

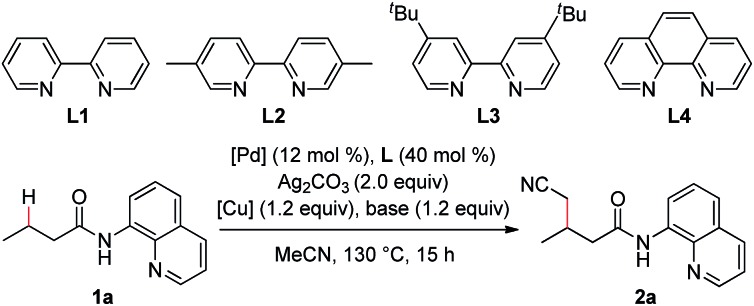
 The direct coupling of unactivated sp3 C–H bonds in aliphatic amides with acetonitrile was achieved via palladium catalysis.
The direct coupling of unactivated sp3 C–H bonds in aliphatic amides with acetonitrile was achieved via palladium catalysis.
Abstract
The direct cyanomethylation of unactivated sp3 C–H bonds of aliphatic amides was achieved via palladium catalysis assisted by a bidentate directing group with good functional group compatibility. This process represents the first example of the direct cross-coupling of sp3 C–H bonds with acetonitrile. Considering the importance of the cyano group in medicinal and synthetic organic chemistry, this reaction will find broad application in chemical research.
Introduction
The cyanomethylation of organic molecules is of great research interest to organic and medicinal chemists due to the wide presence of the cyano group in biologically active molecules and the facile conversion of the cyano group into many other functional groups, such as amides, esters, aldehydes, and primary amines.1 A variety of different synthetic strategies have been developed for the selective introduction of the cyanomethyl group.2 Among these methods, transition metal-catalyzed cross-couplings with acetonitrile as the coupling partner3 have attracted considerable attention in recent years due to the avoidance of prefunctionalized substrates such as haloacetonitrile,4 trimethylsilylacetonitrile,5 cyanoacetate salts6 and cyanomethyltributyltin.7 In 2002, Culkin and Hartwig reported the first cross-coupling reaction of acetonitrile and aryl bromides via palladium catalysis.8 In another study by You and Verkade, aryl chlorides were also demonstrated as effective substrates for this transformation.9 Furthermore, the direct cross-coupling of benzene with acetonitrile was developed with a palladium catalyst hybridized with a titanium dioxide photocatalyst.10 However, to date, the direct cross-coupling of sp3 C–H bonds with acetonitrile has not been discovered. Considering the literature support for the Pd-catalyzed alkylation of unactivated C(sp3)–H bonds11 and the reductive elimination of dialkyl palladium(ii) species,11g–i,12 it is envisaged that this process should be feasible if the cyanomethyl group could effectively replace the anion of an alkyl palladium(ii) species.
Results and discussion
In the previous reports, it was found that an alkyl arylpalladium(ii) species could be formed by the treatment of an arylpalladium(ii) species with a cyanomethyl anion in the presence of a ligand. On the basis of these results, the palladium-catalyzed direct cyanomethylation of N-(quinolin-8-yl)butyramide (1a) with acetonitrile was examined using 2,2′-bipyridine as the ligand, under basic conditions (Table 1, entry 1). Unfortunately, no desired product was observed. A copper(ii) salt was then added into the reaction system, because the copper-promoted C–H bond activation of acetonitrile13 and the transmetalation of an organocopper species onto an organopalladium(ii) species14 have been well documented. As shown in Table 1, copper carboxylates were found to be effective, with Cu(O2CnPr)2 providing the best result (entry 3). A series of mono and bidentate ligands15 were then screened, and it was found out that the reaction yield was improved with 5,5′-dimethyl-2,2′-bipyridine (L2) (entry 6). Furthermore, the effect of the palladium catalyst was examined with Pd(OPiv)2 giving the optimal result (entry 9). Further optimization showed that this reaction was significantly improved with CsOPiv as the base (entry 15). In addition, the use of acetonitrile and heptane as the co-solvent could further increase the yield (entry 18). It was also noted that the reaction yield was dramatically decreased in the absence of the ligand, indicating that the ligand plays a role in stabilizing the dialkyl palladium(ii) species or the in situ generated Pd metal (entry 20). To our delight, the reaction yield could be further improved by increasing the load of palladium catalyst (entry 21).
Table 1. Optimization of the reaction conditions a .
| |||||
| Entry | Pd source | Cu source | Base | L | Yield b (%) |
| 1 c | Pd(OAc)2 | — | NaHMDS | L1 | 0 |
| 2 | Pd(OAc)2 | Cu(OAc)2 | — | L1 | 11 |
| 3 | Pd(OAc)2 | Cu(O2CnPr)2 | — | L1 | 20 |
| 4 | Pd(OAc)2 | Cu(acac)2 | — | L1 | Trace |
| 5 | Pd(OAc)2 | CuOAc | — | L1 | 11 |
| 6 | Pd(OAc)2 | Cu(O2CnPr)2 | — | L2 | 30 |
| 7 | Pd(OAc)2 | Cu(O2CnPr)2 | — | L3 | 23 |
| 8 | Pd(OAc)2 | Cu(O2CnPr)2 | — | L4 | 21 |
| 9 | Pd(OPiv)2 | Cu(O2CnPr)2 | — | L2 | 34 |
| 10 | PdCl2 | Cu(O2CnPr)2 | — | L2 | 22 |
| 11 | Pd(MeCN)2Cl2 | Cu(O2CnPr)2 | — | L2 | 26 |
| 12 | Pd(OPiv)2 | Cu(O2CnPr)2 | K3PO4 | L2 | 25 |
| 13 | Pd(OPiv)2 | Cu(O2CnPr)2 | KOAc | L2 | 49 |
| 14 | Pd(OPiv)2 | Cu(O2CnPr)2 | KOPiv | L2 | 52 |
| 15 | Pd(OPiv)2 | Cu(O2CnPr)2 | CsOPiv | L2 | 56 |
| 16 d | Pd(OPiv)2 | Cu(O2CnPr)2 | CsOPiv | L2 | 69 |
| 17 e | Pd(OPiv)2 | Cu(O2CnPr)2 | CsOPiv | L2 | 73 |
| 18 f | Pd(OPiv)2 | Cu(O2CnPr)2 | CsOPiv | L2 | 76(72) g |
| 19 h | Pd(OPiv)2 | Cu(O2CnPr)2 | CsOPiv | L2 | 68 |
| 20 f | Pd(OPiv)2 | Cu(O2CnPr)2 | CsOPiv | — | 9 |
| 21 i | Pd(OPiv)2 | Cu(O2CnPr)2 | CsOPiv | L2 | 85(80) g |
aReaction conditions: 1a (0.3 mmol), Pd source (0.036 mmol), L (0.12 mmol), Cu source (0.36 mmol), Ag2CO3 (0.6 mmol), base (0.36 mmol), MeCN (3.0 mL), air (1 atm), 130 °C, 15 h unless other noted.
bYields are based on 1a, determined by 1H-NMR using dibromomethane as the internal standard.
cNaHMDS (1 M in THF, 1.5 mL) was used.
dMeCN/toluene (1.5 mL/1.5 mL).
eMeCN/hexane (1.5 mL/1.5 mL).
fMeCN/heptane (1.5 mL/1.5 mL).
gIsolated yield.
hMeCN/cyclohexane (1.5 mL/1.5 mL).
iPd(OPiv)2 (0.045 mmol).
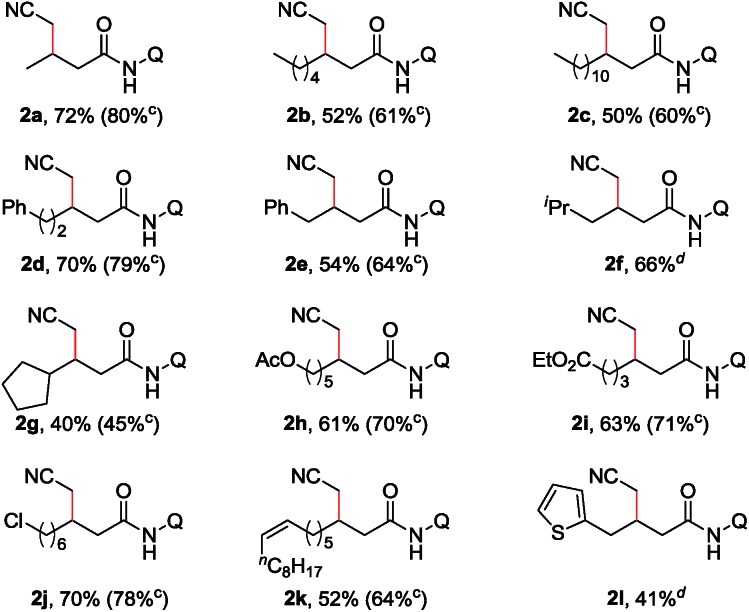
With the optimized reaction conditions in hand, a substrate scope study on linear aliphatic amides was then carried out. As shown in Table 2, the direct cyanomethylation of unbranched amides provided the desired products in moderate to good yields (2a–f). In addition, a variety of functional groups, such as the alkenyl, chloro, ester, phenyl and thienyl groups, were well tolerated under the catalytic system, allowing for the further manipulation of the original products. Furthermore, there is an apparent steric effect for this reaction because a lower yield was obtained with substrates bearing a substituent on γ-carbon (2g).
Table 2. Scope of the linear aliphatic amides a , b .
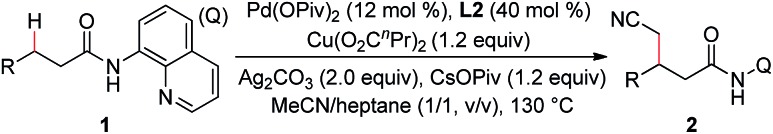
|
|
aReaction conditions: 1 (0.3 mmol), Pd(OPiv)2 (0.036 mmol), L2 (0.12 mmol), Cu(O2CnPr)2 (0.36 mmol), Ag2CO3 (0.6 mmol), CsOPiv (0.36 mmol), MeCN (1.5 mL), heptane (1.5 mL), and air (1 atm), 130 °C, 15 h.
bIsolated yield.
cPd(OPiv)2 (0.045 mmol).
dPd(OPiv)2 (0.06 mmol). Q = 8-quinolinyl.
Furthermore, the scope of α-substituted aliphatic amides was studied under the modified reaction conditions (Table 3). As expected, propanamides bearing a linear, branched, or cyclic alkyl group were shown to be effective substrates (3a–h). It is worth mentioning that this reaction showed high site-selectivity by favouring the sp3 C–H bonds of the methyl group over those of the methylene groups, including that of the relatively reactive benzylic sp3 C–H bond (3c). Furthermore, the cyclic sp3 C–H bond could also be functionalized, albeit with a moderate yield (3k). Amides with α-tertiary carbon (3l) were inappropriate substrates and could be quantitatively recovered under current conditions.
Table 3. Scope of the α-substituted aliphatic amides a , b .
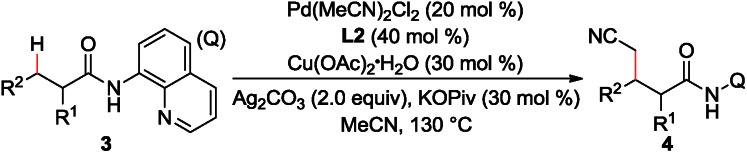
|
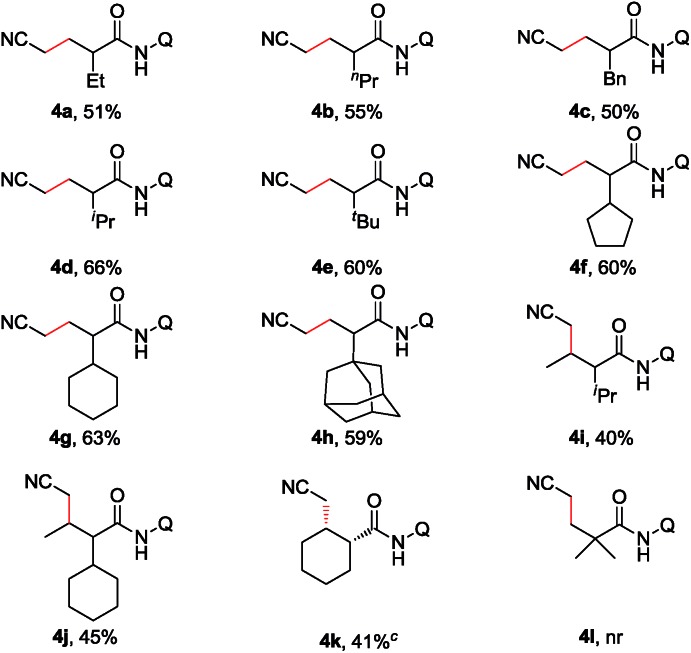
|
aReaction conditions: 1 (0.3 mmol), Pd(MeCN)2Cl2 (0.06 mmol), L2 (0.12 mmol), Cu(OAc)2·H2O (0.09 mmol), Ag2CO3 (0.6 mmol), KOPiv (0.09 mmol), MeCN (2.0 mL), air (1 atm), 130 °C, 1 h.
bIsolated yield.
cPd(OPiv)2 (0.06 mmol), L2 (0.12 mmol), Cu(O2CnPr)2 (0.36 mmol), Ag2CO3 (0.6 mmol), CsOPiv (0.36 mmol), MeCN (1.5 mL), heptane (1.5 mL), air (1 atm), 130 °C, 15 h. Q = 8-quinolinyl.
To provide some insights into the catalytic cycle, we carried out mechanistic studies into this process. It has been reported that aliphatic esters and nitriles could undergo dehydrogenation to form the corresponding α,β-unsaturated derivatives.16 Therefore, a sequential dehydrogenation/1,4-addition process could potentially occur in this reaction and could provide the desired products. To clarify this, N-(quinolin-8-yl)acrylamide (5) was prepared and subjected to the reaction conditions (Scheme 1). It turned out that no desired product (2m) was obtained, and thus the dehydrogenation/1,4-addition process could be excluded.
Scheme 1. Control experiments on the reaction mechanism. Condition A: 1m or 5 (0.3 mmol), Pd(OPiv)2 (0.036 mmol), L2 (0.12 mmol), Cu(O2CnPr)2 (0.36 mmol), Ag2CO3 (0.6 mmol), CsOPiv (0.36 mmol), MeCN (1.5 mL), heptane (1.5 mL), air (1 atm), 130 °C, 15 h. Condition B: 1m or 5 (0.3 mmol), Pd(MeCN)2Cl2 (0.06 mmol), L2 (0.12 mmol), Cu(OAc)2·H2O (0.09 mmol), Ag2CO3 (0.6 mmol), KOPiv (0.09 mmol), MeCN (2.0 mL), air (1 atm), 130 °C, 1 h.

To further probe the reaction mechanism, a series of deuterium-labelling experiments were carried out. As shown in Scheme 2, no apparent H/D exchange was observed with deuterium-labelled 2,3-dimethyl-N-(quinolin-8-yl)butanamide (D3-3d) (Scheme 2a), indicating that the sp3 C–H bond cleavage is an irreversible step under the current reaction conditions. Furthermore, no obvious kinetic isotope effect was observed for 3d (vs. D3-3d) based on the early relative rate of the parallel reactions (Scheme 2b), whereas a primary isotope effect with regard to acetonitrile (MeCN vs. CD3CN) was obtained (Scheme 2c), suggesting that the sp3 C–H bond cleavage of acetonitrile is the rate-limiting step in the catalytic process.
Scheme 2. Deuterium-labelling experiments.

On the basis of the abovementioned observations and the previous studies,11–14 a plausible reaction mechanism is proposed (Scheme 3), involving the coordination of amide 1 or 3 to a PdII species, followed by a ligand exchange process, giving rise to the palladium intermediate A. Irreversible sp3 C–H bond activation of this intermediate under basic conditions generates the cyclometalated palladium(ii) complex B. Transmetalation of the complex B with the cyanomethyl copper(ii) species, possibly from a silver-promoted process of acetonitrile, affords the dialkyl palladium intermediate C, which provides the final product 2 or 4 upon reductive elimination.
Scheme 3. Proposed reaction mechanism.

To further broaden the synthetic application of this methodology, removal of the 8-quinolylamino directing group of 4b was carried out based on the reported two-step process,11l and the C–N bond of amide was selectively cleaved to deliver the desired acid product 6b in a 65% yield without affecting the cyano group (Scheme 4).
Scheme 4. Removal of the directing group.

Conclusions
In summary, a highly regioselective cyanomethylation of aliphatic amides with an 8-aminoquinolinyl group as the directing moiety was developed via a palladium-catalyzed cross dehydrogenative coupling process. This process exhibited a predominant preference for methyl C–H bonds over methylene C–H bonds with good functional group tolerance. Mechanistic studies were carried out that excluded the possibly sequential dehydrogenation/Michael addition process. Detailed mechanistic studies of this reaction and expansion of the substrate scope17 are currently ongoing in our laboratory.
Supplementary Material
Acknowledgments
The authors gratefully acknowledge financial support for this research from NSF CHE-1350541 and Indiana University-Purdue University Indianapolis. Ke Yang is also grateful for financial support from NSFC: (No. 21332005) and the Jiangsu Innovation Programs (P. R. China).
Footnotes
†Electronic supplementary information (ESI) available: Experimental details including characterization data, copies of 1H, 13C NMR and NOESY spectra. See DOI: 10.1039/c5sc04066c
References
- For selected reviews, see: ; for a selected book, see: ; (a) Fleming F. F. Nat. Prod. Rep. 1999;16:597–606. [Google Scholar]; (b) Fleming F. F., Yao L., Ravikumar P. C., Funk L., Shook B. C. J. Med. Chem. 2010;53:7902–7917. doi: 10.1021/jm100762r. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) López R., Palomo C. Angew. Chem., Int. Ed. 2015;54:13170–13184. doi: 10.1002/anie.201502493. [DOI] [PubMed] [Google Scholar]; (d) Rappoport Z., The Chemistry of the Cyano Group, Wiley-Interscience, London, 1970. [Google Scholar]
- (a) Khanapure S. P., Biehl E. R. J. Org. Chem. 1990;55:1471–1475. [Google Scholar]; (b) Kumagai N., Matsunaga S., Shibasaki M. J. Am. Chem. Soc. 2004;126:13632–13633. doi: 10.1021/ja0450509. [DOI] [PubMed] [Google Scholar]; (c) Anxionnat B., Pardo D. G., Ricci G., Cossy J. Org. Lett. 2011;13:4084–4087. doi: 10.1021/ol2015972. [DOI] [PubMed] [Google Scholar]; (d) Velcicky J., Soicke A., Steiner R., Schmalz H. J. Am. Chem. Soc. 2011;133:6948–6951. doi: 10.1021/ja201743j. [DOI] [PubMed] [Google Scholar]; (e) Wu T., Mu X., Liu G. Angew. Chem., Int. Ed. 2011;50:12578–12581. doi: 10.1002/anie.201104575. [DOI] [PubMed] [Google Scholar]; (f) Wang G.-W., Zhou A.-X., Wang J.-J., Hu R.-B., Yang S.-D. Org. Lett. 2013;15:5270–5273. doi: 10.1021/ol402494e. [DOI] [PubMed] [Google Scholar]; (g) Chakraborty S., Patel Y. J., Krause J. A., Guan H. Angew. Chem., Int. Ed. 2013;52:7523–7526. doi: 10.1002/anie.201302613. [DOI] [PubMed] [Google Scholar]; (h) Shen J., Yang D., Liu Y., Qin S., Zhang J., Sun J., Liu C., Liu C., Zhao X., Chu C., Liu R. Org. Lett. 2014;16:350–353. doi: 10.1021/ol403555n. [DOI] [PubMed] [Google Scholar]; (i) Bunescu A., Wang Q., Zhu J. Chem.–Eur. J. 2014;20:14633–14636. doi: 10.1002/chem.201405102. [DOI] [PubMed] [Google Scholar]; (j) Lindsay-Scott P. J., Clarke A., Richardson J. Org. Lett. 2015;17:476–479. doi: 10.1021/ol503479g. [DOI] [PubMed] [Google Scholar]; (k) Mamuye A. D., Castoldi L., Azzena U., Holzera W., Pace V. Org. Biomol. Chem. 2015;13:1969–1973. doi: 10.1039/c4ob02398f. [DOI] [PubMed] [Google Scholar]
- For a selected review, see: Culkin D. A., Hartwig J. F., Acc. Chem. Res., 2003, 36 , 234 –245 . [DOI] [PubMed] [Google Scholar]
- For selected examples, see: ; (a) Frejd T., Klingstedt T. Synthesis. 1987:40–42. [Google Scholar]; (b) Nortcliffe A., Botting N. P., O'Hagan D. Org. Biomol. Chem. 2013;11:4657–4671. doi: 10.1039/c3ob41047a. [DOI] [PubMed] [Google Scholar]; (c) Ali O. M., Amr A. E.-G. E., Mostafa E. E. Res. Chem. Intermed. 2014;40:1545–1556. [Google Scholar]
- For selected examples, see: ; (a) Diaba F., Houerou C. L., Grignon-Dubois M., Gerval P. J. Org. Chem. 2000;65:907–910. [Google Scholar]; (b) Wu L., Hartwig J. F. J. Am. Chem. Soc. 2005;127:15824–15832. doi: 10.1021/ja053027x. [DOI] [PubMed] [Google Scholar]; (c) Mukaiyama T., Michida M. Chem. Lett. 2007;36:1244–1245. [Google Scholar]; (d) Verrier C., Oudeyer S., Dez I., Levacher V. Tetrahedron Lett. 2012;53:1958–1960. [Google Scholar]; (e) Fan Y.-C., Du G.-F., Sun W.-F., Kang W., He L. Tetrahedron Lett. 2012;53:2231–2233. [Google Scholar]
- (a) Shang R., Ji D.-S., Chu L., Fu Y., Liu L. Angew. Chem., Int. Ed. 2011;50:4470–4474. doi: 10.1002/anie.201006763. [DOI] [PubMed] [Google Scholar]; (b) Yeung P. Y., Chung K. H., Kwong F. Y. Org. Lett. 2011;13:2912–2915. doi: 10.1021/ol2009522. [DOI] [PubMed] [Google Scholar]
- Kosugi M., Ishiguro M., Negishi Y., Sano H., Migita T. Chem. Lett. 1984:1511–1512. [Google Scholar]
- Culkin D. A., Hartwig J. F. J. Am. Chem. Soc. 2002;124:9330–9331. doi: 10.1021/ja026584h. [DOI] [PubMed] [Google Scholar]
- (a) You J., Verkade J. G. Angew. Chem., Int. Ed. 2003;42:5051–5053. doi: 10.1002/anie.200351954. [DOI] [PubMed] [Google Scholar]; (b) You J., Verkade J. G. J. Org. Chem. 2003;68:8003–8007. doi: 10.1021/jo034779h. [DOI] [PubMed] [Google Scholar]
- Yoshida H., Fujimura Y., Yuzawa H., Kumagai J., Yoshida T. Chem. Commun. 2013;49:3793–3795. doi: 10.1039/c3cc41068d. [DOI] [PubMed] [Google Scholar]
- For selected reviews, see: ; for selected examples, see: ; (a) Chen X., Engle K. M., Wang D.-H., Yu J.-Q. Angew. Chem., Int. Ed. 2009;48:5094–5115. doi: 10.1002/anie.200806273. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Lyons T. W., Sanford M. S. Chem. Rev. 2010;110:1147–1169. doi: 10.1021/cr900184e. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Li H., Lia B.-J., Shi Z.-J. Catal. Sci. Technol. 2011;1:191–206. [Google Scholar]; (d) Rouquet G., Chatani N. Angew. Chem., Int. Ed. 2013;52:11726–11743. doi: 10.1002/anie.201301451. [DOI] [PubMed] [Google Scholar]; (e) Daugulis O., Roane J., Tran L. D. Acc. Chem. Res. 2015;48:1053–1064. doi: 10.1021/ar5004626. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Chen Z., Wang B., Zhang J., Yu W., Liu Z., Zhang Y. Org. Chem. Front. 2015;2:1107–1295. [Google Scholar]; (g) Chen X., Goodhue C. E., Yu J.-Q. J. Am. Chem. Soc. 2006;128:12634–12635. doi: 10.1021/ja0646747. [DOI] [PubMed] [Google Scholar]; (h) Wang D.-H., Wasa M., Giri R., Yu J.-Q. J. Am. Chem. Soc. 2008;130:7190–7191. doi: 10.1021/ja801355s. [DOI] [PubMed] [Google Scholar]; (i) Shi B.-F., Maugel N., Zhang Y.-H., Yu J.-Q. Angew. Chem., Int. Ed. 2008;47:4882–4886. doi: 10.1002/anie.200801030. [DOI] [PubMed] [Google Scholar]; (j) Tran L. D., Daugulis O. Angew. Chem., Int. Ed. 2012;51:5188–5191. doi: 10.1002/anie.201200731. [DOI] [PMC free article] [PubMed] [Google Scholar]; (k) Zhang S.-Y., He G., Nack W. A., Zhao Y.-S., Li Q., Chen G. J. Am. Chem. Soc. 2013;135:2124–2127. doi: 10.1021/ja312277g. [DOI] [PubMed] [Google Scholar]; (l) Zhang S.-Y., Li Q., He G., Nack W. A., Chen G. J. Am. Chem. Soc. 2013;135:12135–12141. doi: 10.1021/ja406484v. [DOI] [PubMed] [Google Scholar]; (m) Chen K., Hu F., Zhang S.-Q., Shi B.-F. Chem. Sci. 2013;4:3906–3911. [Google Scholar]; (n) Nadres E. T., Santos G. I. F., Shabashov D., Daugulis O. J. Org. Chem. 2013;78:9689–9714. doi: 10.1021/jo4013628. [DOI] [PMC free article] [PubMed] [Google Scholar]; (o) Chen K., Shi B.-F. Angew. Chem., Int. Ed. 2014;53:11950–11954. doi: 10.1002/anie.201407848. [DOI] [PubMed] [Google Scholar]; (p) Zhu R.-Y., He J., Wang X.-C., Yu J.-Q. J. Am. Chem. Soc. 2014;136:13194–13197. doi: 10.1021/ja508165a. [DOI] [PMC free article] [PubMed] [Google Scholar]; (q) Wang B., Wu X., Jiao R., Zhang S.-Y., Nack W. A., He G., Chen G. Org. Chem. Front. 2015;2:1318–1321. [Google Scholar]
- For selected examples, see: ; (a) Netherton M. R., Dai C., Neuschütz K., Fu G. C. J. Am. Chem. Soc. 2001;123:10099–10100. doi: 10.1021/ja011306o. [DOI] [PubMed] [Google Scholar]; (b) Kirchhoff J. H., Dai C., Fu G. C. Angew. Chem., Int. Ed. 2002;41:1945–1947. [PubMed] [Google Scholar]; (c) Kirchhoff J. H., Netherton M. R., Hills I. D., Fu G. C. J. Am. Chem. Soc. 2002;124:13662–13663. doi: 10.1021/ja0283899. [DOI] [PubMed] [Google Scholar]; (d) Zhou J., Fu G. C. J. Am. Chem. Soc. 2003;125:12527–12530. doi: 10.1021/ja0363258. [DOI] [PubMed] [Google Scholar]
- For selected examples, see: ; (a) Suto Y., Kumagai N., Matsunaga S., Kanai M., Shibasaki M. Org. Lett. 2003;5:3147–3150. doi: 10.1021/ol035206u. [DOI] [PubMed] [Google Scholar]; (b) Bunescu A., Wang Q., Zhu J. Angew. Chem., Int. Ed. 2015;54:3132–3135. doi: 10.1002/anie.201411657. [DOI] [PubMed] [Google Scholar]; (c) Chatalova-Sazepin C., Wang Q., Sammis G. M., Zhu J. Angew. Chem., Int. Ed. 2015;54:5443–5446. doi: 10.1002/anie.201412357. [DOI] [PubMed] [Google Scholar]; (d) Suto Y., Tsuji R., Kanai M., Shibasaki M. Org. Lett. 2005;7:3757–3760. doi: 10.1021/ol051423e. [DOI] [PubMed] [Google Scholar]
- For selected reviews, see: ; for selected examples, see: ; (a) Espinet P., Echavarren A. M. Angew. Chem., Int. Ed. 2004;43:4704–4734. doi: 10.1002/anie.200300638. [DOI] [PubMed] [Google Scholar]; (b) Cordovilla C., Bartolomé C., Martínez-Ilarduya J. M., Espinet P. ACS Catal. 2015;5:3040–3053. [Google Scholar]; (c) Farina V., Kapadia S., Krishnan B., Wang C., Liebeskind L. S. J. Org. Chem. 1994;59:5905–5911. [Google Scholar]; (d) Eckhardt M., Fu G. C. J. Am. Chem. Soc. 2003;125:13642–13643. doi: 10.1021/ja038177r. [DOI] [PubMed] [Google Scholar]; (e) Mee S. P. H., Lee V., Baldwin J. E. Angew. Chem., Int. Ed. 2004;43:1132–1136. doi: 10.1002/anie.200352979. [DOI] [PubMed] [Google Scholar]
- With monodentate ligands (80 mol%), such as pyridine, 2,6-lutidine and quinolone, the reactions give only trace amount of product 2a under similar conditions as entry 7 in Table 1
- Chen Y., Romaire J. P., Newhouse T. R. J. Am. Chem. Soc. 2015;137:5875–5878. doi: 10.1021/jacs.5b02243. [DOI] [PubMed] [Google Scholar]
- Attempt to use higher nitriles with secondary α-carbon such as propionitrile, butyronitrile and benzyl cyanide as coupling partners was unsuccessful under current conditions. Besides, the coupling of sp2 C–H of benzamides with CH3CN using 8-quinolylamino as the directing group was not achieved under similar conditions
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.