

Abstract
Thiomuracin is a thiopeptide antibiotic with potent activity towards Gram-positive drug-resistant bacteria. Thiomuracin is biosynthesized from a precursor peptide, TbtA, by a complex array of posttranslational modifications. One of several intriguing transformations is the C-methylation of thiazole, occurring at an unactivated sp2 carbon. Herein, we report the in vitro reconstitution of TbtI, the responsible radical S-adenosyl-methionine (rSAM) C-methyltransferase, which catalyzes the formation of 5-methylthiazole at a single site. Our studies demonstrate that a linear hexazole-bearing intermediate of TbtA is a substrate for TbtI whereas macrocyclized thiomuracin GZ is not. In determining the minimal substrate for TbtI, we found that the enzyme is functional when most of the leader peptide has been removed. The in vitro reconstitution of TbtI, a class C rSAM methyltransferase, further adds to the chemical versatility of rSAM enzymes, and informs on the complexity of thiomuracin biosynthesis.
Graphical abstract
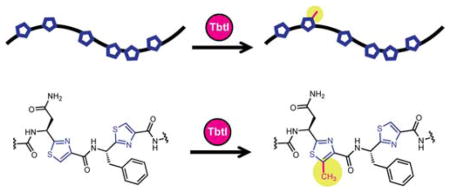
Thiopeptides are structurally complex natural products that are classified within the ribosomally synthesized and posttranslationally modified peptide (RiPP) family.1 Thiopeptides are renowned for their antibiotic activity, which arises from the inhibition of either elongation factor thermo-unstable (EF-Tu) or the 50S subunit of the bacterial ribosome.2 Usually, RiPP precursor peptides consist of a leader peptide (LP), important for recognition by select biosynthetic proteins, and a core peptide where the posttranslational modifications take place.1,3,4 Since the precursor peptide is of ribosomal origin, RiPPs offer a high degree of malleability for biosynthetic engineering.5,6 Recently, we reported the in vitro reconstitution of the core scaffold of thiomuracin GZ (4) and studied the substrate specificity and timing of various enzymes encoded in the biosynthetic gene cluster (BGC, Figure 1).7,8 Thiomuracin biosynthesis begins with the cyclodehydration (TbtFG) and dehydrogenation (TbtE) of six Cys residues within the precursor peptide (TbtA, 1) to yield a hexathiazole intermediate (2). Subsequently, four Ser residues are dehydrated through a glutamylation/elimination sequence (TbtBC) to form a tetradehydrated hexazole peptide (3). Finally, two of the alkenes in 3 undergo a formal [4+2] cycloaddition (TbtD), with the elimination of water and the LP to yield 4 (Figure 1).7,8
Figure 1.

(A) Thiomuracin GZ (4) biosynthesis. (B) Structure of thiomuracin A1 (5). (C) Structure of GE2270A (6). LP, leader peptide. Red, C-methylation sites.
These transformations convert an inactive precursor peptide into a macrocyclic antibiotic with potent activity towards Gram-positive pathogens.7,9 In both thiomuracin A1 (5) and GE2270A (6) the thiazole (Thz) arising from Cys4 is C-methylated; in 6, the Thz at position 6 is also C-methylated (Figure 1).2 These transformations are remarkable in that they occur on unactivated sp2 carbon centers. Inspection of the BGC of 5 identifies a single gene, tbtI, annotated as a rSAM enzyme (Figure S1).9 Prior studies on 6 implicate TbtI in thiazole C-methylation of 5. The BGC for 6 encodes a nearly identical rSAM protein, PbtM2, in addition to a paralog, PbtM3 (Figure S1). Gene deletion experiments demonstrated that PbtM2 and PbtM3 C-methylate Thz4 and Thz6 of 6, respectively.10 In this work, we investigated the timing and substrate scope of thiazole C-methylation by TbtI in vitro.7,9
rSAM enzymes catalyze chemically difficult reactions in primary metabolism and natural product biosynthesis.11–14 A subset of rSAM proteins catalyze methylation of non-nucleophilic acceptors, such as unactivated sp2 or sp3 carbons.15–17 Currently, there are four classes of rSAM methyltransferases.18 Class A includes the RNA-modifying enzymes RlmN and Cfr.19,20 Class B comprises cobalamin-dependent methyltransferases, such as TsrM,21,22 Fom3,23,24 GenK,25 CysS,26 and PoyC.27 Class C are cobalamin-independent and found in a variety of natural product BGCs.28,29 Although class A and B enzymes have been extensively studied,16,19 class C methyltransferases have received much less attention.15,18 The only examples of partially characterized class C methyltransferases are ChuW and NosN.30,31 Class D includes a methylenetetrahydrofolate-dependent enzyme involved in methanopterin biosynthesis.32
To confirm the previous gene deletion experiments implicating rSAMs in thiazole C-methylation, we recombinantly expressed and purified TbtI from Escherichia coli (Figure S2).33 Using the ferrozine and methylene blue assays we found that it contained 1.6 ± 0.2 Fe and 1.9 ± 0.3 sulfides per monomer. 34,35 Fe-S cluster reconstitution with recombinant cysteine desulfurase (IscS), Fe(II), Cys, and dithiothreitol yielded 3.7 ± 0.3 Fe and 4.1 ± 0.3 sulfides per protein, near the expected values for a single [4Fe-4S] cluster. UV-Vis analysis of TbtI also supported the proper incorporation of the Fe-S cluster (Figure S3).
To determine the timing of thiazole C-methylation, we first evaluated whether 4 was a substrate. Under anaerobic conditions, 4 was treated with TbtI in the presence of SAM and dithionite. The reaction mixture was analyzed by matrix-assisted laser desorption ionization time-of-flight mass spectrometry (MALDI-TOF-MS) and no mass shift (+14 Da) was observed (Figure S4). To investigate if methylation occurred at an earlier point in the biosynthesis, we tested the TbtA hexazole 2 and the downstream tetradehydrate 3 as substrates for TbtI. Analysis by MALDI-TOF-MS showed a +14 Da species consistent with a single methylation after the reaction with 2 (Figure S5). Control experiments lacking SAM, TbtI, or dithionite did not result in a mass shift. Under our reaction conditions, 3 was not a substrate for TbtI (Figure S6). Based on the structures of 5 and 6 (Figure 1) and previous gene deletion experiments,10 methylation of 2 likely occurred at Thz4. To test this hypothesis, the pentazole-containing versions of TbtA-C4A and -C4S were prepared and tested as substrates for TbtI. No mass shift was observed in either case, implicating Thz4 as a critical moiety for TbtI (Figure S7).
A prominent feature of RiPP biosynthesis is the role of the LP in substrate recognition by the modifying enzymes.1 Although TbtI lacks an identifiable RiPP Recognition Element (RRE),36 an indicator of LP dependence, TbtD also lacks an RRE yet is LP-dependent;7 further, the RRE-containing TbtB is LP-independent.8 To assess the LP-dependence of TbtI, 2 was treated with the endoproteinase GluC to generate 7, which retains Val-Gly-Ala from the LP. In testing 7 as a TbtI substrate, we observed full conversion to the methylthiazole 8 (Figure 2). Control experiments omitting TbtI, SAM, or dithionite did not result in observable modification of 7. The site of methylation in 8 was confirmed as Thz4 by high-resolution electrospray ionization tandem MS analysis (HR-ESI-MS/MS; Figure 3). These data indicate that most of the LP (31 out of 34 residues) is dispensable for TbtI activity and that the enzyme recapitulates in vitro the specificity observed in the natural product.
Figure 2.

(A) MALDI-TOF mass spectra of 7 treated as indicated. (B) Reaction catalyzed by TbtI to convert 7 to 8.
Figure 3.

MS/MS of the TbtI reaction product 8. The c5, c7, and y8 ions localize the site of C-methylation to the Thz4. Predicted masses and associated errors are in Table S2.
We next addressed the regioselectivity determinants of TbtI, given that Thz4 undergoes selective C-methylation despite the presence of five additional thiazoles. We hypothesized that TbtI recognizes a substructure of 2 adjacent to Thz4. Inspection of 5 and 6 shows that the methylthiazole is preceded in both cases by an Asn3, which in 6 undergoes additional N-methylation.10 It has been reported that Asn3 forms a key contact with EF-Tu and that substitution of Asn3 results in loss of antibiotic activity.37,38 To investigate whether Asn3 governs the regioselectivity of TbtI, we replaced this residue in TbtA with Ala, Asp, and Gln, followed by hexazole production in E. coli.8 The N3A-, N3D- and N3Q-bearing variants of 7 were then anaerobically treated with TbtI, prior to MALDI-TOF-MS analysis. Under the conditions used, the reaction with substrate 7 went to completion. In contrast, no methylation was observed with any Asn3 variant (Figure S8). These data demonstrate that Asn3 in TbtA is critical for TbtI activity.
We hypothesized that introducing an Asn N-terminal to one of the other thiazoles might redirect the location of methylation. Thus, four TbtA double-substituted variants of 7 were constructed by inserting Asn at a position preceding Thz 2, 6, 9 and 12 and replacing Cys4 with Ala. These four GluC-treated pentazole variants (S1N/C4A, C4A/F5N, C4A/I8N, and C4A/S11N) were then tested as TbtI substrates. No methylation was observed, indicating that other factors contribute to the regioselectivity (Figures S9–S10).
To determine if additional residues in the core peptide influence binding, we created additional TbtA variants. For these assays, five Cys residues in 7 (Cys2, 6, 9, 10, 12) were replaced with Ala, while Cys4 was retained. Upon reaction with the thiazole synthetase (TbtEFG), the corresponding Cys4 monoazole was obtained. After reaction with TbtI, MS analysis indicated that it was not a substrate (Figure S11), which suggests that substrate binding requires other thiazole moieties. We therefore constructed all possible combinations of TbtA diazoles (i.e. Thz2/4, Thz4/6, Thz4/9, Thz4/10, and Thz4/12) and tested them as TbtI substrates. We observed that the 2/4 diazole was not accepted by TbtI (Figure S12); however, the 4/6 diazole was fully converted to the methylated product (Figure 4). In contrast, the 4/9 diazole was incompletely processed while the 4/10 and 4/12 diazole-containing peptides were not detectably processed (Figures S13–S15). These data suggested that -Asn-Thz-Phe-Thz- is the minimal substrate recognition motif for TbtI.
Figure 4.

MALDI-TOF-MS of (A) TbtA-Cys4/6 precursor, (B) reaction of TbtA-Cys4/6 with TbtEFG, and (C) reaction of the TbtA-4/6 diazole with TbtI. Laser-induced deamination artifacts are denoted by a red asterisk.
To test this hypothesis, the motif was installed at positions 1–4. This double-substituted variant, TbtA-S1N/N3F, was reacted with TbtEFG and subsequently TbtI. Thiazole formation was observed, but methylation was not (Figure S16), suggesting that while an N-terminal flanking Asn and a downstream thiazole are necessary, they are not sufficient for TbtI methylation. Other, currently unknown structural aspects further contribute to the substrate selectivity of TbtI. All results are summarized in Table 1.
Table 1.
TbtA precursor peptide variants tested for methylation by TbtI. TbtA sequence: Leader-SCNCFCYICCSCSSA
| Variant | Outcome |
|---|---|
| wild-type hexazole | Fully processed |
| wild-type hexazole core (three res. leader) | Fully processed |
| C4A pentazole | Not processed |
| C4S pentazole | Not processed |
| N3A hexazole | Not processed |
| N3D hexazole | Not processed |
| N3Q hexazole | Not processed |
| S1N/C4A pentazole | Not processed |
| C4A/F5N pentazole | Not processed |
| C4A/I8N pentazole | Not processed |
| C4A/S11N pentazole | Not processed |
| Cys4 monoazole | Not processed |
| Cys2/4 diazole | Not processed |
| Cys4/6 diazole | Fully processed |
| Cys4/9 diazole | Partially processed |
| Cys4/10 diazole | Not processed |
| Cys4/12 diazole | Not processed |
| S1N/N3Fhexazole | Not processed |
Sequence analysis of TbtI did not reveal significant homology to class A rSAM methyltransferases outside of the conserved [4Fe-4S]- and SAM-binding motifs. TbtI is clearly distinguished from the cobalamin-dependent class B methyltransferases.18 Class C methyltransferases are homologous to HemN/HemZ, mechanistically unrelated rSAM decarboxylases from heme biosynthesis.15 Analysis of the 20,000 top-scoring BLAST-P results retrieved using TbtI as the query revealed that the vast majority of the class C enzymes belong to the HemN/HemZ family (Figure S17).39,40 Inspection of the remaining enzymes revealed that 35 are encoded within suspected natural product BGCs while others may represent divergent heme metabolic enzymes. Of the 35 with anticipated involvement in natural product biosynthesis, 19 are located in discernable thiopeptide BGCs, including proteins responsible for thiazole C-methylation and Trp C-methylation, including the recently characterized NosN involved in nosiheptide biosynthesis.18,30,41,42 Besides these, no other class C rSAM methyltransferase involved in natural product biosynthesis (e.g. ChuW, YtkT, Jaw5) appears in the top 20,000 BLAST hits, indicating a more distant relationship to the thiopeptide modifying enzymes. Other BGCs that encode homologs of TbtI include non-ribosomal peptide synthetase-polyketide synthase hybrids involved in bleomycin biosynthesis, as well as uncharacterized linear azole-containing peptides that we predict will also bear C-methylated thiazoles (Figure S18). Phylogenetic analysis of non-HemN homologs of TbtI, visualized by a maximum likelihood tree, revealed an enrichment of homologs in actinomycetes and proteobacteria. Examples from the actinomycetes further showed an enrichment towards suspected enzymes involved in natural product biosynthesis (Figures S19–S20).
In summary, we successfully reconstituted the activity of the class C rSAM methyltransferase TbtI of the thiomuracin biosynthetic pathway. This is the first example of in vitro reconstitution of a class C methyltransferase reaction that results in a methylthiazole, setting the stage for detailed mechanistic investigations. Our data demonstrate that TbtI recognizes specific residues upstream and downstream of Thz4 to carry out a regioselective C-methylation reaction. These studies will facilitate generation of novel analogs of thiomuracin with desirable pharmacological properties.
Supplementary Material
Acknowledgments
This work was supported in part by the National Institutes of Health (GM097142 to D.A.M.) and the Howard Hughes Medical Institute (to W.A.V.). We also thank Prof. Tadhg Begley (Texas A&M University) for the pSUF plasmid and Prof. Jim Imlay (University of Illinois, Urbana-Champaign) for IscS.
Footnotes
Notes
The authors declare no competing financial interest.
Experimental details and supporting figures. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Arnison PG, Bibb MJ, Bierbaum G, Bowers AA, Bugni TS, Bulaj G, Camarero JA, Campopiano DJ, Challis GL, Clardy J, et al. Nat Prod Rep. 2013;30:108. doi: 10.1039/c2np20085f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bagley MC, Dale JW, Merritt EA, Xiong X. Chem Rev. 2005;105:685. doi: 10.1021/cr0300441. [DOI] [PubMed] [Google Scholar]
- 3.Yang X, van der Donk WA. Chem - Eur J. 2013;19:7662. doi: 10.1002/chem.201300401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Oman TJ, van der Donk WA. Nat Chem Biol. 2010;6:9. doi: 10.1038/nchembio.286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sardar D, Lin Z, Schmidt EW. Chem Biol. 2015;22:907. doi: 10.1016/j.chembiol.2015.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li CX, Zhang FF, Kelly WL. Mol BioSyst. 2011;7:82. doi: 10.1039/c0mb00129e. [DOI] [PubMed] [Google Scholar]
- 7.Hudson GA, Zhang Z, Tietz JI, Mitchell DA, van der Donk WA. J Am Chem Soc. 2015;137:16012. doi: 10.1021/jacs.5b10194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang Z, Hudson GA, Mahanta N, Tietz JI, van der Donk WA, Mitchell DA. J Am Chem Soc. 2016;138:15511. doi: 10.1021/jacs.6b08987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Morris RP, Leeds JA, Naegeli HU, Oberer L, Memmert K, Weber E, LaMarche MJ, Parker CN, Burrer N, Esterow S, Hein AE, Schmitt EK, Krastel P. J Am Chem Soc. 2009;131:5946. doi: 10.1021/ja900488a. [DOI] [PubMed] [Google Scholar]
- 10.Tocchetti A, Maffioli S, Iorio M, Alt S, Mazzei E, Brunati C, Sosio M, Donadio S. Chem Biol. 2013;20:1067. doi: 10.1016/j.chembiol.2013.07.005. [DOI] [PubMed] [Google Scholar]
- 11.Sofia HJ, Chen G, Hetzler BG, Reyes-Spindola JF, Miller NE. Nucleic Acids Res. 2001;29:1097. doi: 10.1093/nar/29.5.1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vey JL, Drennan CL. Chem Rev. 2011;111:2487. doi: 10.1021/cr9002616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Broderick JB, Duffus BR, Duschene KS, Shepard EM. Chem Rev. 2014;114:4229. doi: 10.1021/cr4004709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mehta AP, Abdelwahed SH, Mahanta N, Fedoseyenko D, Philmus B, Cooper LE, Liu Y, Jhulki I, Ealick SE, Begley TP. J Biol Chem. 2015;290:3980. doi: 10.1074/jbc.R114.623793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang Q, van der Donk WA, Liu W. Acc Chem Res. 2012;45:555. doi: 10.1021/ar200202c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chan KKJ, Thompson S, O’Hagan D. Chem Bio Chem. 2013;14:675. [Google Scholar]
- 17.Fujimori DG. Curr Opin Chem Biol. 2013;17:597. doi: 10.1016/j.cbpa.2013.05.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bauerle MR, Schwalm EL, Booker SJ. J Biol Chem. 2015;290:3995. doi: 10.1074/jbc.R114.607044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Grove TL, Benner JS, Radle MI, Ahlum JH, Landgraf BJ, Krebs C, Booker SJ. Science. 2011;332:604. doi: 10.1126/science.1200877. [DOI] [PubMed] [Google Scholar]
- 20.Grove TL, Livada J, Schwalm EL, Green MT, Booker SJ, Silakov A. Nat Chem Biol. 2013;9:422. doi: 10.1038/nchembio.1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pierre S, Guillot A, Benjdia A, Sandstroem C, Langella P, Berteau O. Nat Chem Biol. 2012;8:957. doi: 10.1038/nchembio.1091. [DOI] [PubMed] [Google Scholar]
- 22.Blaszczyk AJ, Silakov A, Zhang B, Maiocco SJ, Lanz ND, Kelly WL, Elliott SJ, Krebs C, Booker SJ. J Am Chem Soc. 2016;138:3416. doi: 10.1021/jacs.5b12592. [DOI] [PubMed] [Google Scholar]
- 23.Allen KD, Wang SC. Arch Biochem Biophys. 2014;543:67. doi: 10.1016/j.abb.2013.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Woodyer RD, Li G, Zhao H, van der Donk WA. Chem Commun. 2007:359. doi: 10.1039/b614678c. [DOI] [PubMed] [Google Scholar]
- 25.Kim HJ, McCarty RM, Ogasawara Y, Liu Y-n, Mansoorabadi SO, LeVieux J, Liu H-w. J Am Chem Soc. 2013;135:8093. doi: 10.1021/ja312641f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang Y, Schnell B, Baumann S, Muller R, Begley TP. J Am Chem Soc. 2017;139:1742. doi: 10.1021/jacs.6b10901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Parent A, Guillot A, Benjdia A, Chartier G, Leprince J, Berteau O. J Am Chem Soc. 2016;138:15515. doi: 10.1021/jacs.6b06697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Huang W, Xu H, Li Y, Zhang F, Chen XY, He QL, Igarashi Y, Tang GL. J Am Chem Soc. 2012;134:8831. doi: 10.1021/ja211098r. [DOI] [PubMed] [Google Scholar]
- 29.Hiratsuka T, Suzuki H, Kariya R, Seo T, Minami A, Oikawa H. Angew Chem, Int Ed. 2014;53:5423. doi: 10.1002/anie.201402623. [DOI] [PubMed] [Google Scholar]
- 30.Ding W, Li Y, Zhao J, Ji X, Mo T, Qianzhu H, Tu T, Deng Z, Yu Y, Chen F, Zhang Q. Angew Chem Int Ed. 2017 doi: 10.1002/anie.201609948. [DOI] [PubMed] [Google Scholar]
- 31.LaMattina JW, Nix DB, Lanzilotta WN. Proc Natl Acad Sci. 2016;113:12138. doi: 10.1073/pnas.1603209113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Allen KD, Xu H, White RH. J Bacteriol. 2014;196:3315. doi: 10.1128/JB.01903-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jang S, Imlay JA. Mol Microbiol. 2010;78:1448. doi: 10.1111/j.1365-2958.2010.07418.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hidalgo E, Bollinger JM, Jr, Bradley TM, Walsh CT, Demple B. J Biol Chem. 1995;270:20908. doi: 10.1074/jbc.270.36.20908. [DOI] [PubMed] [Google Scholar]
- 35.Brumby PE, Miller RW, Massey V. J Biol Chem. 1965;240:2222. [PubMed] [Google Scholar]
- 36.Burkhart BJ, Hudson GA, Dunbar KL, Mitchell DA. Nat Chem Biol. 2015;11:564. doi: 10.1038/nchembio.1856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Just-Baringo X, Albericio F, Alvarez M. Mar Drugs. 2014;12:317. doi: 10.3390/md12010317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Young TS, Dorrestein PC, Walsh CT. Chem Biol. 2012;19:1600. doi: 10.1016/j.chembiol.2012.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Parish T, Schaeffer M, Roberts G, Duncan K. Tuberculosis. 2005;85:197. doi: 10.1016/j.tube.2005.01.002. [DOI] [PubMed] [Google Scholar]
- 40.Homuth G, Rompf A, Schumann W, Jahn D. J Bacteriol. 1999;181:5922. doi: 10.1128/jb.181.19.5922-5929.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wei M, Deng J, Wang S, Liu N, Chen Y. Biotechnol Lett. 2011;33:585. doi: 10.1007/s10529-010-0460-0. [DOI] [PubMed] [Google Scholar]
- 42.Yu Y, Duan L, Zhang Q, Liao R, Ding Y, Pan H, Wendt-Pienkowski E, Tang G, Shen B, Liu W. ACS Chem Biol. 2009;4:855. doi: 10.1021/cb900133x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.