

Abstract
Background:
Alternative splicing (AS) is a posttranscriptional process that produces differ-ent transcripts from the same gene and is important to produce diverse protein products in response to environmental stimuli. AS occurs at specific sites on the mRNA sequence, some of which have been de-fined. Multiple bioinformatics tools have been developed to detect AS from experimental data.
Objectives:
The goal of this review is to help researchers use specific tools to aid their research and to develop new AS detection tools based on these previously established tools.
Method:
We selected 15 AS detection tools that were recently published; we classified and delineated them on several aspects. Also, a performance comparison of these tools with the same starting input was conducted.
Result:
We reviewed the following categorized features of the tools: Publication information, working principles, generic and distinct workflows, running platform, input data requirement, sequencing depth dependency, reads mapped to multiple locations, isoform annotation basis, precise detected AS types, and performance benchmarks.
Conclusion:
Through comparisons of these tools, we provide a panorama of the advantages and short-comings of each tool and their scopes of application.
Keywords: Alternative splicing, Splice junctions detection, RNA-seq, Spliceosome, Read-split-run, Read-split-walk
1. INTRODUCTION
Alternative splicing (AS) is an important posttranscriptional process that enables a single gene to produce multiple distinct transcripts, namely isoforms, and thereby increases proteome diversity [1]. These isoforms carry different biological properties that are different in catalytic ability, subcellular localization, or protein interaction [2]. AS was first discovered in 1977 when the 5' end mRNA sequences of some adenovirus 2 (Ad2) were found to be various [3]. More than 90% of genes produce multiple isoforms in humans, and dysregulation of gene splicing is the cause of many diseases [4]. In retinitis pigmentosa, mutations in the splicing factors PRP8 and PRPF31/U4-61k lead to autosomal dominant types of this particular disease [2]. Rapid discovery of AS events in a clinical patient setting is conducive to deriving future therapeutic value of the AS events [5]. Splicing differences, when used as powerful biomarkers, can potentially discriminate tissues and improve stratification methods for the diagnosis of cancer patients as well as specific treatments [6].
The general process of AS is to remove introns from the nuclear pre-mRNAs in eukaryotes by a specific mechanism involving the spliceosome. The mechanism can recognize short consensus sequences that are conserved within the intron and at exon-intron boundaries. There are functionally equivalent pairs of splice site sequences immediately adjacent to the exon-intron boundaries.
The spliceosome recognizes conserved dinucleotides located at the last two and the first two positions of introns in pre-mRNAs [7]. The major U2 type spliceosome removes the majority of introns with a 5’ end consensus sequence GT and 3’ end consensus sequence AG, whereas the minor U12 type spliceosome removes a minority of introns with a 5’ end consensus sequence AT and 3’ end consensus sequence AC. In rare cases, the pre-mRNA splicing takes place at 5’- and 3’- ends of splice sites in the GC–AG or GT–GG pattern [8]. Besides the 5’ end splice sites and 3’ end splice sites, the splice apparatus also recognizes a poorly conserved sequence within the intron called the branch site [7].
The splicing process, at molecular level, consists of two steps. Firstly, a cut at the 5’ splice site is made to generate a linear left exon and a right intron-exon sequence that forms a branched structure called lariat. The generated 5’ terminus of the intron end is linked to the 2’ position of a target base A at the branch site within the intron. Secondly, the released linear left exon has a free 3’-OH that attacks the 3’ splice site bond and cleaves the lariat at the 3’ splice site. Afterwards, an excised intron is produced and quickly degraded, and then the left exons and right exons are eventually concatenated together [7].
The regulation of AS is through a combination of cis-factor found within sequences of the pre-mRNA, and trans-factors binding to these cis-factors. Cis-factors include intron splicing silencers (ISSs), intron splicing enhancers (ISEs), exon splicing silencers (ESSs), and exon splicing enhancers (ESEs). Most of the trans-factors are RNA-binding proteins that regulate spliceosome activity [1a].
As previously mentioned, AS reaction requires the spliceosome: a large splicing apparatus consisting of a complex of proteins and ribonucleoproteins [7]. The core of the major spliceosome is formed by 5 small nuclear RNAs [1b]. During the AS process, the pre-mRNAs extensively and specifically interact with 5 small nuclear RNAs, namely U1, U2, U4, U5, and U6, via base pairing [9]. Consensus sequences of the splice sites in higher eukaryotes are very degenerate, and thereby are insufficient for genome-wide accurate recognition by only interacting with the spliceosome core [1b]. Accordingly, protein complexes form on the pre-mRNA and aid in recognizing exon-intron boundaries [2]. In these complexes, the majority of these splicing regulatory proteins are classified into two major classes, namely serine/arginine-rich (SR) proteins and RNA-binding heterogeneous nuclear ribonucleoproteins (hnRNPs). These proteins possess domains to bind to each other as well as to single-stranded pre-mRNA with a low specificity [1b]. For example, SR proteins tend to bind to ESEs, while hnRNP typically bind to ESSs or ISSs [1a]. Exons are then recognized with high fidelity through the combined multiple weak interactions between RNA to RNA, protein to RNA, and protein to protein [2].
Splice site selection is also influenced by a number of other factors such as secondary structures of the pre-mRNA [10], small nucleolar RNAs (snoRNA)s [11], cellular signal transduction pathways [12], histone modifications [13], and DNA methylation [14]. RNA binding proteins preferentially interact with single-stranded RNA. Accordingly, the splicing regulatory sequences prefer to take the form of single-stranded conformation [2]. snoRNA HBII-52 regulates AS via binding to one alternative exon in the serotonin receptor gene. HBII-52 is not expressed in patients suffering from Prader–Willi syndrome; this may be the cause of this syndrome [11]. Phosphorylation events in cellular signal transduction pathways can target exon-recognition protein complexes on pre-mRNA and influence splicing junction (SJ) selections [12]. Phosphorylation is reversible. For example, several kinases can phosphorylate SR-proteins, while protein phosphatase can dephosphorylate splicing regulatory proteins. Phosphatase activity modulation both in vitro and in vivo impacts on exon usage in AS events [2]. Histone modifications also play a direct role in AS. For example, histone modification (H3-K27m3) impacts the recruitment of splice regulators through a chromatin-binding protein on many genes in humans such as PKM2, TPM1, TPM2, and FGFR2 [13]. The methylation of DNA is linked to splicing by CTCF-promoted RNA polymerase II pausing, demonstrating the developmental regulation of AS via heritable epigenetic DNA methylation [14].
Based on the splicing location on the gene, some types of splicing events are historically defined as classical splicing, such as cassette exon (also called exon skipping), mutually exclusive exons, intron retention, alternative 5’ splicing site (5’ss), alternative 3’ splicing site (3’ss), etc. (see Fig. 1). Furthermore, different AS events could occur in a combinatorial fashion; a single exon might even experience more than one AS processing at the same time [1a]. Non-canonical splicing events include intraexonic deletions, trans-splicing, and variations affecting multiple exons [8]. Short deletions in intron regions can also be classified as non-canonical splicing events [15]. A representative example of this specific splicing is IRE1α-targeted Xbp1 mRNA splicing. Unlike U2-type and U12-dependent AS events occurring within the nucleus, the splicing of IRE1α-targeted Xbp1 transcript happens within the cytosol and involves unique recognition site [15].
Fig. (1).
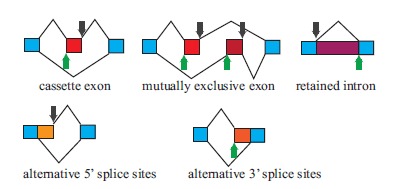
Examples of classical AS events: the alternatively spliced exons are denoted by boxes filled with various colors. Constitutive exons are denoted by blue boxes. Green arrows point out alternative 3’ splice site position. Black arrows point out the 5’ splice sites position. The figure is adapted from [16]. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this paper.)
Some tools have been developed to predict AS events. GeneSplicer is a computational tool, which predicts canonical splice sites by analyzing genomic sequences [17]. It is not reliable, however, to estimate AS merely from known genomic regulatory motifs, because the existence of known AS motifs does not ensure the presence of the corresponding AS events [1a]. Therefore, the AS patterns are mainly determined via experimental methods and bioinformatics using transcript data. For a specific gene, the reverse transcription polymerase chain reaction (RT-PCR) can be performed on a cDNA library. Thereafter, high-throughput transcriptome technology renders genome-wide AS patterns detectable. Nowadays, the largest accumulations of transcriptome data for AS detection include expressed sequence tags (ESTs), SJ microarrays, and RNA sequencing (RNA-Seq) [1a].
RNA-Seq is a revolutionary experimental protocol for sequencing messenger RNAs [4]. In a single run, RNA-Seq yields millions of short sequence reads [18], which facilitate accurate and comprehensive measurement of gene expression levels, discovery of novel transcribed regions, identification of novel and known isoforms [1a], and relative changes of isoform expression under different conditions [19]. The continuously accumulated RNA-seq data in higher depth provide ever more opportunities to detect low-frequency AS events, development-specific AS events, and tissue-specific AS events [1a]. However, in RNA-Seq there are also shortcomings, such as the relatively short read length of 50 bp or less, compared to the roughly 1000 bp read length in first generation sequencing. The relatively short reads limit the capacity of detecting AS events in a single transcript and need to become longer in the future [1a, 20].
2. TOOL SELECTION AND PERFORMANCE COMPARISON SETUP
2.1. Tool Selection
AS analysis of big data and complex information remains a challenging task [19]. In response, there have been many AS tools developed to work with RNA-Seq data. There are tens of AS event detection tools based on RNA-Seq data. After examining literature, we found that some tools had already been reviewed or compared to other tools. For example, NCBI Magic, r-make (which uses STAR), and Subread were compared by the SEQC consortium [21]; GSNAP, TopHat2, STAR, OLego, and SOAP were compared by Alberto, et al. [22]. Furthermore, some tools that we reviewed are based on prior famous tools, like the FineSplice based on TopHat2 [22]. Due to the rapid development of the next generation sequencing, we did not review tools that use microarray technology. Therefore, we selected 15 tools to review, which had not been systematically reviewed or compared before. Their common and/or different features, when categorized, will render the AS detection algorithms conceptually straightforward, thereby facilitating the evaluation and comparison of these tools for their advantages and shortcomings [22].
2.2. Tools’ Publication Information
The latest released version and date are summarized in Table 1. If the program can still be downloaded, we then define this tool as being supported. Most of the citations were collected from the Web of Science database. RSR has only 1 citation, based on a search query of the Google Scholar, as it is relatively new. It was just formally published on 2016-08-23. By comparing the latest released date and the publication date, users can know whether the programs are still being developed.
Table 1.
Publication information of the published tools.
| Tools | Publication Date |
Latest
Released Date |
Latest Version | Support | Citation Number | Reference | Website for Download |
|---|---|---|---|---|---|---|---|
| Alt Event Finder | 2012-12-17 | 2013-01-22 | 0.1 | yes | 6 | [4] | http://compbio.iupui.edu/group/6/pages/alteventfinder |
| SpliceMap | 2010-04-05 | 2010-10-23 | 3.3.5.2 | yes | 125 | [23] | https://web.stanford.edu/group/wonglab/SpliceMap/ |
| FineSplice | 2014-02-25 | 2014-04-01 | 0.2.2 | yes | 6 | [22] | http://sourceforge.net/projects/finesplice/ |
| RSW | 2014-07-03 | 2014-01-22 | N/A | yes | 2 | [15] | http://isu.indstate.edu/ybai2/RSW/index.html |
| RSR | 2016-08-23 | 2016-01-06 | N/A | yes | 1 | [5] | https://github.com/xuric/read-split-run |
| PASTA | 2013-04-04 | 2012-05-07 | 0.95 | yes | 10 | [24] | http://www.biotech.ufl.edu/cores/bioinformatics/dibig/dibig-software/pasta/ |
| rMATS | 2014-12-05 | 2016-08-18 | 3.25 | yes | 22 | [25] | http://rnaseq-mats.sourceforge.net/ |
| SOAPsplice | 2011-07-07 | 2013-04-24 | 1.1 | yes | 44 | [26] | http://soap.genomics.org.cn/soapsplice.html |
| SplicePie | 2015-03-23 | 2014-08-27 | N/A | yes | 3 | [27] | https://github.com/pulyakhina/splicing_analysis_pipeline |
| SplicingCompass | 2013-02-28 | undownloadable | N/A | no | 21 | [6] | http://www.ichip.de/software/SplicingCompass.html |
| TopHat | 2009-03-06 | 2016-02-23 | 2.1.1 | yes | 3307 | [18] | https://github.com/infphilo/tophat |
| TrueSight | 2012-12-18 | 2012-09-15 | 0.06 | yes | 12 | [28] | http://bioen-compbio.bioen.illinois.edu/TrueSight/ |
| NSMAP | 2011-05-16 | December, 2010 | 0.1.0 | yes | 14 | [29] | https://sites.google.com/site/nsmapforrnaseq/ |
| rSeqDiff | 2013-11-18 | 2013-09-10 | 0.1 | yes | 4 | [30] | http://www-personal.umich.edu/~jianghui/rseqdiff/ |
| rSeqNP | 2015-02-24 | 2015-01-01 | 1 | yes | 0 | [31] | http://www-personal.umich.edu/~jianghui/rseqnp/ |
2.3. Running Platform
The reviewed tools are free command-line tools for GNU/ Linux systems, R package, or commercial MatLab package, which are listed in Table 2. All the tools are flexible in parameter settings to different extents. In addition, RSW and RSR provide web interfaces that allow users to use the web form to upload their data to the server and graphically input their own parameter combinations on the server [15].
Table 2.
Working principles, aligners, prior separately running tools, and running platform of reviewed tools.
| Tool | Working Principle |
Running
Platform |
Prior Separately
Running Tools |
Aligner |
|---|---|---|---|---|
| Alt Event Finder | Combines read alignment tool and transcript isoform reconstruction tool to produce transcript isoform annotations. | Linux | BFAST/TopHat + Cufflinks/Scripture | BFAST/ TopHat |
| SpliceMap | Pins down one end of the SJ by matching the read spanning a junction longer than its half-length and then uses the matched part as a seeding to locate the other end. | Linux | no need | Bowtie/ Eland/ Seq Map |
| FineSplice | Uses a semi-supervised anomaly detection approach of a logistic regression model to estimate the SJs from the TopHat2 alignment results. | Linux | TopHat2 | TopHat2 |
| RSW | Uses Bowtie to do initial read alignment, splits IUM reads into halves, and then uses Bowtie to align the split read halves back to the reference genome. | Linux | no need | Bowtie |
| RSR | Similar to RSW, but improved via a linear regression equation in the Generalized Linear Model. | Linux | no need | Bowtie |
| PASTA | Uses heuristic patterned alignments and a logistic regression statistical model to detect exon-intron junctions. | Linux | no need | Bowtie |
| rMATS | Utilizes the hierarchical framework to simultaneously calculate variability among replicates and estimate uncertainty of isoform fraction within individual replicates. | Linux | no need | STAR |
| SOAPsplice | Reports SJ candidates from both spliced and intact alignment and filters out false positives | Linux | no need | BWT |
| SplicePie | Detects AS events by capturing pre-mRNAs at different splicing stages: pre-, intermediate-, and post-splicing stages. | Linux | GSNAP + SAMtools | GSNAP |
| SplicingCompass | Calculates geometric angles between the multiple- dimension exon read counts vectors to detect differential AS events. | R package | TopHat + BEDTools | TopHat |
| TopHat | Mapped regions are computed to yield initial consensus. Initially unmapped reads are indexed and mapped to the potential SJs. | Linux | no need | Bowtie/ Bowtie2 |
| TrueSight | Forms a unified model that uses adaptive training of iterative logistic regression to identify novel SJs and rule out unreliable SJs. | Linux | no need | Bowtie |
| NSMAP | Identifies the structures of expressed isoforms and estimates the expression levels of known and novel expressed isoforms at the same time | MatLab package | TopHat | TopHat |
| rSeqDiff | Uses an extended linear Poisson model to identify differential isoform expressions in multiple RNA-seq samples | R package | rSeq | SeqMap/ Eland/ BWA/ Bowtie/ bowtie2 |
| rSeqNP | Executes a non-parametric approach to test the differential expression (DE) and differential splicing (DS) using RNA-seq data. | R package | rSeq/RSEM | SeqMap/ Eland/ BWA/ Bowtie/ bowtie2 |
2.4. Input Data Requirement
Based on the different working mechanisms and aligners, different tools have different requirements of input files and corresponding annotation files. Most of the tools can start from the fastq files of the RNA-seq data. These tools always required the aligner index files and specific annotation files. By comparison, some tools rely on other upstream tools to produce the input files. In other words, these tools cannot start from the fastq files, so users have to separately run other tools to generate the input files, while learning other tools is time-consuming. All the reviewed tools’ aligners and the prior separately running tools are listed in Table 2.
2.5. Performance Comparison Setup
The best way to compare the performance of the tools is to start the tools with the same input and then benchmark their features with regard to the availability, speed, sensitivity, and so on. We run the tools using their default parameters, except for changes when necessary, to compare them on the same page.
The tools were first classified based on their main functions. One class of tools can detect splice junctions, whereas some other tools could quantify differential isoforms within one condition or between two conditions. There is one tool, NSMAP, which could do both.
For the tools that detect splice junctions, we start with an ENCODE data sample [32]. Its experiment accession identifier is ENCSR368QPC and the file accession is ENCFF002EZM. The fastq file has 1 million reads of 100 bp segments, is 266 MB, and contains only single-ended data.
For the tools that can quantify differential isoforms, including NSMAP, we used two fastq files representing different conditions: Dtt_Het.txt and Dtt_KO.txt. These two mouse RNA-seq were used in the RSW study [15]. Dtt_Het.txt has 23.4 million reads and is 4.8G. Dtt_KO.txt has 23.6 million reads and is 4.8 G. Both files are single-ended and contain 79 bp reads.
Out of the 15 tools that we reviewed, we did not run RSW, SplicePie, SplicingCompass, NSMAP, or rSeqNP. RSW is the precursor of RSR. Since we ran RSR, there is no need to run the RSW. SplicePie has a lot of bugs in its source code, which rendered us unable to properly run it. The authors of SpicePie don’t respond to the query about the bugs. SplicingCompass is no longer supported. NSMAP depends on a commercial license of MatLab, which we did not have access to. rSeqNP requires at least 4-5 replicates for each condition, which we did not have for any available datasets.
3. TOOLS’ MECHANISMS AND FEATURES
3.1. Generic Workflows
All of the selected AS event detection tools utilize algorithms to initially map the relatively short RNA-seq reads to a reference genome or reference transcriptome. Then they predict spliced junctions (SJs) based on the mapping results [26]. Reads entirely located within exons can be correctly aligned, whereas reads overlapping the junctions between two exons generally cannot be mapped to the reference genome and thereby are regarded as initially unmapped reads (IUM reads) [24]. In other words, some of the reads that span the exon boundary won’t be contiguously mapped [18]. Hence, most of AS detection tools use IUM reads to deduce the exact location of exon boundaries [24]. As a whole, the differences between tools lie in the mapping process, the mapping result reliability determination, the algorithm to predict the SJ locations, and the criteria to estimate the positive and negative false rates, etc. [26].
3.2. Detailed Differences of Workflows
Alt Event finder first takes mixed RNA isoforms identified from Cufflinks or Scripture, from which the tool pulls out “minimum non-overlapping exon regions”. It then calculates the expression units by splitting exon region unions into the smallest units that do not overlap genomically with each other. Second, Alt Event Finder projects the input individual transcript isoforms to non-overlapping exon expression units and then counts the number of isoforms that contain each expression unit. Special strings of counts patterns are then used to output a GFF3 format list of appropriate cassette exon events [4].
In SpliceMap, half of every read is aligned at a time using a short-read aligner like SeqMap or ELAND, followed by the base by base extension to the other half until the extension cannot further proceed. The remaining read part, if long enough, is subject to the same processing. SpliceMap contains four main steps: mapping of half-read, selection of seeding, search of junction, and filtering of paired-end. The step of mapping of half-read is to align with high probability the half-length (25 bp) of sensibly long reads (50 bp) to the reference genome. Then the half-read mapped hits, called seeding, are examined for seeding selection: excluding local duplicated mapped hits, and narrowing the search ranges of the junction. For every identified seeding, search of junction is used to extend base by base the alignment on the genome; this catches the perfectly mapped corresponding residual sequence of the split read at the partner splicing point within a customer-specified distance. Paired-end reads, if available, can help filter out false positives by considering the mapping direction as well as the positional order of the two hits on the reference genome [23].
The FineSplice pipeline consists of seven steps. First, using TopHat2 [33], the reads are aligned to the reference genome. Non-uniquely mapped reads that straddle multiple SJs are provisionally put away. Second, the set of split read overhangs across the splice sites is calculated. Third, a subset of likely false positives is then calculated to distinguish abnormal junctions influenced by frequent mismatches and systematically shorter overhangs. Forth, mismatched overhangs at the first mismatch position are trimmed, and feature vectors are created according to the difference between the observed counts of reads that span the specific position and the counts of expected reads. Fifth, a logistic regression model is applied to the likely false positives subset against all the remaining detectable SJs. Sixth, SJs that have a higher posterior probability of residing in the false positive subset are considered as spurious and discarded. Seventh, multiple mapped reads are rescued by being assigned to the accepted candidate SJs, if they have a unique location after filtering. Finally, a confident set of SJs and their corresponding counts are output by the program [22].
In RSW, first, Bowtie [34] is used to align reads against the reference genome with known SJ boundaries. IUM reads are split into halves. Second, all split read halves are mapped to the reference genome by Bowtie with identical parameters, except that no mismatch is allowed. Following the second alignment, the split read pairs are selected if the two ends of the split read can both be mapped within a distance (e.g. the average gene length) on the same strand of the same chromosome. Third, all of the selected split read ends of every single read are merged into a uniquely spliced region, as long as their splice lengths are identical and within a certain threshold value of the genomic boundaries. The region between the minimal lower border and the maximal upper border is defined as the candidate spliced region of a gene, if the region is supported by reads with the same splice lengths. Fourth, a novel candidate SJ is defined as regions supported by at least 2 individual reads and at least one of its boundaries has not been annotated in the University of California, Santa Cruz (UCSC) knownGene reference database. Otherwise, a spliced region is considered as a candidate known canonical region [15]. RSR is an improved version of RSW; in RSR, a modified General Linear Model for RSR considers three variables/parameters: read mapping region boundary buffer size (BB), maximum candidate distance (MD), and minimum split size (MS). RSR uses Bowtie [34] to align the reads. IUM reads are put aside as likely candidates of splicing results. Second, each IUM read sequence is split into pairs in multiple ways to ensure that both parts are at least some MS. Bowtie is called again to map each sub-sequence of split IUM reads generated from the first alignment. Alignments of the sub-sequences are then scanned to find sub-sequences that are derived as the split pairs from the same IUM read, and that are aligned on the same chromosome within MD. All the found pairs of alignments satisfying the conditions are saved and named “matched pairs.” Third, RSR scans all the matched pairs and tries to determine the number of partner pairs of each mapped pair that likely arises from the same spliced region. The SJs with the highest number of matched pairs that support them are the most confident [5].
PASTA (Patterned Alignments for Splicing and Transcriptome Analysis) aligns reads to the reference genome using Bowtie or Bowtie2. Then PASTA goes through three steps. The first step is patterned alignments, which split each IUM read at different cutoff points successively and produces two sets of “patterned” sub-sequences. The second step is using organism-specific logistic regression models, which are based on biological context such as canonical splice signals, regulatory elements, and the expected distribution of intron sizes. The model assigns scores to all putative introns, whenever a set of reads engenders more than one putative SJ. The putative SJ that generates the intron with the highest scores is considered as the predicted junction. The third step is junction identification, which determines the position of the predicted junctions when there are several putative junctions supported by multiple reads that are aligned to the same general genomic region. Eventually, the output files list all the identified SJs as well as the positions of all matched reads [24].
The rMATS (replicate MATS) hierarchical framework is based on the fact that the variability within a sample set can represent the differences of levels of exon inclusion among replicates. For replicates unpaired between different sample sets, rMATS utilizes the binomial distribution model to calculate the number of reads mapped to the exon inclusion isoforms while considering the total read number and the effective lengths of the exon inclusion isoforms in every individual replicate; rMATS utilizes a logit-normal distribution model to calculate the variations among replicates within the sample set. For paired replicates between two sample sets, every replicate in the first sample set is paired with another replicate in the second sample set. rMATS utilizes the bivariate normal distribution model to calculate the variations among the replicates within the sample set and utilizes a covariance structure to calculate the exon-specific correlation for each exon between paired replicates. For both unpaired and paired replicate models, the likelihood-ratio test is adopted to ensure that a user-defined threshold of differences between the variance and mean of the levels of exon inclusion in the two sample sets are not exceeded [25].
The SOAPsplice workflow consists of three steps and two strategies to exclude false positives. First, SOAPsplice uses the Burrows Wheeler Transformation to align reads to the genome. Secondly, SOAPsplice maps IUM reads to the genome by dividing IUM reads into two parts. SOAPsplice aligns the longest 5’ end part to the reference genome, and then aligns the remaining part. Third, for IUM reads longer than 50 nt, SOAPsplice splits them into subreads (no greater than 50 nt in length). Afterwards, SOAPsplice executes the first step and the second step to these subreads. The first strategy guarantees that mate-pair reads are aligned to the proper positions, following their paired-end relationship. The second strategy specifies the threshold number of reads that are greater than 50 nt in length and support a specific type of SJ. In this specific type, sub-reads are not capable of being mapped compatibly back to the reference genome. Finally, SOAPsplice outputs the identified junction sites, the strand, and the supporting reads numbers [26].
SplicePie is based on the fact that the pre-mRNA is present in the nucleolus and chromatin, whereas mature mRNAs are present in nucleoplasm, and the fact that total RNA contains mostly mature mRNA, whereas nuclear RNA possesses (partially) spliced mRNA. Thereby, RNA-seq fastq files are generated from total RNA and nuclear RNA samples, respectively. Then SplicePie uses GSNAP, a tool that can split every read end into multiple parts, to align the paired-end reads to the reference genome. Afterwards, reads are classified based on their mapping destinations: within intron, within exon, exon-exon SJ, or intron-exon boundary. Recursive splicing events are addressed using a combination of medians of coverage of intron and exon and Splice Site Index (SSI) values. SSI is calculated for both 5’ end and 3’ end per intron, using the reads mapped to exon-exon SJ representing post-splicing and mapped to intron-exon boundary representing pre-splicing. The non-sequential splicing is addressed using two approaches. The coverage-based method calculates the difference between the medians of the coverage of introni+1 and introni. The read-based approach calculates the splice-ratio representing the proportion of reads that support sequential splicing of two adjacent introns [27].
In SplicingCompass, for every gene, a union transcript is defined by combining every exon in each corresponding isoform annotated in the UCSC Consensus CDS coding sequences (CCDS). Afterwards, the gene expression is represented as a vector of read counts. Each component in the vector corresponds to the count of reads that are mapped uniquely to a specific exon in that union transcript. Thereby, every vector combines the expression values of all isoforms of that gene. The geometric angles between the vectors are used to measure differences in AS. To identify differentially spliced genes with multiple samples between two different conditions like two specific tissues or two specific stages, all pairwise angles are calculated in an equation and statistically tested to see whether the splicing angles in each condition are significantly smaller than the angles between different conditions. If the proportions of each isoform of a gene are constant between conditions, that gene will have the read count vectors that are approximately parallel between different conditions and there will be a low angle between the vectors, even if there are differences in overall gene expression levels. Thereby, the method can inherently distinguish differences between overall gene expression levels and differential AS [6].
TopHat first runs Bowtie to align all reads against the reference genome, consequentially mapping non-junction reads within-exons and setting aside those reads that do not map to the genome as IUM reads. Then a program Maq is used to compute an initial consensus from the mapped regions. Second, TopHat joins sequences that flank all likely donor or acceptor splice sites that are within nearby regions to predict potential SJs. Then, Tophat utilizes a seeding-and-extension strategy to index and align those IUM reads to the joined sequences. Finally, the Tophat reports alignments that are subsequently used to construct a set of non-redundant SJs [18].
TrueSight utilizes Bowtie to map RNA-seq reads to the reference genome. Remaining IUM reads are passed on to a new algorithm for gapped alignment to identify reads spanning SJs, irrespective of the expression level of their corresponding transcripts. Gapped alignment utilizes an anchor-and-extension strategy that is also used in EST mapping [28].
In the pipeline of NSMAP, first, the TopHat is used to construct the exons based on the detected SJs from the RNA-seq data. By means of combining these detected exons, all the likely isoforms are enumerated. Then, NSMAP tries to identify authentic expressed isoforms out of the large pool of the candidate isoforms and calculate the expression levels of the isoforms using a sparsity control term to limit the amount of expressed isoforms, because only as few isoforms as possible are supposed to be selected to better rationalize the observed counts of reads that are mapped to each exon of a gene. Eventually, the proper model is selected to choose the solution that better compromises the fitting of the counts and observations of the expressed isoforms [29].
In the rSeqDiff, first, the linear Poisson model for one RNA-seq sample is extended to multiple RNA-seq samples to estimate the isoform abundance. Each gene is classified as three situations: (1) no differential expression (DE), model 0, (2) DE without differential splicing (DS), model 1, (3) DS, model 2. Then, the maximum likelihood estimation is used to estimate the parameters of every model. The model is then selected via likelihood ratio test. When there are two biological conditions to be compared, a ranking of genes that are differentially spliced is generated [30].
In the pipeline of rSeqNP, expression estimates of all the genes and their isoforms in each sample need to be obtained by processing the raw RNA-seq reads using tools such as rSeq [35], Cuffdiff2 [36], or RSEM [37]. Then, based on the ranks of expression values, a non-parametric statistical approach is applied to test the DE of genes and isoforms. Then, the DE and DS of genes are jointly tested. Gene level differential score is then calculated and estimated via P-value and FDR using a permutation plug-in method [31].
3.3. Dealing with Reads Mapped to Multiple Locations
Spanning of reads over more than one SJ occurs frequently when the reads are greater than 100 bp in length, considering that about 30 percent of humans’ exons have lengths shorter than 100 bp [28]. TrueSight, SpliceMap, and FineSplice can map such spanning reads.
SpliceMap can identify multiple SJs from a single long read by adding a filter for post-processing of long-read data [23]. SpliceMap predicts the alignment of split reads by significance of tag mapping; if one side of a read is capable of being mapped to more locations, it has a smaller tag significance. Nevertheless, tag significance does not aid in determining the right candidate, and thereby a read might be aligned to the genome in various gap sizes, which means the tag on one side is likely to be mapped to several homologous locations by the algorithm. Therefore, SpliceMap is not good at handling the split reads aligned to multiple homologous locations, and SpliceMap ignores hits that fall too close together [26]. Consequently, the locations predicted by SpliceMap might be incorrect [28]. By comparison, TrueSight can sensitively and specifically detect SJs based on junction-spanning reads, particularly in SJs with low coverage reads and in the situation that a read might be aligned to the reference genome with different gap size [28]. The “parent” program of FineSplice, TopHat2, has high sensitivity and mapping accuracy but produces a lot of false positive gapped alignments, especially when handling reads with low-quality ends and reads that span multiple splice sites. That’s why TopHat2’s downstream tool, FineSplice, was developed [22].
3.4. Sequencing Depth Dependency
Some tools’ performances are highly dependent on the depth of sequencing, because if the depth of sequencing is low, many reads at SJs might be ignored, and low-expressed exons might be accidentally split or disconnected [4]. Moreover, tools that use a mapping and extension approach generally cannot effectively address reads that possess sequencing errors, resulting in a decrease of the call rate, especially when it comes to low expression levels [26]. Some tools claimed to have high specificity and sensitivity on low sequencing depth RNA-seq data. However, only some of them provide their sequencing depth threshold values in their publications, which are summarized in Table 3. The sequencing depth dependency values arise from the corresponding publications of the tools. If the sequencing depth is lower than the proposed thresholds, the sensitivities of the tools drop significantly.
Table 3.
Sequencing depth dependency and isoform annotation basis of the reviewed tools.
| Tool | Sequencing Depth Threshold | Isoform Annotation Dependency |
|---|---|---|
| Alt Event Finder | N/A | no |
| SpliceMap | 50X | no |
| FineSplice | N/A | no |
| RSW | N/A | no |
| RSR | N/A | no |
| PASTA | N/A | no |
| rMATS | N/A | yes |
| SOAPsplice | 10X | no |
| SplicePie | N/A | no |
| SplicingCompass | N/A | yes |
| TopHat | 20X | no |
| TrueSight | N/A | no |
| NSMAP | N/A | no |
| rSeqDiff | N/A | yes |
| rSeqNP | N/A | yes |
Note: Since there is no clear declaration of sequencing depth threshold values mentioned in some of the tool papers, the sequencing depth information in the depth threshold column is marked as “N/A”.
3.5. Isoform Annotation Basis and AS Types Detected
Some tools align reads relying on known splice junctions of exons or putative exons. Therefore, they cannot discover de novo AS events. By contrast, other tools for de novo splice junction detection are independent of isoform annotations [26]. All the reviewed tools are summarized in (Table 3). As an efficient downstream pipeline of TopHat2, FineSplice can align the unmapped or the potentially misaligned reads to the reference genome. FineSplice thereby can detect de novo AS events, utilizing the known isoform annotations but not exclusively relying on them [33].
3.6. Precise Detected AS Events by Tools
Most of the reviewed tools are designed to detect AS events, although some have a different focus on differentially spliced isoforms within one condition or between two conditions, which are summarized in Table 4. Some tools have unique features worth mentioning. SOAPsplice requires the intron boundaries to possess the pattern of “GT-AG”, “GC-AG”, or “AT-AC”. Thereby, SOAPsplice can not detect novel AS patterns, though it can detect novel splice junctions possessing these patterns [26]. By analysis of pre-mRNA processing mechanisms, SplicePie can detect AS events such as exon skipping, intron retention, and novel exons. Besides resolving the splicing order and recursive splicing events, SplicePie can detect non-sequentially spliced introns [27].
Table 4.
Detected AS types or differentially spliced isoforms of these tools. If the tools can detect the splice junctions, the precise AS events are listed in the right column.
| Tools | Can Quantify Differential Expression of Isoforms | Can Detect Splice Junctions | SE | MXE | RI | A5SS | A3SS | AFE | ALE | NSS&RS | CS | DNSJ | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Within One Condition | Between Two Conditions | ||||||||||||
| Alt Event Finder | no | no | yes | yes | no | no | no | no | no | no | no | no | yes |
| SpliceMap | no | no | yes | yes | yes | yes | yes | yes | yes | yes | no | no | yes |
| FineSplice | no | no | yes | yes | yes | yes | yes | yes | yes | yes | no | no | yes |
| RSW | no | no | yes | yes | yes | yes | yes | yes | yes | yes | no | yes | yes |
| RSR | no | no | yes | yes | yes | yes | yes | yes | yes | yes | no | yes | yes |
| PASTA | no | no | yes | yes | yes | yes | yes | yes | yes | yes | no | no | yes |
| rMATS | no | yes | yes | yes | yes | yes | yes | yes | no | no | no | no | no |
| SOAPsplice | no | no | yes | yes | yes | yes | yes | yes | yes | yes | no | no | yes |
| SplicePie | no | no | yes | yes | no | yes | no | no | no | no | yes | no | yes |
| SplicingCompass | no | yes | no | no | no | no | no | no | no | no | no | no | no |
| TopHat | no | no | yes | yes | yes | yes | yes | yes | yes | yes | no | no | yes |
| TrueSight | no | no | yes | yes | yes | yes | yes | yes | yes | yes | no | no | yes |
| NSMAP | yes | no | yes | yes | yes | yes | yes | yes | yes | yes | no | no | yes |
| rSeqDiff | no | yes | no | no | no | no | no | no | no | no | no | no | no |
| rSeqNP | no | yes | no | no | no | no | no | no | no | no | no | no | no |
The abbreviations are as follows: cassette exon (also called exon skipping or skipped exon): SE; mutually exclusive exons: MXE; retention of intron: RI; alternative 5’ splicing site (5’ss): A5SS; alternative 3’ splicing site (3’ss): A3SS; alternative first exon: AFE; alternative last exon: ALE; non-sequential splicing and recursive splicing: NSS&RS; cytosol splicing: CS; de novel splice junction: DNSJ.
3.7. Approaches to Improve Accuracy and Specificity
Certain tools adopt their unique approaches to improve accuracy and specificity. For example, SOAPsplice improves the mapping process by trimming reads exhibiting higher sequencing errors at the 3’ terminus. Afterwards, SOAPsplice repeats the complete mapping procedure for remaining segments of unaligned reads. To achieve more accurate alignments of IUM reads, TrueSight considers the genomic motifs, like the canonical GT-AG pattern, and uses an expectation maximization algorithm to do the logistic regression. rMATS utilizes paired replicate data between sample sets and thus can diminish individual specific variation and increase the statistical power. RSW and RSR use different parameter settings for Bowtie to achieve better sensitivity for detection of the 26 bp non-canonical spliced region in Xbp1 mRNA. Out of the 15 reviewed tools, only SpliceMap was geared towards the mammal genomes; all other tools work with all the eukaryotic organisms.
As for the tools that require prior separately running programs, the running time, maximum memory, and maximum CPU of the prior separately running programs and their own performance results are added together. These tools include AltEventFinder (prior running of TopHat and Cufflinks), FineSplice (prior running of TopHat2), rSeqDiff (prior running of rSeq), and rSeqNP (prior running of rSeq).
Huang, Zhang et al., based on the literature and actual running of the tools, showed that many junctions can be identified by only one tool but not by the other tools. This suggests room for improvement in AS detection algorithms [26].
4. PERFORMANCE COMPARISON RESULTS
Based on the foregoing performance comparison setup, we ran the tools and listed results in Table 5. Of the tools that detect splice junctions, we have demonstrated that TopHat and its downstream tool, FineSplice, are the fastest tools, whereas PASTA is the slowest program. We find that AltEventFinder detects the highest number of junctions, and RSR detects the lowest number of junctions; splice junctions detected by other tools (e.g., TopHat) are likely to be false positive ones [5]. Of the two tools that detect differentially spliced isoforms, rMATS is faster than the rSeqDiff but detects less differentially spliced isoforms than rSeqDiff.
Table 5.
Running speed, accuracy, and specificity of AS detection tools.
| Tool | Data Source | Running Time (Minutes) |
Maximum
Memory (GB) |
Maximum CPU (%) | Number of SJs |
Number of
Differentially Spliced Isoforms |
|---|---|---|---|---|---|---|
| Alt Event Finder | ENCODE | 12 | 1.364 | 100 | 30569 | N/A |
| SpliceMap | ENCODE | 42 | 3.1 | 99.9 | 11882 | N/A |
| FineSplice | ENCODE | 2 | 1.364 | 100 | 8577 | N/A |
| RSW | N/A | N/A | N/A | N/A | N/A | N/A |
| RSR | ENCODE | 24 | 3.968 | 100 | 3143 | N/A |
| PASTA | ENCODE | 350 | 2.17 | 101 | 14675 | N/A |
| rMATS | mouse used in RSW study | 44 | 26.536 | 274 | N/A | 17 |
| SOAPsplice | ENCODE | 123 | 5.332 | 99.7 | 10381 | N/A |
| SplicePie | N/A | N/A | N/A | N/A | N/A | N/A |
| SplicingCompass | N/A | N/A | N/A | N/A | N/A | N/A |
| TopHat | ENCODE | 1.75 | 1.364 | 100 | 9619 | N/A |
| TrueSight | ENCODE | 229 | 2.914 | 571 | 12360 | N/A |
| NSMAP | N/A | N/A | N/A | N/A | N/A | N/A |
| rSeqDiff | mouse used in RSW study | 115 | 0.186 | 119 | N/A | 203 |
| rSeqNP | N/A | N/A | N/A | N/A | N/A | N/A |
CONCLUSION
In this paper, we summarized the achievements, drawbacks, and scopes of application of these AS detection tools developed in recent years. We reviewed the outstanding features of these tools by categories, including their publication information, working principle, generic and distinct workflows, running platform, input data requirement, sequencing depth dependency, reads mapped to multiple locations, isoform annotation basis, precise detected AS types, and performance benchmarks.
Our categorization and performance comparison will be conducive to the development of new AS detection tools and selection of these tools by various researchers according to their need in speed or precise detected alternative splicing events.
ACKNOWLEDGEMENTS
We thank The Center for Genomic Advocacy (TCGA) at Indiana State University for computing servers, Yihua Bai and Aaron Cox for assistance with tool debugging; Dr. Gary Stuart for his critical reading on this manuscript.
CONTRIBUTION
Dr. Yongsheng Bai designed and guided the study. Lizhong Ding read literature, wrote the manuscript, and coordinated the performance comparison. Lizhong Ding and Ethan Rath ran the tools and collected the performance data.
CONFLICT OF INTEREST
The authors confirm that this article content has no conflict of interest.
REFERENCES
- 1.(a) Chen L., Tovar-Corona J.M., Urrutia A.O. Alternative Splicing: A potential source of functional innovation in the eukaryotic genome. Int. J. Evol. Biol. 2012 doi: 10.1155/2012/596274. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Roy B., Haupt L.M., Griffiths L.R. Review: Alternative Splicing(AS) of genes as an approach for generating protein complexity. Curr. Genomics. 2013;14(3):182–94. doi: 10.2174/1389202911314030004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tazi J., Bakkour N., Stamm S. Alternative splicing and disease. Biochim. Biophys. Acta. 2009;1792(1):14–26. doi: 10.1016/j.bbadis.2008.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chow L.T., Gelinas R.E., Broker T.R., Roberts R.J. An amazing sequence arrangement at the 5' ends of adenovirus 2 messenger RNA. Cell. 1977;12(1):1–8. doi: 10.1016/0092-8674(77)90180-5. [DOI] [PubMed] [Google Scholar]
- 4.Zhou A., Breese M.R., Hao Y., Edenberg H.J., Li L., Skaar T.C., Liu Y. Alt Event Finder: A tool for extracting alternative splicing events from RNA-seq data. BMC Genomics. 2012;13(Suppl. 8):S10. doi: 10.1186/1471-2164-13-S8-S10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bai Y., Kinne J., Donham B., Jiang F., Ding L., Hassler J.R., Kaufman R.J. Read-Split-Run: An improved bioinformatics pipeline for identification of genome-wide non-canonical spliced regions using RNA-Seq data. BMC Genomics. 2016;17(7):107–117. doi: 10.1186/s12864-016-2896-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Aschoff M., Hotz-Wagenblatt A., Glatting K-H., Fischer M., Eils R., König R. SplicingCompass: differential splicing detection using RNA-Seq data. Bioinformatics. 2013;29:1141–1148. doi: 10.1093/bioinformatics/btt101. [DOI] [PubMed] [Google Scholar]
- 7.Krebs J.E., Goldstein E.S., Kilpatrick S.T. 2013. Lewin's essential genes. In Jones Bartlett Learning titles in biological science, third edition ed.; Jones and Bartlett Learning: Burlington, [Google Scholar]
- 8.Dubrovina A.S., Kiselev K.V., Zhuravlev Y.N. The Role of Canonical and Noncanonical Pre-mRNA Splicing in Plant Stress Responses. 2013. [DOI] [PMC free article] [PubMed]
- 9.Valadkhan S., Mohammadi A., Wachtel C., Manley J.L. Protein-free spliceosomal snRNAs catalyze a reaction that resembles the first step of splicing. RNA. 2007;13(12):2300–2311. doi: 10.1261/rna.626207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Buratti E., Baralle F.E. Influence of RNA secondary structure on the Pre-mRNA splicing process. Mol. Cell. Biol. 2004;24(24):10505–10514. doi: 10.1128/MCB.24.24.10505-10514.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kishore S., Stamm S. The snoRNA HBII-52 regulates alternative splicing of the serotonin receptor 2C. Science. 2006;311(5758):230–232. doi: 10.1126/science.1118265. [DOI] [PubMed] [Google Scholar]
- 12.Stamm S. Regulation of alternative splicing by reversible protein phosphorylation. J. Biol. Chem. 2008;283(3):1223–1227. doi: 10.1074/jbc.R700034200. [DOI] [PubMed] [Google Scholar]
- 13.Luco R.F., Pan Q., Tominaga K., Blencowe B.J., Pereira-Smith O.M., Misteli T. Regulation of alternative splicing by histone modifications. Science. 2010;327(5968):996–1000. doi: 10.1126/science.1184208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shukla S., Kavak E., Gregory M., Imashimizu M., Shutinoski B., Kashlev M., Oberdoerffer P., Sandberg R., Oberdoerffer S. CTCF-promoted RNA polymerase II pausing links DNA methylation to splicing. Nature. 2011;479(7371):74–79. doi: 10.1038/nature10442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bai Y., Hassler J., Ziyar A., Li P., Wright Z., Menon R., Omenn G.S., Cavalcoli J.D., Kaufman R.J., Sartor M.A. Novel Bioinformatics Method for Identification of Genome-Wide Non-Canonical Spliced Regions Using RNA-Seq Data. PLoS One. 2014;9:e100864. doi: 10.1371/journal.pone.0100864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Stamm S., Ben-Ari S., Rafalska I., Tang Y., Zhang Z., Toiber D., Thanaraj T.A., Soreq H. Function of alternative splicing. Gene. 2005;344:1–20. doi: 10.1016/j.gene.2004.10.022. [DOI] [PubMed] [Google Scholar]
- 17.Pertea M., Lin X., Salzberg S.L. GeneSplicer: a new computational method for splice site prediction. Nucleic Acids Res. 2001;29:1185–1190. doi: 10.1093/nar/29.5.1185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Trapnell C., Pachter L., Salzberg S.L. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009;25:1105–1111. doi: 10.1093/bioinformatics/btp120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Alamancos G., Agirre E., Eyras E. Methods to Study Splicing from High-Throughput RNA Sequencing Data. In: Hertel K.J., editor. Spliceosomal Pre-mRNA Splicing. Vol. 1126. Humana Press; 2014. pp. 357–397. [DOI] [PubMed] [Google Scholar]
- 20.Min F., Wang S., Zhang L. Survey of Programs Used to Detect Alternative Splicing Isoforms from Deep Sequencing Data In Silico. Biomed. Res. Int. 2015. [DOI] [PMC free article] [PubMed]
- 21.SEQC/MAQC-III_Consortium, A comprehensive assessment of RNA-seq accuracy, reproducibility and information content by the Sequencing Quality Control Consortium. Nat. Biotechnol. 2014;32(9):903–914. doi: 10.1038/nbt.2957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gatto A., Torroja-Fungairiño C., Mazzarotto F., Cook S.A., Barton P.J., Sánchez-Cabo F., Lara-Pezzi E. FineSplice, enhanced splice junction detection and quantification: A novel pipeline based on the assessment of diverse RNA-Seq alignment solutions. Nucleic Acids Res. 2014;42:e71. doi: 10.1093/nar/gku166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Au K.F., Jiang H., Lin L., Xing Y., Wong W.H. Detection of splice junctions from paired-end RNA-seq data by Splice Map. Nucleic Acids Res. 2010;38:4570–4578. doi: 10.1093/nar/gkq211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tang S., Riva A. PASTA: splice junction identification from RNA-sequencing data. BMC Bioinformatics. 2013;14:116. doi: 10.1186/1471-2105-14-116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shen S., Park J.W., Lu Z-X., Lin L., Henry M.D., Wu Y.N., Zhou Q., Xing Y. rMATS: Robust and flexible detection of differential alternative splicing from replicate RNA-Seq data. Proc. Natl. Acad. Sci. USA. 2014;111:E5593–E5601. doi: 10.1073/pnas.1419161111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Huang S., Zhang J., Li R., Zhang W., He Z., Lam T-W., Peng Z., Yiu S-M. SOAPsplice: Genome-Wide ab initio Detection of Splice Junctions from RNA-Seq Data. Front. Genet. 2011;2:46. doi: 10.3389/fgene.2011.00046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pulyakhina I., Gazzoli I., 't Hoen P.A., Verwey N., Dunnen J.d., Aartsma-Rus A., Laros J.F. SplicePie: A novel analytical approach for the detection of alternative, non-sequential and recursive splicing. Nucleic Acids Res. 2015:gkv242. doi: 10.1093/nar/gkv242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li Y., Li-Byarlay H., Burns P., Borodovsky M., Robinson G.E., Ma J. TrueSight: a new algorithm for splice junction detection using RNA-seq. Nucleic Acids Res. 2013;41:e51. doi: 10.1093/nar/gks1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Xia Z., Wen J., Chang C-C., Zhou X. NSMAP: A method for spliced isoforms identification and quantification from RNA-Seq. BMC Bioinformatics. 2011;12(1):1–13. doi: 10.1186/1471-2105-12-162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shi Y., Jiang H. rSeqDiff: Detecting differential isoform expression from RNA-Seq data using hierarchical likelihood ratio test. PLoS One. 2013;8(11):e79448. doi: 10.1371/journal.pone.0079448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shi Y., Chinnaiyan A.M., Jiang H. rSeqNP: a non-parametric approach for detecting differential expression and splicing from RNA-Seq data. Bioinformatics. 2015;31(13):2222–2224. doi: 10.1093/bioinformatics/btv119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.ENCODE_Project_Consortium, An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489(7414):57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kim D., Pertea G., Trapnell C., Pimentel H., Kelley R., Salzberg S.L. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013;14(4):1–13. doi: 10.1186/gb-2013-14-4-r36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Langmead B., Trapnell C., Pop M., Salzberg S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10(3):1–10. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jiang H., Wong W.H. Statistical inferences for isoform expression in RNA-Seq. Bioinformatics. 2009;25(8):1026–1032. doi: 10.1093/bioinformatics/btp113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Trapnell C., Hendrickson D.G., Sauvageau M., Goff L., Rinn J.L., Pachter L. Differential analysis of gene regulation at transcript resolution with RNA-seq. Nat. Biotechnol. 2013;31(1):46–53. doi: 10.1038/nbt.2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li B., Dewey C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics. 2011;12(1):1–16. doi: 10.1186/1471-2105-12-323. [DOI] [PMC free article] [PubMed] [Google Scholar]