

Abstract
Val-boroPro (talabostat, PT-100), a nonselective inhibitor of post-proline cleaving serine proteases, stimulates mammalian immune systems through an unknown mechanism of action. Despite this lack of mechanistic understanding, Val-boroPro has attracted significant interest as a potential anticancer agent, reaching Phase III trials in humans. Here we show that Val-boroPro stimulates the immune system by triggering a proinflammatory form of cell death in monocytes and macrophages known as pyroptosis. We demonstrate that the inhibition of two serine proteases, DPP8 and DPP9, activates the proprotein form of caspase-1 independent of the inflammasome adaptor ASC. Activated pro-caspase-1 does not efficiently process itself or IL-1β, but does cleave and activate gasdermin D to induce pyroptosis. Mice lacking caspase-1 do not show immune stimulation after treatment with Val-boroPro. Our data identifies the first small molecule that induces pyroptosis and reveals a new checkpoint that controls the activation of the innate immune system.
INTRODUCTION
The immune system can recognize and destroy cancer cells, but cancer cells can find ways to evade or block the immune system and render the host immunologically tolerant1,2. In recent years, a number of therapeutic strategies that re-activate the immune system to attack cancer cells have achieved unprecedented responses in the clinic, and cancer immunotherapy has become a new pillar of cancer therapy3,4. As promising as the results have been with these agents so far, however, only a fraction of patients respond (typically < 50%), and continued development of new immunotherapy agents with improved or complementary mechanisms of action is clearly needed5,6.
Val-boroPro (Fig. 1a) is a nonselective inhibitor of the S9 family of post-proline cleaving serine proteases, including DPP4, DPP7, DPP8, DPP9, and FAP, and has been demonstrated to induce dramatic tumor regressions in multiple mouse models of cancer7,8. The responses are immune-mediated, as the efficacy of Val-boroPro is dramatically attenuated in lymphocyte-deficient mice and Val-boroPro-treated mice that have rejected their tumors become immune to rechallenge7. Val-boroPro is known to stimulate the transcriptional upregulation several cytokines, including IL-1β, IL-6, G-CSF, and CXCL1/KC, in both tumors and tumor-draining lymph nodes7, and to increase the mouse serum protein levels of several of these cytokines, including G-CSF and CXCL1/KC7,8. Bone marrow-derived cells appear to be key mediators of the antitumor immune response, as Val-boroPro has been reported to induce cytokines from bone marrow stromal cells9 and from co-cultures of THP-1 macrophages and fibroblasts in vitro9,10, and clodronate depletion of phagocytic cells significantly reduces Val-boroPro’s antitumor activity in vivo8. However, the molecular and cellular mechanisms controlling Val-boroPro-induced immune activation, including the key serine protease target(s), signaling pathways, and the cell types affected by Val-boroPro, have not been established.
Figure 1. Val-boroPro is cytotoxic to monocytes and macrophages.
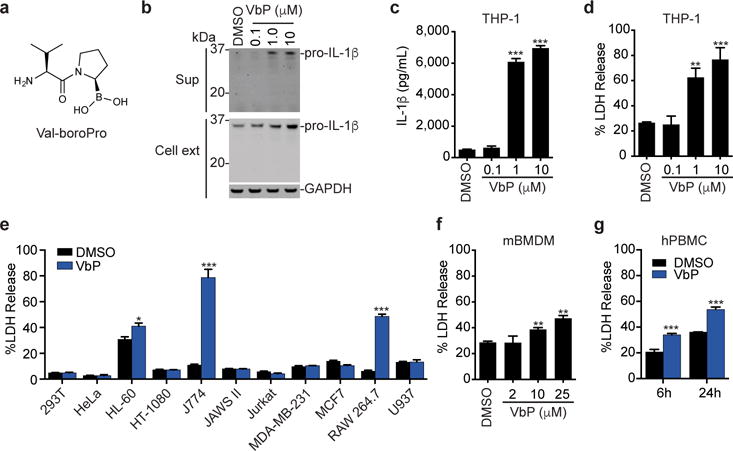
a, The structure of Val-boroPro. b,c, The effects of Val-boroPro (2 μM, 24 h) on IL-1β expression and secretion in THP-1 macrophages as determined by immunoblotting (b) and ELISA (c). Full gel images are shown in Supplementary Figure 12. d, Val-boroPro induces the release of LDH from THP-1 macrophages. e, Profiles of the sensitivities of various cell lines to Val-boroPro. f, g, Val-boroPro induces LDH release from primary mouse BMDMs (f) and primary human PBMCs (g). In c-g, data are means ± SEM of three biological replicates. * p < 0.05, ** p < 0.01, ***p < 0.001 by two-sided Student’s t-test for DMSO versus Val-boroPro-treated cells. ext, extract; sup, supernatant; VbP, Val-boroPro.
Here, we show that Val-boroPro activates the immune system by triggering pyroptosis selectively in monocytes and macrophages. We demonstrate that the inhibition of two related serine proteases, DPP8 and DPP9, activates the proprotein form of pro-caspase-1 without autoproteolysis, and that active pro-caspase-1 then cleaves gasdermin D (GSDMD) to induce pyroptosis. This pathway is required for Val-boroPro-mediated immune activation, as caspase-1-deficient mice do not show cytokine elevation after Val-boroPro administration. Overall, these data identify the first small molecule that induces pyroptosis and uncover a new pathway regulating the innate immune system.
RESULTS
Val-boroPro induces monocytes and macrophage cell death
To study Val-boroPro’s mechanism of action, we first wanted to identify several cell culture model systems that respond to Val-boroPro. As Val-boroPro was preliminarily reported in a meeting abstract to activate IL-1β in co-cultures of THP-1 macrophages and MM46T fibrosarcoma cells10, we first investigated Val-boroPro’s impact on IL-1β in THP-1 macrophages (Fig. 1b). We found that Val-boroPro only modestly increased the levels of intracellular pro-IL-1β (~2-fold at 10 μM), but dramatically increased the amount of pro-IL-1β secreted into the supernatant (Fig. 1 b,c). Similarly, Val-boroPro did not increase intracellular protein levels of IL-1α (which was not detected in DMSO- or Val-boroPro-treated cells) or pro-IL-18, and only pro-IL-18 was secreted into the supernatant (Supplementary Results, Supplementary Fig. 1a). Our results are plainly inconsistent with the meeting abstract7, which reported that Val-boroPro induced the secretion of cleaved, mature IL-1β in the co-culture system. We speculate that a factor secreted from MM64T cells led to the activation of IL-1β, but we were unable to confirm this hypothesis as ATCC has discontinued sales of the MM46T cell line. Instead, as IL-1β is thought to be released from cells by a lytic form of programmed cell death known as pyroptosis11,12, our results suggest that Val-boroPro’s primary effect might be to induce pyroptosis and release the already present intracellular contents, including pro-IL-1β and pro-IL-18 that happen to be endogenously expressed, and not to stimulate the expression and activation of these cytokines. Indeed, we observed concurrent release of the cytoplasmic enzyme lactate dehydrogenase (LDH) with pro-IL-1β (Fig. 1c,d).
We next evaluated the specificity of this response by treating various cell lines with Val-boroPro. We found that THP-1 cells do not have to be differentiated to respond to Val-boroPro, but that undifferentiated THP-1 monocytes released less LDH than PMA-differentiated THP-1 macrophages (Supplementary Fig. 1 b,c). Moreover, out of diverse panel of cell lines tested, only HL-60, J774, and RAW 264.7 cells released LDH (Fig. 1e, Supplementary Fig. 1 d–g), suggesting that Val-boroPro’s effects might be restricted to the monocyte/macrophage lineage. Consistent with this premise, primary mouse bone marrow derived macrophages (mBMDMs) (Fig. 1f) and primary human peripheral blood mononuclear cells (hPBMCs) (Fig. 1g) also released LDH upon treatment with Val-boroPro. It is currently unclear why U937 cells, which are monocytic, do not release LDH after Val-boroPro treatment, but these cancer cells do not always behave like normal human monocytes. For example, U937 cells, unlike primary human monocytes, are insensitive to cytosolic bacterial flagellin13, and we demonstrate here that they do not undergo cell death following treatment with bacterial lipopolysaccharide (LPS) plus nigericin like other monocytes and macrophages (Supplementary Fig. 1 b,c, g–i).
DPP8/9 inhibition induces cell death
We next sought to identify the key target or targets of Val-boroPro whose inhibition causes cell death. Val-boroPro is known to potently inhibit the post-proline cleaving serine proteases DPP4, DPP8, and DPP9 (IC50 values of <4, 4, and 11 nM, respectively), and to more weakly inhibit DPP7, FAP, and PREP14 (Supplementary Table 1). We confirmed that Val-boroPro is cell penetrant and therefore can inhibit intracellular targets like DPP8 and DPP9 in living systems (Supplementary Fig. 2). To assess the selectivity of Val-boroPro more broadly across the serine hydrolase family, we profiled Val-boroPro across ~100 serine hydrolase enzymes using EnPlex15 and by competitive activity-based protein profiling in THP-1 and RAW 264.7 lysates (Fig. 2a, Supplementary Fig. 3). This analysis revealed only one previously unknown target, SCPEP1, inhibited by Val-boroPro at 10 μM.
Figure 2. DPP8/9 inhibition induces cell death.
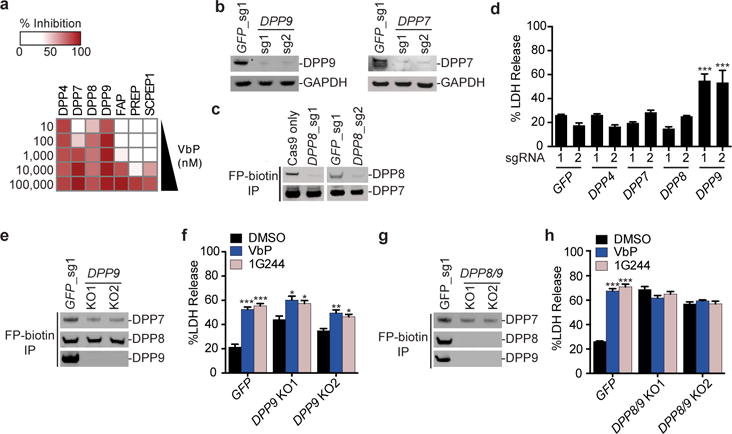
a, The serine hydrolase targets of Val-boroPro identified by EnPlex (10 nM to 100 μM, 10-fold dilution series). The percent inhibition at each concentration relative to DMSO controls is shown. b, Confirmation of DPP7 and DPP9 knockout in THP-1 cells by immunoblotting. c, Enrichment of active serine hydrolases in THP-1 cells with the FP-biotin activity-based probe enables DPP8 to be observed by immunoblotting and confirms DPP8 knockout. d, DPP9 knockout THP-1 cells secrete higher levels of LDH. e, Activity-based probe enrichment of serine hydrolases in THP-1 cells confirms that no active DPP9 was expressed in DPP9 knockout cells. f, DPP9 knockout cells release additional LDH after treatment with Val-boroPro (2 μM) and 1G244 (10 μM) for 24 h. g, Confirmation of DPP8/9 double knockouts in FP-biotin enriched THP-1 cell extracts by immunoblotting. h, DPP8/9 double knockouts do not respond to Val-boroPro or 1G244. Full gel images for b, c, e and g are shown in Supplementary Figure 12. In d, f, and h, data are means ± SEM of three biological replicates. *p<0.05, **p < 0.01, ***p < 0.001 by two-sided Student’s t-test for DMSO versus compound-treated cells.
As we observed Val-boroPro-mediated cytoxicity at 1 μM (Fig. 1d) and only DPP4, DPP7, DPP8, and DPP9 are inhibited at this concentration (Fig. 2a), we reasoned that knockout of one of these targets (or a combination of these targets) in macrophages would result in lytic cell death. We therefore treated Cas9-expressing THP-1 monocytes with single guide RNAs (sgRNAs) targeting DPP4, DPP7, DPP8, and DPP9 (Supplementary Table 2). We confirmed that cells treated with sgRNAs to DPP7 and DPP9 had significantly lower levels of these proteins than controls (Fig. 2b). We could not detect DPP4 or DPP8 in control THP-1 cells by immunoblotting, but we were able to detect DPP8, and confirm CRISPR-mediated protein loss, after enriching cell lysates for active serine proteases using a biotinylated fluorophosphonate (FP-biotin) activity-based probe (Fig. 2c). We were unable to detect DPP4 in THP-1 cells even after activity-based probe enrichment, consistent with previous work that also failed to detect DPP4 protein in THP-1 cells16. Only cells treated with sgRNAs targeting DPP9 released significantly higher levels of LDH and pro-IL-1β (Fig. 2d, Supplementary Fig. 4 a–d), showing that depletion of DPP9 alone results in cell death. However, we also observed that these cells released even more LDH upon treatment with Val-boroPro (Supplementary Fig. 4a). To confirm that this additional LDH release was not due to small remaining amounts of DPP9 (Fig. 2b), we isolated two DPP9 knockout cell lines (Fig. 2e, Supplementary Fig. 4e,f). Val-boroPro was able to induce additional cytotoxicity in DPP9 knockout cells (Fig. 2f), demonstrating that inhibition of a second Val-boroPro target also contributes to cell death.
We hypothesized that the second key target was DPP8, which is 79% similar in protein sequence to DPP9, shares peptide substrate specificity in vitro17,18, and, like DPP9, is cytosolic19–23. Although DPP8 deficient cells released similar basal and Val-boroPro-induced LDH levels as control cells (Fig. 2d, Supplementary Fig. 4b), we speculated that DPP9 might fully compensate for DPP8 loss in these cells. We next treated THP-1 cells with 1G244 (Supplementary Fig. 5), a compound structurally unrelated to Val-boroPro that potently inhibits DPP8 and DPP9 (IC50 values of 14 and 53 nM, respectively), but does not inhibit DPP4 or DPP7 (Supplementary Fig. 3a, Supplementary Table 1)24,25. Consistent with our hypothesis that DPP8 and DPP9 are the key targets, we observed that Val-boroPro and 1G244 induced similar LDH release from wild-type THP-1 cells and RAW 264.7 cells (Supplementary Fig. 1 b,c,f), and 1G244 also induced additional LDH release in DPP9 knockout THP-1 cells (Fig. 2f). Moreover, others have previously observed that 1G244, like Val-boroPro, induces cell death in J774 cells26. We then treated THP-1 monocytes with sgRNAs to both DPP8 and DPP9 and isolated double knockout cells (Fig. 2g, Supplementary Fig. 6). These cells released high levels of LDH upon differentiation and gave no additional LDH increase after treatment with either Val-boroPro or 1G244 (Fig. 2h). Therefore, inhibition of DPP8 and DPP9 causes programmed lytic cell death in macrophages. DPP9 appears to play the major role in this process, as the loss of DPP8 contributes to cytotoxicity only in cells lacking DPP9.
Val-boroPro induced pyroptosis requires caspase-1
Pyroptosis is a proinflammatory mode of cell death mediated by caspases-1, -4, or -5 that serves as key part of the innate immune response against infectious organisms27. In canonical pyroptosis, certain microbial structures (termed pathogen-associated molecular patterns, or ‘PAMPs’) trigger the formation of large, cytosolic multiprotein complexes called inflammasomes that recruit and promote the autoproteolysis and activation of caspase-127. In noncanonical pyroptosis, intracellular LPS directly activates caspases-4/528. To further confirm our hypothesis that DPP8/9 inhibition induces pyroptosis and to examine the mechanism in more detail, we first imaged RAW 264.7 cells treated with Val-boroPro or 1G244 (Supplementary Videos 1–4). The time-lapse videos show that both compounds induce a lytic cell death consistent with pyroptosis and inconsistent with apoptotic cell death. To determine which, if any, inflammatory caspase mediates Val-boroPro-induced pyroptosis, we treated Cas9-expressing THP-1 monocytes with sgRNAs to caspase-1, -4, or -5 and validated knockout of capases-1 and -4 (Fig. 3a). We, like others previously28, could not detect caspase-5 protein in THP-1 cells. As expected, only the caspase-1 knockout cells were resistant to the cytotoxic effects of LPS plus nigericin (Fig. 3b), which activates the NLRP3 inflammasome and induces canonical caspase-1-dependent pyroptosis. Caspase-1 knockout THP-1 cells were also resistant to Val-boroPro and 1G244 (Fig. 3b, Supplementary Fig. 7a), showing that pyroptosis induced by DPP8/9 inhibition also requires caspase-1. To confirm that caspase-1 is also required in RAW 264.7 cells, we generated RAW 264.7 cells lacking caspase-1 and demonstrated that these cells were also resistant to Val-boroPro-induced cell death (Supplementary Fig. 7 b–f).
Figure 3. DPP8/9 inhibitor cytotoxicity is caspase-1-dependent.
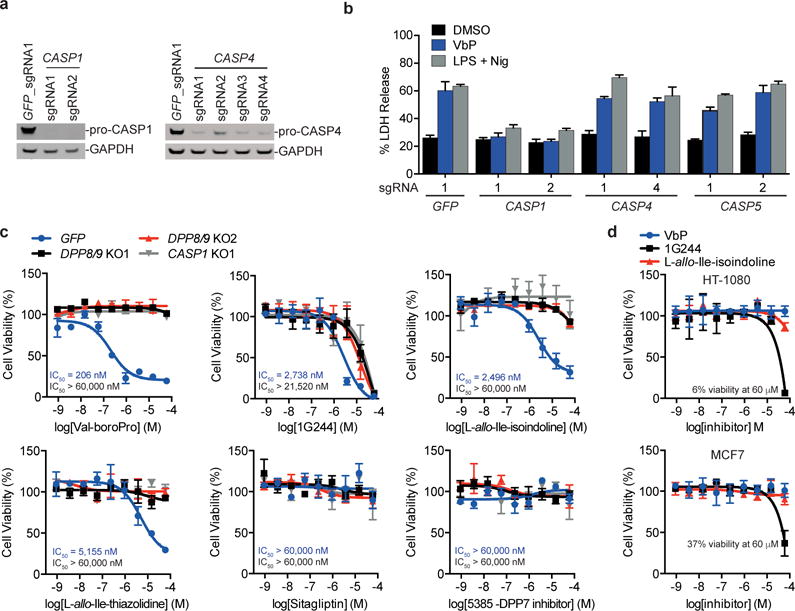
a, Confirmation of caspase-1 and caspase-4 knockout in THP-1 cells by immunoblotting. Full gel images are shown in Supplementary Figure 12. b, LDH induced by Val-boroPro and LPS plus nigericin in THP-1 macrophages treated with sgRNAs to CASP1, CASP4, and CASP5. c, Cell viability of THP-1 monocytes after treatment with the indicated compounds for 48 h relative to DMSO as determined by CellTiter-Glo. The IC50 values for control THP-1 cell lines (treated with an sgRNA to GFP, values in blue) and for DPP8/9 KO1 THP-1 (values in black) cells are shown. d, Viability of HT-1080 and MCF7 cells after treatment with Val-boroPro, 1G244, or L-allo-Ile-isoindoline for 48 h relative to DMSO treatment was determined by CellTiter-Glo. In b–d, data are means ± SEM of three biological replicates.
Before further investigating the mechanism of action of Val-boroPro, we wanted to evaluate the relative potencies and specificities of Val-boroPro, 1G244, and other known DPP inhibitors for the induction of caspase-1-dependent cell death (Supplementary Table 1, Supplementary Fig. 5). Here, we treated control, DPP8/9 knockout, and CASP1 knockout THP-1 cells with these inhibitors and monitored cell viability using CellTiter-Glo, which measures cellular ATP levels as a surrogate for cell number and is higher throughput than LDH assays. Importantly, the potency of Val-boroPro measured by CellTiter-Glo (Fig. 3c, IC50 = 206 nM) closely matched that observed with the LDH assay in control THP-1 cells (Fig. 1d), and no additional cell death compared to DMSO controls was observed in the DPP8/9 or CASP1 knockout cell lines. Also as expected, we observed no cytotoxicity with the selective DPP4 inhibitor sitagliptin or the selective DPP7 inhibitor 5385. By contrast, the DPP8/9 inhibitors L-allo-Ile-isoindoline14 (a selective DPP8/9 inhibitor), L-allo-Ile-thiazolidine14 (a DPP4/8/9 inhibitor), and saxagliptin29 and vildagliptin30 (potent DPP4 inhibitors that also inhibit DPP8/9) all induced cell death in control THP-1 cells (with IC50 values ranging from 2.5 to 18.7 μM), but not in DPP8/9 or CASP1 knockouts (Fig. 3c, Supplementary Fig. 8a). We confirmed that these DPP8/9 inhibitors, but not sitagliptin or compound 5385, also induced LDH release from THP-1 cells (Supplementary Fig. 8b). These data further show that DPP8/9 inhibition induces cell death and highlights Val-boroPro as both the most potent DPP8/9 inhibitor and the most potent inducer of this form of cell death.
Importantly, we discovered that 1G244, unlike all the other inhibitors, not only killed control THP-1 cells (IC50 = 2.7 μM), but surprisingly also killed DPP8/9 and CASP1 knockout cells at slightly higher concentrations (IC50 = 21.5 μM for DPP8/9 KO1). 1G244 is considered one of the most potent and selective DPP8/9 inhibitors available, but these results unequivocally show that 1G244 interacts with an unknown off-target to induce cell death in THP-1 cells at doses >10 μM. We further demonstrated that 1G244 induces cell death via this off-target mechanism in other cell types, as 1G244, but not Val-boroPro or L-allo-Ile-isoindoline, kills HT-1080, MCF7, and caspase-1 knockout RAW 264.7 cells (Fig. 3d, Supplementary Fig. 7 d,f). Our data indicates that the cell death induced by 1G244 at low concentrations (≤10 μM) is primarily due to the inhibition of DPP8/9, but that results with this compound must be interpreted cautiously, particularly at higher doses. For example, 1G244 injection in rats at 30 mg/kg caused startle reflex, convulsions and cyanosis, and injection at ≥60 mg/kg caused immediate death25. As 1G244 achieved concentrations >10 μM in these animals, these toxic effects cannot be unambiguously attributed to DPP8/9 inhibition. Moreover, we note that 1G244 off-target-mediated cell death may even occur in some systems at doses ≤10 μM, as we saw small amounts of caspase-1-independent cell death induced in mouse RAW 264.7 cells with only 10 μM 1G244 (Supplemental Fig. 7d). Similarly, a previous study observed that treatment of U937 cells and monocyte-derived macrophages with 8 μM 1G244 induced a form of cell death that appeared to be apoptosis, as evidenced by an increase in caspase-3 activity and annexin V positive, propidium iodide negative staining31. We believe it is possible that these cytotoxic responses are due to the off-target activity of 1G244, or a combination of the off-target activity and DPP8/9 inhibition. Regardless, our results show that 1G244 has an off-target interaction that was not previously known and that this interaction induces cell death in all cell lines tested. 1G244 should therefore be used with caution and with genetic controls (e.g., DPP8/9 or caspase-1 knockout cell lines).
DPP8/9 inhibition activates pro-caspase-1
During canonical pyroptosis, mature, activated caspase-1 cleaves and activates proinflammatory cytokines, including IL-1β and IL-18, as well as proteins that induce pyroptosis, including GSDMD11,32. Although Val-boroPro-induced cell death was dependent on caspase-1, we immediately recognized that this cytotoxic response was atypical, as only pro-IL-1β was released from Val-boroPro-treated THP-1 cells (Fig. 1b). Moreover, Val-boroPro induced cell death in RAW 264.7 cells (Fig. 1e), which do not express the essential inflammasome component ASC33. We confirmed that ASC is not essential for Val-boroPro’s cytotoxicity in THP-1 cells either, as ASC-knockout THP-1 cells responded equivalently as control THP-1 cells (Fig. 4a, b). As a control, we confirmed that induction of pyroptosis by LPS plus nigericin, which activates caspase-1 via the NLRP3 inflammasome, is blocked in ASC-knockout THP-1 cells (Fig. 4b). Therefore, the active form of caspase-1 stimulated by DPP8/9 inhibition does not involve the formation of a canonical ASC-containing inflammasome.
Figure 4. DPP8/9 inhibition activates pro-caspase-1 without autoproteolysis.
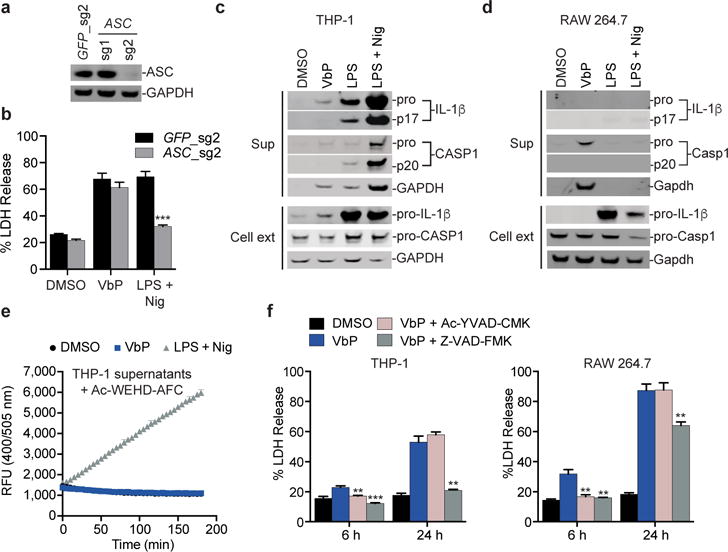
a, Confirmation of ASC knockout in THP-1 cells by immunoblotting. b, ASC-knockout THP-1 macrophages release LDH in response to Val-boroPro but not to LPS plus nigericin. Data are means ± SEM of three biological replicates. *** p < 0.001 by two-sided Student’s t-test for GFP control versus ASC knockout cells treated with LPS plus nigericin. c,d, Immunoblots of cell extracts and supernatants from THP-1 (c) or RAW 264.7 (d) macrophages treated with Val-boroPro or LPS plus nigericin. Val-boroPro elicits exclusively pro-caspase-1. Unlike THP-1 cells, RAW 264.7 cells do not endogenously produce IL-1β and therefore no IL-1β is detected in the supernatant of Val-boroPro-treated RAW 264.7 cells. RAW 264.7 cells do not release any intracellular contents after treatment with LPS plus nigericin because these cells are ASC-deficient and are therefore unable to activate the NLRP3 inflammasome. e, Cleavage of Ac-WEHD-AFC (50 μM) was monitored in supernatants from treated THP-1 macrophages. Data are means ± SEM of four independent experiments. f, Pre-treatment (30 min) of THP-1 (left) or RAW 264.7 (right) macrophages with Ac-YVAD-CMK or Z-VAD-FMK (50 μM) blocks Val-boroPro-induced cytotoxicity at 6 h. Data are means ± SEM of three biological replicates. ** p < 0.01, ***p < 0.001 by two-sided Student’s t-test for Val-boroPro alone versus Val-boroPro plus caspase inhibitor-treated cells. Full gel images for a, c and d are shown in Supplementary Figure 12.
We next wanted to investigate the active form of caspase-1 induced by Val-boroPro. Intriguingly, we found that Val-boroPro-treated THP-1 and RAW 264.7 cells secreted exclusively pro-caspase-1 (Fig. 4c,d), a finding supported by the observation that supernatants from these cells are unable to efficiently cleave the fluorgenic caspase-1 substrate Ac-WEHD-AFC (Fig. 4e, Supplementary Fig. 9a) and consistent with an ASC-independent mechanism. Similarly, Val-boroPro induced pro-caspase-1 secretion from primary mBMDMs and J774 cells (Supplementary Fig. 9b–e). In contrast to Val-boroPro, LPS plus nigericin-treated THP-1 cells secrete cleaved caspase-1, cleaved IL-1β, and cleaved IL-18 (Fig. 4c, Supplementary Fig. 1a), and supernatants from these cells robustly cleave the tetrapeptide substrate (Fig. 4e). These data indicate that pro-caspase-1 mediates Val-boroPro-induced pyroptosis.
We next tested whether the catalytic activity of pro-caspase-1 was required for pyroptosis by incubating THP-1 and RAW 264.7 macrophages with the specific caspase-1 inhibitor Ac-YVAD-CMK or the pan caspase inhibitor Z-VAD-FMK before adding Val-boroPro. Both inhibitors completely blocked Val-boroPro-induced pyroptosis in both cell lines at 6 h (Fig. 4f). However, Ac-YVAD-CMK had no impact on the cumulative LDH release over 24 hours in either cell line, and Z-VAD-FMK only partially reduced LDH release in RAW 264.7 cells at 24 hours. These results indicate that the catalytic activity of pro-caspase-1 is required, but that the duration of DPP8/9 inhibition outlasts the duration of caspase-1 inhibition. Consistent with this premise, re-administration of Ac-YVAD-CMK after four and eight hours lengthened the blockade of LDH release from RAW 264.7 cells (Supplementary Fig. 9f). Collectively, these results suggest that Val-boroPro induces an active form of pro-caspase-1 that can sufficiently cleave a substrate (or substrates) that mediates pyroptosis, but that does not efficiently cleave IL-1β or the tetrapeptide Ac-WEHD-AFC.
GSDMD mediates Val-boroPro-induced pyroptosis
GSDMD was recently identified as the key executor of pyroptosis11,32. After GSDMD is cleaved by caspases-1, -4, or -5, its N-terminal fragment promotes rapid cell death. GSDMD is required for noncanonical pyroptosis, but these early reports differ as to whether GSDMD knockout prevents or delays caspase-1-dependent canonical pyroptosis. Interestingly, several groups have recently reported that activators of the pathogenic sensor proteins NLRP1b (e.g., anthrax lethal toxin) and NLRC4 (e.g., S. typhimurium and P. aeruginosa) can generate activated forms of full-length pro-caspase-1 in ASC-deficient cells34–36. In particular, the activated form of pro-caspase-1 induced by NLRC4 ligands can cleave GSDMD and promote rapid cell death, but is unable to process IL-1β efficiently34,37. Our data above indicates that Val-boroPro might generate a similar form of activated pro-caspase-1. Indeed, THP-1 cells treated with Val-boroPro, 1G244, and other DPP8/9 inhibitors showed significant cleavage of GSDMD, but, as expected, no cleavage of the apoptotic caspases-3 or -7 or the apoptotic caspase substrate PARP (Fig. 5a, Supplementary Fig. 10). Similarly, GSDMD cleavage was also observed in RAW 264.7 cells, J774 cells, and hPBMCs after treatment with Val-boroPro (Fig. 5b). RAW 264.7 cells, which do not activate caspase-1 in response to LPS plus nigericin, do not cleave GSDMD after this treatment.
Figure 5. GSDMD is cleaved after DPP8/9 inhibition and contributes to cell death.
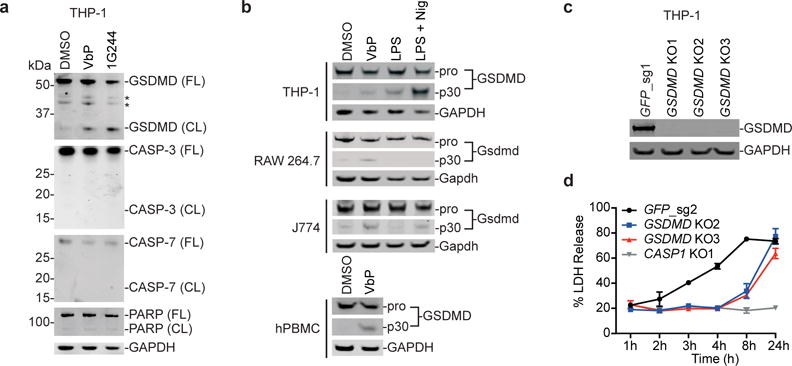
a, GSDMD is cleaved in THP-1 macrophage extracts after treatment with Val-boroPro or 1G244 for 24 h, but CASP-3, CASP-7, and PARP are not. FL, full-length; CL, cleaved; * denotes non-specific bands. b, GSDMD is cleaved after Val-boroPro treatment of THP-1, RAW 264.7, J774, and primary human PBMCs as determined by immunoblotting. RAW 264.7 do not cleave Gsdmd after LPS plus nigericin treatment, as expected. c, d, The pyroptotic response in GSDMD-deficient THP-1 macrophages, which were validated by immunoblotting (c), was delayed, but not entirely prevented (d). Full gel images for a–c are shown in Supplementary Figure 12. In d, data are means ± SEM of three biological replicates.
To investigate whether GSDMD mediates Val-boroPro-induced pyroptosis, we next isolated GSDMD knockout THP-1 cells and treated these cells with Val-boroPro (Fig. 5c,d). We observed that the onset of Val-boroPro-induced cell death is significantly delayed in GSDMD knockout cells, consistent with previous results that GSDMD knockout delays, but does not entirely prevent, caspase-1-mediated cell death38.
Caspase-1 knockout mice are insensitive to Val-boroPro
We hypothesized that the induction of pyroptosis in monocytes and macrophages by Val-boroPro might be a key factor for the stimulation of the immune response in mammals. Our data predict that monocytes and macrophages in Casp1−/− knockout mice will not undergo pyroptosis and could be used to test this hypothesis. We first confirmed that Val-boroPro (100 μg/mouse), but not sitagliptin (1000 μg/mouse), dramatically induces serum G-CSF and CXCL1/KC in wild-type mice (Supplementary Fig. 11). We next treated wild-type and Casp1−/− knockout C57BL/6NJ mice, which also harbor an intrinsic deficiency in caspase-1138, with Val-boroPro (100 μg/mouse) and assayed their serum G-CSF and CXCL1/KC levels after 6 h. Indeed, the Casp1−/− knockout mice showed no increase in serum G-CSF (Fig. 6a) or CXCL1/KC (Fig. 6b), confirming that caspase-1 is a key mediator of immune stimulation in vivo.
Figure 6. Val-boroPro does not induce cytokines in caspase-1-knockout mice.
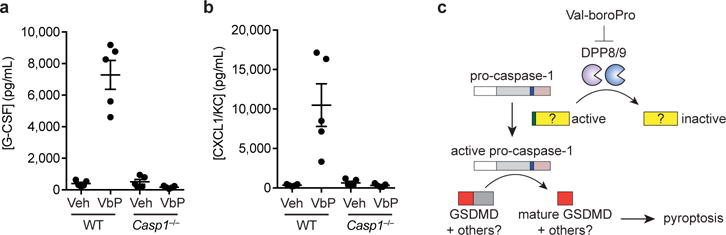
a,b Val-boroPro (100 μg/mouse) induces high levels of serum G-CSF (a) and CXCL1/KC (b) after 6 h in wild-type (WT), but not Casp1−/−, mice as measured by ELISA. p <0.0001 for G-CSF and p <0.01 for CXLCL1/KC in Val-boroPro-treated wild-type versus Val-boroPro-treated Casp1−/− mice. Data are means ± SEM. n = 5 mice/group. Veh, vehicle. c, Model for the induction of pyroptosis by DPP8/9 inhibition.
DISCUSSION
How Val-boroPro stimulates the immune system has remained enigmatic for more than a decade. Here we show that the inhibition of DPP8/9 by Val-boroPro results in the conversion of pro-caspase-1 into an activated form of pro-caspase-1, which then induces pyroptosis in monocytes and macrophages and stimulates the immune system (Fig. 6c). The absolute dependence on caspase-1, as demonstrated by the results in caspase-1 knockout cells and mice, is in itself sufficient to establish the cytotoxic effects as pyroptosis. However, we also show that several other hallmarks of pyroptosis are present. These include: 1) the cleavage of GSDMD, the key effector of pyroptosis; 2) that knockout of GSDMD delays the cytotoxic response; 3) that the apoptotic caspases-3 and -7 and the apoptotic caspase substrate PARP are not cleaved; and 4) that the cell death appears lytic and not apoptotic by microscopy. Significantly, Val-boroPro’s cytotoxic effects appear to be completely selective to monocytes and macrophages.
It is noteworthy that Val-boroPro is now the first small molecule demonstrated to have the ability to induce pyroptosis. In addition, while it is known that certain pathogenic stimuli can generate a pyroptosis-inducing form of pro-caspase-1 in systems lacking the key inflammasome adapter ASC34–36, DPP8/9 inhibition activates pro-caspase-1 regardless of whether or not ASC is present. Although the molecular mechanisms governing the ASC-independent activation of pro-caspase-1 remain unclear, our results demonstrate that the DPP8/9 cleavage and inactivation of some yet unidentified substrate which, unless held in check by DPP8/9 cleavage, goes on to induce pyroptosis (Fig. 6c). Therefore, DPP8/9 represent a new checkpoint that holds pro-caspase-1, and consequently the innate immune and ultimately the adaptive immune systems, at bay. A recent study attempted to discover DPP8/9 substrates on a proteome-wide scale using the N-terminomics approach called terminal amine isotopic labeling of substrates (TAILS) in a human ovarian cancer cell line39. In addition, a separate study profiled mouse embryonic fibroblasts (MEFs) from DPP9−/− mice for DPP9 substrates by two-dimensional differential in-gel electrophoresis40. While these studies together identified over one hundred potential substrates of DPP8/9, it is unclear which, if any, mediates the pyroptotic response we have discovered here. As such, the identification of the critical DPP8/9 substrates remains an important research problem, and our work suggests performing similar profiling experiments in monocytes may enable the capture of the biologically relevant substrates.
During the clinical development of DPP4 inhibitors, off-target inhibition of DPP8/9 was proposed to be responsible for a variety of toxicities, including alopecia, thrombocytopenia, and gastrointestinal toxicity14, but some investigators have challenged this proposal and it remains unsettled25,41,42. Our discovery here that 1G244, currently considered the most potent and selective DPP8/9 inhibitor available, induces cell death through an off-target mechanism shows that 1G244 cannot be unreservedly used to interrogate the biological effects of DPP8/9 inhibition and that studies already in the literature using 1G244 should be interpreted cautiously. It will now be important to develop more selective DPP8/9 inhibitors to determine if DPP8/9 inhibition mediates both the toxicities and the anticancer immune effects of Val-boroPro, or whether yet unknown factors are involved in either or both processes, in order to guide how this new immune checkpoint can best be harnessed for therapeutic benefit.
ONLINE METHODS
Cloning
sgRNAs were designed using the Broad Institute’s web portal43 (http://www.broadinstitute.org/rnai/public/analysis-tools/sgrna-design) and cloned into the lentiGuide-Puro vector (Addgene #52963) as described previously44. The sgRNA sequences are listed in Supplementary Table 2. To create double knockouts, we generated a version of this vector that contains a hygromycin selectable marker.
Reagents and antibodies
LPS was purchased from Santa Cruz Biotechnology, nigericin, and vildagliptin and Ac-YVAD-CMK from the Cayman Chemical Company, PMA and sitagliptin from Sigma, Ala-Pro-AFC from Bachem, saxagliptin from Toronto Research Chemicals, and Z-VAD-FMK and etoposide from Enzo Life Sciences. Val-boroPro45, 1G24424, FP-biotin15, L-allo-Ile-isoindoline14, and L-allo-Ile-thiazolidine14 were synthesized according to previously published protocols. For cell culture experiments, Val-boroPro was resuspended in DMSO containing 0.1% TFA to prevent compound cyclization. Antibodies used include: human caspase-1 (#2225, Cell Signaling Technology), mouse caspase-1 (clone Casper-1, Adipogen), caspase-3 (clone 8G10, Cell Signaling Technology), human caspase-4 (clone 4B9, Santa Cruz), human caspase-5 (clone D3G4W, Cell Signaling Technology), caspase-7 (clone D2Q3L, Cell Signaling Technology), human IL-1β (Clone 2805, R&D Systems), mouse IL-1β (clone D4T2D, Cell Signaling Technology), IL-1α (#AF-200, R&D Systems), IL-18 (#AF2548, R&D Systems), GAPDH (clone 14C10, Cell Signaling Technology), DPP7 (Clone 398024, R&D Systems), DPP8 (ab42076, Abcam), DPP9 (ab42080, Abcam), PARP (#9542, Cell Signaling Technology), GSDMD (NBP2-33422, Novus Biologicals), DPP4 (#11D7, GeneTex), FAP (ABT11, Millipore), and SCPEP1 (SAB2700267, Sigma).
Cell culture
HEK 293T, HeLa, HT-1080, J774, JAWS II, Jurkat, MDA-MB-231, MCF7, RAW 264.7, THP-1, U937 and hPBMCs were purchased from ATCC. HL-60 cells were a gift from A. Kentsis and verified using STR genotyping (Genetica). HEK 293T, HeLa, HT-1080, J774, PBMCs, and RAW 264.7 cells were grown in Dulbecco’s modified Eagle’s medium (DMEM) with 10% fetal bovine serum (FBS). MDA-MB-231, MCF-7, Jurkat, THP-1 and U937 were grown in RPMI-1640 medium with 10% FBS. HL-60 cells were grown in Iscove’s Modified Dulbecco’s Medium with 20% FBS. JAWS II cells were grown in alpha minimum essential medium with ribonucleosides, deoxyribonucleosides, 4 mM L-glutamine, 1 mM sodium pyruvate and 5 ng/mL murine GM-CSF (80%) supplemented with 20% FBS. All cells were grown at 37 °C in a 5% CO2 incubator. Cell lines were tested for mycoplasma by the PCR method (Universal Mycoplasma Detection Kit, ATCC).
LDH cytotoxicity and IL-1β ELISA assays
For experiments involving THP-1 macrophages, THP-1 monocytes were seeded at 1 × 106 cells/well in 6-well plates in RPMI-1460 with 10% FBS and 50 ng/mL PMA and differentiated for 3 d. U937 cells were seeded at 1 × 106 cells/well in 6-well plates in RPMI with 10% FBS and 100 ng/mL PMA for 2 d. Primary mBMDM were isolated from the femurs and tibias of four 6 month old 6 B6D2F1 mice, and 1 × 107 cells were plated on 10 cm petri dishes and differentiated in DMEM with 10% FBS and 25 ng/mL recombinant M-CSF (PeproTech) for 6 days. mBMDM were then scraped and reseeded at 0.5 × 106 cells/well in 6-well plates in Opti-MEM and incubated overnight before drug treatment. This animal protocol was reviewed and approved by the Memorial Sloan Kettering Cancer Center Institutional Animal Care and Use Committee (IACUC). All other cells were plated at 0.5 × 106 cells in standard gro h medium 24 h prior to drug treatment. Cells were treated with Val-boroPro (2 μM), 1G244 (10 μM), or LPS (10 μg/mL) for 24 h, unless indicated otherwise. After 24 h, LPS-primed cells were treated with nigericin (20 μM) for 45 minutes. Supernatants were then harvested and analyzed for LDH activity using an LDH cytotoxicity assay kit (Pierce) and for IL-1β levels using an IL-1β ELISA kit (R&D Systems) according to the manufacturers’ instructions. The reported t-test values are two-sided.
CellTiter-Glo cell viability assays
Cells were plated (2000 cells/well) in white, 384-well clear bottom plates (Corning) using a EL406 Microplate Washer/Dispenser (BioTek) in 25 μL final volume of media. Compounds were added using a pintool (CyBio). RAW 264.7 cells were incubated for 24 h and THP-1 monocytes were incubated for 48 h at 37 °C. Assay plates were then removed from the incubator and allowed to equilibrate to room temperature on the bench top before addition of 10 μL of CellTiter-Glo reagent (Promega) according to the manufacturer’s instructions. Assay plates were shaken on an orbital shaker for 2 minutes and incubated at room temperature on the bench top for 10 minutes. Luminescence was then read using a Cytation 5 Cell Imaging Multi-Mode Reader (BioTek).
Imaging of cell death by microscopy
To examine cell death morphology, RAW 264.7 cells were plated at 1.5 × 104 cells on 8 chambered slides (Nunc Lab-Tek II Chambered Coverglass) and treated with DMSO, Val-boroPro (2 μM), 1G244 (10 μM), or etoposide (50 μM). Static bright field images were captured using Zeiss AxioObserver inverted microscope with a 10×/0.45NA objective every 5 minutes over 24 h. Images were processed using the Zeiss ZEN 2 Blue Edition software. The data shown is representative of 4 randomly selected fields.
Caspase-1 activity assays
THP-1 cells (7 × 106 cells/plate) were differentiated in 10 cm dishes in complete medium with 50 ng/mL PMA for 3 d. RAW 264.7 and J774 cells were plated at 3 × 106 cells per 10 cm plate in complete medium and incubated overnight. The cells were washed with 2 × 5 mL PBS before the addition of 10 mL of serum free media. Cells were then treated with Val-boroPro (2 μM) or LPS (10 μg/mL) for 24 h. After 24 h, LPS-treated cells were stimulated with nigericin (20 μM) for 45 minutes. The supernatants were then harvested, spun at 400g to remove debris, and concentrated ~20-fold using Amicon Ultra Centrifugal Filter Units (EMD Millipore, 10 kDa molecular weight cutoff). Concentrated supernatants were assayed for cleavage of the tetrapeptide substrate Ac-WEHD-AFC using the caspase-1 fluorometric activity assay kit (R&D Systems) according to the manufacturer’s instructions. The concentrated supernatants were also used for immunoblotting.
Intracellular DPP8/9 inhibition assay
HEK 293T cells express low levels of endogenous DPP8/9, but not DPP4, DPP7, or FAP46,47, and therefore can be used to assess intracellular inhibition of DPP8/9 without interference from background DPP activity. HEK 293T cells were washed with PBS, resuspended in RPMI 1640, and plated (1 × 105 cells/well) in a 96-well black clear-bottomed plate (100 μL final concentration). The cells were incubated overnight before the addition of Val-boroPro incubation for an additional 2 h. Ala-Pro-AFC substrate (250 μM final concentration) was then added to each well and fluorescence was monitored (excitation 400 nm, emission 505 nm). Similarly, THP-1 cells were washed with PBS, resuspended in Opti-MEM, and plated (1 × 105 cells/well) in a 384-well black clear-bottomed plate (100 μL final volume). As THP-1 cells express endogenous DPP8/9 and DPP7, but not DPP4 or FAP, cells were incubated for 30 min with the selective DPP7 inhibitor 5385 (20 μM) to assess intracellular inhibition of DPP8/9 without interference from background DPP7 activity. The cells were then incubated with the indicated inhibitors for 30 minutes before the addition of the Ala-Pro-AMC substrate (250 μM final concentration). Fluorescence was then monitored (excitation 380 nm, emission 460 nm).
FP-biotin enrichment of serine proteases
2 × 107 THP-1 cells were pelleted at 400g, resuspended in 1 mL of PBS, and lysed by sonication. Protein concentrations were determined using the DC Protein Assay kit (Bio-Rad) and adjusted to 1 mg/mL. 1 mL of 1 mg/mL lysate was reacted with the FP-biotin probe (5 μM) for 1 h at 25 °C. Triton-X100 was then added to a final concentration of 1% and samples were incubated for 1 h while rotating at 4 °C to solubilize membranes. Lysates were then separated from free probe by passage over a Sephadex G-25M column (GE Healthcare). SDS was added to a final concentration of 0.5% (in 3.5 mL total volume) and samples were boiled for 10 m at 95 °C to denature proteins. Samples were then diluted in PBS to 11 mL final volume before the addition of 200 μL of NeutrAvidin-Agarose resin (Pierce) and incubation for 1 h while rotating at 25 °C. Samples were then washed with 4 × 10 mL PBS and eluted by resuspending in 150 mL 2X sample loading dye and boiling at 95 °C for 10 min.
Gel-based competitive ABPP assays
Cell lysates (1 mg/mL) in PBS were incubated with DMSO or compound for 30 min at 25 °C (25 μL total reaction volume). FP-biotin was then added at a final concentration of 1 μM. After 1 h, the reactions were quenched with 2× SDS-PAGE loading buffer (reducing), separated by SDS-PAGE (4–20% acrylamide), and transferred to a nitrocellulose membrane. Blots were probed using the a streptavidin-coupled IR dye following manufacturers’ instructions, and were visualized and quantified using the Odyssey Imaging System (Li-Cor).
EnPlex assay
Selectivity profiles were determined by EnPlex as described previously15.
In vitro enzyme IC50 assays for compound 5385
In vitro inhibition of DPP4, DPP7, DPP8, DPP9, and FAP were determined as described previously.45,48
Knockout cell lines
Constructs were packaged into lentivirus in HEK 293T cells using the Fugene HD transfection reagent (Promega) and 2 μg of the vector, 2 μg psPAX2, and 0.2 μg pHEF-VSV-G. THP-1 or RAW 264.7 cells were spinfected with virus for 2 h at 1000g at 30 °C supplemented 8 μg/mL polybrene. After 2 days, cells were selected for stable expression of S. pyogenes Cas9 (Addgene #52962) using blasticidin (5 μg/mL for THP-1, 1 μg/mL for RAW 264.7) and for stable expression of sgRNAs using puromycin (0.5 μg/mL for THP-1, 5 μg/mL for RAW 264.7) or hygromycin (100 μg/mL for THP-1). After 10 d, the cells were harvested for immunoblotting or experiments. To confirm allelic modifications, genomic DNA was harvested using Blood and Tissue Culture Kit (Qiagen). A 600 base pair region containing the sgRNA cut site was then PCR amplified and subcloned using the CloneJET PCR Cloning Kit (ThermoFisher). Single bacterial colonies were isolated and sequenced.
Cytokine stimulation in mice
9 week old male caspase-1 knockout (B6N.129S2-Casp1tm1Flv/J), C57BL/6NJ and C57BL/6 mice were obtained from the Jackson Laboratory, and BALB/c mice were obtained from Charles River Laboratories. Mice were fasted for two hours prior to oral treatment with 100 μg Val-boroPro or 1000 μg sitagliptin in pH 2.0 water (0.01N HCl). Serum was collected at indicated time after dosing via cardiac puncture. G-CSF and CXCL1/KC levels were measured by Quantikine ELISA (R&D Systems). Sample size was based on the statistical analysis of previous experiments with vehicle treated mice versus Val-boroPro treated mice7. In these experiments, a sample size of 5 allows detection of increased G-CSF and CXCL1/KC levels with 80% power and a significance level below p = 0.05. No animals were excluded. The experiments were not randomized and the investigators were not blinded. This animal protocol was reviewed and approved by the Tufts University Medical Center Institutional Animal Care and Use Committee (IACUC).
Synthesis of dipeptide boronate cyclohexyl-Gly-boroNva (Compound 5385)
To a solution of Boc-1-aminocyclohexane-carboxylic acid (0.4 mmol) in DMF (3ml) was added DIPEA (0.14 ml, 0.83 mmol) at 0°C, followed by HATU (150 mg, 0.41 mmol), L-boroNva-pn.HCl (160 mg, 0.41 mmol). The resulting mixture was stirred at room temperature for 1h and concentrated in vacuo under 30°C. The residue was re-dissolved in EtOAc (25 ml), washed with 0.1N KHSO4 (3 ´ 5 ml), saturated aq. NaHCO3 (2 ´ 5 ml), brine (5 ml) and then dried over MgSO4, filtered, and concentrated in vacuo to yield the crude product. Without any purification further, the coupling compound was dissolved in dry CH2Cl2 (4 ml) and cooled to -78°C. BCl3 (4 ml of 1M solution in CH2Cl2, 4mmol) was added dropwise. The mixture was stirred at −78°C for 1h, and then was concentrated in vacuo. Water (5 ml) and ether (5 ml) were added, the aqueous phase was separated and washed with ether (2 ´ 5 ml). The aqueous phase was then concentrated in vacuo and purified by semi-preparative RP-HPLC to afford the target compound. 1H NMR (D2O): δ 0.84 (t, J = 7.3 Hz, 3H, CH3CH2CH2), 1.22 – 1.64 (m, 10H, CH3CH2CH2 and protons at cyclohexyl), 1.85 – 2.03 (m, 4H, protons at cyclohexyl), 2.74 (t, J = 7.4 Hz, 1H, NHCHB). MS (ESI+) for C11H23BN2O3 m/z (rel intensity): 449.3 ([2 ´ (M – H2O) + H]+, 100), 225.2 ([M – H2O + H]+, 75).
Supplementary Material
Acknowledgments
We thank S. Fujisawa for microscopy assistance, E. De Stanchina and B. Qeriqi for assistance harvesting mBMDMs, C. Taabazuing for helpful comments, and A. Kentsis and F. Brown for the HL-60 cell line. This work was supported by Josie Robertson Foundation (D.A.B), the MSKCC Core Grant (P30 CA008748), the NCI (grant no. U54CA112962 to T.R.G.), HHMI (T.R.G.), and NIH (CA174008-01A1 to W.W.B.).
Footnotes
AUTHOR CONTRIBUTIONS
D.A.B. conceived and directed the project, performed experiments, analyzed data, and wrote the paper; M.O., D.C.J., R.S., E.B.G., A.J.C, and M.S.W. performed experiments and analyzed data; S.E.P. performed the in vivo mouse experiments and the intracellular DPP8/9 inhibition experiment; W.W.B. and D.G.S. directed the in vivo mouse experiments; W.W., Y.L., and J.H.L. synthesized Val-boroPro, 1G244, L-allo-Ile-isoindoline, L-allo-Ile-thiazolidine, compound 5385 and FP-biotin; M.O.A. characterized compound 5385. T.R.G. and W.W.B. helped plan the study.
COMPETING FINANCIAL INTERESTS
William W. Bachovchin is a co-founder, advisor and Board member of Arisaph Pharmaceuticals, a biotechnology company interested in developing boronic acid-based inhibitors of serine proteases as therapeutics.
References
- 1.Zitvogel L, Tesniere A, Kroemer G. Cancer despite immunosurveillance: immunoselection and immunosubversion. Nat Rev Immunol. 2006;6:715–27. doi: 10.1038/nri1936. [DOI] [PubMed] [Google Scholar]
- 2.Dunn GP, Bruce AT, Ikeda H, Old LJ, Schreiber RD. Cancer immunoediting: from immunosurveillance to tumor escape. Nat Immunol. 2002;3:991–8. doi: 10.1038/ni1102-991. [DOI] [PubMed] [Google Scholar]
- 3.Topalian SL, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. 2012;366:2443–54. doi: 10.1056/NEJMoa1200690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wolchok JD, et al. Nivolumab plus ipilimumab in advanced melanoma. N Engl J Med. 2013;369:122–33. doi: 10.1056/NEJMoa1302369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sharma P, Allison JP. Immune checkpoint targeting in cancer therapy: toward combination strategies with curative potential. Cell. 2015;161:205–14. doi: 10.1016/j.cell.2015.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Couzin-Frankel J. Breakthrough of the year 2013. Cancer immunotherapy. Science. 2013;342:1432–3. doi: 10.1126/science.342.6165.1432. [DOI] [PubMed] [Google Scholar]
- 7.Adams S, et al. PT-100, a small molecule dipeptidyl peptidase inhibitor, has potent antitumor effects and augments antibody-mediated cytotoxicity via a novel immune mechanism. Cancer Res. 2004;64:5471–80. doi: 10.1158/0008-5472.CAN-04-0447. [DOI] [PubMed] [Google Scholar]
- 8.Walsh MP, et al. Val-boroPro accelerates T cell priming via modulation of dendritic cell trafficking resulting in complete regression of established murine tumors. PLoS One. 2013;8:e58860. doi: 10.1371/journal.pone.0058860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jones B, et al. Hematopoietic stimulation by a dipeptidyl peptidase inhibitor reveals a novel regulatory mechanism and therapeutic treatment for blood cell deficiencies. Blood. 2003;102:1641–8. doi: 10.1182/blood-2003-01-0208. [DOI] [PubMed] [Google Scholar]
- 10.Jesson MI, et al. Immune mechanism of action of talabostat: a dipeptidyl peptidase targeted antitumor agent. Proceedings of the 98th Annual Meeting of the American Association for Cancer Research Vol Abstract #. 2007;1984 [Google Scholar]
- 11.Kayagaki N, et al. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature. 2015;526:666–71. doi: 10.1038/nature15541. [DOI] [PubMed] [Google Scholar]
- 12.Broz P. Immunology: Caspase target drives pyroptosis. Nature. 2015;526:642–3. doi: 10.1038/nature15632. [DOI] [PubMed] [Google Scholar]
- 13.Kortmann J, Brubaker SW, Monack DM. Cutting Edge: Inflammasome Activation in Primary Human Macrophages Is Dependent on Flagellin. J Immunol. 2015;195:815–9. doi: 10.4049/jimmunol.1403100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lankas GR, et al. Dipeptidyl peptidase IV inhibition for the treatment of type 2 diabetes: potential importance of selectivity over dipeptidyl peptidases 8 and 9. Diabetes. 2005;54:2988–94. doi: 10.2337/diabetes.54.10.2988. [DOI] [PubMed] [Google Scholar]
- 15.Bachovchin DA, et al. A high-throughput, multiplexed assay for superfamily-wide profiling of enzyme activity. Nat Chem Biol. 2014;10:656–63. doi: 10.1038/nchembio.1578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ohnuma K, et al. CD26 up-regulates expression of CD86 on antigen-presenting cells by means of caveolin-1. Proc Natl Acad Sci U S A. 2004;101:14186–91. doi: 10.1073/pnas.0405266101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lee HJ, et al. Investigation of the dimer interface and substrate specificity of prolyl dipeptidase DPP8. J Biol Chem. 2006;281:38653–62. doi: 10.1074/jbc.M603895200. [DOI] [PubMed] [Google Scholar]
- 18.Tang HK, et al. Biochemical properties and expression profile of human prolyl dipeptidase DPP9. Arch Biochem Biophys. 2009;485:120–7. doi: 10.1016/j.abb.2009.02.015. [DOI] [PubMed] [Google Scholar]
- 19.Ajami K, Abbott CA, McCaughan GW, Gorrell MD. Dipeptidyl peptidase 9 has two forms, a broad tissue distribution, cytoplasmic localization and DPIV-like peptidase activity. Biochim Biophys Acta. 2004;1679:18–28. doi: 10.1016/j.bbaexp.2004.03.010. [DOI] [PubMed] [Google Scholar]
- 20.Abbott CA, et al. Cloning, expression and chromosomal localization of a novel human dipeptidyl peptidase (DPP) IV homolog, DPP8. Eur J Biochem. 2000;267:6140–50. doi: 10.1046/j.1432-1327.2000.01617.x. [DOI] [PubMed] [Google Scholar]
- 21.Zhang H, Chen Y, Keane FM, Gorrell MD. Advances in understanding the expression and function of dipeptidyl peptidase 8 and 9. Mol Cancer Res. 2013;11:1487–96. doi: 10.1158/1541-7786.MCR-13-0272. [DOI] [PubMed] [Google Scholar]
- 22.Waumans Y, Baerts L, Kehoe K, Lambeir AM, De Meester I. The Dipeptidyl Peptidase Family, Prolyl Oligopeptidase, and Prolyl Carboxypeptidase in the Immune System and Inflammatory Disease, Including Atherosclerosis. Front Immunol. 2015;6:387. doi: 10.3389/fimmu.2015.00387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wagner L, Klemann C, Stephan M, von Horsten S. Unravelling the immunological roles of dipeptidyl peptidase 4 (DPP4) activity and/or structure homologue (DASH) proteins. Clin Exp Immunol. 2016;184:265–83. doi: 10.1111/cei.12757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jiaang WT, et al. Novel isoindoline compounds for potent and selective inhibition of prolyl dipeptidase DPP8. Bioorg Med Chem Lett. 2005;15:687–91. doi: 10.1016/j.bmcl.2004.11.023. [DOI] [PubMed] [Google Scholar]
- 25.Wu JJ, et al. Biochemistry, pharmacokinetics, and toxicology of a potent and selective DPP8/9 inhibitor. Biochem Pharmacol. 2009;78:203–10. doi: 10.1016/j.bcp.2009.03.032. [DOI] [PubMed] [Google Scholar]
- 26.Waumans Y, et al. The Dipeptidyl Peptidases 4, 8, and 9 in Mouse Monocytes and Macrophages: DPP8/9 Inhibition Attenuates M1 Macrophage Activation in Mice. Inflammation. 2016;39:413–24. doi: 10.1007/s10753-015-0263-5. [DOI] [PubMed] [Google Scholar]
- 27.Lamkanfi M, Dixit VM. Mechanisms and functions of inflammasomes. Cell. 2014;157:1013–22. doi: 10.1016/j.cell.2014.04.007. [DOI] [PubMed] [Google Scholar]
- 28.Shi J, et al. Inflammatory caspases are innate immune receptors for intracellular LPS. Nature. 2014;514:187–92. doi: 10.1038/nature13683. [DOI] [PubMed] [Google Scholar]
- 29.Augeri DJ, et al. Discovery and preclinical profile of Saxagliptin (BMS-477118): a highly potent, long-acting, orally active dipeptidyl peptidase IV inhibitor for the treatment of type 2 diabetes. J Med Chem. 2005;48:5025–37. doi: 10.1021/jm050261p. [DOI] [PubMed] [Google Scholar]
- 30.Villhauer EB, et al. 1-[[(3-hydroxy-1-adamantyl)amino]acetyl]-2-cyano-(S)-pyrrolidine: a potent, selective, and orally bioavailable dipeptidyl peptidase IV inhibitor with antihyperglycemic properties. J Med Chem. 2003;46:2774–89. doi: 10.1021/jm030091l. [DOI] [PubMed] [Google Scholar]
- 31.Matheeussen V, et al. Dipeptidyl peptidases in atherosclerosis: expression and role in macrophage differentiation, activation and apoptosis. Basic Res Cardiol. 2013;108:350. doi: 10.1007/s00395-013-0350-4. [DOI] [PubMed] [Google Scholar]
- 32.Shi J, et al. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature. 2015;526:660–5. doi: 10.1038/nature15514. [DOI] [PubMed] [Google Scholar]
- 33.Pelegrin P, Barroso-Gutierrez C, Surprenant A. P2X7 receptor differentially couples to distinct release pathways for IL-1beta in mouse macrophage. J Immunol. 2008;180:7147–57. doi: 10.4049/jimmunol.180.11.7147. [DOI] [PubMed] [Google Scholar]
- 34.Broz P, von Moltke J, Jones JW, Vance RE, Monack DM. Differential requirement for Caspase-1 autoproteolysis in pathogen-induced cell death and cytokine processing. Cell Host Microbe. 2010;8:471–83. doi: 10.1016/j.chom.2010.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Van Opdenbosch N, et al. Activation of the NLRP1b inflammasome independently of ASC-mediated caspase-1 autoproteolysis and speck formation. Nat Commun. 2014;5:3209. doi: 10.1038/ncomms4209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Guey B, Bodnar M, Manie SN, Tardivel A, Petrilli V. Caspase-1 autoproteolysis is differentially required for NLRP1b and NLRP3 inflammasome function. Proc Natl Acad Sci U S A. 2014;111:17254–9. doi: 10.1073/pnas.1415756111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.He WT, et al. Gasdermin D is an executor of pyroptosis and required for interleukin-1beta secretion. Cell Res. 2015;25:1285–98. doi: 10.1038/cr.2015.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kayagaki N, et al. Non-canonical inflammasome activation targets caspase-11. Nature. 2011;479:117–21. doi: 10.1038/nature10558. [DOI] [PubMed] [Google Scholar]
- 39.Wilson CH, et al. Identifying natural substrates for dipeptidyl peptidases 8 and 9 using terminal amine isotopic labeling of substrates (TAILS) reveals in vivo roles in cellular homeostasis and energy metabolism. J Biol Chem. 2013;288:13936–49. doi: 10.1074/jbc.M112.445841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang H, et al. Identification of novel dipeptidyl peptidase 9 substrates by two-dimensional differential in-gel electrophoresis. FEBS J. 2015;282:3737–57. doi: 10.1111/febs.13371. [DOI] [PubMed] [Google Scholar]
- 41.Burkey BF, et al. Adverse effects of dipeptidyl peptidases 8 and 9 inhibition in rodents revisited. Diabetes Obes Metab. 2008;10:1057–61. doi: 10.1111/j.1463-1326.2008.00860.x. [DOI] [PubMed] [Google Scholar]
- 42.Rosenblum JS, Liu Y, Wu J, Kozarich JW. American Diabletes Association Conference. Chicago, IL: 2007. The Case against Toxicity from DPP8/9 Inhibition. [Google Scholar]
- 43.Doench JG, et al. Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nat Biotechnol. 2016;34:184–91. doi: 10.1038/nbt.3437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sanjana NE, Shalem O, Zhang F. Improved vectors and genome-wide libraries for CRISPR screening. Nat Methods. 2014;11:783–4. doi: 10.1038/nmeth.3047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Coutts SJ, et al. Structure-activity relationships of boronic acid inhibitors of dipeptidyl peptidase IV. 1. Variation of the P2 position of Xaa-boroPro dipeptides. J Med Chem. 1996;39:2087–94. doi: 10.1021/jm950732f. [DOI] [PubMed] [Google Scholar]
- 46.Danilova O, Li B, Szardenings AK, Huber BT, Rosenblum JS. Synthesis and activity of a potent, specific azabicyclo[3.3.0]-octane-based DPP II inhibitor. Bioorg Med Chem Lett. 2007;17:507–10. doi: 10.1016/j.bmcl.2006.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang XM, Yu DM, McCaughan GW, Gorrell MD. Fibroblast activation protein increases apoptosis, cell adhesion, and migration by the LX-2 human stellate cell line. Hepatology. 2005;42:935–45. doi: 10.1002/hep.20853. [DOI] [PubMed] [Google Scholar]
- 48.Poplawski SE, et al. Identification of selective and potent inhibitors of fibroblast activation protein and prolyl oligopeptidase. J Med Chem. 2013;56:3467–77. doi: 10.1021/jm400351a. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.