957.He J, Jiang H, Takise R, Zhu RY, Chen G, Dai HX, Dhar TGM, Shi J, Zhang H, Cheng PTW, Yu JQ. Angew Chem, Int Ed. 2016;55:785. doi: 10.1002/anie.201509996.Gurak JA, Jr, Yang KS, Liu Z, Engle KM. J Am Chem Soc. 2016;138:5805. doi: 10.1021/jacs.6b02718.Zhang XG, Dai HX, Wasa M, Yu JQ. J Am Chem Soc. 2012;134:11948. doi: 10.1021/ja305259n.Shang R, Ilies L, Nakamura E. J Am Chem Soc. 2015;137:7660. doi: 10.1021/jacs.5b04818.Matsubara T, Ilies L, Nakamura E. Chem - Asian J. 2016;11:380. doi: 10.1002/asia.201501095.Yokota A, Aihara Y, Chatani N. J Org Chem. 2014;79:11922. doi: 10.1021/jo501697n.Fruchey ER, Monks BM, Cook SP. J Am Chem Soc. 2014;136:13130. doi: 10.1021/ja506823u.Roane J, Daugulis O. Org Lett. 2013;15:5842. doi: 10.1021/ol402904d.Nishino M, Hirano K, Satoh T, Miura M. Angew Chem, Int Ed. 2013;52:4457. doi: 10.1002/anie.201300587.
- 6.For reviews on amides as directing groups in ortho-metalation, see: Whisler MC, MacNeil S, Snieckus V, Beak P. Angew Chem, Int Ed. 2004;43:2206. doi: 10.1002/anie.200300590.Snieckus V. Chem Rev. 1990;90:879.Beak P, Snieckus V. Acc Chem Res. 1982;15:306.
- 7.(a) Rosen BM, Quasdorf KW, Wilson DA, Zhang N, Resmerita AM, Garg NK, Percec V. Chem Rev. 2011;111:1346. doi: 10.1021/cr100259t. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Tasker SZ, Standley EA, Jamison TF. Nature. 2014;509:299. doi: 10.1038/nature13274. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Mesganaw T, Garg NK. Org Process Res Dev. 2013;17:29. [Google Scholar]; (d) Ananikov VP. ACS Catal. 2015;5:1964. [Google Scholar]
- 8.For recent breakthroughs (non-nickel), see the following and references cited therein: Zhou S, Junge K, Addis D, Das S, Beller M. Angew Chem, Int Ed. 2009;48:9507. doi: 10.1002/anie.200904677.Das S, Addis D, Zhou S, Junge K, Beller M. J Am Chem Soc. 2010;132:1770. doi: 10.1021/ja910083q.Nakatani N, Hasegawa JY, Sunada Y, Nagashima H. Dalton Trans. 2015;44:19344. doi: 10.1039/c5dt02767e.Tinnis F, Volkov A, Slagbrand T, Adolfsson H. Angew Chem, Int Ed. 2016;55:4562. doi: 10.1002/anie.201600097.Das S, Join B, Junge K, Beller M. Chem Commun. 2012;48:2683. doi: 10.1039/c2cc17209g.Das S, Addis D, Junge K, Beller M. Chem - Eur J. 2011;17:12186. doi: 10.1002/chem.201101143.Cabrero-Antonino JR, Alberico E, Junge K, Junge H, Beller M. Chem Sci. 2016;7:3432. doi: 10.1039/c5sc04671h.Reeves JT, Tan Z, Marsini MA, Han ZS, Xu Y, Reeves DC, Lee H, Lu BZ, Senanayake CH. Adv Synth Catal. 2013;355:47.Xie W, Zhao M, Cui C. Organometallics. 2013;32:7440.Pelletier G, Bechara WS, Charette AB. J Am Chem Soc. 2010;132:12817. doi: 10.1021/ja105194s.Sunada Y, Kawakami H, Imaoka T, Motoyama Y, Nagashima H. Angew Chem, Int Ed. 2009;48:9511. doi: 10.1002/anie.200905025.Fernandes AC, Romão CC. J Mol Catal A: Chem. 2007;272:60.Zhang T, Zhang Y, Zhang W, Luo M. Adv Synth Catal. 2013;355:2775.Xiang SH, Xu J, Yuan HQ, Huang PQ. Synlett. 2010:1829.Huang PQ, Lang QW, Wang YR. J Org Chem. 2016;81:4235. doi: 10.1021/acs.joc.6b00572.Huang PQ, Geng H. Org Chem Front. 2015;2:150.
- 9.Beller’s report includes one example pertaining to the reduction of a secondary amide (N-Me-benzamide).
- 10.(a) Smith ME, Adkins H. J Am Chem Soc. 1938;60:407. [Google Scholar]; (b) Ovakimian G, Christman CC, Kuna M, Levene PA. J Biol Chem. 1940;134:151. [Google Scholar]; (c) Witkop B. J Am Chem Soc. 1949;71:2559. [PubMed] [Google Scholar]; (d) Riebsomer JL. J Org Chem. 1950;15:68. [Google Scholar]; (e) Robba M, Rault S, Sevricourt CD. Heterocycles. 1979;12:1009. [Google Scholar]
- 11.For the reduction of imides using Raney nickel and H2, see: Adkins H, Cramer HI. J Am Chem Soc. 1930;52:4349.Paden JH, Adkins H. J Am Chem Soc. 1936;58:2487.Arévalo A, Ovando-Segovia S, Flores-Alamo M, García JJ. Organometallics. 2013;32:2939.
- 12.Mamillapalli NC, Sekar G. Chem Commun. 2014;50:7881. doi: 10.1039/c4cc02744b. [DOI] [PubMed] [Google Scholar]
- 13.For a discussion regarding deuterated drugs, see: Mullard A. Nat Rev Drug Discovery. 2016;15:219. doi: 10.1038/nrd.2016.63.
- 14.Attempts to reduce primary amides, such as benzamide, led to decomposition of the starting material.
- 15.Performing the reaction at lower temperature and in solvents other than toluene led to a decreased yield of the reduced product.
- 16.Use of 2 equiv of PhSiH3 enabled the reduction of most substrates and was therefore used in most experiments. Altering the silane (e.g., Ph2SiH2, Ph3SiH, PHMDS, Et3SiH) proved detrimental to the yield of amine 4.
- 17.Numerous nickel(II) precatalysts (e.g., NiBr2, NiCl2, Ni(acac)2, NiCl2·H2O, NiCl2(PPh3)2) were screened in the reduction of 3 with no appreciable decrease in yield obverved.
- 18.We recently reported the Ni-catalyzed esterification of amides using nonbenzamide substrates, albeit with N-Boc groups for activation (ref 1f).
- 19.The reduction of 3 was performed in the presence of galvinoxyl (1.0 equiv). The formation of the corresponding amine 4 was observed in 70% yield, which may indicate that a radical mechanism is not operative. Any further mechanistic conjecture would be premature at this stage.
- 20.Nitriles were deemed incompatible with our methodology. Attempts to employ nitriles led to nonspecific decomposition.
- 21.For select examples of lactam reduction in total synthesis, see: Bonazzi S, Cheng B, Wzorek JS, Evans DA. J Am Chem Soc. 2013;135:9338. doi: 10.1021/ja404673s.Petronijevic FR, Wipf P. J Am Chem Soc. 2011;133:7704. doi: 10.1021/ja2026882.Mewald M, Medley JW, Movassaghi M. Angew Chem, Int Ed. 2014;53:11634. doi: 10.1002/anie.201405609.Mercado-Marin EV, Sarpong R. Chem Sci. 2015;6:5048. doi: 10.1039/c5sc01977j.Beadle JD, Powell NH, Raubo P, Clarkson GJ, Shipman M. Synlett. 2016;27:169.
- 22.The reduction of amino acid derivatives has previously been achieved using lithium aluminum deuteride, which led to global reduction and the formation of α-deutero amino alcohols; see: Franchi P, Lucarini M, Mezzina E, Pedulli GF. J Am Chem Soc. 2004;126:4343. doi: 10.1021/ja049713y.
- 23.For examples of deuterium in pharmaceuticals, see: Foster AB. Trends Pharmacol Sci. 1984;5:524.Kushner DJ, Baker A, Dunstall TG. Can J Physiol Pharmacol. 1999;77:79.Nelson SD, Trager WF. Drug Metab Dispos. 2003;31:1481. doi: 10.1124/dmd.31.12.1481.Sharma R, Strelevitz TJ, Gao H, Clark AJ, Schildknegt K, Obach RS, Ripp SL, Spracklin DK, Tremaine LM, Vaz ADN. Drug Metab Dispos. 2012;40:625. doi: 10.1124/dmd.111.042770. references cited therein.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.