

Abstract
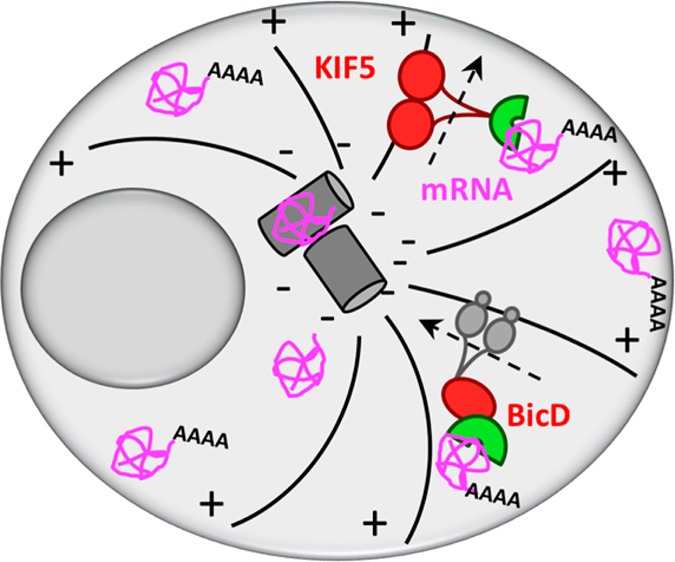
Localization of mRNA is important in a number of cellular processes such as embryogenesis, cellular motility, polarity, and a variety of neurological processes. A synthetic device that controls cellular mRNA localization would facilitate investigations on the significance of mRNA localization in cellular function and allow an additional level of controlling gene expression. In this work, we developed the PUF (Pumilio and FBF homology domain)-assisted localization of RNA (PULR) system, which utilizes a eukaryotic cell’s cytoskeletal transport machinery to reposition mRNA within a cell. Depending on the cellular motor used, we show ligand-dependent transport of mRNA toward either pole of the microtubular network of cultured cells. In addition, implementation of the reprogrammable PUF domain allowed the transport of untagged endogenous mRNA in primary neurons.
Keywords: mRNA transport, kinesin, dynein, RNA-binding proteins (RBP), Pumilio and fem3 mRNA-binding factor (PUF)
Mounting evidence suggests ubiquitous subcellular mRNA localization across the domains of life.1−4 Subcellular mRNA localization is important in asymmetric organization of cells, and hence is crucial in biological processes such as cell division, embryonic patterning, and cell migration.5 Trafficking and local translation of mRNA is particularly relevant in highly polarized cells like neurons, where the phenomenon has been implicated in a variety of developmental and regenerative processes, including axon outgrowth, synapse formation and plasticity, and neuron regeneration.6 Development of a molecular tool for controlling mRNA transport would enable spatial regulation of gene expression, which in turn would facilitate investigations on causality between mRNA localization and cellular organization and function.
Previously, a tool was engineered for mRNA transport during the yeast budding process;7 however, it required prior tagging of the target RNA and was limited to yeast. Here, by linking a designer PUF (Pumilio and FBF homology) domain to a cellular motor, we developed the PUF-assisted Localization of RNA (PULR) system, which is capable of cytoplasmic redistribution of mRNA in animal cells. Depending on the motor protein used, the system directionally transports the mRNA toward either cellular periphery or perinuclear region. In addition, our system takes advantage of the reprogrammability of the PUF domain, which enables switching the system’s specificity toward an RNA sequence of choice, and allowing the transport of tagged as well as untagged mRNA.
PULR is designed to function via reprogrammable RNA recognition in combination with chemical-induced dimerization with cellular motors (Figure 1). For RNA recognition and binding, we used the PUF RNA-binding domain (residues 828–1176) of human Pumilio 1 protein. The domain is composed of 8 imperfectly repeated 36 amino acid motifs (PUF repeats) and flanking N- and C-terminal regions, which all pack together to form a crescent-shaped right-handed superhelix.8−10 RNA is bound to the concave surface of the domain, where each repeat interacts with a single RNA base in the sequence of 8-base RNA target.10 The PUF domain is oriented toward RNA in such a way that the N-terminal PUF repeat (R1) interacts with the 3′ base of the RNA sequence (N8), and vice versa, the C-terminal PUF repeat (R8) interacts with the 5′ base of the RNA sequence (N1).10 Each repeat establishes base-specific hydrogen bonds with a Watson–Crick edge of an RNA base via amino acid side chains at positions 12 and 16, while the amino acid side chains at position 13 in each repeat form stacking interactions between adjacent RNA bases.10
Figure 1.
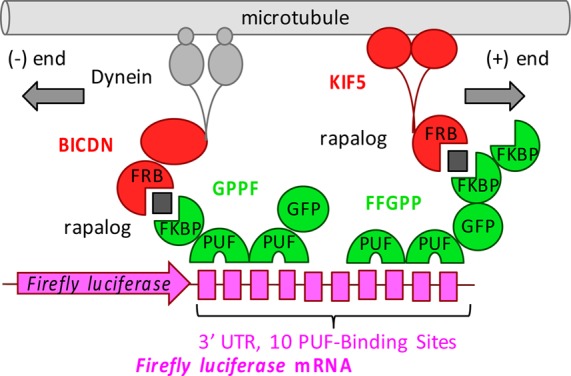
Schematic overview of the PULR system. In the presence of rapalog (black squares), FFGPP/KIF5 or GPPF/BICDN protein combinations transport reporter mRNA to the plus (+) or minus (−) ends of microtubules, respectively. FFGPP and GPPF constructs are denominated based on the order of their domains from amino to the carboxyl termini, depicted right (N-t) to left (C-t). For PUF modifications/specificities introduced in this work, as well as Firefly luciferase 3′UTR sequences used in this work, see Tables S1 and S2, respectively.
The appeal to using the PUF domain for RNA recognition is its modular repeat-base recognition mode. Through a combination of inferences from the crystal structure,10 engineering,11,12 and binding assays,11,13 the RNA-recognition code for the PUF domain was established, where amino acid combination N12Q16 recognizes uracil, C12Q16 or S12Q16 recognizes adenine, S12E16 recognizes guanine, and S12R16 recognizes cytosine. Elucidation of this RNA recognition code by the PUF domain allows reprogramming the RNA-binding domain for recognition of unaltered, endogenous RNA, consequently alleviating the need of tagging and potentially disturbing the metabolism of endogenous mRNA. We have previously developed a modular assembly strategy for facilitated introduction of mutations in the key PUF domain amino acid positions,14 and used the strategy for all the modifications of the PUF domain in this work.
In order to prevent the PUF domain from binding to hundreds of its natural mRNA targets in the transcriptome,15 we modified its repeats 6 and 7 so that its specificity is switched from the sequence 5′-UGUAnAUA-3′ to the sequence 5′-UUGAnAUA-3′ (Table S1),13 which is predicted to be less abundant in the human transcriptome (Supplemental Note S1). To anchor the PUF domains to the transport machinery, two PUF domains were fused with one or two consecutive FKBP (FK506-binding protein) domains and the enhanced green fluorescent protein (eGFP) via flexible GGGS linkers in different polypeptide chain variations that differed in the order of the domains. The constructs were named based on the order of the domains from the N-terminus to the C-terminus.
For controlling the localization of mRNA, we utilized a eukaryotic cell’s transport system, by which molecular motors such as dyneins and kinesins carry cellular cargos along the network of microtubules. For retrograde mRNA transport, we utilized the N-terminal portion of Bicaudal D2 (amino acid residues 1–594, hereafter referred to as BICDN), which induces dynein-mediated cargo transport.16 For anterograde mRNA transport, we generated truncated kinesin-1 heavy chain KIF5B without the cargo binding tail domain (amino acid residues 1–807).17 To anchor the transport machinery to mRNA, we fused BICDN or KIF5 to a modified FRB (FKBP and rapamycin binding) domain, which heterodimerizes with the FKBP domain upon addition of rapalog18 (Figure 1).
Since HeLa cells contain radial microtubule arrays pointing from the centrosome to the cell periphery, we expected KIF5 to transport the PUF constructs toward the cell periphery (plus-ends of microtubules) (Figure 2a). Live-cell fluorescent imaging of HeLa cells treated with rapalog revealed that the PUF construct FFGPP relocalized to the cell periphery in the presence of KIF5-FRB, but remained diffuse in the absence thereof (Figure S1). To determine PULR’s effectiveness in mRNA transport, we implemented a firefly luciferase (FLuc) mRNA as a reporter. In the 3′ untranslated region (UTR) of FLuc mRNA, we placed 2, 10, or no cognate PUF-binding sites (PUF-BS) (Table S2). In subsequent imaging of fixed cells, RNA fluorescence in situ hybridization (RNA-FISH) using antisense probes against the FLuc transcript and simultaneous immunofluorescence of the proteins revealed that FLuc mRNA tagged with 10xPUF-BS was strongly redistributed to the cell periphery and colocalized both with FFGPP and KIF5-FRB after rapalog treatment (Figure 2b). The cytoplasmic redistribution of mRNA was observed as intensification of a number of speckles along the cell outline, although the central region that encompasses the nucleus and its adjacent cytoplasmic area retained some untransported Fluc RNA. Quantification of the RNA-FISH signal intensity in the regions of brightest FFGPP immunofluorescence across 20 cells confirmed that FLuc mRNA tagged with 10xPUF-BS was strongly enriched in the cell periphery compared to an adjacent proximal cytoplasmic region (Figure 2c). This sequence of FLuc mRNA tagged with 10x(UUGAUAUA) was previously shown to not be significantly recognized by other PUF variants, such as the WT PUF domain and a PUF mutant carrying the mutations in repeat 1(S12E16),14 suggesting that RNA binding to this PUF construct is sequence specific. In contrast, no enrichment of Random FLuc mRNA or FLuc mRNA tagged with 2xPUF-BS was observed in peripheral regions of cells (Figure 2b,c), suggesting that the FFGPP function is sequence-specific; however, anterograde transport of mRNA tagged with only 2 PUF-BS is not enough for efficient relocalization in Hela cells. This observation is consistent with our previous finding that the number of PUF-BS in the 3′ UTR of targeted mRNA is a limiting factor in PUF-effector fusion protein’s functional effect on cognate RNA in live cells. Our previously reported PUF fused to a translational regulator showed marginal effect on the efficiency of translation of mRNA tagged with a single repetition of PUF-BS.14 We also observed that both Kif5-FRB and FFGPP were intracellularly redistributed to the same extent regardless of the sequence of mRNA (Figure S2), further suggesting that redistribution of mRNA is sequence-specific.
Figure 2.

Transport of reporter mRNA to the distal ends of microtubules in HeLa cells. (a) Schematic overview of KIF5-mediated mRNA transport. Microtubules are represented as black solid arrows pointing from their (+) to (−) ends. (b) Transport of FLuc mRNA to the cell periphery by KIF5-FRB and the PUF construct FFGPP. Hoechst, nuclear stain. White solid lines, cell outlines. Quantified areas: C, cytoplasm (light blue dashed lines); P, periphery (yellow dashed lines). PUF-BS, PUF-binding sites. Enriched spots indicated with arrows. Scale bar: 20 μm. (c) Quantitation of mRNA translocation to the cell periphery. RNA-FISH intensity at the region coinciding with the brightest fluorescent region of KIF5 immunofluorescence was normalized to an adjacent proximal region of the same area. n = 20 cells in 3 biological replicates. ***P < 0.001. Mean ± SEM.
BICDN, on the other hand, was expected to transport the PUF constructs toward the centrosome (minus-ends of microtubules) (Figure 3a). In live cells, we observed that the PUF construct GPPF was redistributed to the perinuclear region in the presence of BICDN-FRB, but remained diffuse in the absence thereof (Figure S1). In fixed cells, immunofluorescence against BicD-FRB revealed rapalog-dependent aggregation of BicD-FRB to the perinuclear region (Figure 3b). This observation is consistent with our previous finding that overexpressed BICDN inhibits dynein function, preventing dynein from binding to cargo and also association from microtubules.16,19 However, artificial attachment of cargo reverses the inhibition possibly through a conformational change of BICDN.17 Co-immunofluorescence against GPPF also revealed that in response to rapalog, both BICDN-FRB and GPPF proteins were intracellularly redistributed to the perinuclear region to a similar extent regardless of the sequence of mRNA (Figure S3). RNA-FISH, in turn, revealed that only FLuc mRNA tagged with 10xPUF-BS strongly accumulated at the perinuclear region and colocalized with GPPF and BICDN-FRB (Figure 3b,c). FLuc mRNA carrying no or 2 PUF-BS were not redistributed, (Figure 3b,c), reminiscent of what was observed with the Kif5-FRB/FFGPP combination.
Figure 3.

Transport of reporter mRNA to the proximal ends of microtubules in HeLa cells. (a) Schematic overview of BICDN-mediated mRNA transport. Microtubules are represented as black solid arrows pointing from their (−) to (+) ends. (b) Transport of FLuc mRNA to the perinuclear region by BICDN-FRB and the PUF construct GPPF. Hoechst, nuclear stain. White lines, cell outlines. Quantified areas: C, cytoplasm (light blue dashed lines); Cen, centrosome (yellow dashed lines). PBS, PUF-binding sites. Enriched spots indicated with arrows. Scale bar: 20 μm. (c) Quantitation of mRNA transport to the perinuclear region. RNA-FISH intensity at the region coinciding with the brightest fluorescent region of BICDN immunofluorescence was normalized to an adjacent distal region of the same area. n = 20 cells in 3 biological replicates. ***P < 0.001. Mean ± SEM.
We next questioned whether the PULR system is capable of (1) transporting untagged, endogenous mRNA, and (2) transporting mRNA to greater distances. We therefore examined whether β-actin mRNA, whose axonal localization and biological importance are well studied,6 could be relocalized using our tool in cultured neuronal cells. Although we did not observe redistribution of reporter mRNA tagged with 2 PUF-BS in HeLa cells, we anticipated that interaction of FFGPP with only 2 PUF-BS in the untagged endogenous mRNA would still mediate significant mRNA redistribution in the context of a neuronal cell. We reasoned that greater transport distances in neurites would diminish the overall effect of mRNA diffusion, which must be a significant factor in relatively small cells such as HeLa.
β-actin mRNA, the subcellular localization of which is one of the best studied, is known to be localized to cell bodies as well as to growth cones in neurons.20 However, zipcode-binding protein 1 (ZBP1), a protein responsible for axonal localization of β-actin mRNA,21 was found in limited levels in adult sensory neurons.22 Specifically, it was shown that expression of exogenous ZBP1 in neurons from ZBP1+/ZBP1+ mice led to increased axonal β-actin mRNA levels,22 suggesting that β-actin mRNA levels in adult sensory neurons are not saturated. Assuming that such β-actin mRNA distribution is typical for most neurons, we expected the Kif5/FFGPP-mediated increase in axonal β-actin mRNA levels of embryonic rat hippocampal neurons.
To showcase the effectiveness of PULR independent of ZBP1, we designed the PUF domain to recognize a sequence in the β-actin mRNA 3′ UTR other than the “RNA zipcode”, which is recognized by ZBP1.21,23 To prepare functional PUF fusion constructs for endogenous β-actin mRNA transport in rat hippocampal neurons, we picked 8 potential target regions in the 3′ UTR of rat β-actin mRNA. Each of these 8 target regions contained 2 PUF-BS that satisfied two criteria: (1) are 3–30 bp apart and (2) would require minimal mutagenesis in repeats 6–8 that recognize the conserved UGU triplet in the consensus target sequence24 (Table S3). For recognition of each of these target regions, we assembled individual PUF variants from a library of repeat units using the PUF assembly kit that we previously developed,14 and assembled them pairwise into 8 GPPF constructs (GPPF act1-act8, Table S4). To determine the most potent PUF variant combination, we assayed each GPPF against its target sequence in the context of BICDN transport to the centrosome, since it is easier to quantify transport to a single centrosomal locus in HeLa cells. Consistent with Figure 3b and c, redistribution of reporter mRNA containing only 2 PUF-BS was marginal (Figure S4). However, construct GPPF-act6 displayed increased fluorescence compared to the remaining 7 constructs (data not shown), which was likely due to its superior soluble expression. We therefore subsequently reassembled this combination of PUF variants in the FFGPP configuration and used for anterograde transport of endogenous β-actin mRNA in primary hippocampal neurons.
Since the microtubules are arranged in the axon with their positive ends pointing toward the growth cones, we expected the FFGPP/Kif5-FRB protein complex to redistribute endogenous β-actin mRNA toward axonal growth cones (Figure 4a). Coexpressing KIF5-FRB and FFGPP targeting β-actin mRNA (FFGPP-Act-6) or unrelated sequence (FFGPP-control, Table S1) in embryonic rat hippocampal neurons, we observed a marked enrichment of both FFGPP-Act-6 and FFGPP-control in axonal growth cones in response to rapalog (Figure S5 and S6). Quantification (Supplemental Note S2) corroborated that FFGPP-act and FFGPP-control were strongly enriched in the growth cones compared to the adjacent axon (Figure 4b,c,), suggesting that Kif5/FFGPP is transported to axonal growth cones independent of its RNA recognition properties. We next assessed whether FFGPP-act cotransports β-actin mRNA. Using antisense probes against β-actin mRNA, we found that β-actin mRNA levels in the growth cones of neurons expressing KIF5-FRB and FFGPP-act increased on average 1.9-fold after rapalog treatment (Figure 4b, d). No significant change in β-actin mRNA levels was observed in neurons expressing KIF5-FRB/FFGPP-control (Figure 4b, d). These results demonstrate that untagged endogenous mRNA can be transported and enriched in axonal growth cones beyond their normal levels. Moreover, our data suggest that enrichment of β-actin mRNA in the growth cones is sequence- and ligand-dependent.
Figure 4.

Transport of endogenous β-actin mRNA to axonal growth cones in primary neurons. (a) Schematic of endogenous mRNA transport in hippocampal neurons. Microtubules are represented as black solid arrows pointing from their (−) to (+) ends. (b) Effect of FFGPP-Act-6 or FFGPP-control on abundance of β-actin mRNA in axonal growth cones. FFGPP-act-6 PUF domains recognize β-actin mRNA and FFGPP-control PUF domains recognize unrelated 5′UUGAnAUA3′. BFP was transfected to track the neuron outline. Quantified regions: GC, growth cone (yellow dashed lines); A, axon (light blue dashed lines). Scale bar: 5 μm. (c) Quantitation of FFGPP redistribution. FFGPP fluorescence in a growth cone was normalized to fluorescence measured along a 10–20 μM line following the axon approximately 10–20 μM away from the growth cone. (d) Quantitation of β-actin mRNA in axonal growth cones in the presence of FFGPP-act or FFGPP-control. n = 20 cells in 3 biological replicates. Mean ± SEM. ***P < 0.001. **P < 0.01. *P < 0.05.
In this work, we developed a molecular tool for chemical-controlled, sequence-specific transport of mRNA along microtubules in animal cells. The PULR system proved effective in ligand-dependent redistribution of tagged reporter mRNA toward distal or proximal poles of microtubular network in HeLa cells. In addition, it showcased the possibility of transporting untagged endogenous mRNA such as β-actin mRNA toward the axonal growth cones of primary neurons. Targeting and relocalization of endogenous mRNA was made possible by utilization of a programmable RNA-binding domain PUF, suggesting that other endogenous mRNA could be targeted in a similar manner.
The current study opens the prospect of further investigations into controlled local translation of mRNA and resulting phenotypical changes, which are timely now with increasing interest in mRNA localization and local mRNA translation. We are similarly intrigued with the possibility of this work inspiring alternative and/or supplementary approaches to regulating mRNA localization in the cytoplasm, such as locally entrapping mRNA or locally protecting against degradation.
Methods
Cell Culture and Transfection
HeLa cells were maintained in modified Eagle’s Medium (MEM) supplemented with 10% Fetal Bovine Serum (FBS). HeLa cells were plated a day before transfection at a density of 1 × 104 cells/well in an 8-well poly-l-lysine-coated μ-slide (Ibidi) in 300 μL of media. The next day, transfection mixtures were prepared by mixing 15 μL OptiMEM (Life Technologies), 300 ng DNA, and 0.9 μL Fugene HD reagent (Promega). The DNA mixture contained 100 ng of pCMV5-KIF5B-FRB or pCMV5-BICDN-FRB, 100 ng of PUF fusion expression plasmid, and 100 ng of FLuc mRNA-transcribing plasmid. The transfection mixtures were incubated for 15 min at room temperature and added to the cells. The cells were allowed to grow for 48 h, after which they were treated with 1 μM rapalog (commercial name A/C heterodimerizer, Clontech Laboratories) for 1 h at 37 °C before live cell imaging or fixation.
Primary rat hippocampal neurons were prepared from embryonic day 18 (E18) rat brains as described previously.25 Briefly, cells were plated on coverslips coated with poly-l-lysine (30 μg/mL) and laminin (2 μg/mL) at a density of 75 000/well in a 12-well plate. Hippocampal cultures were grown in Neurobasal medium (NB) supplemented with B27, 0.5 mM glutamine, 12.5 μM glutamate, and penicillin-streptomycin. For subsequent RNA-FISH treatment, the neuron culturing was modified as following: the cells were plated at a density of 20 000/well in a 24-well plate, and cultured in NB supplemented with B27, 0.5 mM glutamine, 12.5 μM glutamate, penicillin-streptomycin, and 2 μM cytosine β-d-arabinofuranoside hydrochloride (Sigma-Aldrich). Half of conditioned media volume was replaced with equal amount of fresh media twice a week. Hippocampal neurons cultured in 12-well plates were transfected at DIV8 (8 days in vitro) with Lipofectamine 2000 (Invitrogen). DNA (1.8 μg/well) was mixed with 3.3 mL Lipofectamine 2000 in 200 μL NB, incubated for 30 min, and then added to the neurons in NB at 37 °C in 5% CO2 for 45 min. For transfection in 24-well plates, the reagents were scaled down by a factor of 0.5. Next, neurons were washed with NB and transferred to the original medium at 37 °C in 5% CO2. At DIV11, rapalog was added to the media to a final concentration of 0.1 μM for 24 h 37 °C in 5% CO2 before fixation of the cells.
RNA-FISH and Immunofluorescence
For combined RNA-FISH and immunofluorescence, HeLa cells were washed twice with phosphate buffer saline (PBS) and fixed in 4% formaldehyde (Polysciences) in PBS buffer for 10 min at room temperature (RT). The cells were washed twice in PBS and permeabilized with 70% ethanol at 4 °C overnight, after which they were washed in a washing buffer containing 10% formamide (Sigma) in 2× saline-sodium-citrate (SSC) for 5 min at RT. The cells were incubated in 170 nM Stellaris Quasar 670-tagged 48 probe mix (Biosearch Technologies) against firefly luciferase transcript, 1 μg/mL mouse monoclonal anti-HA tag IgG antibody (Abcam) (for HA-tagged motor protein binding), and 5 μg/mL rabbit monoclonal anti-GFP IgG Abfinity antibody (Invitrogen) (for PUF-GFP-FKBP binding) in the FISH-IF buffer (10% dextran sulfate, 10% formamide, 2× SSC, 2 mM vanadyl ribonucleoside complex, 0.02% RNase-free bovine serum albumin (BSA)) in a humidified chamber overnight at 37 °C in the dark. The next day, the cells were washed three times in the washing buffer and incubated with 4 μg/mL Alexa-Fluor568-tagged antimouse IgG antibodies and 10 μg/mL Alexa-Fluor488-tagged antirabbit antibody (Invitrogen) in the FISH-IF buffer for 1 h at RT in the dark, after which they were subsequently washed three times in the washing buffer. The cells were next counterstained in 130 ng/mL Hoechst 33342 (Pierce) for 5 min at RT, and washed three times in 2× SSC buffer. After washing in the oxygen scavenger (GLOX) buffer (2× SSC, 0.4% glucose, 10 mM Tris-HCl pH 8) for 5 min at RT, the cells were imaged in GLOX solution (37 μg/mL glucose oxidase (Sigma) and 100 μg/mL catalase (Sigma) in GLOX buffer). No/negligible cross-signal was observed between anti-HA and anti-GFP immunofluorescent staining and RNA-FISH (Figure S7).
For RNA-FISH in rat hippocampal neurons, the cells were fixed in 4% formaldehyde supplemented with 4% sucrose in PBS for 10 min at RT, washed 3× with PBS, and permeabilized in 0.1% Triton X-100 in PBS. The coverslips were next washed in the washing buffer for 5 min at RT. RNA-FISH was performed by incubation with 170 nM Stellaris Quasar 670-tagged 48 probe mix against rat actb transcript (Biosearch Technologies) in the FISH-IF buffer in a humidified chamber overnight at 37 °C in the dark. The next day, the cells were washed three times in the washing buffer and imaged in the GLOX solution as above.
Image Acquisition of Hela Cells
HeLa cell fluorescent images were acquired using the Zeiss Axiovert 200 M widefield fluorescence microscope equipped with a 40× NA 1.4 oil objective, Andor iXon DV887-BV camera, and DAPI (Zeiss, 49 DAPI shift free (E)), FITC (Semrock, Brightline FF01–494/20–25), Rhodamine (Semrock, Brightline SpOr-B-CSC-ZERO), and Cy5 (Semrock, Brightline Cy5-4040B-CSC-ZERO) excitation/emission filter sets. Care was taken not to oversaturate the fluorescence measurements, and to use the same imaging conditions, including the exposure time, across all image acquisitions for quantifying samples to be compared.
Quantification of mRNA Transport in HeLa Cells
Quantification
of the RNA-FISH signal from the Quasar 670-tagged probes in HeLa cells
was performed using the ImageJ software (http://rsb.info.nih.gov/ij/index.html). RNA-FISH signal (cell periphery/cytoplasm), was quantified as
follows: 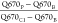 ; where
Q670P is the mean gray
value of Q670 fluorescence detected at the region coinciding with
the brightest immunofluorescence spot of FFGPP at the cell periphery,
Q670B is the mean gray value of background fluorescence
detected in a region of the same area but at an adjacent extracellular
region, and Q670C1 is fluorescence detected at an adjacent
proximal cytoplasmic region of the same area. RNA-FISH signal (centrosome/cytoplasm),
was quantified as follows:
; where
Q670P is the mean gray
value of Q670 fluorescence detected at the region coinciding with
the brightest immunofluorescence spot of FFGPP at the cell periphery,
Q670B is the mean gray value of background fluorescence
detected in a region of the same area but at an adjacent extracellular
region, and Q670C1 is fluorescence detected at an adjacent
proximal cytoplasmic region of the same area. RNA-FISH signal (centrosome/cytoplasm),
was quantified as follows: 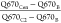 ; where Q670Cen is the mean gray
value of Q670 fluorescence detected at the region coinciding with
the brightest immunofluorescence spot of GPPF at the perinuclear region,
and Q670C2 is fluorescence detected at an adjacent distal
cytoplasmic region of the same area. The normalized RNA-FISH signal
values were averaged over 20–30 cells in which both of the
immunofluorescence signals as well as the RNA-FISH signal were observed
above background. The statistical analysis was performed with Student’s t test assuming a two-tailed and unequal variation.
; where Q670Cen is the mean gray
value of Q670 fluorescence detected at the region coinciding with
the brightest immunofluorescence spot of GPPF at the perinuclear region,
and Q670C2 is fluorescence detected at an adjacent distal
cytoplasmic region of the same area. The normalized RNA-FISH signal
values were averaged over 20–30 cells in which both of the
immunofluorescence signals as well as the RNA-FISH signal were observed
above background. The statistical analysis was performed with Student’s t test assuming a two-tailed and unequal variation.
Image Acquisition of Hippocampal Neurons
Whole neuron images were acquired using a Nikon Eclipse 80i microscope equipped with a Plan Fluor 20× N.A. 1.40 oil objective, Chroma ET-DAPI (49000), Chroma ET-GFP (49002), and a Photometrics CoolSNAP HQ2 CCD camera. Neuron images subjected to RNA-FISH were acquired using the above-mentioned Zeiss Axiovert 200 M widefield fluorescence microscope.
Quantification of mRNA Transport in Hippocampal Neurons
Quantification of the BFP, GFP, or Quasar 670-tagged RNA-FISH probe fluorescence in the growth cones was performed using the ImageJ software.
FFGPP redistribution (growth cone/axon) was quantified as follows: 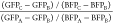 ; where GFPC is the mean gray
value of GFP fluorescence detected within the area of a growth cone,
GFPB is the mean gray value of background GFP fluorescence
detected at the adjacent extracellular region, BFPC is
the mean gray value of BFP fluorescence detected within the area of
a growth cone, BFPB is the mean gray value of background
BFP fluorescence detected at the adjacent extracellular region, GFPA is the mean gray value of GFP fluorescence detected along
a 10–20 μM line following the axon approximately 10–20
μM away from the growth cone, and BFPA is the same
value measured for BFP. Both of the regions of interest were delimitated
based on the blue fluorescent protein (BFP) distribution in the axon.
; where GFPC is the mean gray
value of GFP fluorescence detected within the area of a growth cone,
GFPB is the mean gray value of background GFP fluorescence
detected at the adjacent extracellular region, BFPC is
the mean gray value of BFP fluorescence detected within the area of
a growth cone, BFPB is the mean gray value of background
BFP fluorescence detected at the adjacent extracellular region, GFPA is the mean gray value of GFP fluorescence detected along
a 10–20 μM line following the axon approximately 10–20
μM away from the growth cone, and BFPA is the same
value measured for BFP. Both of the regions of interest were delimitated
based on the blue fluorescent protein (BFP) distribution in the axon.
FFGPP or RNA-FISH distribution was quantified similarly, by measuring background-subtracted integrated density values of fluorescence signal within the area of a growth cone and normalizing it to mean gray value of fluorescence signal detected in a stretch (20–40 μm) of an axon 10–50 μm away from the growth cone. (exposure time was kept consistent for acquisition of fluorescence from 670-tagged RNA-FISH probe). Approximately 20 growth cones in 3 biological replicates were analyzed and averaged, and a statistical analysis was performed with Student’s t test assuming a two-tailed and unequal variation.
Acknowledgments
We thank Dr. Hee Jung Chung and John Paul Cavaretta at the University of Illinois for kindly sharing the rat hippocampal neurons for the RNA-FISH experiments. We also thank the Core Facilities at the Carl R. Woese Institute for Genomic Biology and the Biology Imaging Center at the Utrecht University for their help with fluorescence microscopy. This work was financially supported by the Centennial Chair Professorship in the Department of Chemical and Biomolecular Engineering at the University of Illinois at Urbana–Champaign (H.Z.), The Netherlands Organization for Scientific Research (NWO-ALW-VICI, C.C.H.), and FP7 EU Marie Curie postdoctoral fellowship to L.F.G.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acssynbio.7b00025.
Supplemental figures, tables, notes, methods, and references (PDF)
Author Contributions
Z.A. conceived the project, carried out the experiments, and analyzed the data. Z.A., L.F.G., and C.C.H. designed the experiments and interpreted the data. C.C.H. and H.Z. oversaw the research.
The authors declare no competing financial interest.
Supplementary Material
References
- Muench D. G.; Zhang C.; Dahodwala M. (2012) Control of cytoplasmic translation in plants. Wiley Interdiscip. Rev. RNA 3, 178–194. 10.1002/wrna.1104. [DOI] [PubMed] [Google Scholar]
- Haag C.; Steuten B.; Feldbrugge M. (2015) Membrane-Coupled mRNA Trafficking in Fungi. Annu. Rev. Microbiol. 69, 265–281. 10.1146/annurev-micro-091014-104242. [DOI] [PubMed] [Google Scholar]
- Govindarajan S.; Nevo-Dinur K.; Amster-Choder O. (2012) Compartmentalization and spatiotemporal organization of macromolecules in bacteria. FEMS Microbiol. Rev. 36, 1005–1022. 10.1111/j.1574-6976.2012.00348.x. [DOI] [PubMed] [Google Scholar]
- Holt C. E.; Bullock S. L. (2009) Subcellular mRNA localization in animal cells and why it matters. Science 326, 1212–1216. 10.1126/science.1176488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cody N. A.; Iampietro C.; Lecuyer E. (2013) The many functions of mRNA localization during normal development and disease: from pillar to post. Wiley Interdiscip. Rev. Dev. Biol. 2, 781–796. 10.1002/wdev.113. [DOI] [PubMed] [Google Scholar]
- Jung H.; Yoon B. C.; Holt C. E. (2012) Axonal mRNA localization and local protein synthesis in nervous system assembly, maintenance and repair. Nat. Rev. Neurosci. 13, 308–324. 10.1038/nrn3254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belmont B. J.; Niles J. C. (2012) Inducible control of subcellular RNA localization using a synthetic protein-RNA aptamer interaction. PLoS One 7, e46868. 10.1371/journal.pone.0046868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards T. A.; Pyle S. E.; Wharton R. P.; Aggarwal A. K. (2001) Structure of Pumilio reveals similarity between RNA and peptide binding motifs. Cell 105, 281–289. 10.1016/S0092-8674(01)00318-X. [DOI] [PubMed] [Google Scholar]
- Wang X.; Zamore P. D.; Hall T. M. (2001) Crystal structure of a Pumilio homology domain. Mol. Cell 7, 855–865. 10.1016/S1097-2765(01)00229-5. [DOI] [PubMed] [Google Scholar]
- Wang X.; McLachlan J.; Zamore P. D.; Hall T. M. (2002) Modular recognition of RNA by a human pumilio-homology domain. Cell 110, 501–512. 10.1016/S0092-8674(02)00873-5. [DOI] [PubMed] [Google Scholar]
- Dong S.; Wang Y.; Cassidy-Amstutz C.; Lu G.; Bigler R.; Jezyk M. R.; Li C.; Hall T. M.; Wang Z. (2011) Specific and modular binding code for cytosine recognition in Pumilio/FBF (PUF) RNA-binding domains. J. Biol. Chem. 286, 26732–26742. 10.1074/jbc.M111.244889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filipovska A.; Razif M. F.; Nygard K. K.; Rackham O. (2011) A universal code for RNA recognition by PUF proteins. Nat. Chem. Biol. 7, 425–427. 10.1038/nchembio.577. [DOI] [PubMed] [Google Scholar]
- Cheong C. G.; Hall T. M. (2006) Engineering RNA sequence specificity of Pumilio repeats. Proc. Natl. Acad. Sci. U. S. A. 103, 13635–13639. 10.1073/pnas.0606294103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abil Z.; Denard C. A.; Zhao H. (2014) Modular assembly of designer PUF proteins for specific post-transcriptional regulation of endogenous RNA. J. Biol. Eng. 8, 7. 10.1186/1754-1611-8-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galgano A.; Forrer M.; Jaskiewicz L.; Kanitz A.; Zavolan M.; Gerber A. P. (2008) Comparative analysis of mRNA targets for human PUF-family proteins suggests extensive interaction with the miRNA regulatory system. PLoS One 3, e3164. 10.1371/journal.pone.0003164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoogenraad C. C.; Wulf P.; Schiefermeier N.; Stepanova T.; Galjart N.; Small J. V.; Grosveld F.; de Zeeuw C. I.; Akhmanova A. (2003) Bicaudal D induces selective dynein-mediated microtubule minus end-directed transport. EMBO J. 22, 6004–6015. 10.1093/emboj/cdg592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapitein L. C.; Schlager M. A.; van der Zwan W. A.; Wulf P. S.; Keijzer N.; Hoogenraad C. C. (2010) Probing intracellular motor protein activity using an inducible cargo trafficking assay. Biophys. J. 99, 2143–2152. 10.1016/j.bpj.2010.07.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollock R.; Issner R.; Zoller K.; Natesan S.; Rivera V. M.; Clackson T. (2000) Delivery of a stringent dimerizer-regulated gene expression system in a single retroviral vector. Proc. Natl. Acad. Sci. U. S. A. 97, 13221–13226. 10.1073/pnas.230446297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoogenraad C. C.; Akhmanova A.; Howell S. A.; Dortland B. R.; De Zeeuw C. I.; Willemsen R.; Visser P.; Grosveld F.; Galjart N. (2001) Mammalian Golgi-associated Bicaudal-D2 functions in the dynein-dynactin pathway by interacting with these complexes. EMBO J. 20, 4041–4054. 10.1093/emboj/20.15.4041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bassell G. J.; Zhang H.; Byrd A. L.; Femino A. M.; Singer R. H.; Taneja K. L.; Lifshitz L. M.; Herman I. M.; Kosik K. S. (1998) Sorting of beta-actin mRNA and protein to neurites and growth cones in culture. J. Neurosci. 18, 251–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross A. F.; Oleynikov Y.; Kislauskis E. H.; Taneja K. L.; Singer R. H. (1997) Characterization of a beta-actin mRNA zipcode-binding protein. Mol. Cell. Biol. 17, 2158–2165. 10.1128/MCB.17.4.2158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donnelly C. J.; Willis D. E.; Xu M.; Tep C.; Jiang C.; Yoo S.; Schanen N. C.; Kirn-Safran C. B.; van Minnen J.; English A.; Yoon S. O.; Bassell G. J.; Twiss J. L. (2011) Limited availability of ZBP1 restricts axonal mRNA localization and nerve regeneration capacity. EMBO J. 30, 4665–4677. 10.1038/emboj.2011.347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kislauskis E. H.; Zhu X.; Singer R. H. (1994) Sequences responsible for intracellular localization of beta-actin messenger RNA also affect cell phenotype. J. Cell Biol. 127, 441–451. 10.1083/jcb.127.2.441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickens M.; Bernstein D. S.; Kimble J.; Parker R. (2002) A PUF family portrait: 3′UTR regulation as a way of life. Trends Genet. 18, 150–157. 10.1016/S0168-9525(01)02616-6. [DOI] [PubMed] [Google Scholar]
- Kapitein L. C.; Schlager M. A.; Kuijpers M.; Wulf P. S.; van Spronsen M.; MacKintosh F. C.; Hoogenraad C. C. (2010) Mixed microtubules steer dynein-driven cargo transport into dendrites. Curr. Biol. 20, 290–299. 10.1016/j.cub.2009.12.052. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.