

Abstract
Prostate cancer (PC) is the most common uro-oncological disease in the global population and still requires a more efficient laboratory diagnosis. Point mutations of oncogenes and tumor sup-pressor genes are the most frequent molecular genetic events in carcinogenesis. The mutations are re-sponsible, to a great extent, for the clonal evolution of cancer and can be considered as primary candi-date molecular markers of PC. Using next-generation sequencing to analyze the mutations in PC, the main molecular PC subtypes were identified, which depended on the presence of fusion genes and FOXA1, CHD1, and SPOP point mutations; other driver mutations responsible for the progression of PC subclones were also characterized. This review summarizes the data on early PC genetic markers (an mtDNA deletion, and TMPRSS2:ERG expression), as well as these somatic mutations at later stages of PC. Emphasis is placed on a switch in AR synthesis to a constitutively active variant and the point muta-tions that facilitate PC transition to a castration-refractory state that is resistant to new AR inhibitors. Based on the current whole-exome sequencing data, the frequencies and localizations of the somatic mu-tations that may provide new genetic diagnostic markers and drug targets are described.
Keywords: Oncogene, Somatic mutation, Clonal evolution, Prostate cancer, Diagnostics, Targeted therapy
1. INTRODUCTION
More than 1.8 million adults are diagnosed with uro-oncological diseases annually, of which prostate cancer (PC) is the most prevalent; thus, PC is a pressing problem of modern oncology [1]. PC originates from various molecular genetic alterations that arise in somatic cells, including point mutations, extended deletions (loss of heterozygosity) in tumor suppressor gene loci, oncogene amplifications, aberrant DNA methylation patterns, and changes in the expression patterns of regulatory RNAs and many structural genes [2]. Certain alterations can be considered as diagnostic or prognostic markers, often occurring in early carcinogenesis and determining the tumor progression and/or sensitivity to antitumor drugs [3].
New generations of malignant cells originating from an initial clone accumulate different sets of molecular genetic alterations during carcinogenesis. Several populations of morphologically identical tumor cells arise as a result, and some of them may have a selective advantage over the others, leading to tumor progression. Of considerable importance in this process is the influence of the stromal microenvironment, the intensity of the tumor vascularization, the tumor nodule size, the speed of cell proliferation in the tumor, and other factors [4]. At the same time, genetic alterations, including point mutations of the coding and regulatory regions of oncogenes and tumor suppressor genes, have important roles in carcinogenesis, and are the most common molecular genetic events in carcinogenesis. Although some PC-related mutations were studied as early as the 20th century, it was not until recently that whole exome or whole genome mutation profiling of tumors became possible due to high-throughput microarray and next-generation sequencing (NGS) [5]. This review summarizes the data obtained by routine molecular genetic methods and NGS related to the somatic mutations that arise at various steps of carcinogenesis and are of importance for diagnosing PC and testing its sensitivity to targeted therapy.
2. DNA MARKERS IN THE FIELD CANCERIZATION AREA
Early laboratory diagnosis of PC is generally based on measuring the prostate-specific antigen (PSA) levels in the blood. PSA is an organ-specific, but not cancer-specific, marker, and its concentration is elevated in hyperplasia, adenoma of the prostate and prostatitis. In addition to PSA, the concentration of p2PSA (PSA precursor containing the N-terminal peptide) is measured in the laboratory practice, and the Prostate Health Index (PHI) is calculated as (p2PSA/fPSA) x (tPSA)½, where the concentrations of р2PSA, free (f) and total (t) PSA should be considered. The accuracy of the PHI is higher than the accuracy of the tPSA or fPSA fractions (the areas under curve, AUC, are 0.73, 0.55 and 0.6, respectively) [6]. An analysis of pro- and retrospective studies also indicates that AUC for p2PSA and PHI generally vary from 0.7 to 0.8, whereas the AUC for tPSA ranges from 0.55 to 0.6. However, the reported thresholds of p2PSA and PHI vary widely among publications, and these tests are not as widely used as the traditional PSA test [7].
Increased PSA levels that are confirmed by a repeat test are an indication for biopsy. Morphological examinations of prostate neoplasms are rather difficult because of their heterogeneous and multifocal nature. The glandular structures of benign prostatic hyperplasia (BPH) often neighbor prostatic intraepithelial neoplasia (PIN) foci and multiple PC foci, which vary in the extent of cell differentiation [8]. Hence, repeat biopsy is recommended when a negative result is obtained in the primary biopsy and a higher PSA level persists; however, the result of the repeat biopsy is similarly negative in 70-80% of cases [9].
Therefore, it is important to identify the genetic alterations that would be detectable not only in the tumor but also in the area of field cancerization in its immediate vicinity in primary biopsy material, providing a better foundation for repeat biopsy. Field cancerization is a phenomenon where cells of an epithelium area accumulate more mutations during ontogeny compared with cells of neighboring regions such that the risk of carcinogenesis is the highest in the field cancerization area. As observed in previous genome-wide studies, the tumor-adjacent epithelium from the field cancerization area contains a 3.4-kb deletion of the mitochondrial DNA (3.4mtΔ) in 80% of cases [10]. The sensitivity and specificity of 3.4mtΔ testing in PC was estimated at approximately 80% and 70%, respectively, and the deletion was detected in 10-15% of BPH cases [11]. Despite of its relatively low specificity, PCR for 3.4mtΔ is efficient at detecting false positive results of the primary biopsy; i.e., the deletion is promising for use as a criterion to repeat biopsy [10, 12, 13]. The pathogenic role of 3.4mtΔ is still a matter of discussion.
Sequencing of the cell exome and mitochondrial genome in 64 PC samples showed that mutations of mtDNA genes occur at a 55-fold higher frequency compared with mutations of the coding regions of the nuclear genome. Although little is known about the fate of mtDNA mutations within the clonal evolution of tumors, several studies provide evidence for their selection. An investigation of mtDNA mutations in 14 tumor types, including 19 PC samples, reported positive selection of clones with deletions that damaged the oxidative phosphorylation genes and contributed to the manifestation of the Warburg effect [14]. In another study, sequencing of the mtDNA derived from normal prostate tissues, primary tumors, soft tissue metastases and bone metastases of 9 PC patients showed that subclones with the Thr114Ala mutation in the ND3 gene are predominant in bone metastases compared with other sites [15]. At the same time, the majority of mtDNA point mutations in a variety of tumors are passenger mutations that are not subject to clonal selection [16]. High genetic heterogeneity was reported for primary tumors from different patients in studies of the nuclear genome, as well as for multiple tumor foci from one patient in studies using laser microdissection and exome sequencing [17-19]. The vast majority of somatic mutations detected in the field cancerization area and primary tumors by exome sequencing occur at low frequencies, preventing the use of any of the mutations as an individual marker for PC diagnosis. However, common genetic alterations (e.g. PCA3 and TMPRSS2:ERG) were observed and characterized in PC before the NGS technology was developed.
3. TMPRSS2:ERG AND PCA3 EXPRESSION IN PRIMARY TUMORS
The PCA3 (prostate cancer gene 3) cDNA, which is overexpressed in PC, was identified by differential display approximately 15 years ago. The PCA3 product is a noncoding RNA that has several isoforms [20]. PC cells desquamate into the lumen of the urinary tract and are present in the urine sediment, allowing noninvasive diagnosis. In some studies, PCA3 expression in the urine sediment obtained after prostate massage was studied by real-time PCR with TaqMan probes. The results were calibrated with KLK3, which encodes PSA and is expressed in a prostate-specific manner [21]. Another approach based on transcription-mediated amplification (TMA) uses a “Progensa” kit to analyze PCA3 and KLK3 expression [22]. PCA3 is upregulated in the tumor compared with the normal tissue, and thus a problem arises as to what threshold should be set for the “Progensa” PCA3-Score. A score of 35 is commonly accepted, whereas a score of 20-25 is used in some studies to increase the diagnostic sensitivity of the assay [23, 24]. Based on a PCA3-Score of 35, the assay has a sensitivity of 52-67%, a specificity of 72-80%, and a diagnostic accuracy of 70-77%, according to several estimates [25-28]. The highest (85%) diagnostic accuracy is achieved with PCA3-Score of 20, according to the most recent meta-analysis [29]. PCA3 was characterized as a marker whose diagnostic accuracy is higher than the accuracies of total PSA and the free-to-total PSA ratio [25, 30, 31]. Studies of more than 3,000 patients showed that the urine-based test for PCA3 expression aids not only in the decision for repeat prostate biopsy in patients with elevated PSA levels but also in the primary PC diagnosis [32].
It is possible to assay PCA3 in a multiplex analysis with other genes that are specifically expressed in PC to improve the diagnostic accuracy. PC is often associated with fusion genes that result from fusion of the 5’-untranslated region of TMPRSS2 with genes of the ETS transcription factor family. TMPRSS2:ERG is the fusion gene that is detected in the vast majority of cases; it is detected in 50% of PC cases and is not detected in normal tissue [33]. TMPRSS2:ERG generation is an early event in carcinogenesis because the fusion gene is identified in 11% of high-grade PIN foci. The specificity of the marker is usually no more than 90%, based on this finding. However, there is evidence that high-grade PIN more frequently progresses to PC in the presence of TMPRSS2:ERG than in its absence, which may be explained by the coexistence of the high grade PIN with the already shaped adenocarcinoma foci, although this result has not been confirmed by all investigators [34, 35]. The results of the relevant studies indicate that the PCA3 and TMPRSS2:ERG mRNAs should be simultaneously assayed in the urine sediment to reduce the number of false negative results without appreciably increasing the number of false positive result [36, 37]. A multicenter study showed that the combination of PCA3 and TMPRSS2:ERG increases the diagnostic accuracy to 84% in urine tests for PC [38].
The above genetic markers (the mtDNA deletion, PCA3 overexpression, and TMPRSS2:ERG fusion) are characteristics of PC initiation and are found in early tumors. These markers can be considered as clinical markers, and commercial tests for their detection exist (“Progensa”, which is approved by the FDA, Prostate Core Mitomic Test, and Mi-Prostate Score) [6]. Other somatic mutations arise in PC during further clonal evolution, determining the variant of tumor that develops and the set of potential targets for targeted therapy.
4. MUTATIONS AND CLONAL EVOLUTION OF CASTRATION-REFRACTORY PROSTATE CANCER
The androgen receptor (AR) signaling pathway is a key mechanism regulating prostate cell proliferation in both normal and pathological conditions [39]. Anti-androgen therapy has come to occupy a main place among drug treatments for PC. Anti-androgens significantly increase patient survival, but the tumors become refractory to hormonal therapy in several years, and the disease progresses [40]. The AR signaling pathway remains active in these tumors, although testosterone synthesis is suppressed (castration). The AR gene is amplified or acquires an activating mutation in the tumor. Consequently, AR synthesis switches to a constitutively active AR variant as a result of alternative splicing of the AR mRNA, and intratumoral/extragonadal androgen production occurs [41]. Constitutively active receptors lacking the ligand-binding domain can be expressed from the AR gene. Approximately ten alternative AR mRNAs have been detected in PC. AR variant 7 (AR-V7) is the most common, arising when cryptic exon 3b is included in the mRNA in place of exon 3 during splicing [42]. The greatest increase in AR-V7 is observed for a short period immediately after the patient starts androgen deprivation therapy, presumably resulting from compensatory overexpression of both the normal and minor AR mRNA variants. Therefore, it is expedient to block the pathological AR splice variants while using AR antagonists in therapy for PC. It should be noted that both alternative splicing and complex point mutations of AR are theoretically capable of generating AR-Vs and that both mechanisms were experimentally detected in tumor cells [43]. This circumstance explains the rapid acquisition of a castration-refractory character by the tumor and the subsequent selection and proliferation of the cell population in which the character is fixed both functionally and structurally. Based on the mechanisms known to underlie the development of castration-refractory prostate cancer (CRPC), drugs that block the AR signaling pathway at various levels are currently used in medicine. The first is abiraterone, which acts as a 17-α-hydroxylase and 17,20-liase inhibitor and thereby inhibits CYP17A1, a key androgen biosynthesis enzyme; enzalutamide is another drug that acts as a high-affinity AR antagonist [44, 45]. However, these new drugs are only effective for a limited period. Further clonal evolution generates cancer cell populations with missense mutations that affect AR and certain other genes and abolish the effect of the antagonists. For instance, clonal evolution of the tumor during enzalutamide treatment sometimes yields clones with AR containing the F876L missense mutation, which structurally alters the ligand-binding domain of the receptor such that enzalutamide exerts an effect that is virtually opposite to the expected effect, acting as an agonist of AR, rather than an antagonist [46-48]. The T877A mutation adapts the ligand-binding domain for AR activation by estrogens and progesterone; the L701H mutation, allows AR to be activated by cortisol. Other mutations characterized in CRPC include G142V, D221H, L179R, and W435L, which increase AR activity; E225K, which alters the E3-ubiquitin ligase-binding site to increase the AR lifetime in the nucleus; and T575A, which allows AR to activate nonspecific palindromic androgen response elements [49, 50]. Some tumors with acquired abiraterone resistance were found to carry the N367T somatic mutation of HSD3B1; the mutation protects the protein product of the gene from ubiquitin-dependent proteolysis and allows dihydrotestosterone synthesis to bypass testosterone [51, 52].
In addition to the androgen-independent constitutive activation of the AR signaling pathway, the tumor may evolve toward neuroendocrine differentiation and suppression of the pathway. Neuroendocrine differentiation (NED) occurs in 10-25% of CRPC tumors, producing a genuine hormone-refractory tumor, i.e., the tumor no longer expresses AR and no longer depends on activation of the AR signaling pathway [53]. NED can be considered a qualitatively new stage of CRPC clonal evolution. The largest-scale genetic study in PC with NED was performed by transcriptome sequencing on the GA II Illumina NGS platform and included 7 adenocarcinomas with NED, 30 adenocarcinomas without NED, and 6 BPH samples. Amplification and subsequent overexpression of the AURKA and MYCN oncogenes was identified as the most prominent molecular genetic feature of NED, occurring in 40% of tumors with NED and only 5% of primary PC tumors [54]. It has been shown that AURKA and MYCN induce the expression of neuroendocrine markers, and the MYCN gene product (N-MYC transcription factor) directly binds to promoters of the genes encoding neuron-specific enolase, synaptophysin and AR. The REST gene may be considered a marker whose expression is reduced by NED. REST undergoes silencing in 50% of CRPC with NED and in only 3% of CRPC without NED. In this context, it is interesting to consider the results obtained for small cell CRPC with NED by exome sequencing and array-based SNP genotyping. Tumors were often found to carry TP53, RB1, CHD1, and CDKN1B deletions and TP53, RB1, FOXA1, and CCAR1 point mutations. On one hand, the results support the idea that TP53 and RB1 inactivation are crucial in PC with NED; on the other hand, mutations of the AR cofactor genes (FOXA1 and CCAR1) are implicated in the generation of AR-independent subclones [55]. Clonal evolution of primary PC to CRPC, including CRPC with NED, can be outlined as shown in Fig. (1). The above examples point again to the increasingly important role NGS plays in studies of PC and its molecular genetic markers.
Fig. (1).
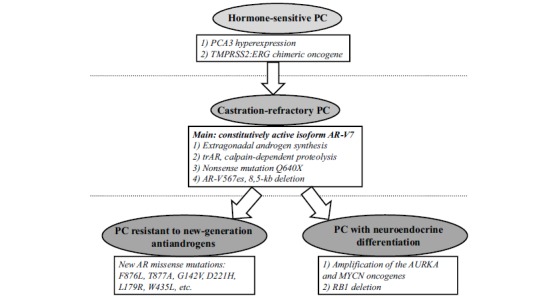
Key molecular genetic events in PC progression.
5. DRIVER MUTATIONS IDENTIFIED AS NEW MARKERS BY NEXT GENERATION SEQUENCING
Each NGS study identifies an original set of genes that are often affected by mutations, but only few of the genes are similarly identified in other such studies and are of interest as new PC markers. For instance, the candidate gene set included FOXA1, MED12, PTPN11, TSC2, and FGFR1 in a study of PC, and a role in carcinogenesis was emphasized for FOXA1 [56]. FOXA1 acts as a cofactor of the estrogen receptor and AR, facilitating the opening of the chromatin structure and assembly of the transcription complex. FOXA1 missense mutations are observed in 5% of PC and breast cancer cases. Codons 226 and 253 are identified as “hot spots” based on the mutation spectrum, indicating that FOXA1 analysis may provide a relatively simple test for PCR-diagnostics. In addition to mutations, FOXA1 amplification was detected in several estrogen- and androgen-dependent tumors [57]. Exome sequencing in 112 PC samples revealed point mutations of SPOP in 13% of cases. Tumors with SPOP driver mutations lack TMPRSS2:ERG and vice versa, providing further evidence for divergent clonal evolution in cancer. In fact, a separate subgroup of tumors with a “SPOP mutations/no TMPRSS2:ERG” status can be isolated [58-60]. SPOP encodes an AR cofactor that is involved in transcription complex assembly. The set of SPOP mutations detected in tumors by NSG includes Y87C, Y87N, F102C, S119N, F125V, W131G, F133L, and F133V [61]. Clustering within a region of approximately 200 bp, the SPOP mutations provide a convenient target for routine DNA diagnosis.
In another study, targeted sequencing of the key carcinogenesis-related genes in 45 samples of localized, metastatic hormone-sensitive, and hormone-refractory PC confirmed again that TMPRSS2:ERG occurs at a high frequency (44%) in PC and that AR plays an important role in both hormone-sensitive PC and CRPC (AR amplification or point mutations were observed in 24% and 20% of cases, respectively). The study additionally detected PTEN (44%) and RB1 (28%) deletions, TP53 mutations (40%), MYC amplification (12%), and ATM (8%) and PIK3CA (4%) mutations [62]. SPOP, which often mutates during PC progression according to earlier data, was not included in the NGS study design, but a fusion gene involving the BRAF proto-oncogene was observed. A BRAF mutation (T599_V600insHT) and a CYP11B2 amplification were found to arise in PC during ketoconazole treatment to compensate for the cytostatic effect of the drug [63]. A subsequent study revealed slightly higher frequencies of the mutations in AR (63%), the ETS family (57%), TP53 (53%) and PTEN (41%). In this study the majority of samples with ТР53 point mutations were compound heterozygotes, in which one of the mutations was represented by a deletion, and another by a missense mutation, possibly with a dominant negative effect [64].
In general, NGS and earlier experiments showed that the main features that allow the identification of molecular subtypes in PC include fusion oncogenes, TP53/RB1 inactivating mutations, AURKA/MYCN amplification, PTEN deletions, AR mutations and alternatively spliced variants, and CHD1 and SPOP mutations. Subgroups isolated on the basis of these alterations differ in the activation of the AR, PI3K/PTEN/AKT, and RTK/RAS/MAPK signaling pathways and require different approaches to anticancer drug therapy [64]. According to the NGS data, the most common chromosomal rearrangements responsible for the progression of primary PC include AR amplification, homozygous PTEN and NKX3.1 deletions, CHD1 copy number alterations, and fusion genes. Regarding the common missense and nonsense point mutations that play a substantial role in the clonal evolution of the tumor, the most common are AR point mutations, which are found in 50% of primary PC subclones and more than 95% of metastases after hormonal therapy; NCOA2 mutations, which occur at frequencies of 8% in primary PC and 37% in metastases; and mutations and deletions of the TP53 tumor suppressor gene. A far greater number of genes display driver point mutations with a frequency of 1-15%; several tens of such mutations are described in metastatic low-differentiated PC and CRPC [65, 66] (Table 1).
Table 1.
DNA mutations and RNA gene expression as genetic markers of PС.
| Gene (locus) | Target | Routine Method Suitable for Target Testing | Interpretation |
|---|---|---|---|
| Early PC diagnostic markers | |||
| mtDNA | 3.4mtΔ deletion | RT PCR | Associated with primary PC and the field cancerization area adjacent to the tumor |
| PCA3 | Expression of exons 3 and 4 | -//- | Overexpression is characteristic of PC |
| TMPRSS2:ERG | mRNA expression | -//- | Occurs in 50% of PC cases and 10% of high-grade PIN cases with a poor prognosis |
| SPOP | Point mutations of the MATH domain | RT PCR, PCR after laser microdissection with subsequent pyrosequencing or Sanger sequencing, TS | Mutations characteristic of primary PC subgroups |
| FOXA1 | Point mutations of the DNA-binding or C-terminal domain | -//- | -//- |
| CHD1 | Point mutations/deletions | MLPA and/or TS | -//- |
| PTEN | -//- | -//- | -//- |
| Markers of CRPC and anticancer drug resistance | |||
| AR | 1) isoforms AR-V7 and AR-V567es, 2) missense mutations, amplification |
1) RT PCR 2) MLPA and/or TS |
1) associated with resistance to hormonal therapy 2) F876L is a contraindication to enzalutamide treatment; T877A and L701H confer resistance to new-generation antiandrogens; G142V, D221H, L179R, W435L, E225K, and T575A suggest an adjustment of therapy (the mutations increase AR activity) |
| HSD3B1 | Missense mutations | TS | Abiraterone resistance in CRPC |
| ZBTB16 | Homozygous deletions | MLPA or RT PCR | Associated with enzalutamide resistance |
| IDH1 | Missense mutations of codon 132 | RT PCR, PCR with SNaPshot | Expedient use of IDH inhibitors |
| Markers of metastatic PC, CRPC progression, and NED | |||
| TP53 | Deletions and inactivating point mutations | MLPA, TS | Frequency of more than 50% in CRPC with NED, increases with PC progression Frequency of up to 85% in small cell PC with NED and CRPC |
| RB1 | -//- | -//- | -//- |
| AURKA | Amplification and/or overexpression | RT PCR and/or MLPA | Frequency of 5% in primary PC vs 40-60% in NED and up to 80% in metastases of HRPC with NED |
| MYCN | -//- | -//- | -//- |
| NCOA1 | Somatic point mutations | TS | Frequency of 8% in primary PC vs 37% in metastases |
| PTK2 | -//- | -//- | Frequency of 1% in localized PC vs 35% in CRPC |
| YWHAZ | -//- | -//- | Frequency of 3% in localized PC vs 48% in CRPC |
RT PCR – real-time polymerase chain reaction, TS – targeted resequencing on a benchtop-NGS platform, MLPA – multiple ligation probe amplification.
Among the main signaling pathways involved in the PC–CRPC transition, components of the Wnt pathway are overexpressed to a greater extent, and somatic mutations of these genes occur in CRPC more often than in hormone-sensitive PC [67, 68]. In particular, mutations or amplification of CTNNB1, which is involved in the Wnt pathway, are detected in the cancer cell population possibly as an adaptive response to hormonal therapy. A mutation of IDH1 at codon 132 arises in PC subpopulations and has potential therapeutic significance, because IDH inhibitors have already been used in clinical trials [69]. Whole-exome sequencing on the SOLiD platform revealed amplification of PTK2 (1% in localized PC vs 35% in CRPC) and YWHAZ (3% in localized PC vs 48% in CRPC). Approximately 5-7% of PCs acquire a homozygous ZBTB16 deletion, which is associated with a transition to CRPC and enzalutamide resistance [70, 71]. In addition to abiraterone and enzalutamide, which target amplified and overexpressed AR in CRPC, other agents can be considered as potential treatments based on the above high mutation frequencies, including PARP inhibitors (ATM is mutated in 8-20% of CRPC cases) and, in certain cases, inhibitors of the Akt (PIK3CA) or BRAF signaling pathways [67, 69-72]. Thus, NGS studies made it possible to expand the list of mutations responsible for the development and progression of PC and to identify potential targets for the design of new drugs.
CONCLUSION
Studies of the specific molecular genetics of PC enabled considerable progress in our understanding of the mechanisms of prostate carcinogenesis. PC is already multifocal and genetically heterogeneous at early stages, as determined using conventional genetic methods and NGS. The earliest PC markers are the Δ3.4 mtDNA deletion, PCA3 overexpression, and expression of the TMPRSS2:ERG fusion oncogene; PCA3 and TMPRSS2:ERG are suitable for noninvasive PC diagnosis by urine sediment analysis. The point mutations identified in SPOP and FOXA1 by NGS are of particular interest among the early DNA markers of PC. NGS has made it possible to isolate the main molecular subtypes of PC that depend on the presence or absence of chromosome aberrations producing TMPRSS2:ERG or TMPRSS2:ETV1 and mutations of CHD1 or SPOP. The mechanisms that mediate a switch to alternative splicing or the acquisition of new missense mutations, oncogene amplifications, and deletions of tumor suppressor genes may in turn act during androgen-deprivation therapy or PC progression to confer a selective advantage to the mutant CRPC clones. The first event is a switch to constitutively active AR-V7 or the acquisition of the AR missense mutations that increase AR activity or protect AR from proteolysis. Treatment with new-generation antiandrogens may lead to the selection of clones with the point mutations that affect AR or other key genes that activate AR through atypical ligands or abolish the antiandrogen effect. In a number of PC cases, the AR signaling pathway is inactivated and NED develops, accompanied by amplification of the AURKA and MYCN oncogenes and inactivating mutations of TP53 and RB1, leading to a genuine castration-refractory character of the tumor, and suggesting a poor prognosis. At each step of its development, the tumor evolves to produce a new cancer cell population and to evade the effects of the anticancer treatments. Driver mutations responsible for the progression of PC subclones were characterized in FOXA1, MED12, ATM, PIK3CA, TP53, RB1, MYC, AR, PTEN, PTK2, CDKN1B, and other genes. Molecular diagnosis based on the data of mutations acquired during clonal evolution may substantially improve the accuracy of PC diagnosis and the efficiency of targeted therapy in the foreseeable future.
ACKNOWLEDGEMENTS
Declared none.
CONFLICT OF INTEREST
The authors confirm that this article content has no conflict of interest.
REFERENCES
- 1.Ferlay J., Soerjomataram I., Dikshit R., Eser S., Mathers C., Rebelo M., Parkin D.M., Forman D., Bray F. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int. J. Cancer. 2015;136(5):E359–E386. doi: 10.1002/ijc.29210. [DOI] [PubMed] [Google Scholar]
- 2.Mendelsohn J., Howley P.M., Israel M.A., Gray J.W., Thompson C.B. The Molecular Basis of Cancer. 4th ed. Philadelphia, USA: Saunders; 2015. [Google Scholar]
- 3.Tan D., Lynch H.T. Principles of Molecular Diagnostics and Personalized Cancer Medicine. 4th ed. Philadelphia, USA: Lippincott Williams & Wilkins; 2013. [Google Scholar]
- 4.Greaves M., Maley C.C. Clonal evolution in cancer. Nature. 2012;481(7381):306–313. doi: 10.1038/nature10762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schott A.F., Perou C.M., Hayes D.F. Genome medicine in cancer: What's in a name? Cancer Res. 2015;75(10):1930–1935. doi: 10.1158/0008-5472.CAN-15-0174. [DOI] [PubMed] [Google Scholar]
- 6.Sartori D.A., Chan D.W. Biomarkers in prostate cancer: what’s new? Curr. Opin. Oncol. 2014;26(3):259–264. doi: 10.1097/CCO.0000000000000065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Huang Y.Q., Sun T., Zhong W.D., Wu C.L. Clinical performance of serum -2(pro)PSA derivatives, %p2PSA and PHI, in the detection and management of prostate cancer. Am. J. Clin. Exp. Urol. 2014;2(4):343–350. [PMC free article] [PubMed] [Google Scholar]
- 8.Paltsev M.A., Zaletaev D.V. Systems of genetic and epigenetic markers in diagnostics of the oncological diseases. Moscow, Russia: Medicine; 2009. [Google Scholar]
- 9.Park Y.H., Lee J.K., Jung J.W., Lee B.K., Lee S., Jeong S.J., Hong S.K., Byun S.S., Lee S.E. Prostate cancer detection rate in patients with fluctuating prostate-specific antigen levels on the repeat prostate biopsy. Prostate Int. 2014;2(1):26–30. doi: 10.12954/PI.13037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Maki J., Robinson K., Reguly B., Alexander J., Wittock R., Aguirre A., Diamandis E.P., Escott N., Skehan A., Prowse Q., Thayer R.E., Froberg M.K., Wilson M.J., Maragh S., Jakupciak J.P., Wagner P.D., Srivastava S., Dakubo G.D., Parr R.L. Mitochondrial genome deletion aids in the identification of false- and true-negative prostate needle core biopsy specimens. Am. J. Clin. Pathol. 2008;129:57–66. doi: 10.1309/UJJTH4HFEPWAQ78Q. [DOI] [PubMed] [Google Scholar]
- 11.Yu J.J., Yan T. Effect of mtDNA mutation on tumor malignant degree in patients with prostate cancer. Aging Male. 2010;13(3):159–165. doi: 10.3109/13685530903536668. [DOI] [PubMed] [Google Scholar]
- 12.Robinson K., Creed J., Reguly B., Powell C., Wittock R., Klein D., Maggrah A., Klotz L., Parr R.L., Dakubo G.D. Accurate prediction of repeat prostate biopsy outcomes by a mitochondrial DNA deletion assay. Prostate Cancer Prostatic Dis. 2010;13:126–131. doi: 10.1038/pcan.2009.64. [DOI] [PubMed] [Google Scholar]
- 13.Maragh S., Veltri R.W., Lund S.P., Mangold L., Isharwal S., Christudass C.S., Partin A.W., Humphreys E.B., Sorbara L., Srivastava S., Wagner P.D. Evaluation of two mitochondrial DNA biomarkers for prostate cancer detection. Cancer Biomark. 2015;15(6):763–773. doi: 10.3233/CBM-150518. [DOI] [PubMed] [Google Scholar]
- 14.Stewart J.B., Alaei-Mahabadi B., Sabarinathan R., Samuelsson T., Gorodkin J., Gustafsson C.M., Larsson E. Simultaneous DNA and RNA mapping of somatic mitochondrial mutations across diverse human cancers. PLoS Genet. 2015;11(6):e1005333. doi: 10.1371/journal.pgen.1005333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Arnold R.S., Fedewa S.A., Goodman M., Osunkoya A.O., Kissick H.T., Morrissey C., True L.D., Petros J.A. Bone metastasis in prostate cancer: recurring mitochondrial DNA mutation reveals selective pressure exerted by the bone microenvironment. Bone. 2015;78:81–86. doi: 10.1016/j.bone.2015.04.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ju Y.S., Alexandrov L.B., Gerstung M., Martincorena I., Nik-Zainal S., Ramakrishna M., Davies H.R., Papaemmanuil E., Gundem G., Shlien A., Bolli N., Behjati S., Tarpey P.S., Nangalia J., Massie C.E., Butler A.P., Teague J.W., Vassiliou G.S., Green A.R., Du M.Q., Unnikrishnan A., Pimanda J.E., Teh B.T., Munshi N., Greaves M., Vyas P., El-Naggar A.K., Santarius T., Collins V.P., Grundy R., Taylor J.A., Hayes D.N., Malkin D., Foster C.S., Warren A.Y., Whitaker H.C., Brewer D., Eeles R., Cooper C., Neal D., Visakorpi T., Isaacs W.B., Bova G.S., Flanagan A.M., Futreal P.A., Lynch A.G., Chinnery P.F., McDermott U., Stratton M.R., Campbell P.J., ICGC Breast Cancer Group. ICGC Chronic Myeloid Disorders Group. ICGC Prostate Cancer Group Origins and functional consequences of somatic mitochondrial DNA mutations in human cancer. eLife. 2014;3 doi: 10.7554/eLife.02935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lindberg J., Mills I.G., Klevebring D., Liu W., Neiman M., Xu J., Wikstrom P., Wiklund P., Wiklund F., Egevad L., Gronberg H. The mitochondrial and autosomal mutation landscapes of prostate cancer. Eur. Urol. 2013;63(4):702–708. doi: 10.1016/j.eururo.2012.11.053. [DOI] [PubMed] [Google Scholar]
- 18.van der Weele D.J., Brown C.D., Taxy J.B., Gillard M., Hatcher D.M., Tom W.R., Stadler W.M., White K.P. Low-grade prostate cancer diverges early from high grade and metastatic disease. Cancer Sci. 2014;105(8):1079–1085. doi: 10.1111/cas.12460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lindberg J., Klevebring D., Liu W., Neiman M., Xu J., Wiklund P., Wiklund F., Mills I.G., Egevad L., Gronberg H. Exome sequencing of prostate cancer supports the hypothesis of independent tumour origins. Eur. Urol. 2013;63(2):347–353. doi: 10.1016/j.eururo.2012.03.050. [DOI] [PubMed] [Google Scholar]
- 20.Clarke R.A., Zhao Z., Guo A.Y., Roper K., Teng L., Fang Z.M., Samaratunga H., Lavin M.F., Gardiner R.A. New genomic structure for prostate cancer specific gene 3 (PCA3) within BMMC1: implication for prostate cancer detection and progression. PLoS One. 2009;4(3):e4995. doi: 10.1371/journal.pone.0004995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ng C.F., Yeung R., Chiu P., Lam N.Y., Chow J., Chan B. The role of urine prostate cancer antigen 3 mRNA levels in the diagnosis of prostate cancer among Hong Kong Chinese patients. Hong Kong Med. J. 2012;18:459–465. [PubMed] [Google Scholar]
- 22.Durand X., Moutereau S., Xylinas E., de la Taille A. Progensa™ PCA3 test for prostate cancer. Expert Rev. Mol. Diagn. 2011;11(2):137–144. doi: 10.1586/erm.10.122. [DOI] [PubMed] [Google Scholar]
- 23.Capoluongo E., Zambon C.F., Basso D., Boccia S., Rocchetti S., Leoncini E., Palumbo S., Padoan A., Albino G., Todaro A., Prayer-Galetti T., Zattoni F., Zuppi C., Plebani M. PCA3 score of 20 could improve prostate cancer detection: results obtained on 734 Italian individuals. Clin. Chim. Acta. 2014;429:46–50. doi: 10.1016/j.cca.2013.10.022. [DOI] [PubMed] [Google Scholar]
- 24.Pepe P., Fraggetta F., Galia A., Skonieczny G., Aragona F. PCA3 score and prostate cancer diagnosis at repeated saturation biopsy. Which cut-off: 20 or 35? Int. Braz. J. Urol. 2012;38(4):489–495. doi: 10.1590/s1677-55382012000400008. [DOI] [PubMed] [Google Scholar]
- 25.Ochiai A., Okihara K., Kamoi K., Oikawa T., Shimazui T., Murayama S., Tomita K., Umekawa T., Uemura H., Miki T. Clinical utility of the prostate cancer gene 3 (PCA3) urine assay in Japanese men undergoing prostate biopsy. BJU Int. 2013;111(6):928–933. doi: 10.1111/j.1464-410X.2012.11683.x. [DOI] [PubMed] [Google Scholar]
- 26.Bradley L.A., Palomaki G.E., Gutman S. Comparative effectiveness review: prostate cancer antigen 3 testing for the diagnosis and management of prostate cancer. J. Urol. 2013;190(2):389–398. doi: 10.1016/j.juro.2013.02.005. [DOI] [PubMed] [Google Scholar]
- 27.Ramos C.G., Valdevenito R., Vergara I., Anabalon P., Sanchez C., Fulla J. PCA3 sensitivity and specificity for prostate cancer detection in patients with abnormal PSA and/or suspicious digital rectal examination. First Latin American experience. Urol. Oncol. 2013;31(8):1522–1526. doi: 10.1016/j.urolonc.2012.05.002. [DOI] [PubMed] [Google Scholar]
- 28.Crawford E.D., Rove K.O., Trabulsi E.J., Qian J., Drewnowska K.P., Kaminetsky J.C., Huisman T.K., Bilowus M.L., Freedman S.J., Glover W.L., Bostwick D.G. Diagnostic performance of PCA3 to detect prostate cancer in men with increased prostate specific antigen: a prospective study of 1,962 cases. J. Urol. 2012;188(5):1726–1731. doi: 10.1016/j.juro.2012.07.023. [DOI] [PubMed] [Google Scholar]
- 29.Luo Y., Gou X., Huang P., Mou C. The PCA3 test for guiding repeat biopsy of prostate cancer and its cut-off score: a systematic review and meta-analysis. Asian J. Androl. 2014;16(3):487–492. doi: 10.4103/1008-682X.125390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Goode R.R., Marshall S.J., Duff M., Chevli E., Chevli K.K. Use of PCA3 in detecting prostate cancer in initial and repeat prostate biopsy patients. Prostate. 2013;73(1):48–53. doi: 10.1002/pros.22538. [DOI] [PubMed] [Google Scholar]
- 31.Ferro M., Bruzzese D., Perdona S., Marino A., Mazzarella C., Perruolo G., D'Esposito V., Cosimato V., Buonerba C., di Lorenzo G., Musi G., de Cobelli O., Chun F.K., Terracciano D. Prostate Health Index (Phi) and Prostate Cancer Antigen 3 (PCA3) significantly improve prostate cancer detection at initial biopsy in a total PSA range of 2-10 ng/ml. PLoS One. 2013;8(7):e67687. doi: 10.1371/journal.pone.0067687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chevli K.K., Duff M., Walter P., Yu C., Capuder B., Elshafei A., Malczewski S., Kattan M.W., Jones J.S. Urinary PCA3 as a predictor of prostate cancer in a cohort of 3,073 men undergoing initial prostate biopsy. J. Urol. 2014;191(6):1743–1748. doi: 10.1016/j.juro.2013.12.005. [DOI] [PubMed] [Google Scholar]
- 33.Chan S.W., Nguyen P.N., Violette P., Brimo F., Taguchi Y., Aprikian A., Chen J.Z. Early detection of clinically significant prostate cancer at diagnosis: a prospective study using a novel panel of TMPRSS2:ETS fusion gene markers. Cancer Med. 2013;2(1):63–75. doi: 10.1002/cam4.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Park K., Dalton J.T., Narayanan R., Barbieri C.E., Hancock M.L., Bostwick D.G., Steiner M.S., Rubin M.A. TMPRSS2:ERG gene fusion predicts subsequent detection of prostate cancer in patients with high-grade prostatic intraepithelial neoplasia. J. Clin. Oncol. 2014;32(3):206–211. doi: 10.1200/JCO.2013.49.8386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liu H., Shi J., Wilkerson M., Yang X.J., Lin F. Immunohistochemical evaluation of ERG expression in various benign and malignant tissues. Ann. Clin. Lab. Sci. 2013;43(1):3–9. [PubMed] [Google Scholar]
- 36.Tomlins S.A. Urine PCA3 and TMPRSS2:ERG using cancer-specific markers to detect cancer. Eur. Urol. 2014;65(3):543–545. doi: 10.1016/j.eururo.2012.12.001. [DOI] [PubMed] [Google Scholar]
- 37.Robert G., Jannink S., Smit F., Aalders T., Hessels D., Cremers R., Mulders P.F., Schalken J.A. Rational basis for the combination of PCA3 and TMPRSS2:ERG gene fusion for prostate cancer diagnosis. Prostate. 2013;73(2):113–120. doi: 10.1002/pros.22546. [DOI] [PubMed] [Google Scholar]
- 38.Leyten G.H., Hessels D., Jannink S.A., Smit F.P., de Jong H., Cornel E.B., de Reijke T.M., Vergunst H., Kil P., Knipscheer B.C., van Oort I.M., Mulders P.F., Hulsbergen-van de Kaa C.A., Schalken J.A. Prospective multicentre evaluation of PCA3 and TMPRSS2-ERG gene fusions as diagnostic and prognostic urinary biomarkers for prostate cancer. Eur. Urol. 2014;65(3):534–542. doi: 10.1016/j.eururo.2012.11.014. [DOI] [PubMed] [Google Scholar]
- 39.Augello M.A., Den R.B., Knudsen K.E. AR function in promoting metastatic prostate cancer. Cancer Metastasis Rev. 2014;33(2-3):399–411. doi: 10.1007/s10555-013-9471-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sun Y., Niu J., Huang J. Neuroendocrine differentiation in prostate cancer. Am. J. Transl. Res. 2009;1(2):148–162. [PMC free article] [PubMed] [Google Scholar]
- 41.Sharifi N. Mechanisms of androgen receptor activation in castration-resistant prostate cancer. Endocrinology. 2013;154(11):4010–4017. doi: 10.1210/en.2013-1466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ware K.E., Garcia-Blanco M.A., Armstrong A.J., Dehm S.M. Biologic and clinical significance of androgen receptor variants in castration resistant prostate cancer. Endocr. Relat. Cancer. 2014;21:T87–T103. doi: 10.1530/ERC-13-0470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sprenger C.C., Plymate S.R. The link between androgen receptor splice variants and castration-resistant prostate cancer. Horm. Cancer. 2014;5(4):207–217. doi: 10.1007/s12672-014-0177-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ryan C.J., Smith M.R., de Bono J.C., Molina A., Logothetis C.J., de Souza P., Fizazi K., Mainwaring P., Piulats J.M., Ng S., Carles J., Mulders P.F., Basch E., Small E.J., Saad F., Schrijvers D., van Poppel H., Mukherjee S.D., Suttmann H., Gerritsen W.R., Flaig T.W., George D.J., Yu E.Y., Efstathiou E., Pantuck A., Winquist E., Higano C.S., Taplin M.E., Park Y., Kheoh T., Griffin T., Scher H.I., Rathkopf D.E. COU-AA-302 Investigators. Abiraterone in metastatic prostate cancer without previous chemotherapy. N. Engl. J. Med. 2013;368(2):138–148. doi: 10.1056/NEJMoa1209096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Golshayan A.R., Antonarakis E.S. Ensalutamide: an evidence-based review of its use in the treatment of prostate cancer. Core Evid. 2013;8:27–35. doi: 10.2147/CE.S34747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Boudadi K., Antonarakis E.S. Resistance to novel antiandrogen therapies in metastatic castration-resistant prostate cancer. Clin. Med. Insights Oncol. 2016;10(S1):1–9. doi: 10.4137/CMO.Ss34534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Joseph J.D., Lu N., Qian J., Shao G., Brigham D., Moon M., Maneval E.C., Chen I., Darimont B., Hager J.H. A clinically relevant androgen receptor mutation confers resistance to second-generation antiandrogens enzalutamide and ARN-509. Cancer Discov. 2013;3(9):1020–1029. doi: 10.1158/2159-8290.CD-13-0226. [DOI] [PubMed] [Google Scholar]
- 48.Tan M.H., Li J., Xu H.E., Melcher K., Yong E.L. Androgen receptor: structure, role in prostate cancer and drug discovery. Acta Pharmacol. Sin. 2015;36(1):3–23. doi: 10.1038/aps.2014.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Eisermann K., Wang D., Jing Y., Pascal L.E., Wang Z. Androgen receptor gene mutation, rearrangement, polymorphism. Transl. Androl. Urol. 2013;2(3):137–147. doi: 10.3978/j.issn.2223-4683.2013.09.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chen E.J., Sowalsky A.G., Gao S., Cai C., Voznesensky O., Schaefer R., Loda M., True L.D., Ye H., Troncoso P., Lis R.L., Kantoff P.W., Montgomery R.B., Nelson P.S., Bubley G.J., Balk S.P., Taplin M.E. Abiraterone treatment in castration-resistant prostate cancer selects for progesterone responsive mutant androgen receptors. Clin. Cancer Res. 2015;21(6):1273–1280. doi: 10.1158/1078-0432.CCR-14-1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Stein M.N., Patel N., Bershadskiy A., Sokoloff A., Singer E.A. Androgen synthesis inhibitors in the treatment of castration-resistant prostate cancer. Asian J. Androl. 2014;16(3):387–400. doi: 10.4103/1008-682X.129133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chang K.H., Ercole C.E., Sharifi N. Androgen metabolism in prostate cancer: from molecular mechanisms to clinical consequence. Br. J. Cancer. 2014;111:1249–1254. doi: 10.1038/bjc.2014.268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Conteduca V., Aieta M., Amadori D., de Giorgi U. Neuroendocrine differentiation in prostate cancer: current and emerging therapy strategies. Crit. Rev. Oncol. Hematol. 2014;92(1):11–24. doi: 10.1016/j.critrevonc.2014.05.008. [DOI] [PubMed] [Google Scholar]
- 54.Beltran H., Rickman D.S., Park K., Chae S.S., Sboner A., MacDonald T.Y., Wang Y., Sheikh K.L., Terry S., Tagawa S.T., Dhir R., Nelson J.B., de la Taille A., Allory Y., Gerstein M.B., Perner S., Pienta K.J., Chinnaiyan A.M., Wang Y., Collins C.C., Gleave M.E., Demichelis F., Nanus D.M., Rubin M.A. Molecular characterization of neuroendocrine prostate cancer and identification of new drug targets. Cancer Discov. 2011;1(6):4. doi: 10.1158/2159-8290.CD-11-0130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Li Q., Zhang C.S., Zhang Y. Molecular aspects of prostate cancer with neuroendocrine differentiation. Chin. J. Cancer Res. 2016;28(1):122–129. doi: 10.3978/j.issn.1000-9604.2016.01.02. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kim T.M., Jung S.H., Baek I.P., Lee S.H., Choi Y.J., Lee J.Y., Chung Y.J., Lee S.H. Regional biases in mutation screening due to intratumoural heterogeneity of prostate cancer. J. Pathol. 2014;233(4):425–435. doi: 10.1002/path.4380. [DOI] [PubMed] [Google Scholar]
- 57.Robinson J.L., Holmes K.A., Carroll J.S. FOXA1 mutations in hormone-dependent cancers. Front. Oncol. 2013;3:20. doi: 10.3389/fonc.2013.00020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sunkel B., Wang Q. Mapping mutations in prostate cancer exomes. Asian J. Androl. 2012;14(6):801–802. doi: 10.1038/aja.2012.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Barbieri C.E., Baca S.C., Lawrence M.S., Demichelis F., Blattner M., Theurillat J.P., White T.A., Stojanov P., van Allen E., Stransky N., Nickerson E., Chae S.S., Boysen G., Auclair D., Onofrio R.C., Park K., Kitabayashi N., MacDonald T.Y., Sheikh K., Vuong T., Guiducci C., Cibulskis K., Sivachenko A., Carter S.L., Saksena G., Voet D., Hussain W.M., Ramos A.H., Winckler W., Redman M.C., Ardlie K., Tewari A.K., Mosquera J.M., Rupp N., Wild P.J., Moch H., Morrissey C., Nelson P.S., Kantoff P.W., Gabriel S.B., Golub T.R., Meyerson M., Lander E.S., Getz G., Rubin M.A., Garraway L.A. Exome sequencing identifies recurrent SPOP, FOXA1 and MED12 mutations in prostate cancer. Nat. Genet. 2012;44(6):685–689. doi: 10.1038/ng.2279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Schrecengost R.S., Knudsen K.E. Molecular pathogenesis and progression of prostate cancer. Semin. Oncol. 2013;40(3):244–258. doi: 10.1053/j.seminoncol.2013.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lai J., Batra J. Speckle-type POZ protein mutations interrupt tumor suppressor function of speckle-type POZ protein in prostate cancer by affecting androgen receptor degradation. Asian J. Androl. 2014;16(5):659–660. doi: 10.4103/1008-682X.133323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Beltran H., Yelensky R., Frampton G.M., Park K., Downing S.R., MacDonald T.Y., Jarosz M., Lipson D., Tagawa S.T., Nanus D.M., Stephens P.J., Mosquera J.M., Cronin M.T., Rubin M.A. Targeted next-generation sequencing of advanced prostate cancer identifies potential therapeutic targets and disease heterogeneity. Eur. Urol. 2013;63(5):920–926. doi: 10.1016/j.eururo.2012.08.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Grasso C.S., Cani A.K., Hovelson D.H., Quist M.J., Douville N.J., Yadati V., Amin A.M., Nelson P.S., Betz B.L., Liu C-J., Knudsen K.E., Cooney K.A., Feng F.Y., McDaniel A.S., Tomlins S.A. Integrative molecular profiling of routine clinical prostate cancer specimens. Ann. Oncol. 2015;26(6):1110–1118. doi: 10.1093/annonc/mdv134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Robinson D., Van Allen E.M., Wu Y.M., Schultz N., Lonigro R.J., Mosquera J.M., Montgomery B., Taplin M.E., Pritchard C.C., Attard G., Beltran H., Abida W., Bradley R.K., Vinson J., Cao X., Vats P., Kunju L.P., Hussain M., Feng F.Y., Tomlins S.A., Cooney K.A., Smith D.C., Brennan C., Siddiqui J., Mehra R., Chen Y., Rathkopf D.E., Morris M.J., Solomon S.B., Durack J.C., Reuter V.E., Gopalan A., Gao J., Loda M., Lis R.T., Bowden M., Balk S.P., Gaviola G., Sougnez C., Gupta M., Yu E.Y., Mostaghel E.A., Cheng H.H., Mulcahy H., True L.D., Plymate S.R., Dvinge H., Ferraldeschi R., Flohr P., Miranda S., Zafeiriou Z., Tunariu N., Mateo J., Perez-Lopez R., Demichelis F., Robinson B.D., Schiffman M., Nanus D.M., Tagawa S.T., Sigaras A., Eng K.W., Elemento O., Sboner A., Heath E.I., Scher H.I., Pienta K.J., Kantoff P., de Bono J.S., Rubin M.A., Nelson P.S., Garraway L.A., Sawyers C.L., Chinnaiyan A.M. Integrative clinical genomics of advanced prostate cancer. Cell. 2015;161(5):1215–1228. doi: 10.1016/j.cell.2015.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Yadav S.S., Li J.L., Lavery H.L., Yadav K.K., Tech M., Tewari A.K. Next-generation sequencing technology in prostate cancer diagnosis, prognosis, and personalized treatment. Urol. Oncol. 2015;33(6):e267. doi: 10.1016/j.urolonc.2015.02.009. [DOI] [PubMed] [Google Scholar]
- 66.Lonigro R.J., Grasso C.S., Robinson D.R., Jing X., Wu Y.M., Cao X., Quist M.J., Tomlins S.A., Pienta K.J., Chinnaiyan A.M. Detection of somatic copy number alterations in cancer using targeted exome capture sequencing. Neoplasia. 2011;13(11):1019–1025. doi: 10.1593/neo.111252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Spans L., Clinckemalie L., Helsen C., Vanderschueren D., Boonen S., Lerut E., Joniau S., Claessens F. The genomic landscape of prostate cancer. Int. J. Mol. Sci. 2013;14:10822–10851. doi: 10.3390/ijms140610822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Yokoyama N.N., Shao S., Hoang B.H., Mercola D., Zi X. Wnt signaling in castration-resistant prostate cancer: implications for therapy. Am. J. Clin. Exp. Urol. 2014;2(1):27–44. [PMC free article] [PubMed] [Google Scholar]
- 69.Kumar A., White T.A., MacKenzie A.P., Clegg N., Lee C., Dumpit R.F., Coleman I., Ng S.B., Salipante S.J., Rieder M.J., Nickerson D.A., Corey E., Lange P.H., Morrissey C., Vessella R.L., Nelson P.S., Shendure J. Exome sequencing identifies a spectrum of mutation frequencies in advanced and lethal prostate cancers. Proc. Natl. Acad. Sci. USA. 2011;108(41):17087–17092. doi: 10.1073/pnas.1108745108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hovelson D.H., McDaniel A.S., Cani A.K., Johnson B., Rhodes K., Williams P.D., Bandla S., Bien G., Choppa P., Hyland F., Gottimukkala R., Liu G., Manivannan M., Schageman J., Ballesteros-Villagrana E., Grasso C.S., Quist M.J., Yadati V., Amin A., Siddiqui J., Betz B.L., Knudsen K.E., Cooney K.A., Feng F.Y., Roh M.H., Nelson P.S., Liu C.J., Beer D.G., Wyngaard P., Chinnaiyan A.M., Sadis S., Rhodes D.R., Tomlins S.A. Development and validation of a scalable next-generation sequencing system for assessing relevant somatic variants in solid tumors. Neoplasia. 2015;17(4):385–399. doi: 10.1016/j.neo.2015.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Menon R., Deng M., Ruenauver K., Queisser A., Peifer M., Offermann A., Boehm D., Vogel W., Scheble V., Fend F., Kristiansen G., Wernert N., Oberbeckmann N., Biskup S., Rubin M.A., Shaikhibrahim Z., Perner S. Somatic copy number alterations by whole-exome sequencing implicates YWHAZ and PTK2 in castration-resistant prostate cancer. J. Pathol. 2013;231(4):505–516. doi: 10.1002/path.4274. [DOI] [PubMed] [Google Scholar]
- 72.Hsieh C.L., Botta G., Gao S., Li T., van Allen E.M., Treacy D.J., Cai C., He H.H., Sweeney C.J., Brown M., Balk S.P., Nelson P.S., Garraway L.A., Kantoff P.W. PLZF, a tumor suppressor genetically lost in metastatic castration-resistant prostate cancer, is a mediator of resistance to androgen deprivation therapy. Cancer Res. 2015;75(10):1944–1948. doi: 10.1158/0008-5472.CAN-14-3602. [DOI] [PMC free article] [PubMed] [Google Scholar]