

Abstract
Nanoscale metal–organic frameworks (nMOF) materials represent an attractive tool for various biomedical applications. Due to the chemical versatility, enormous porosity, and tunable degradability of nMOFs, they have been adopted as carriers for delivery of imaging and/or therapeutic cargos. However, the relatively low stability of most nMOFs has limited practical in vivo applications. Here we report the production and characterization of an intrinsically radioactive UiO-66 nMOF (89Zr-UiO-66) with incorporation of positron-emitting isotope zirconium-89 (89Zr). 89Zr-UiO-66 was further functionalized with pyrene-derived polyethylene glycol (Py–PGA-PEG) and conjugated with a peptide ligand (F3) to nucleolin for targeting of triple-negative breast tumors. Doxorubicin (DOX) was loaded onto UiO-66 with a relatively high loading capacity (1 mg DOX/mg UiO-66) and served as both a therapeutic cargo and a fluorescence visualizer in this study. Functionalized 89Zr-UiO-66 demonstrated strong radiochemical and material stability in different biological media. Based on the findings from cellular targeting and in vivo positron emission tomography (PET) imaging, we can conclude that 89Zr-UiO-66/Py–PGA-PEG-F3 can serve as an image-guidable, tumor-selective cargo delivery nanoplatform. In addition, toxicity evaluation confirmed that properly PEGylated UiO-66 did not impose acute or chronic toxicity to the test subjects. With selective targeting of nucleolin on both tumor vasculature and tumor cells, this intrinsically radioactive nMOF can find broad application in cancer theranostics.
Keywords: metal–organic frameworks (MOF), positron emission tomography, intrinsic radiolabel, nucleolin, cancer targeting, image-guided drug delivery
Graphical Abstract
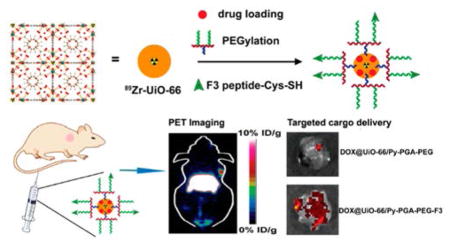
As an emerging category of multifunctional materials, metal–organic frameworks (MOF, also known as porous coordination polymers) are built from metal ions/clusters bridged by organic linkers (via coordination bonds).1 They have been utilized in a variety of applications, such as molecule (particularly in a gaseous form) storage,2,3 separation,4 catalysis,5,6 sensing,7 among many others. Compared with other conventional nanomaterials, nanoscale MOF (nMOF) possesses multiple advantages: superior structure and component tunability (allowing for production of nMOFs with different shapes, sizes, and chemical properties),8 more adaptable degradability, and better chemical modification capability.9 These obvious benefits permit nMOFs to be used as attractive cancer theranostic tools and to be loaded with various therapeutic (drug) and imaging cargos.10,11
There are plenty of studies involving the utilization of nMOF materials as in vivo cancer treatment agents. For example, a hafnium-chlorin based nMOF was reported to possess good photodynamic therapy (PDT) efficacy of colon cancer.12 More recently, hafnium-tetrakis (4-carboxyphenyl) porphyrin-based nMOF was PEGylated and used successfully for combinational PDT and radiation therapy in a murine breast cancer model.13 The optimal cargo-loading capacity of nMOFs allows them to be used for combinational therapies in different cancer types. Various therapeutic combinations, such as small interfering RNA (siRNA)/chemo-drug,14 dual chemotherapy drugs (e.g., gemcitabine and oxaliplatin),15 or chemotherapy drug/PDT,16 have been attempted for in vivo tumor therapies with encouraging data obtained. On the other hand, although compelling evidence has been collected to prove that nMOFs can be readily used in multiple imaging techniques, such as computed tomography (CT),17 magnetic resonance imaging (MRI),18,19 or optical imaging,20 very limited studies were carried out to date using nMOFs as in vivo tumor imaging agents.21–23
Compared with other imaging techniques, positron emission tomography (PET) imaging has superior detection sensitivity (down to picomolar range), deeper signal penetration (especially when compared with optical imaging techniques), and better quantitative capacity,24 thus gaining more widespread use in both preclinical and clinical scenarios. Here, our goal is to develop a biocompatible nMOF platform applicable in PET imaging and tumor targeting, preparing it for future PET-guided cargo delivery to cancer. From the current library of nMOFs, zirconium-containing UiO-66 nMOF (with 1,4-benzene-dicarboxylate (BDC) and benzoic acid (BA) as bridging linkers) was selected as a template material since they are well-known for their optimal surface areas, along with a linker independent, exceptional stability.25,26 Moreover, due to the existing Zr6O4(OH)4 connecting clusters,27 PET isotope zirconium-89 (89Zr, t1/2 = 78.4 h)28 can be seamlessly incorporated into UiO-66 structure during the synthesis (Scheme 1). Surface engineering with pyrene-derived poly-ethylene glycol (PEG)29 could not only improve the stability and dispersity of UiO-66 in biological media,30 but also provide further functionalization sites for integration of tumor-targeting molecules. At the same time, the highly porous structure of UiO-66 enables good accommodation of both hydrophilic and hydrophobic drugs, and their sustained (up to 30 days), pH-dependent drug release profile has been revealed.27,31
Scheme 1.

(a) Schematic Synthesis Route of 89Zr-UiO-66/Py–PGA-PEG-F3 Conjugates; (b) Schematic Crystal Structure of 89Zr-UiO-66
F3 peptide (KDEPQRRSARLSAKPAPPKPEPKPKKAPAKK) was chosen as tumor-targeting ligand in this study, since it exhibited potent binding to both tumor cells and activated endothelial cells in tumor vasculatures.32 One previous study has demonstrated that F3 peptide selectively binds to nucleolin on tumor (and activated endothelial) cell surfaces and can be subsequently transported into nucleus and cytoplasm.33 Elevated expression of nucleolin on cell surface is an important signature for various cancer cells, especially breast cancer.34 A cysteine residue was incorporated into the C-terminus of F3 peptide to facilitate its conjugation onto UiO-66 nMOF (via thiol maleimide reaction with pyrene-PEG-maleimide).35
After attachment of F3 onto UiO-66 (final conjugates named UiO-66/Py–PGA-PEG-F3), in vitro assays (e.g., flow cytometry, confocal fluorescence microscopy) were carried out to validate the targeting specificity of UiO-66/Py–PGA-PEG-F3 against cellular nucleolin. Subsequently, in vivo PET imaging, organ distribution, and histology studies were performed in mice bearing orthotopic MDA-MB-231 tumors to confirm tumor targeting capacity of UiO-66/Py–PGA-PEG-F3. Doxorubicin (DOX) was loaded onto UiO-66 conjugates to serve as both a model anticancer drug and a fluorophore to define the location of these nanoconjugates. A proof-of-principle in vivo drug delivery study was also executed to validate the enhanced tumor-targeted delivery efficiency of DOX post intravenous injection of DOX-loaded UiO-66 conjugates. With proper functionalization from Py–PGA-PEG, these UiO-66 conjugates did not impose acute or chronic toxicity to Balb/c mice, validated by both histological staining and serum biochemical assays. Deduced from these experimental data, intrinsically radioactive UiO-66 conjugates showed great potential to be used for future image-guided therapeutic delivery and targeted cancer therapy.
RESULTS AND DISCUSSION
Material Characterization
As-synthesized UiO-66 nMOFs were composed of octahedron-shape nanocrystals with the individual size of 50–90 nm based on the transmission electron microscopy (TEM) measurement (Figures 1a and S1) and scanning electron microscopy (SEM) images (Figure S2), consistent with the previous report.27 Powder X-ray diffraction (PXRD) analysis confirmed that UiO-66 type nMOF was successfully acquired (Figure 1e). The size of UiO-66 aggregates (most stable form in aqueous solution) increased slightly after surface modifications with poly(γ-glutamic acid)-pyrene(30%)-poly(ethylene glycol)-maleimide (Py–PGA-PEG-Mal) and F3 peptide, from ca. 220 nm (UiO-66) to ca. 250 nm (UiO-66/Py–PGA-PEG-F3) (Figures 1a and S1). The successful surface engineering was further validated in dynamic light scattering (DLS) and ζ-potential measurements, shown in Figure 1b and Table S1. Compared with as-synthesized UiO-66, significant change of surface charge was observed after Py–PGA-PEG-Mal coating (ζ-potential: from 37.6 ± 0.5 mV to −5.18 ± 0.05 mV) and also after attachment of the F3 peptide (ζ-potential: from −5.18 ± 0.05 mV to −0.9 ± 0.4 mV).
Figure 1.

Characterization of Py–PGA-PEG functionalized UiO-66 nMOFs. (a) Representative TEM images of UiO-66, UiO-66/Py–PGA-PEG-F3, and DOX@UiO-66/Py–PGA-PEG-F3. Scale bar: 50 nm. (b) DLS measurement of UiO-66, UiO-66/Py–PGA-PEG-F3, and DOX@ UiO-66/Py–PGA-PEG-F3. (c) The correlation of UiO-66 concentration with radioactivity concentration obtained from PET for 89Zr-UiO-66/Py–PGA-PEG-F3. (d) Stability evaluation of 89Zr-UiO-66/Py–PGA-PEG-F3 in various media. (e) PXRD analysis of UiO-66 conjugates over different pH values, post DOX loading, or post serum incubation (for 5 days).
In addition, FT-IR and 1H NMR spectra of UiO-66/Py–PGA-PEG-F3 were also recorded to evaluate the PEGylation level (Figures S3 and S4). Characteristic IR absorbance peaks from UiO-66 (at ~1370 cm−1)36 and Py–PGA-PEG (two major peaks at ~1200 cm−1 and ~1050 cm−1 for C–C and CO stretching) confirmed the successful Py–PGA-PEG conjugation with UiO-66 (Figure S3), consistent with 1H NMR findings (Figure S4). Calculated from thermogravimetric analysis (TGA) results of UiO-66/Py–PGA-PEG-F3 (Figure S5), there is ~1.3 mg of Py–PGA-PEG-F3 loaded per mg of UiO-66. The UiO-66/Py–PGA-PEG-F3 conjugates could be well dispersed in physiological media (water, saline, Dulbecco’s modified eagle medium [DMEM] and fetal bovine serum [FBS]) without observable aggregation for up to 2 weeks, while significant amount of precipitates could be seen in naked UiO-66 distributed in saline within 2 days (Figures S6–S8), which further confirmed the successful PEGylation. The hydrodynamic diameter of UiO-66/Py–PGA-PEG-F3 was found to be 255 ± 10 nm (Figure 1b).
With the incorporation of 89Zr into UiO-66 structure, the specific activity of 89Zr-UiO-66 could reach up to ~13.9 MBq/mg, and the decay-corrected radiochemical yield was 85.7 ± 6.2% (n = 3). A phantom study was performed during PET imaging to examine the detectability of 89Zr-UiO-66/Py–PGA-PEG in PET. PET image of a 96-well plate containing various concentrations of 89Zr-UiO-66/Py–PGA-PEG (0–20 μg/mL, 100 μL per well) was shown in Figure 1c. The good linear correlation between UiO-66 concentration and calculated radioactivity concentration confirmed that quantification data acquired from PET truly reflected the amount of 89Zr-UiO-66/Py–PGA-PEG conjugates.
Stability Evaluation
Serum stability analysis was conducted to evaluate both stability of radioisotope on UiO-66 and the structural integrity of UiO-66/Py–PGA-PEG-F3. More than 99.99% of 89Zr was found to stay intact on 89Zr-UiO-66/Py–PGA-PEG-F3 after 120 h of incubation with full mouse serum (Figure 1d). At the same time, no significant morphological alteration of UiO-66/Py–PGA-PEG-F3 was identified post fetal bovine serum (FBS) incubation based on DLS measurements (Figure S7). Since PET imaging localizes the radiolabel (herein 89Zr) instead of UiO-66 conjugates, excellent stability is the prerequisite that the signal from PET accurately reflects the distribution profile of UiO-66 conjugates.
To fully prepare a nanomaterial for in vivo applications, proper functionalization is critical to optimize its performance inside the test subjects. We chose Py–PGA-PEG as a coating/functionalization agent for UiO-66 since it can establish strong π–π interactions with the bridging organic molecules (e.g., BDC and BA) inside UiO-66. Although certain ligands were reported to coordinate with metal ions inside nMOF structure,37,38 we feel that this may not be the case for Py–PGA-PEG. Judging from the structure, Py–PGA-PEG(-F3) is not a strong Lewis base to coordinate with metal ions (Zr) inside the UiO-66, especially when the majority of carboxyl groups on the PGA backbone have already been reacted with amine groups on PEG. The abundant pyrene rings on Py–PGA-PEG(-F3) makes π–π stacking more reasonable for PEG functionalization. One thing we have to point out here is that some Py–PGA-PEG(-F3) can enter and occupy the pores (at least partially) of UiO-66 instead of just serving as “brushes” on the external surface, since it is molecularly flexible. This can be deduced from the high amount of Py–PGA-PEG(-F3) loaded onto UiO-66 (1.3 mg PEG/mg UiO-66 from TGA analysis). To make the PEGylation process more controllable, one possible solution is to use covalent modification method instead of π–π interaction. For example, functional groups such as azides can be introduced on BDC during UiO-66 synthesis to undertake “click chemistry” with dibenzylcyclooctyne (DBCO)-modified PEG molecules.30
PXRD analyses revealed that, after PEGylation with Py–PGA-PEG, the structural integrity of UiO-66 was maintained post various pH treatment (5.0 and 7.4), serum treatment, or cargo (DOX) loading (Figure 1e). Although UiO-66 nMOFs have already been used as drug carrier27,31 and imaging contrast agent,39 no functionalization has been done for this type of nMOF in these studies since no in vivo administration was involved. Our results confirmed from different angles that UiO-66/Py–PGA-PEG conjugates maintained adequate stability in various biological media, which encouraged us to proceed and study their distribution profiles in living animals.
DOX Loading and Release
UiO-66 nMOF possesses relatively high specific surface areas of 988.625 m2/g, as measured by nitrogen adsorption–desorption isotherm (Figure S9), which confirms that it is suitable for loading of various cargos. The surface area decreases to 438.978 m2/g post functionalization by Py–PGA-PEG-F3 (Figure S9). The amount of DOX loadable into UiO-66 was calculated to be 1 mg DOX/mg UiO-66 based on the absorbance measurement at 488 nm for DOX. The DOX release profile from DOX@UiO-66/Py–PGA-PEG-F3 was tested under simulated physiological condition with the pH of 5.0, 6.8, and 7.4 at 37 °C. As shown in Figure 2c, medium pH imposes an impact on the release rate of DOX from UiO-66 conjugates. At the pH of 7.4, 27.73% of DOX (0.27 mg DOX/mg UiO-66) could be released after 2 weeks, which suggested that loaded DOX within UiO-66 structure was relatively stable under physiological conditions. When exposed to the pH of 6.8, which mimics extracellular pH in a tumor, the release percentage of DOX was elevated to 32.65% (0.33 mg DOX/mg UiO-66) at the same time frame. When media pH was further decreased to 5.0 (mimics endocytic compartments where the pH ranges from 4.5 to 6.5.40), the amount of released DOX increased to approximately 37.06% (0.37 mg DOX/mg UiO-66) after 2 weeks. These numbers confirmed that the DOX release from DOX@ UiO-66/Py–PGA-PEG-F3 was sustained and pH-dependent, which is consistent with a recent report.22
Figure 2.

In vitro evaluation of UiO-66/Py–PGA-PEG conjugates. (a) Representative confocal fluorescence microscopy images of MDA-MB-231 (nucleolin+) and L929 cells (nucleolin−) incubated with DOX@UiO-66/Py–PGA-PEG-F3 and DOX@UiO-66/Py–PGA-PEG (both containing 50 μg/mL of DOX). Scale bar: 20 μm. (b) Flow cytometry analysis of DOX@UiO-66/Py–PGA-PEG-F3 and DOX@UiO-66/Py–PGA-PEG in MDA-MB-231 and L929 cells (incubation time: 0.5 h). (c) DOX release profile from UiO-66/Py–PGA-PEG-F3 at pH of 5.0, 6.8, and 7.4. (d) Cell viability of MDA-MB-231 cells incubated with different concentration of DOX@UiO-66/Py–PGA-PEG-F3, DOX@UiO-66/Py–PGA-PEG, and Py–PGA-PEG-F3, respectively.
One critical question here is the loading position of DOX on UiO-66 conjugates. The molecular size of DOX is 15.3 × 11.9 Å,19 which seems to be very difficult to fit in the micropores of UiO-66 (~11 Å octahedral and ~8 Å tetrahedral).27 However, a relatively high DOX loading capacity (1 mg DOX/mg UiO-66) was still achieved in this study. The successful DOX loading can be also confirmed by a significant decrease in surface area for DOX@UiO-66/Py–PGA-PEG-F3 (17.4975 m2/g, Figure S9). The π–π interaction between DOX and organic linkers (e.g., BDC) in UiO-66 can partially contribute to this good loading efficiency. Zhao et al. revealed that in a core–shell Fe3O4@UiO-66 nanoplatform, DOX loading capacity increased over the thickness of UiO-66 shell.22 Their observation suggested that DOX may not merely stay on the external surface of UiO-66. One possibility is that DOX can undergo some molecular distortion to enter the UiO-66 pores, but further experimental evidence will be needed to prove this point, which will be one of our future research goals.
At the same time, we have to admit, although UiO-66/Py–PGA-PEG-F3 can partially prevent premature release of DOX in the bloodstream (pH 7.4), and act as a continuous drug release source to kill cancer cells once they arrive at the extracellular region of tumors, the cargo releasing speed/efficiency needs to be further improved. Also, ~ 19% of loaded DOX was released within 30 min postincubation (all pH groups). This initial “burst” release, possibly due to the leakage of DOX from the external surface of UiO-66 (more susceptible to environment changes), can be disadvantageous for a drug delivery platform. To solve this dilemma, a more protective “capping” system (e.g., with lipids or other stimuli-responsive polymers) can be necessary to further prevent the premature DOX release, as used in other porous nanomaterials or other types of nMOF.41,42
In Vitro Tumor Cell Targeting
Two cell lines were used for in vitro evaluation of UiO-66 conjugates: MDA-MB-231 triple-negative breast cancer cells (nucleolin+) and L929 fibroblasts (nucleolin−). Western blot was conducted to confirm the expression profiles of nucleolin in these two cells (Figure S10). Here DOX loaded onto UiO-66/Py–PGA-PEG-F3 or UiO-66/Py–PGA-PEG served as a fluorophore to indicate the location of these nanoconjugates (Figure 1d). As demonstrated in both flow cytometry and confocal microscopy examination, the fluorescence intensity from DOX@UiO-66/Py–PGA-PEG-F3 was substantially stronger than that from DOX@UiO-66/Py–PGA-PEG in MDA-MB-231 cells, and fluorescence from both UiO-66 conjugates distributed primarily in the cytoplasm (Figure 2a,b). On the contrary, both DOX@ UiO-66/Py–PGA-PEG-F3 and DOX@UiO-66/Py–PGA-PEG exhibited very minimal nonspecific binding with L929 cells (Figure 2a,b). In order to track the fate of UiO-66/Py–PGA-PEG-F3 after internalization by MDA-MB-231 cells, cellular lysosomes were stained by the lysotracker (Thermo-Fisher) and examined under microscopy. Since the fluorescence spectrum from DOX and lysotracker is not separable in the fluorescence microscope, fluorescein isothiocyanate (FITC) was used as fluorescence cargo here instead of DOX. As shown in Figure S11, the distribution pattern of lysotracker is well overlapped with that from FITC, indicating that the majority of the UiO-66/Py–PGA-PEG-F3 entered lysosomes after cellular internalization. UiO-66/Py–PGA-PEG-F3 alone did not show obvious cytotoxicity to MDA-MB-231 cells at the tested concentration range (0–50 μg/mL), while both of DOX@ UiO-66/Py–PGA-PEG and DOX@UiO-66/Py–PGA-PEG-F3 inhibited the growth of MDA-MB-231 cells (Figure 2d), from the release of DOX. DOX@UiO-66/Py–PGA-PEG-F3 possessed stronger inhibition effects to MDA-MB-231 cells than that from DOX@UiO-66/Py–PGA-PEG.
To further clarify the dynamic interactions between MDA-MB-231 cells and UiO-66 conjugates, 89Zr-UiO-66/Py–PGA-PEG-F3 was incubated with MDA-MB-231 cells. As revealed in Figure S12, the cell internalization of 89Zr-UiO-66/Py–PGA-PEG-F3 plateaued within 0.25 h, and there was almost equal amount of 89Zr-UiO-66/Py–PGA-PEG-F3 binding to the surface of MDA-MB-231 cells without internalization. In the meantime, once internalized into the cells, less than 30% of 89Zr-UiO-66/Py–PGA-PEG-F3 can escape within the time frame of 24 h. These facts again confirmed specific binding between UiO-66/Py–PGA-PEG-F3 and MDA-MB-231 (likely via nucleolin on the cell surface), which warranted its further in vivo investigation.
In Vivo Tumor Targeting and PET Imaging
Taking advantage of the long half-life from 89Zr, we can fully examine the distribution/clearance profile of 89Zr-UiO-66 conjugates for up to 120 h postinjection (p.i.). Although 89Zr has not been approved by US Food and Drug Administration (FDA) for clinical diagnostic utilization, it is an isotope with good clinical potential and suitable for long-term monitoring of different metabolic processes. To date, there are 11 active, recruiting, or completed clinical trials (search from clinicaltrials.gov) using 89Zr-labeled antibodies.43 The time points of 0.5, 2, 20, 24, 48, 72, 96, and 120 h p.i. were chosen for serial PET scans and the coronal slides containing the center of MDA-MB-231 tumor are shown in Figures 3 and S13. Simultaneously, we used serial blood sampling method in nude mice (n = 4) to determine that the circulation half-life of 89Zr-UiO-66/Py–PGA-PEG is around 118.8 min (Figure S14). Here the relatively short circulation time of these 89Zr-UiO-66/Py–PGA-PEG conjugates again indicated that surface PEG density on UiO-66 may not be very high by using Py–PGA-PEG functionalization via π–π interaction, thus more controllable and efficient surface engineering method of UiO-66 is under our current investigation.
Figure 3.

Representative coronal PET images of MDA-MB-231 tumor bearing mice at different time points postinjection of 89Zr-UiO-66/Py–PGA-PEG-F3, 89Zr-UiO-66/Py–PGA-PEG, and 89Zr-UiO-66/Py–PGA-PEG-F3 with excessive amount of F3 peptide blocking. The location of tumors was identified by dashed circles.
Quantitative organ distribution data obtained from region-of-interest (ROI) analysis of PET results are shown in Figure 4 and Table S2. Most importantly, 89Zr-UiO-66/Py–PGA-PEG-F3 demonstrated a fast accumulation in MDA-MB-231 tumors, which was clearly visible at 0.5 h p.i. (7.4 ± 0.9%ID/g, n = 4) and kept stable at 2 h p.i. (8.2 ± 0.3%ID/g, n = 4). In contrast, tumor uptake of 89Zr-UiO-66/Py–PGA-PEG without the attachment of F3 peptide was found to be significantly lower than that of 89Zr-UiO-66/Py–PGA-PEG-F3 at all time points examined (n = 4, Figures 3, 4a,b, and Table S2). At late time points (>48 h p.i.), the uptake of 89Zr-UiO-66/Py–PGA-PEG-F3 in MDA-MB-231 tumors gradually decreased over time. We suspected that the decreased uptake of 89Zr-UiO-66/Py–PGA-PEG-F3 in MDA-MB-231 tumors over the time was caused by two major factors. First, the size of 89Zr-UiO-66/Py–PGA-PEG-F3 (~250 nm in diameter) is relatively large, which can prevent some 89Zr-UiO-66/Py–PGA-PEG-F3 from effective internalization into tumor (vasculature) cells, resulting in their “wash-out” from MDA-MB-231 tumors, as also evidenced in Figure S12a. Thus, smaller and more uniform UiO-66 nMOF will be more desirable and will be investigated in vivo for our future studies. Second, the efflux rate of 89Zr-UiO-66/Py–PGA-PEG-F3 (~30% within 24 h, Figure S12b) can also contribute to the reduction of tumor accumulation. The interactions between 89Zr-UiO-66/Py–PGA-PEG-F3 and proteins in charge of drug/cargo removal44 need to be elucidated to diminish the loss of UiO-66 conjugates from tumors. We believe that the decreased tumor uptake over the time is not caused by degradation of 89Zr-UiO-66 nMOF since we have evidence that it is relatively stable over different pH (down to 5.0) and serum treatment (Figures 1e and S7) within 5 days (same as the PET imaging time).
Figure 4.

Quantitative region-of-interest (ROI) analysis of PET data and organ distribution data. Time-activity curves of the liver, MDA-MB-231 tumor, blood, and muscle were shown postinjection of (a) 89Zr-UiO-66/Py–PGA-PEG-F3, (b) 89Zr-UiO-66/Py–PGA-PEG, or (c) 89Zr-UiO-66/Py–PGA-PEG-F3 + F3 peptide blocking. (d) Organ distribution data obtained by tissue gamma-counting were also given at and 120 h postinjection of 89Zr-UiO-66 conjugates. ** P < 0.01.
To further confirm the in vivo nucleolin specificity of 89Zr-UiO-66/Py–PGA-PEG-F3, blocking study with excessive amount of F3 peptide was adopted. It was found that administration of a blocking dose (~10 mg/kg) of F3 peptide at 5 min before 89Zr-UiO-66/Py–PGA-PEG-F3 injection could cause a substantial long-term tumor uptake reduction to 89Zr-UiO-66/Py–PGA-PEG-F3 (n = 3, Figure 4c and Table S2), demonstrating that nucleolin targeting may be one of the leading reasons for the enhanced tumor uptake of 89Zr-UiO-66/Py–PGA-PEG-F3 in vivo. Radioactivity captured in other major organs (e.g., liver, blood etc.) was not significantly affected by the blocking dose of F3 peptide, as shown in Figure 4. Based on the uptake of 89Zr-UiO-66 conjugates in MDA-MB-231 tumors from all three groups (targeted, untargeted, and blocking) over time, 89Zr-UiO-66/Py–PGA-PEG-F3 shows the highest tumor uptake throughout the study period (P < 0.01 in all cases, n = 4). Good image contrast for tumors can be obtained since the tumor-to-muscle ratio for 89Zr-UiO-66/Py–PGA-PEG-F3 can reach as high as 76.3 ± 2.9 at 2 h postinjection (n = 4).
From PET images, liver and spleen are the organs with the most significant capture of 89Zr-UiO-66 conjugates, and their uptakes are similar in all three groups. 89Zr-UiO-66 conjugates have slow clearance from the liver, as liver uptake decrease gradually over time from ~50%ID/g at 0.5 h p.i. to ~40%ID/g at 120 h p.i. No radioactivity deposition was found in the bones or kidneys during the whole imaging time frame, which once again confirmed the superior in vivo stability of 89Zr-UiO-66 conjugates, since 89Zr4+ ion has a strong affinity for phosphate and is readily incorporated into hydroxyapatite structures in bone.45 Overall, except for differences in tumor uptake, conjugation of F3 peptide or administration of a blocking dose of F3 peptide did not change the in vivo kinetics of these UiO-66 conjugates in breast tumor-bearing mice, which indicated that nucleolin targeting is one of the most crucial contributing factors behind the enhanced tumor uptake of 89Zr-UiO-66/Py–PGA-PEG-F3.
Ex Vivo Organ Distribution Studies
Ex vivo organ distribution data of the 89Zr-UiO-66 conjugates at 120 h p.i. (after the last PET scans) are shown in Figure 4d, which matched very well with the PET data. The majority of 89Zr-UiO-66 conjugates was retained in liver and spleen, with no noticeable uptake in kidneys or bone (<2%ID/g). Aside from these organs that are responsible for clearance (e.g., liver or kidney), the tumor uptake (1.5 ± 0.4%ID/g, n = 4) of 89Zr-UiO-66/Py–PGA-PEG-F3 was still higher than all other major organs, thus providing good tumor contrast with a tumor/muscle (T/M) ratio of 19.6 ± 5.4 at 120 h p.i. (n = 4). In comparison, mice injected with 89Zr-UiO-66/Py–PGA-PEG-F3 + F3 blocking demonstrated similar organ distribution pattern except significantly decreased uptake in tumor (T/M ratio: 6.1 ± 2.5, n = 4). Overall, the PET and organ distribution data confirmed nucleolin-targeting specificity of 89Zr-UiO-66/Py–PGA-PEG-F3 in MDA-MB-231 tumors in vivo, and these corroborating results validated that serial PET imaging truly reflected the distribution pattern of UiO-66 conjugates in tumor-bearing mice.
Safety/Toxicity Evaluation of UiO-66 Conjugates
No noticeable signs of toxicity or side effects from the injection of UiO-66/Py–PGA-PEG were found in the Balb/c mice based on the body weight measurement. Two injection doses were used here: 10 mg/kg (representing medium dose, Figure 5a) and 50 mg/kg (representing high dose, Figure S15). Histological assessment was carried out on hematoxylin and eosin (H&E) stained organ slides (heart, liver, spleen, lung, kidney, intestine, and stomach), from which no observable changes were found from acute (1 day p.i.), medium (7 days p.i.), or chronic (30 days p.i.) toxicity caused by UiO-66/Py–PGA-PEG (Figures 5c and S16). To further study the toxicological impact from UiO-66/Py–PGA-PEG, mini chemistry panel test was also carried out to determine the general health status of UiO-66/Py–PGA-PEG injected mice. Inspiringly, all measured biochemical parameters fell within the normal ranges on day 1, day 7, and day 30 postinjection of UiO-66/Py–PGA-PEG (Figure 5b and Table S3). Combining with retention information acquired from PET, it may be reasonable to conclude that UiO-66/Py–PGA-PEG conjugates would not cause significant toxicity to the test subjects.
Figure 5.

Toxicity evaluations of UiO-66 conjugates. (a) Growth chart of mice post-treatment of UiO-66/Py–PGA-PEG (dose: 10 mg/kg) and normal saline. (b) Healthy female Balb/c mice with the intravenous injection of UiO-66/Py–PGA-PEG were sacrificed on 1, 7, and 30 days p.i. for blood collection. Untreated healthy mice were used as the control. Alanine aminotransferase (ALT), alkaline phosphatase (ALP), and aspartate aminotransferase (AST) levels of Balb/c mice injected with saline (control) are similar to those injected with UiO-66/Py–PGA-PEG (dose: 10 mg/kg) at different time points (n = 5). (c) H&E stained major organ slides collected from healthy Balb/c mice and UiO-66/Py–PGA-PEG-injected mice (dose: 10 mg/kg) on day 30 post injection (n = 5). Scale bar: 100 μm.
Enhanced DOX Delivery to Tumor in Vivo
Since DOX loaded on UiO-66 gave strong fluorescence emission under 465 nm excitation (Figure 6a), the feasibility of enhanced tumor targeted drug delivery in vivo using DOX@UiO-66/Py–PGA-PEG-F3 was studied by ex vivo fluorescence imaging. At 2 h after intravenous injection (strongest tumor accumulation based on PET findings) of DOX@UiO-66/Py–PGA-PEG-F3 and DOX@UiO-66/Py–PGA-PEG into MDA-MB-231 tumor-bearing mice (dose: 5 mg of DOX/kg of mouse body weight), the major organs were collected and imaged in an IVIS Spectrum optical imaging system (excitation: 465 nm; emission: 580 nm), shown in Figure 6b. It is important to note that because of different absorption/scattering behaviors of various tissues, optical signal intensities from different organs may not accurately reflect the absolute uptake level of injected DOX@UiO-66 conjugates. For example, although liver and spleen are the dominant organs for DOX@UiO-66/Py–PGA-PEG-F3 accumulation, as evidenced in PET and organ distribution studies, only weak optical signal could be identified in these two organs based on ex vivo optical imaging because of their dark color and strong absorbance of visible DOX fluorescence (Figure 6b). In contrast, due to the much lighter color from tumors, strong optical signal from DOX could be observed for DOX@UiO-66/Py–PGA-PEG-F3 injection group (Figure 6b). Quantitative data from ROI analysis of the tumor tissue showed significantly higher fluorescent signal of DOX for DOX@UiO-66/Py–PGA-PEG-F3 (3.07 × 108 [p/s]/[μW/cm2]), about 4-fold of that for DOX@UiO-66/Py–PGA-PEG (8.58 × 107 [p/s]/[μW/cm2]), clearly demonstrating the feasibility to deliver larger amount of anticancer drugs to the tumor site in vivo by using F3 peptide-conjugated UiO-66 nanoconjugates.
Figure 6.

Enhanced drug delivery into MDA-MB-231 tumors and validation of nucleolin-targeting specificity for DOX@UiO-66/Py–PGA-PEG-F3. (a) Fluorescence image confirmed successful loading of DOX on UiO-66 and that it can be readily detectable in IVIS Spectrum. Left: DOX@UiO-66/Py–PGA-PEG; middle: DOX@UiO-66/Py–PGA-PEG-F3; right: deionized water. (b) Ex vivo fluorescence images of DOX in the major organs/tissues at 2 h after intravenous injection of DOX@UiO-66/Py–PGA-PEG-F3 and DOX@UiO-66/Py–PGA-PEG. (c) Immunohistological staining of MDA-MB-231 tumors from mice injected with DOX@UiO-66/Py–PGA-PEG-F3 and DOX@UiO-66/Py–PGA-PEG. Nucleolin (red) expression profile was compared with DOX (green) distribution pattern from both of the UiO-66 conjugates. Scale bar: 100 μm.
Cargo Delivery in Tumor Is Primarily Mediated from Nucleolin
MDA-MB-231 tumors from mice injected with DOX@UiO-66/Py–PGA-PEG-F3 and DOX@UiO-66/Py–PGA-PEG (5 mg of DOX/kg of mouse body weight) were frozen for histological analysis. The green fluorescence signal indicated the presence of DOX in the MDA-MB-231 tumor, which was substantially stronger in the group of DOX@UiO-66/Py–PGA-PEG-F3 compared with that from DOX@UiO-66/Py–PGA-PEG. Simultaneously, green fluorescence in DOX@UiO-66/Py–PGA-PEG-F3 group exhibited good overlay with nucleolin, indicating that uptake of DOX@UiO-66/Py–PGA-PEG-F3 in MDA-MB-231 tumors was most likely mediated by interactions with nucleolin in vivo. Here we want to note that this “nucleolin mediation” of UiO-66/Py–PGA-PEG-F3 conjugates only happens inside tumors, since the strong accumulation of UiO-66 conjugates inside liver and spleen is more likely the result of MPS capture (e.g., from macrophages).
CONCLUSIONS
In conclusion, an intrinsically radioactive UiO-66 MOF nanomaterial (89Zr-UiO-66) was designed, synthesized, and surface engineered for tumor targeting and enhanced drug delivery. Good radiochemical stability, material integrity, and reliable material functionalization were all confirmed in this study. Relatively high amount of DOX (1 mg DOX/mg UiO-66) can be loaded onto UiO-66 conjugates, however the DOX release behavior needs further optimization. By adopting F3 peptide (targeting nucleolin) as a targeting ligand, specific and significantly enhanced targeting of MDA-MB-231 tumor by 89Zr-UiO-66 conjugates was demonstrated in vivo, which was further confirmed by ex vivo organ distribution and histology studies. Toxicity study with medium and large dose of UiO-66/Py–PGA-PEG-F3 did not show significant in vivo toxicity. Furthermore, enhanced DOX delivery to MDA-MB-231 tumors in vivo was also demonstrated in tumor-bearing mice. We hope that this report can encourage other researchers to use different types of radioactive MOF nanomaterials for cancer theranostic applications. We are currently trying to further optimize their in vivo pharmacokinetics by improving surface engineering methods and investigating the potential of these nanoconjugates for combinational radiation/chemotherapy.
MATERIALS AND METHODS
Materials
Zirconium chloride (ZrCl4), terephthalic acid (BDC), benzoic acid (BA), 1-pyrenemethylamine hydrochloride, triethylamine (TEA), poly(γ-glutamic acid) (γ-PGA), 1-ethyl-3-(3-(dimethylamino)-propyl) carbodiimide hydrochloride (EDC·HCl), and 37% HCl were all purchased from Sigma-Aldrich (St Louis, MO). 89Zr-oxolate (in 1 M oxalic acid) was acquired from University of Wisconsin Cyclotron group. Doxorubicin hydrochloride and tris(2-carboxyethyl)phosphine hydrochloride (TCEP·HCl) were ordered from Thermo Fisher Scientific (Fair Lawn, NJ). Amine-PEG-Mal (i.e., amine PEG maleimide, MW: 5 kDa) was purchased from Creative PEGworks (Chapel Hill, NC). Antinucleolin antibody and anti-β-actin antibody (conjugated with horseradish peroxidase [HRP] for Western blotting) were both purchased from Abcam (Cambridge, MA, USA). Secondary HRP antibodies were purchased from Jackson ImmunoResearch (St. Louis MO, USA). Chelex 100 resin (50–100 mesh) was purchased from Sigma-Aldrich. Buffers used in this study were prepared from Millipore-grade water and pretreated with Chelex 100 resin to ensure that the aqueous solution was free of heavy metals. Size exclusive PD-10 columns were purchased from GE Healthcare (Piscataway, NJ). All other chemicals were purchased from Thermo Fisher Scientific.
Syntheses of UiO-66 Nanoconjugates
Synthesis of UiO-66 nMOF was carried out by a solvothermal method.27 In brief, ZrCl4 (466 mg, 2 mmol), BDC (320 mg, 2 mmol), benzoic acid (2.44 g 20 mmol), and HCl (37%, 12 M; 0.33 mL, 4 mmol) were dissolved in DMF (36 mL) at room temperature. The obtained mixture was sealed and placed in a preheated oven with a temperature of 120 °C for 48 h. After cooling down to room temperature, the white powder of UiO-66 nMOFs was collected, washed by DMF, and dispersed in DMF under stirring for 6 h to remove free BDC. Acetone was then used to disperse UiO-66 to exchange the trapped DMF. The final product was obtained after vacuum drying at 60 °C overnight. For production of 89Zr-UiO-66, 200 μL concentrated HCl was added into 185 MBq of 89Zr-oxalate and heated to 200 °C for 2 h to vaporize all the oxalate. The obtained 89ZrCl4 was then added into the reaction system (same reaction scale as described above) along with nonradioactive ZrCl4 to serve as a radioactive building precursor.
Synthesis of PEG Derivatives
Poly(γ-glutamic acid)-pyrene-(30%)-poly(ethylene glycol) (Py–PGA-PEG) and poly(γ-glutamic acid)-pyrene(30%)-poly(ethylene glycol)-F3 (Py–PGA-PEG-F3) were synthesized according to a previous report.29 The precursor of PGA-Py-COOH was synthesized first. Briefly, γ-PGA (25 mg), 1-pyrenemethylamine hydrochloride (15.4), and EDC·HCl (42.8 mg) were dissolved in 10 mL of DMSO/pyridine/water (8/1/1) mixture and reacted for 24 h. The product of PGA-Py-COOH was obtained after dialysis (Mcut off = 10000) against pure water and lyophilization. For the synthesis of Py–PGA-PEG, PGA-Py-COOH (10 mg), PEG-NH2 (230 mg), and EDC·HCl (19.2 mg) were dissolved in 15 mL of DMSO/pyridine (9/1) mixture and reacted for 24 h. The product of PGA-Py-PEG was obtained after dialysis (Mcut off = 10000) against pure water and lyophilization. For the synthesis of Py–PGA-PEG-F3, the precursor Py–PGA-PEG-MAL was presynthesized. The synthesis of Py–PGA-PEG-MAL is the same as that of PGA-Py-PEG, except the reactant is PEG-MAL (230 mg) rather than PEG-NH2 (230 mg). F3 peptide (30 mg) and TCEP (5 mg) in 5 mL HEPES (pH = 7.0) buffer was reacted for 1 h, then Py–PGA-PEG-MAL (50 mg) was added and reacted for another 1 h. The final product of PGA-Py-PEG-F3 was obtained after dialysis (Mcut off = 10000) against pure water and lyophilization as well.
UiO-66 Functionalization
UiO-66 (5 mg) and Py–PGA-PEG (25 mg) or Py–PGA-PEG-F3 (25 mg) were dispersed in Millipore-grade water under sonication for 1 h. After washing with water three times, the final product was dispersed in desired medium. The functionalized UiO-66/Py–PGA-PEG-F3 and UiO-66/Py–PGA-PEG could remain stable in physiological medium (water, saline, cell culture medium) for over 2 weeks.
Material Characterization
Powder X-ray diffraction (PXRD) patterns of UiO-66 nMOF were recorded on Rigaku R-Axis Spider with a Cu Kα source and an Oxford Instruments Cryostream Plus (80–500 K) over the range of 3–60° (2θ). Approximately 100 mg of UiO-66 samples were dried before PXRD analysis. Nitrogen adsorption–desorption isotherms were measured with a Quantachrome NOVA 4200e surface area and pore size analyzer at 77.35 K. UiO-66 nMOF was degassed in a vacuum at 50 °C overnight before measurements. Surface area and pore volume were calculated by the Brunauer–Emmett–Teller (BET) using the adsorption data. SEM was performed on a FEI Nova 200 Nanolab eletron microscope. The morphology of UiO-66 conjugates was also evaluated by JEM-1400Plus transmission electron microscope (JEOL USA, Peabody, MA) and Zetasizer Nano Z (Malvern Instruments, Westborough, MA). FT-IR spectrum was recorded in a Spectrum BX FTIR spectrometer (PerkinElmer) using the KBr method. Proton nuclear magnetic resonance spectra (1H NMR) were recorded by a Varian MR400 NMR spectrometer. Thermogravimetric analysis (TGA) was carried out in a TGA-7 thermogravimetric analyzer (PerkinElmer) with a scan rate of 5 °C/min.
Drug Loading/Release Study
The DOX was loaded into UiO-66 by a modified nanoprecipitation method. Briefly, 5 μL TEA was added into 1 mL of DOX·HCl aqueous solution (1 mg/mL) and the mixture was under ultrasonication for 3 min and stirring for 1 h. Subsequently, 10 mg of UiO-66 was added and the mixture was under ultrasonication for 10 min and stirring for 1 h. Then the solution was dried by the blow of nitrogen in the dark and the solid was redissolved in HEPES buffer (pH = 7.0) and washed three times. UV–visible spectrometry was used to quantify the unbound DOX in loading/washing solutions. A calibration curve for DOX was obtained by using standard solutions of DOX with the different concentrations (range: 1 nM–1 μM) and the curve was fitted by linear regression. The DOX encapsulation efficiency was calculated from the ratio of the drug amount incorporated into UiO-66 to the total drug amount added. After the acquisition of DOX@UiO-66, further functionalization with Py–PGA-PEG-F3 was carried out. Drug release studies were performed at 37 °C in an acetate buffer (pH = 5.0), a phosphate buffer (pH = 6.8), and a phosphate buffer (pH = 7.4). One mg of DOX@UiO-66@Py–PGA-PEG-F3 was dispersed in 1 mL of medium and placed in a dialysis bag with a molecular weight cutoff of 3 kDa. The dialysis bag was immersed in 25 mL of the release medium and kept in a horizontal laboratory shaker (150 rpm) under constant temperature. Samples of 2 mL volume were periodically removed and the same volume of fresh medium was added. The amount of released DOX was analyzed with a spectrophotometer at 488 nm. The drug release studies were performed in triplicate for each sample.
Flow Cytometry and Confocal Fluorescence Microscopy
MDA-MB-231 (nucleolin+) and L929 (nucleolin−) cells were harvested and suspended in PBS (supplemented with 2% bovine serum albumin) at a concentration of 2 × 106 cells/ml, incubated with DOX@UiO-66/Py–PGA-PEG-F3 or DOX@UiO-66/Py–PGA-PEG (concentration: 10 μg/mL based on DOX) for 30 min at RT, washed three times with cold PBS, and resuspended in PBS at 2 × 106 cells/ml. Afterward, the cells were analyzed using a BD LSR Fortessa four-color analysis cytometer, which is equipped with 488 and 633 nm lasers (Becton-Dickinson, San Jose, CA). Cellular fluorescence was computed via FlowJo (X.0.7, Tree Star, Ashland, OR). The cells were also examined under a Nikon A1 confocal microscope with a magnitude of 200× to validate the flow cytometry results. To clearly visualize the distribution of DOX@UiO-66/Py–PGA-PEG-F3 inside MDA-MB-231 cells and compare with lysosome locations, a magnitude of 600× was used.
PET Imaging and Organ Distributions
All animal studies were conducted under a protocol (PRO00006023) approved by the University Committee on Use and Care of Animals (UCUCA) at University of Michigan. Tumors were established by subcutaneous injection of 2 × 106 of MDA-MB-231 cells suspended in 100 μL of a 1:1 mixture of PBS and Matrigel (BD Biosciences, Franklin Lakes, NJ) into the breast fat pad of female nude mice (Charles River Laboratories). The tumor sizes were monitored every other day and the mice were subjected to imaging studies when the tumor diameter reached 5–8 mm. PET scans were performed using a rodent Inveon microPET/CT scanner (Siemens Medical Solutions USA, Inc.). Each tumor-bearing mouse was injected with 5–10 MBq of 89Zr-UiO-66/Py–PGA-PEG-F3 or 89Zr-UiO-66/Py–PGA-PEG into its tail vein and subjected to static PET scans (40 million events per scan) at various time points postinjection (p.i.). The images were reconstructed using a three-dimensional ordered subset expectation maximization (3D-OSEM) algorithm, with no attenuation or scatter correction. For each microPET scan, Inveon Research Workshop (IRW, v4.2.0.8) was used to superimpose three-dimensional (3D) regions-of-interest (ROIs) on the tumor and major organs in the decay-corrected whole-body images. By adopting a tissue density of 1 g/mL, the radioactivity in each ROI volume was converted to MBq/g using a conversion factor and then divided by the total administered radioactivity to obtain a percentage of injected dose per gram of tissue (%ID/g) for each organ/tissue.
Organ distribution studies by gamma counting were carried out to confirm that the quantitative tracer uptake values based on PET imaging truly represented the radioactivity distribution in tumor-bearing mice. After the last PET scans at 120 h p.i., the mice were euthanized and their blood, tumors, and major organs/tissues were collected and wet-weighed. The radioactivity in each collected sample was measured using a WIZARD2 automatic gamma-counter (PerkinElmer) and recorded as %ID/g (mean ± SD).
Immunostaining
MDA-MB-231 tumor-bearing mice were injected with DOX@UiO-66/Py–PGA-PEG-F3 and DOX@UiO-66/Py–PGA-PEG (5 mg of DOX/kg of mouse body weight) and sacrificed at 2 h p.i. MDA-MB-231 tumors from each injection group were frozen cut into 6-μm slices and incubated with 5 μg/mL antinucleolin antibody (Abcam, Cat # ab22758) at room temperature for 1 h. After that, tumor slices were rinsed repeatedly by PBS and incubated with 2 μg/mL FITC-labeled goat antirabbit IgG (Jackson Immunoresearch) for 1 h at room temperature. Immunostained tumor slices were examined under Nikon A1 confocal microscope with a magnitude of 200×.
Toxicity Study of UiO-66/Py–PGA-PEG
To study the potential toxicity of UiO-66/Py–PGA-PEG-F3, medium (1 mg/kg) and large dose (5 mg/kg) of UiO-66/Py–PGA-PEG was intravenously (i.v.) injected to healthy female BALB/c mice (weight: ~ 18 g, n = 5). The fluctuation of mouse body weight was monitored every other day for 30 days. H&E staining of tissues from heart, liver, spleen, lung, and kidney was conducted after Day 1, 7, and 30 (standing for acute, medium, and long-term toxicity, n = 5 for each time point) to further investigate the potential signs of toxicity (i.e., cellular shrinkage or blebing, steatosis in liver cells, condensation of chromatin, rupture of cell membrane and apoptotic bodies). Mice were sacrificed to collect blood (0.8 mL) for mini chemistry panel test on Day 1, 7, and 30 postinjection of UiO-66/Py–PGA-PEG.
In Vivo Drug Delivery Evaluation
As previously described, DOX was loaded into UiO-66 before PEGylation and F3 peptide conjugation to prepare DOX@UiO-66/Py–PGA-PEG-F3 and DOX@UiO-66/Py–PGA-PEG. For in vivo drug delivery study, DOX@UiO-66/Py–PGA-PEG-F3 and DOX@UiO-66/Py–PGA-PEG (5 mg/kg UiO-66, 5 mg/kg DOX) were injected intravenously into MDA-MB-231 tumor-bearing mice. The mice were sacrificed at 2 h p.i., with tumor and other major organs taken for ex vivo fluorescence imaging in the IVIS Spectrum system (excitation: 465 nm, emission: 580 nm).
Statistical Analysis
A minimum sample number of 3 was employed to ensure statistical power. Differences between groups were analyzed for statistical significance using the unpaired, two-tailed Student’s t test. A confidence interval of 95% was selected and P < 0.05 was considered to be statistically significant.
Acknowledgments
We thank Dr. Gary D. Luker for manuscript proofreading and helpful suggestions in experimental designs. This work is supported, in part, by the University of Michigan Department of Radiology, Else U. Pardee Foundation, NIH/NCI P01 CA085878, the National Natural Science Foundation of China (81201696), and Jiangsu Government Scholarship for Overseas Studies.
Footnotes
Notes
The authors declare no competing financial interest.
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsnano.7b01530.
Detailed material characterization, chemical synthesis methods, PET analysis, and biochemical toxicity evaluation data (PDF)
References
- 1.Lu W, Wei Z, Gu ZY, Liu TF, Park J, Park J, Tian J, Zhang M, Zhang Q, Gentle T, III, Bosch M, Zhou HC. Tuning the Structure and Function of Metal-Organic Frameworks via Linker Design. Chem Soc Rev. 2014;43:5561–5593. doi: 10.1039/c4cs00003j. [DOI] [PubMed] [Google Scholar]
- 2.Suh MP, Park HJ, Prasad TK, Lim DW. Hydrogen Storage in Metal-Organic Frameworks. Chem Rev. 2012;112:782–835. doi: 10.1021/cr200274s. [DOI] [PubMed] [Google Scholar]
- 3.Wu H, Gong Q, Olson DH, Li J. Commensurate Adsorption of Hydrocarbons and Alcohols in Microporous Metal Organic Frameworks. Chem Rev. 2012;112:836–868. doi: 10.1021/cr200216x. [DOI] [PubMed] [Google Scholar]
- 4.Li JR, Sculley J, Zhou HC. Metal-Organic Frameworks for Separations. Chem Rev. 2012;112:869–932. doi: 10.1021/cr200190s. [DOI] [PubMed] [Google Scholar]
- 5.Liu J, Chen L, Cui H, Zhang J, Zhang L, Su CY. Applications of Metal-Organic Fameworks in Heterogeneous Supra-molecular Catalysis. Chem Soc Rev. 2014;43:6011–6061. doi: 10.1039/c4cs00094c. [DOI] [PubMed] [Google Scholar]
- 6.Ranocchiari M, van Bokhoven JA. Catalysis by Metal-Organic Frameworks: Fundamentals and Opportunities. Phys Chem Chem Phys. 2011;13:6388–6396. doi: 10.1039/c0cp02394a. [DOI] [PubMed] [Google Scholar]
- 7.Kreno LE, Leong K, Farha OK, Allendorf M, Van Duyne RP, Hupp JT. Metal-Organic Framework Materials as Chemical Sensors. Chem Rev. 2012;112:1105–1125. doi: 10.1021/cr200324t. [DOI] [PubMed] [Google Scholar]
- 8.He C, Liu D, Lin W. Nanomedicine Applications of Hybrid Nanomaterials Built from Metal-Ligand Coordination Bonds: Nanoscale Metal-Organic Frameworks and Nanoscale Coordination Polymers. Chem Rev. 2015;115:11079–11108. doi: 10.1021/acs.chemrev.5b00125. [DOI] [PubMed] [Google Scholar]
- 9.Li S, Huo F. Metal-Organic Framework Composites: from Fundamentals to Applications. Nanoscale. 2015;7:7482–7501. doi: 10.1039/c5nr00518c. [DOI] [PubMed] [Google Scholar]
- 10.Horcajada P, Gref R, Baati T, Allan PK, Maurin G, Couvreur P, Ferey G, Morris RE, Serre C. Metal-Organic Frameworks in Biomedicine. Chem Rev. 2012;112:1232–1268. doi: 10.1021/cr200256v. [DOI] [PubMed] [Google Scholar]
- 11.Cai W, Chu CC, Liu G, Wang YX. Metal-Organic Framework-Based Nanomedicine Platforms for Drug Delivery and Molecular Imaging. Small. 2015;11:4806–4822. doi: 10.1002/smll.201500802. [DOI] [PubMed] [Google Scholar]
- 12.Lu K, He C, Lin W. A Chlorin-Based Nanoscale Metal-Organic Framework for Photodynamic Therapy of Colon Cancers. J Am Chem Soc. 2015;137:7600–7603. doi: 10.1021/jacs.5b04069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu J, Yang Y, Zhu W, Yi X, Dong Z, Xu X, Chen M, Yang K, Lu G, Jiang L, Liu Z. Nanoscale Metal-Organic Frameworks for Combined Photodynamic & Radiation Therapy in Cancer Treatment. Biomaterials. 2016;97:1–9. doi: 10.1016/j.biomaterials.2016.04.034. [DOI] [PubMed] [Google Scholar]
- 14.He C, Liu D, Lin W. Self-Assembled Nanoscale Coordination Polymers Carrying siRNAs and Cisplatin for Effective Treatment of Resistant Ovarian Cancer. Biomaterials. 2015;36:124–133. doi: 10.1016/j.biomaterials.2014.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Poon C, He C, Liu D, Lu K, Lin W. Self-Assembled Nanoscale Coordination Polymers Carrying Oxaliplatin and Gemcitabine for Synergistic Combination Therapy of Pancreatic Cancer. J Controlled Release. 2015;201:90–99. doi: 10.1016/j.jconrel.2015.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.He C, Liu D, Lin W. Self-Assembled Core-Shell Nano-particles for Combined Chemotherapy and Photodynamic Therapy of Resistant Head and Neck Cancers. ACS Nano. 2015;9:991–1003. doi: 10.1021/nn506963h. [DOI] [PubMed] [Google Scholar]
- 17.deKrafft KE, Xie Z, Cao G, Tran S, Ma L, Zhou OZ, Lin W. Iodinated Nanoscale Coordination Polymers as Potential Contrast Agents for Computed Tomography. Angew Chem, Int Ed. 2009;48:9901–9904. doi: 10.1002/anie.200904958. [DOI] [PubMed] [Google Scholar]
- 18.Taylor KM, Rieter WJ, Lin W. Manganese-Based Nanoscale Metal-Organic Frameworks for Magnetic Resonance Imaging. J Am Chem Soc. 2008;130:14358–14359. doi: 10.1021/ja803777x. [DOI] [PubMed] [Google Scholar]
- 19.Horcajada P, Chalati T, Serre C, Gillet B, Sebrie C, Baati T, Eubank JF, Heurtaux D, Clayette P, Kreuz C, Chang JS, Hwang YK, Marsaud V, Bories PN, Cynober L, Gil S, Ferey G, Couvreur P, Gref R. Porous Metal-Organic-Framework Nanoscale Carriers as A Potential Platform for Drug Delivery and Imaging. Nat Mater. 2010;9:172–178. doi: 10.1038/nmat2608. [DOI] [PubMed] [Google Scholar]
- 20.Taylor-Pashow KM, Della Rocca J, Xie Z, Tran S, Lin W. Postsynthetic Modifications of Iron-Carboxylate Nanoscale Metal-Organic Frameworks for Imaging and Drug Delivery. J Am Chem Soc. 2009;131:14261–14263. doi: 10.1021/ja906198y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yang Y, Liu J, Liang C, Feng L, Fu T, Dong Z, Chao Y, Li Y, Lu G, Chen M, Liu Z. Nanoscale Metal-Organic Particles with Rapid Clearance for Magnetic Resonance Imaging-Guided Photothermal Therapy. ACS Nano. 2016;10:2774–2781. doi: 10.1021/acsnano.5b07882. [DOI] [PubMed] [Google Scholar]
- 22.Zhao HX, Zou Q, Sun SK, Yu C, Zhang X, Li RJ, Fu YY. Theranostic Metal-Organic Framework Core-Shell Composites for Magnetic Resonance Imaging and Drug Delivery. Chem Sci. 2016;7:5294–5301. doi: 10.1039/c6sc01359g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gao X, Zhai M, Guan W, Liu J, Liu Z, Damirin A. Controllable Synthesis of A Smart Multifunctional Nanoscale Metal-Organic Framework for Magnetic Resonance/Optical Imaging and Targeted Drug Delivery. ACS Appl Mater Interfaces. 2017;9:3455–3462. doi: 10.1021/acsami.6b14795. [DOI] [PubMed] [Google Scholar]
- 24.Hong H, Chen F, Zhang Y, Cai W. New Radiotracers for Imaging of Vascular Targets in Angiogenesis-Related Diseases. Adv Drug Delivery Rev. 2014;76:2–20. doi: 10.1016/j.addr.2014.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cavka JH, Jakobsen S, Olsbye U, Guillou N, Lamberti C, Bordiga S, Lillerud KP. A New Zirconium Inorganic Building Brick Forming Metal Organic Frameworks with Exceptional Stability. J Am Chem Soc. 2008;130:13850–13851. doi: 10.1021/ja8057953. [DOI] [PubMed] [Google Scholar]
- 26.Kandiah M, Nilsen MH, Usseglio S, Jakobsen S, Olsbye U, Tilset M, Larabi C, Quadrelli EA, Bonino F, Lillerud KP. Synthesis and Stability of Tagged UiO-66 Zr-MOFs. Chem Mater. 2010;22:6632–6640. [Google Scholar]
- 27.Zhu X, Gu J, Wang Y, Li B, Li Y, Zhao W, Shi J. Inherent Anchorages in UiO-66 Nanoparticles for Efficient Capture of Alendronate and Its Mediated Release. Chem Commun (Cambridge, U K) 2014;50:8779–8782. doi: 10.1039/c4cc02570a. [DOI] [PubMed] [Google Scholar]
- 28.Zhang Y, Hong H, Cai W. PET Tracers Based on Zirconium-89. Curr Radiopharm. 2011;4:131–139. doi: 10.2174/1874471011104020131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Prencipe G, Tabakman SM, Welsher K, Liu Z, Goodwin AP, Zhang L, Henry J, Dai H. PEG Branched Polymer for Functionalization of Nanomaterials with Ultralong Blood Circulation. J Am Chem Soc. 2009;131:4783–4787. doi: 10.1021/ja809086q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Morris W, Briley WE, Auyeung E, Cabezas MD, Mirkin CA. Nucleic Acid-Metal Organic Framework (MOF) Nanoparticle Conjugates. J Am Chem Soc. 2014;136:7261–7264. doi: 10.1021/ja503215w. [DOI] [PubMed] [Google Scholar]
- 31.Orellana-Tavra C, Baxter EF, Tian T, Bennett TD, Slater NK, Cheetham AK, Fairen-Jimenez D. Amorphous Metal-Organic Frameworks for Drug Delivery. Chem Commun (Cambridge, U K) 2015;51:13878–13881. doi: 10.1039/c5cc05237h. [DOI] [PubMed] [Google Scholar]
- 32.Porkka K, Laakkonen P, Hoffman JA, Bernasconi M, Ruoslahti E. A Fragment of the HMGN2 Protein Homes to the Nuclei of Tumor Cells and Tumor Endothelial Cells In Vivo. Proc Natl Acad Sci U S A. 2002;99:7444–7449. doi: 10.1073/pnas.062189599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Christian S, Pilch J, Akerman ME, Porkka K, Laakkonen P, Ruoslahti E. Nucleolin Expressed at the Cell Surface is A Marker of Endothelial Cells in Angiogenic Blood Vessels. J Cell Biol. 2003;163:871–878. doi: 10.1083/jcb.200304132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Berger CM, Gaume X, Bouvet P. The Roles of Nucleolin Subcellular Localization in Cancer. Biochimie. 2015;113:78–85. doi: 10.1016/j.biochi.2015.03.023. [DOI] [PubMed] [Google Scholar]
- 35.Bhojani MS, Ranga R, Luker GD, Rehemtulla A, Ross BD, Van Dort ME. Synthesis and Investigation of a Radioiodinated F3 Peptide Analog As a SPECT Tumor Imaging Radioligand. PLoS One. 2011;6:e22418. doi: 10.1371/journal.pone.0022418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Garibay SJ, Cohen SM. Isoreticular Synthesis and Modification of Frameworks with the UiO-66 Topology. Chem Commun (Cambridge, U K) 2010;46:7700–7702. doi: 10.1039/c0cc02990d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wuttke S, Dietl C, Hinterholzinger FM, Hintz H, Langhals H, Bein T. Turn-On Fluorescence Triggered by Selective Internal Dye Replacement in MOFs. Chem Commun (Cambridge, U K) 2014;50:3599–3601. doi: 10.1039/c3cc46591h. [DOI] [PubMed] [Google Scholar]
- 38.Zimpel A, Preiß T, Röder R, Engelke H, Ingrisch M, Peller M, Radler JO, Wagner E, Bein T, Lächelt U, Wuttke S. Imparting Functionality to MOF Nanoparticles by External Surface Selective Covalent Attachment of Polymers. Chem Mater. 2016;28:3318–3326. [Google Scholar]
- 39.Dekrafft KE, Boyle WS, Burk LM, Zhou OZ, Lin W. Zr- and Hf-Based Nanoscale Metal-Organic Frameworks as Contrast Agents for Computed Tomography. J Mater Chem. 2012;22:18139–18144. doi: 10.1039/C2JM32299D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Geisow MJ, Evans WH. pH in the Endosome. Measurements During Pinocytosis and Receptor-Mediated Endocytosis. Exp Cell Res. 1984;150:36–46. doi: 10.1016/0014-4827(84)90699-2. [DOI] [PubMed] [Google Scholar]
- 41.Argyo C, Weiss V, Brauchle C, Bein T. Multifunctional Mesoporous Silica Nanoparticles as A Universal Platform for Drug Delivery. Chem Mater. 2014;26:435–451. [Google Scholar]
- 42.Wuttke S, Braig S, Preiss T, Zimpel A, Sicklinger J, Bellomo C, Radler JO, Vollmar AM, Bein T. MOF Nanoparticles Coated by Lipid Bilayers and Their Uptake by Cancer Cells. Chem Commun (Cambridge, U K) 2015;51:15752–15755. doi: 10.1039/c5cc06767g. [DOI] [PubMed] [Google Scholar]
- 43.Jauw YW, Menke-van der Houven van Oordt CW, Hoekstra OS, Hendrikse NH, Vugts DJ, Zijlstra JM, Huisman MC, van Dongen GA. Immuno-Positron Emission Tomography with Zirconium-89-Labeled Monoclonal Antibodies in Oncology: What Can We Learn from Initial Clinical Trials? Front Pharmacol. 2016;7:131. doi: 10.3389/fphar.2016.00131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ganoth A, Merimi KC, Peer D. Overcoming Multidrug Resistance with Nanomedicines. Expert Opin Drug Delivery. 2015;12:223–238. doi: 10.1517/17425247.2015.960920. [DOI] [PubMed] [Google Scholar]
- 45.Abou DS, Ku T, Smith-Jones PM. In Vivo Biodistribution and Accumulation of 89Zr in Mice. Nucl Med Biol. 2011;38:675–681. doi: 10.1016/j.nucmedbio.2010.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]