

Abstract
Purpose
While multikinase inhibitors with RET activity are active in RET-rearranged thyroid and lung cancers, objective response rates are relatively low and toxicity can be substantial. The development of novel RET inhibitors with improved potency and/or reduced toxicity is thus an unmet need. RXDX-105 is a small molecule kinase inhibitor that potently inhibits RET. The purpose of the preclinical and clinical studies was to evaluate the potential of RXDX-105 as an effective therapy for cancers driven by RET alterations.
Experimental design
The RET-inhibitory activity of RXDX-105 was assessed by biochemical and cellular assays, followed by in vivo tumor growth inhibition studies in cell line– and patient-derived xenograft models. Antitumor activity in patients was assessed by imaging and Response Evaluation Criteria in Solid Tumors (RECIST).
Results
Biochemically, RXDX-105 inhibited wild-type RET, CCDC6-RET, NCOA4-RET, PRKAR1A-RET, and RET M918T with low to subnanomolar activity while sparing VEGFR2/KDR and VEGFR1/FLT. RXDX-105 treatment resulted in dose-dependent inhibition of proliferation of CCDC6-RET–rearranged and RET C634W-mutant cell lines and inhibition of downstream signaling pathways. Significant tumor growth inhibition in CCDC6-RET, NCOA4-RET, and KIF5B-RET–containing xenografts was observed, with the concomitant inhibition of p-ERK, p-AKT, and p-PLCγ. Additionally, a patient with advanced RET-rearranged lung cancer had a rapid and sustained response to RXDX-105 in both intracranial and extracranial disease.
Conclusions
These data support the inclusion of patients bearing RET alterations in ongoing and future molecularly enriched clinical trials to explore RXDX-105 efficacy across a variety of tumor types.
Introduction
The rearranged during transfection (RET) gene is an established proto-oncogene. It encodes a single-pass transmembrane receptor tyrosine kinase that is required for the development, maturation, and maintenance of a number of tissues and cell types (1). Under normal conditions, the binding of glial cell line–derived neurotrophic factor (GDNF) family ligands to RET on the cell surface (2) leads to dimerization and auto-phosphorylation of intracellular tyrosine residues. This, in turn, results in the activation of downstream RAS–MAPK, PI3K–AKT, and phospholipase Cγ (PLCγ) pathways (3), and increased cell survival and proliferation.
Aberrant ligand-independent RET activation can occur via a variety of mechanisms. Germline gain-of-function RET mutations are identified in patients with multiple endocrine neoplasia type 2 (MEN2) and familial medullary thyroid cancer (MTC). In addition, somatic RET mutations are found in the majority of sporadic MTC (4). Such mutations lead to constitutive receptor activation and are found in either the extracellular or intracellular kinase domains of the protein. Examples of activating RET mutations include C634W, M918T, and the gatekeeper mutations, V804L and V804M.
In contrast, recurrent RET gene rearrangements, resulting in the expression of oncogenic RET fusion proteins, have been detected in papillary thyroid carcinoma (PTC; ref. 5) and other tumor types, including non–small cell lung cancer (NSCLC; refs. 6–8) and colorectal cancer (CRC; refs. 9, 10). A variety of upstream partners (7, 8) provide coiled-coil domains that cause ligand-independent dimerization and constitutive activation of the RET kinase (6). These fusion oncoproteins are transforming in vitro and in vivo in engineered Ba/F3 cells and NIH-3T3 cells (6, 7, 11, 12), and in genetically engineered mouse models (GEMM) in which KIF5B-RET was expressed in lung epithelial cells (13, 14).
RET inhibitors are active in patients with RET-rearranged or RET-mutant solid tumors (15). In thyroid cancers, for example, the multikinase inhibitors (MKI) cabozantinib, vandetanib, and lenvatinib have been approved for treatment based on improvements in response and progression-free survival (PFS; ref. 16). The efficacy of these MKIs is believed to be driven by RET inhibition, but the involvement of other mechanism of action such as antiangiogenesis cannot be excluded. The activity of cabozantinib (17, 18), vandetinib (19), and lenvatinib (20) in patients with RET-rearranged lung cancers treated in phase II trials has been reported. Unfortunately, treatment with these inhibitors is associated with relatively low response rates in comparison with tyrosine kinase inhibitor therapy in epidermal growth factor receptor (EGFR)-mutant, anaplastic lymphoma kinase (ALK)-rearranged and ROS1-rearranged lung cancers. In addition, toxicity can be substantial.
The development of potent new RET inhibitors with an improved efficacy and tolerability profile is thus an unmet need for patients with RET-dependent cancer. RXDX-105 is a clinical stage, potent inhibitor of wild-type RET, RET fusions, and RET activating mutations, with activity also against wild-type BRAF and BRAF V600E mutation. In biochemical kinase assay (21) and cell-based assays (22), RXDX-105 exhibited about 800-fold selectivity against VEGFR2/KDR and VEGFR1/FLT compared with wild-type RET and had no growth inhibitory activity toward HUVEC cells at 1 μmol/L. The VEGFR-sparing profile of RXDX-105 is expected to reduce the hypertensive liabilities in the clinic compared with other MKIs such as cabozantinib (23, 24). In this report, we explore the in vitro and in vivo activity of RXDX-105 in a variety of RET-rearranged and RET-mutant solid tumor models and describe the first proof-of-principle clinical report of a patient with advanced RET-rearranged lung adenocarcinoma who responded to RXDX-105 in an ongoing clinical trial.
Materials and Methods
Compound and cell lines
Good manufacturing practice (GMP)-quality RXDX-105, formerly named CEP-32496, was synthesized at Ignyta. The human lung adenocarcinoma cell line LC-2/ad (Cat. No. 94072247) and the medullary thyroid cancer cell line TT (CRL-1803) were obtained from Sigma-Aldrich and the American Type Culture Collection (ATCC), respectively. Both cell lines were obtained directly, within 6 months of the studies, from the respective cell banks with certificate of short tandem repeat (STR) authentication. Cells were propagated in F-12K or RPMI medium supplemented with 10% (vol/vol) FBS as recommended. All cell lines were maintained in a humidified incubator at 37°C in a 5% CO2 environment. The HBEC3KT-RET cell line was generated by expressing CCDC6-RET and a dominant-negative p53 (c-terminal region of wild-type p53; ref. 25) in HBEC3-KT cells (human bronchial epithelial cells immortalized with CDK4 and hTERT; ref. 26).
3-Dimensional modeling of RXDX-105 binding to RET
The x-ray co-crystal structure of RXDX-105 in complex with RET has not been determined. However, a similar analogue in the same series of RXDX-105 was successfully co-crystalized with RET. This complex structure was determined with a resolution of 1.7 Å and was used for modeling. Glide, as implemented in Schrodinger’s modeling suite, was used for docking of the RXDX-105 analogue into the RET binding site. The docked poses were subjected to further optimization with Prime MMGBSA.
Biochemical kinase assay
RXDX-105 biochemical IC50 values were determined using vendor protocols at the Km level of ATP by the Reaction Biology Corporation using the radioactive HotSpot assay platform.
Western blot analysis and phospho-protein profiling
Cells were seeded at a density of 5 × 105 cells per well in 6-well plates and cultured for 24 hours The cells were then treated with 50 to 5,000 nmol/L of the indicated compounds for 2 hours and harvested/lysed in 1x RIPA buffer containing Halt protease and phosphatase inhibitor cocktail (Thermo Fisher Scientific). Lysates were quantified using the Pierce 660 nmol/L protein assay kit (Thermo Fisher Scientific). Twenty-five to 30 μg of protein was resolved on 8% denaturing SDS-polyacrylamide gels, transferred to PVDF membranes, and blotted with indicated primary antibodies followed by HRP-conjugated secondary antibodies (LI-COR Biotechnology). Bands were detected by enhanced chemiluminescence (GE Healthcare). To make lysate from xenografted tumor, frozen tumor tissue was weighed, and approximately 100 mg tumor tissue was placed in 200 μL RIPA buffer. The tissue was then homogenized in RIPA buffer using FastPrep-24 5G (MP Bio) according to the manufacturer’s protocol. After homogenization, the samples were centrifuged at 14,000 × g for 10 minutes at 4°C. The supernatant was isolated, protein was quantitated, and 30 μg/lane protein was separated by 4% to 20% SDS-PAGE for immunoblotting. All primary antibodies used in these studies were obtained from Cell Signaling Technology and include phospho-RET (Tyr905; #3221), RET (#3220), phospho-MEK1/2 (Ser217/221; #9154), MEK1/2 (#9126), Phospho-ERK (T202/Y204; #9101), ERK (#4695), Phospho-AKT (S473; #4060), AKT (#4691), Phospho-PLCγ (Y783; #2821), PLCγ (#5690), and β-Actin (#3700).
For phosphoprotein profiling, 5 × 106 cells were plated in 10-cm dishes, then deprived of serum for 24 hours. Cells were then treated with 1 μmol/L RXDX-105 for 30 minutes. Protein phosphorylation was determined using a phosphokinase profiling array obtained from R&D Systems, according to the manufacturer’s instructions.
Cell viability assays
LC-2/ad and TT cells were seeded at a density of 5,000 cells per well in 96-well plates in medium containing 10% (vol/vol) FBS. The following day, cells were serum-starved in 0.5% FBS-containing media for 24 hours and then treated with the indicated compounds for an additional 72 hours. Viable cell numbers were determined using the CellTiter-Glo assay kit according to the manufacturer’s protocol (Promega). Each assay consisted of duplicate samples and the experiments were repeated in triplicate at minimum. Data were expressed as relative luciferase activity (percentages relative to control cells). HBEC3KT-RET cells were plated at a density of 7,500 cells per well of 96-well plates and treated as described above, with the exception that complete medium with 10% serum was used throughout the treatment. Viable cell numbers were determined using alamar blue viability dye (Invitrogen), and IC50 values were determined using GraphPad Prism 6.
Apoptosis assay
The cells were split into 10-cm dishes. Twenty-four hours later, the cells were treated with different concentrations of indicated compounds for 24 hours. Cell lysate was prepared in RIPA lysis buffer. The cleaved Caspase-3 (Cell Signaling Technology #9661) and cleaved PARP (Cell Signaling Technology #9541) were measured by standard Western blot method.
Patient- and cell line–derived subcutaneous xenograft models
Studies using colorectal cancer xenograft models CR1520 and CR2518 were carried out at Crown Biosciences. Model CR1520 contains a NCOA4-RET fusion, while model CR2518 contains a CCDC6-RET fusion. No other activating mutations were detected in KRAS, EGFR, PIK3CA, AKT, BRAF, ERK, TP53, PTEN, or CTNNB in these models. To generate study cohorts, tumor fragments from stock mice or cryopreserved stock were inoculated into the right flank of female BALB/c nude mice. Mice were randomly allocated to treatment groups (6 per group) according to their tumor volume when the average tumor size reached about 150 to 200 mm3. The test articles were formulated in 22% 2-hydroxypropyl-beta-cyclodextrin and administered to the tumor-bearing mice for 14 consecutive days (2 weeks) at 10 mg/kg and 30 mg/kg, twice daily (BID). An additional group of 60 mg/kg, once daily (QD) was included in the CR2518 study. Tumor size and body weight were monitored at least 2 times a week.
Additional in vivo studies using a low-passage PDX model of NSCLC harboring KIF5B-RET fusion were carried out at Champions Oncology. Briefly, models CTG-0838 and CTG-1048 were expanded in nu/nu mice to establish study cohorts. Animals were then randomized into treatment groups based on tumor size, and dosing was initiated once tumors reached a volume that fell within the range of 150 to 300 mm3. The test articles were formulated in 22% 2-hydroxypropyl-beta-cyclodextrin and administered to the tumor-bearing mice for 4 consecutive weeks at 30 mg/kg, BID. Tumor size and body weight were monitored at least 2 times a week.
For the HBEC3KT-RET xenograft experiments, cell line xenograft tumors were transplanted from seed mice (nu/nu) to experimental animals as a single subcutaneous tumor. When tumors reached 100 to 150 mm3, mice were randomly assigned to treatment groups (5 mice per group). RXDX-105 was administered by oral gavage at 50 mg/kg and 100 mg/kg, QD, with a total of 5 treatments given in a 7-day period. Tumor size and body weight were monitored at least two times a week.
Data were expressed as mean ± SEM unless indicated otherwise. Statistical significance was determined by analysis of variance (ANOVA) using Dunnett multiple-comparison posttest with GraphPad Prism software unless otherwise noted.
RXDX-105 administration
A treatment-naïve patient with advanced RET-rearranged lung adenocarcinoma received RXDX-105 in an ongoing, open-label, phase I/Ib trial of the drug (NCT01877811). The patient was treated in the phase Ib portion of this study at the provisional recommended phase II dose (RP2D) of RXDX-105 at 350 mg daily in the fed state. The primary objective of the phase Ib portion of the trial was to further assess the safety profile and tolerability of RXDX-105 at the provisional RP2D, with the secondary objectives including an evaluation of the preliminary antitumor activity of the drug as assessed by overall response rate. Antitumor activity was assessed by imaging every eight weeks and Response Evaluation Criteria in Solid Tumors (RECIST) version 1.1. Patients with a variety of advanced solid tumors harboring activating RET alterations were enrolled into predefined baskets. The patient was treated in a treatment-naïve basket of patients with advanced RET-rearranged NSCLCs.
Results
RXDX-105 is a potent inhibitor of RET
The chemical synthesis and characterization of RXDX-105 has been described previously (22, 27, 28). Its structure is depicted in Fig. 1A. Although RXDX-105 was initially identified as an inhibitor of wild-type and V600E-mutated BRAF (28), the kinome profiling of RXDX-105 also indicated potent inhibitory activity against RET (22). As a follow up to these data, we determined the biochemical IC50 values against wild-type RET, as well as a select panel of RET fusions and mutations by a cell-free, radioactive kinase assay platform, performed in multiple repeats. Biochemically, RXDX-105 is a potent wild-type RET inhibitor, with an IC50 of 0.33 nmol/L. RXDX-105 has similar activity against RET fusions tested that contain wild-type kinase domain, including CCDC6-RET (IC50 = 0.33 nmol/L), NCOA4-RET (IC50 = 0.41 nmol/L), and PRKAR1A-RET (IC50 = 0.81 nmol/L). The drug is active against RET M918T (IC50 = 4.34 nmol/L), but displayed reduced activity against the gatekeeper mutations RET V804L (IC50 = 319 nmol/L) and RET V804M (IC50 = 266 nmol/L; ref. 14) in the biochemical assay. In the same assay, the IC50 values of RXDX-105 against VEGFR1/FLT and VEGFR2/KDR are 140.60 nmol/L and 257.60 nmol/L, respectively. The relative selectivity between RET wild-type and VEGFR2/KDR is about 800-fold.
Figure 1.
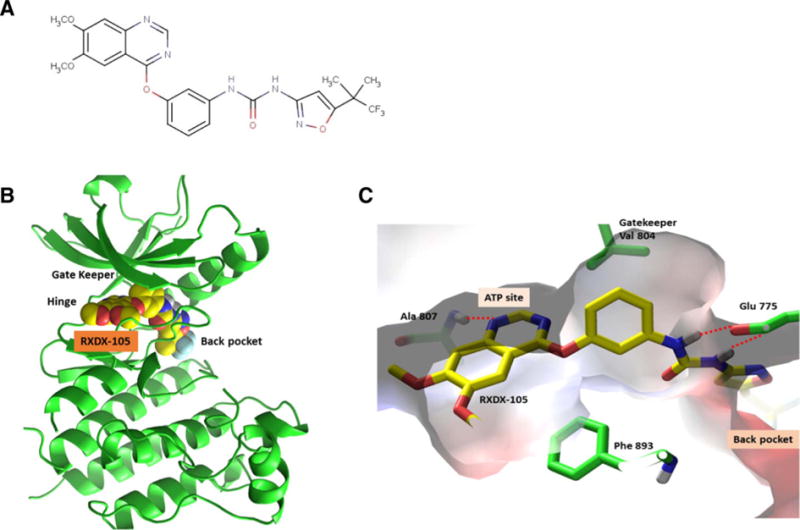
The predicted binding mode of RXDX-105 with RET. A, Chemical structure of RXDX-105. B and C, Predicted binding mode by modeling.
RXDX-105 is predicted to bind to the DFG-out conformation of RET
An analogue of RXDX-105 in the same series was successfully co-crystalized with RET. This complex structure was determined with a resolution of 1.7 Å and was used for modeling. Glide was used for docking of RXDX-105 into the RET binding site and docked poses were subjected to further optimization with Prime MMGBSA. In this model, the RXDX-105 analogue was found to bind to the DFG-out, inactive conformation of RET. The molecule was buried deeply in the ATP binding site as well as a back pocket, interacting extensively with residues in the ATP site, DFG loop, αC- helix (Fig. 1B and C). Specifically, the drug hydrogen bonds with the NH of Ala 807 in the hinge region and Ser 891 in the activation loop, as well as side-chain carboxyl group of Glu 775 in the αC Helix. In addition, the molecule was predicted to make extensive hydrophobic interactions with the gatekeeper residue Val 804, Phe 893 of the DFG loop, as well as side-chains of Val 778, Leu 779, Val 782, Val 787, Leu 865, and Leu 870 in the back pocket.
RXDX-105 is active in vitro against RET-rearranged and RET-mutant models
We first characterized the ability of RXDX-105 to inhibit RET activation in two independent cellular models harboring distinct RET alterations. The LC-2/ad lung adenocarcinoma and the TT medullary thyroid cancer cell lines harbor a CCDC6-RET gene fusion and RET C634W activating point mutation, respectively. Cells were incubated with 50 nmol/L to 5 μmol/L RXDX-105 for 2 hours, then lysed in RIPA buffer containing phosphatase and protease inhibitors. The relative protein and phospho-protein levels were qualitatively determined by Western blot analysis. Treatment with RXDX-105 led to robust decrease in phosphorylation of RET and PLCγ in both cellular models (Fig. 2). In LC-2/ad cells, RXDX-105 activity against RET and PLCγ was comparable with, or slightly higher than that achieved with cabozantinib and alectinib, two clinically approved multikinase inhibitors (for thyroid cancer and ALK-rearranged lung cancer, respectively) that have RET activity (Fig. 2A). However, while cabozantinib was able to decrease phospho-ERK and phospho-MEK at as low as 50 nmol/L, neither RXDX-105 nor alectinib treatment showed detectable change in ERK and MEK phosphorylation. In TT cells, however, RXDX-105 outperformed cabozantinib and alectinib in decreasing phospho-RET, phospho-PLCγ, phospho-ERK, and phospho-MEK (Fig. 2B). Despite the difference in MARK pathway response between LC-2/ad and TT cells, RXDX-105 effectively inhibited both cell lines in the antiproliferative assay. LC-2/ad and TT cells were treated with various doses of RXDX-105, and cell viability was measured after three (LC-2/ad) or four days (TT). In LC-2/ad cells, RXDX-105 (IC50 = 40 nmol/L) out-performed both cabozantinib (IC50 = 98 nmol/L) and alectinib (IC50 = 204 nmol/L). Similarly, RXDX-105 exhibited more potent activity in TT cells, with an IC50 of 11 nmol/L, compared with 77 nmol/L for cabozantinib and 90 nmol/L for alectinib. To further characterize the mechanism of inhibition, apoptosis was measured by the amount of cleaved caspase-3 and cleaved PARP proteins after 24-hour incubation with RXDX-105, cabozantinib, and alectinib. While in LC-2/ad cells (Fig. 2C), all three compounds induced apoptosis at 24-hour time point, no cleavage of caspase-3 and PARP was seen in the TT cells, except for the higher doses of alectinib (Fig. 2D). The reason for the observed difference in apoptosis upon treatment is unclear.
Figure 2. In vitro.

characterization of RET-inhibitory activity of RXDX-105. Inhibition of phosphorylation of RET and downstream pathways by RXDX-105 in (A) LC-2/ad cells (CCDC6-RET) and (B) TT cells (RET C634W). Apoptosis signals, cleaved caspase-3 and cleaved PARP, were measured after 24-hour incubation with indicated compounds in LC-2/ad cells (C) and TT cells (D).
We next examined if treatment with RXDX-105 could pharmacodynamically and functionally inhibit the proliferation of an engineered RET-dependent cell line, HBEC3KT-RET, derived by expressing CCDC6-RET and a dominant-negative p53 mutant in HBEC3-KT cells, human bronchial epithelial cells immortalized with CDK4 and hTERT (25). Treatment with RXDX-105 for 30 minutes inhibited the phosphorylation of RET, AKT, and ERK (Supplementary Fig. S1A). We then extended the study by looking at the effect of RXDX-105 on the phosphorylation of a larger panel on kinases using a phosphokinase profiling array (R&D Systems). Treatment of serum-deprived cells with 1 μmol/L RXDX-105 caused a robust reduction in phosphorylation of AKT, ERK1/2, STAT1, and WNK1 in HBEC3KT-RET cells (Supplementary Fig. S1B). Concurrent with the pathway inhibition, a growth-inhibitory effect (IC50 = 0.44 μmol/L) was observed via an alamar blue cell viability assay in these cells treated with RXDX-105 for 96 hours (Supplementary Fig. S1C). Taken together, these results demonstrate that RXDX-105 is a potent RET inhibitor in vitro that is able to abrogate proliferation of RET-dependent human cancer cell lines and engineered bronchial epithelial cells.
RXDX-105 exhibits dose-dependent inhibition of RET-rearranged xenografts
The in vivo efficacy of RXDX-105 was evaluated in four RET fusion–containing xenograft models, including the HBEC3KT-RET cell line–derived xenograft model, one NSCLC PDX model (CTG-0838, Champions Oncology), and two colorectal cancer (CRC) PDX models (CR2518 and CR 1520; Crown Biosciences).
We first evaluated RXDX-105 in the HBEC3KT-RET xenograft model driven by CCDC6-RET. Treatment of HBEC3KT-RET xenografts with 50 mg/kg or 100 mg/kg BID RXDX-105 resulted in a significant reduction in tumor growth (Supplementary Fig. S2A). At the dose of 100 mg/kg, all tumors shrank more than 30% in size by the end of the study (day 7). The average tumor regression was approximately 50% (Supplementary Fig. S2B). Based on body weight measurement, both 50 mg/kg and 100 mg/kg treatments were well tolerated under the regimen of once daily for 5 consecutive days followed by a 2-day dosing break per 7-day cycle (Supplementary Fig. S2C).
Furthermore, we identified low-passage NSCLC PDX models CTG-0838 and CTG-1048, available at Champions Oncology, both of which contain the KIF5B-RET fusion, the most common RET gene rearrangement observed in lung cancer. Treatment of CTG-0838 with RXDX-105 at 30 mg/kg BID resulted in significant tumor growth inhibition (Fig. 3A), accompanied by inhibition of phosphorylated RET and its downstream pathways of ERK and PLCγ (Fig. 3B). Similarly, treatment of CTG-1048 with RXDX-105 resulted in significant regression in all tumor-bearing mice (Fig. 3C).
Figure 3. In vivo.
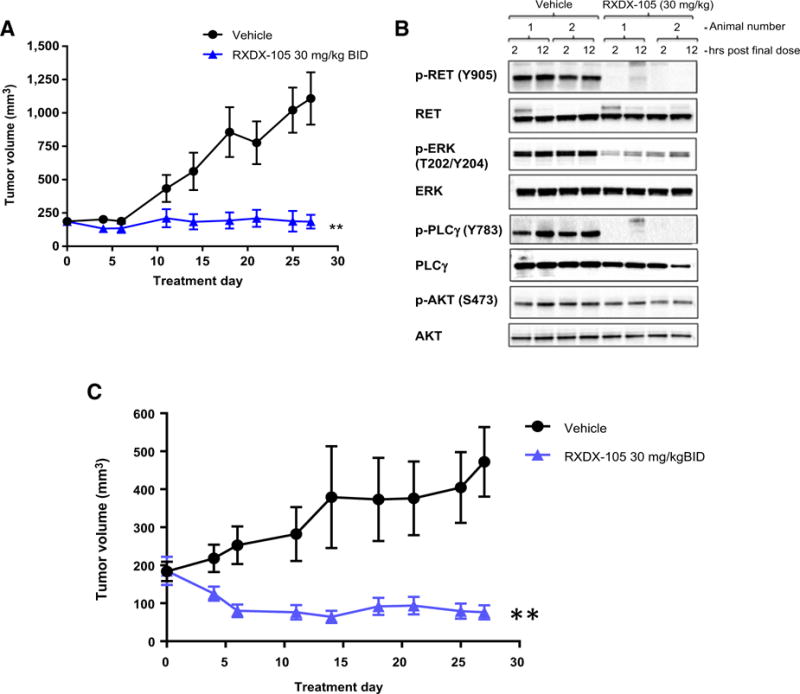
efficacy of RXDX-105 in a PDX model of NSCLC harboring KIF5B-RET fusion. A, Tumor growth inhibition of CTG-0838 by RXDX-105 orally administered BID at 30 mg/kg. Tumor sizes are presented as average ± SEM. (**, P < 0.01). B, At the end of the study (day 27), tumor samples randomly selected from two mice per group were collected two and 12 hours after the final treatment. Western blot was performed using antibodies against phospho- and total RET, ERK, PLCg, and AKT. C, Tumor growth inhibition of CTG-1048 by RXDX-105. Tumor sizes are presented as average ± SEM (**, P < 0.01).
In addition to NSCLC, we further tested RXDX-105 in two CRC PDX models harboring distinct RET rearrangements. Based on sequencing data provided by Crown Biosciences and confirmed in-house at Ignyta, model CR2518 possesses a CCDC6-RET gene fusion, joining exon 1 of CCDC6 to exon 12 of RET; and model CR1520 possesses a NCOA4-RET gene fusion, joining exon 6 of NCOA4 to exon 12 of RET. Both models were dosed with RXDX-105 at 10 and 30 mg/kg BID, and CR2518 was dosed with an additional 60 mg/kg QD dose group. As expected from our in vitro data, treatment with RXDX-105 produced a dose-dependent inhibition of tumor growth, with 30 mg/kg BID inducing tumor regression in all tumors in both models (Figs. 4A and B, 5A and B). In the CR2518 model, treatment with 30 mg/kg BID was equivalent to the 60 mg/kg QD dose group (Fig. 4A and B). Importantly, in these studies, RXDX-105 was well tolerated in all dose groups and did not result in any significant body weight loss over the course of the studies (Fig. 4C; Supplementary Fig. S2C).
Figure 4. In vivo.
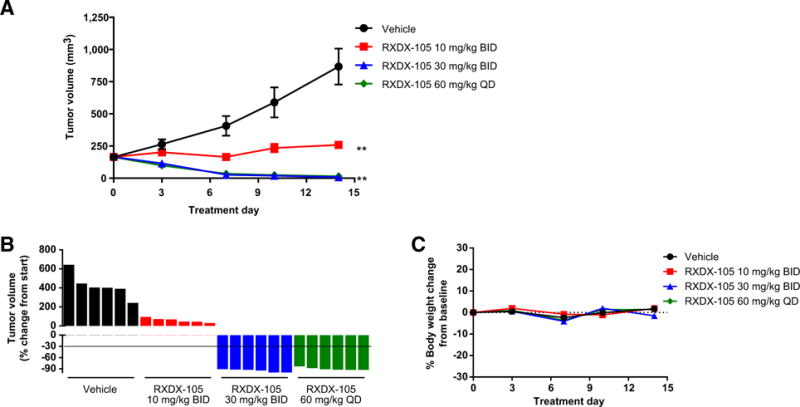
efficacy of RXDX-105 in a PDX model of CRC harboring CCDC6-RET fusion. A, Tumor growth inhibition by RXDX-105 orally administered BID at 10, 30 mg/kg, and QD at 60 mg/kg. Tumor sizes are presented as average ± SEM (**, P < 0.01). B, Plot of percentage change of individual tumor volume compared with baseline. The y-axis of individual tumor responses represents “% of change from start.” C, Plot of percentage body weight change over time during treatment.
Figure 5. In vivo.
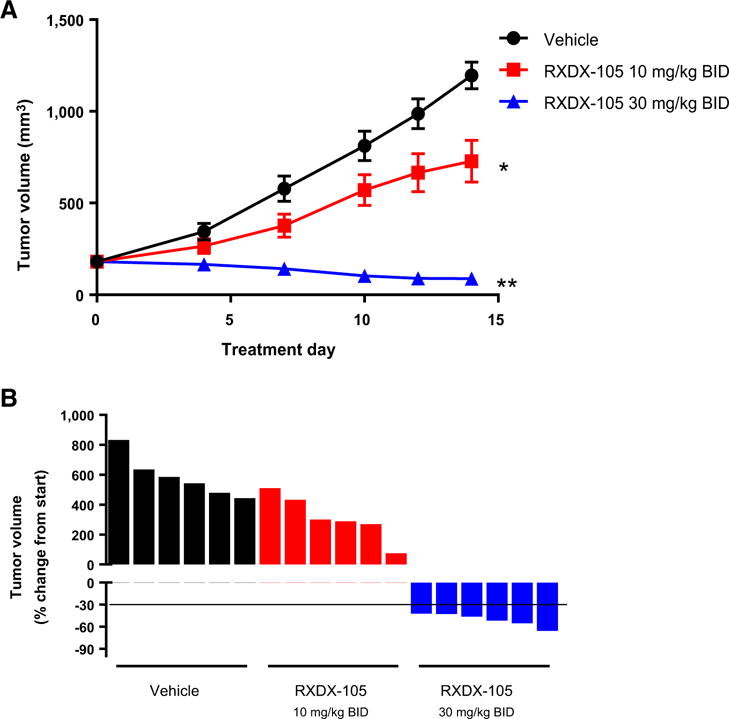
efficacy of RXDX-105 in a PDX model of CRC harboring NCOA4-RET fusion. A, Tumor growth inhibition by RXDX-105 orally administered BID at 10 and 30 mg/kg. Tumor sizes are presented as average ±SEM (*, P < 0.05; **, P < 0.01). B, Plot of percentage change of individual tumor volume compared with baseline. The y-axis of individual tumor responses represents “% of change from start.”
RXDX-105 treatment results in a rapid and sustained response in a patient with advanced RET-rearranged lung cancer with brain metastasis
A 33-year-old female never smoker presented with worsening dyspnea in February of 2016. Workup including computed tomography (CT) and positron emission tomography (PET) imaging revealed a hypermetabolic right perihilar mass, mediastinal adenopathy, and bony metastases. Magnetic resonance imaging (MRI) of the brain identified three small asymptomatic brain metastases. An endobronchial biopsy of the right lung mass and a mediastinal lymph node revealed adenocarcinoma with signet ring features. Immunohistochemistry was positive for TTF-1 and consistent with a lung adenocarcinoma.
A RET rearrangement was detected by dual-color break-apart fluorescence in situ hybridization (FISH) testing. Split red and green signals were detected in 60% of the nuclei scored. Targeted next-generation sequencing on an Ion Torrent PGM instrument (Thermo Fisher Scientific) for specific mutations in 50 genes including EGFR, ERBB2, BRAF, KRAS, MET, PTEN, PIK3CA, and TP53 did not reveal any mutations with known significance. FISH testing for ALK and ROS1 did not reveal recurrent gene rearrangements involving these genes.
The patient was enrolled onto the phase Ib portion of an ongoing phase I/Ib clinical trial of RXDX-105 (NCT01877811). The drug was administered at 350 mg daily. Of note, she had not received any prior chemotherapy, immune checkpoint inhibition, or radiotherapy for her cancer. A rapid partial response was achieved after 3 weeks of therapy with RXDX-105, with 48% shrinkage of the patient’s target lesions by RECIST v1.1. This response was confirmed at 8 weeks of therapy with note of further reduction in tumor burden. Repeat imaging of the brain likewise revealed almost complete resolution of the previously noted three subcentimeter asymptomatic brain metastases. This radiologic response was accompanied by a significant improvement in the patient’s dyspnea. She currently remains on RXDX-105 with continued disease control and a best objective response of a 78% decrease in tumor burden from baseline (Fig. 6). She did develop a grade 1 rash that resulted in dose reduction of the drug to 200 mg daily with subsequent improvement. The patient remains on treatment with RXDX-105 at 6.7 months with no major issues with treatment tolerance.
Figure 6.
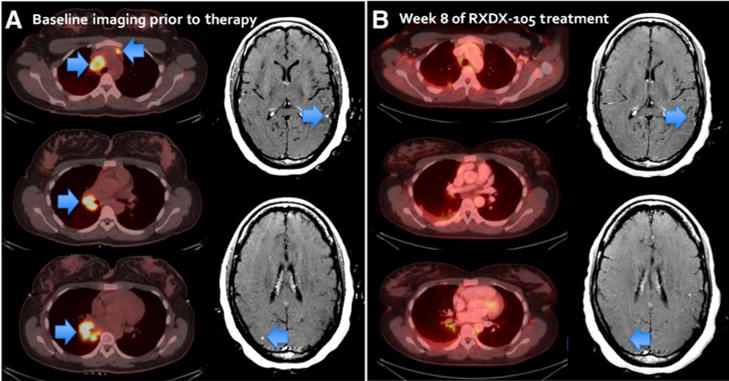
A rapid partial response was achieved after three weeks of therapy with RXDX-105. A, Baseline imaging prior to RXDX-105 treatment. Arrows indicate the lesions. B, Confirmation scan at week 8 with 64% decrease in tumor burden from the baseline, and almost complete resolution of subcentimeter asymptomatic brain metastases.
Discussion
In recent years, the extensive genomic characterization of cancers has led to the discovery of molecular alterations involved in tumorigenesis, metastasis, and drug resistance. The dependency of the tumors on these driver events in various tumor histologies underlies the rationale to pharmacologically target their vulnerability using specific inhibitors (29). RET rearrangements are present in approximately 1% to 2% of lung adenocarcinomas and 20% to 40% of sporadic papillary thyroid cancers and tend to occur in a mutually exclusive fashion with other known driver alterations (30, 31). In addition, RET rearrangements have been identified in other cancer types such as CRC (9), breast cancer (32), Spitz tumors (33), and chronic myelomonocytic leukemia (34). Furthermore, RET mutations are known drivers of oncogenesis in tumors such as medullary thyroid cancer.
RXDX-105 is an orally available, VEGFR-sparing, multikinase inhibitor that is potent against RET in a wide range of in vitro and in vivo models of RET-rearranged and RET-mutant tumors. The drug is predicted to bind to the DFG-out conformation of RET. Biochemically, RXDX-105 potently inhibited wild-type RET and RET fusion proteins with IC50 values below 1 nmol/L. In addition, RXDX-105 was able to inhibit RET M918T, an activating mutation found in the majority of cases of multiple endocrine neoplasia type 2B (35). However, point mutations at V804 site, V804L and V804M, could render the tumor less sensitive to RXDX-105 treatment based on the biochemical activity, but whether this is true in the clinic requires further clinical evidence (14). Inhibition of RET was confirmed in cells by measuring RET pathway phosphorylation, and this correlated with inhibition of cell proliferation. Furthermore, in multiple cell line- and patient-derived NSCLC and CRC xenograft models harboring KIF5B-RET, CCDC6-RET, and NCOA4-RET, RXDX-105 was able to stabilize or cause tumor regression at clinically achievable doses (36). These preclinical studies support the inclusion of patients bearing RET alterations in future clinical trials exploring RXDX-105 efficacy across a variety of tumor types.
While other multikinase inhibitors with RET activity are either approved or currently in clinical development for thyroid and lung cancers, response rates are low even in genomically enriched subgroups, and toxicity can be significant. In RET-rearranged lung cancers, the overall response rate with cabozantinib in a phase II trial was 28% (17). While this activity is comparable with single-agent dabrafenib therapy in BRAF V600E-mutant lung cancers (37), it is much lower than the 60% to 80% response rates that can be achieved with targeted therapy in EGFR-mutant and ALK- and ROS1-rearranged lung cancers (38, 39). In addition, prospective phase II trials of vandetanib and lenvatinib, and a retrospective global series of multiple multikinase inhibitors with RET activity for RET-rearranged lung cancers have confirmed variable, low overall response rates ranging from 17% to 53% (15, 19, 20, 40, 41). Moreover, the incidence of long-term drug-related adverse events with cabozantinib and many other multi-kinase inhibitors can be substantial (42). In the same phase II study of cabozantinib for RET-rearranged lung cancers, approximately two thirds of patients required a dose reduction due to drug-related toxicities including proteinuria, hypertension, and hand–foot syndrome. In this article, we show that RXDX-105 has comparable and potentially improved activity against the CCDC6-RET–rearranged LC-2/ad cell line in vitro. In addition, the drug appears to have less toxicity in phase I trials compared with some of the other multikinase inhibitors in development such as cabozantinib (21, 43). This profile of substantial potency against RET with improved tolerability may result in better RET target inhibition.
Finally, we demonstrate clinical activity of RXDX-105. A rapid and sustained response to RXDX-105 was achieved in a treatment-naïve patient with advanced RET-rearranged NSCLC treated on the phase Ib portion of an ongoing phase I/Ib trial. Response was noted not only in the patient’s extracranial extent of disease, but contraction of small asymptomatic brain metastases was also noted. The patient remains on therapy with continued benefit. The safety and activity of RXDX-105 continues to be explored on this trial (NCT01877811) for patients with RET-rearranged and RET-mutant advanced solid tumors.
Conclusions
RXDX-105 is a potent RET inhibitor that leads to dose-dependent inhibition of downstream signaling events and cell proliferation in a variety of RET-rearranged and RET-mutant models. The drug displayed potent and dose-dependent in vivo antitumor activity with significant tumor regression in cell line– and patient-derived xenograft models harboring RET rearrangements. A rapid and sustained response to therapy was observed in a patient with an advanced RET-rearranged lung cancer. These data support the inclusion of patients whose tumors harbor RET fusions or mutations in ongoing molecularly enriched clinical trials to explore the efficacy of RXDX-105 across a variety of tumor types.
Supplementary Material
Translational Relevance.
To address the relatively low and variable objective response rates and substantial toxicity associated with current therapeutic agents, we are developing a novel RET inhibitor, RXDX-105, for the treatment of molecularly selected patients with RET alterations. Preclinically, RXDX-105 is a potent inhibitor against RET, RET fusions and certain activating RET mutations in the biochemical and cellular assays. In a panel of RET-rearranged, patient-derived xenograft (PDX) models representing different histologies and different fusion partners, RXDX-105 inhibited tumor growth at clinically achievable concentrations. The preclinical observations were then confirmed clinically by a RECIST partial response (PR) of both intracranial and extracranial disease in a RET-rearranged lung cancer patient treated with RXDX-105. Together, these data support the inclusion of patients bearing RET alterations in ongoing and future molecularly enriched clinical trials to explore RXDX-105 efficacy across a variety of tumor types.
Acknowledgments
G.G. Li has ownership interest (including patents) in Ignyta. G.J. Riely is a consultant/advisory board member for Genentech/Roche and Novartis, and reports receiving commercial research support from Ariad, Genentech/Roche, Millennium, Novartis, and Pfizer. A. Drilon reports receiving speaker fees from Ignyta.
Footnotes
Note: Supplementary data for this article are available at Clinical Cancer Research Online (http://clincancerres.aacrjournals.org/).
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed by the other authors.
Authors’ Contributions
Conception and design: G.G. Li, R. Somwar, J. Joseph, R.S. Smith, L. Martin, A. Franovic, E. Martin, G. Wei, P. Multani, A. Drilon
Development of methodology: R. Somwar, R.S. Smith, L. Martin, A. Franovic, J. Harris, S. Yan, G. Wei, P. Multani
Acquisition of data (provided animals, acquired and managed patients, provided facilities, etc.): R. Somwar, J. Joseph, R.S. Smith, T. Hayashi, L. Martin, A. Schairer, G.J. Riely, J. Harris, S. Yan, G. Wei, J.W. Oliver, P. Multani, A. Drilon Analysis and interpretation of data (e.g., statistical analysis, biostatistics, computational analysis): G.G. Li, R. Somwar, J. Joseph, R.S. Smith, T. Hayashi, L. Martin, A. Franovic, A. Schairer, E. Martin, G.J. Riely, J. Harris, S. Yan, J.W. Oliver, P. Multani, A. Drilon
Writing, review, and/or revision of the manuscript: G.G. Li, R. Somwar, J. Joseph, R.S. Smith, L. Martin, A. Franovic, A. Schairer, E. Martin, G.J. Riely, J. Harris, S. Yan, G. Wei, J.W. Oliver, P. Multani, M. Ladanyi, A. Drilon
Administrative, technical, or material support (i.e., reporting or organizing data, constructing databases): L. Martin, S. Yan
Study supervision: G.G. Li, R. Somwar, E. Martin, R. Patel, M. Ladanyi, A. Drilon
References
- 1.Mulligan LM. RET revisited: expanding the oncogenic portfolio. Nat Rev Cancer. 2014;14:173–86. doi: 10.1038/nrc3680. [DOI] [PubMed] [Google Scholar]
- 2.Arighi E, Borrello MG, Sariola H. RET tyrosine kinase signaling in development and cancer. Cytokine Growth Factor Rev. 2005;16:441–67. doi: 10.1016/j.cytogfr.2005.05.010. [DOI] [PubMed] [Google Scholar]
- 3.Phay JE, Shah MH. Targeting RET receptor tyrosine kinase activation in cancer. Clin Cancer Res. 2010;16:5936–41. doi: 10.1158/1078-0432.CCR-09-0786. [DOI] [PubMed] [Google Scholar]
- 4.Krampitz GW, Norton JA. RET gene mutations (genotype and phenotype) of multiple endocrine neoplasia type 2 and familial medullary thyroid carcinoma. Cancer. 2014;120:1920–31. doi: 10.1002/cncr.28661. [DOI] [PubMed] [Google Scholar]
- 5.Santoro M, Melillo RM, Fusco A. RET/PTC activation in papillary thyroid carcinoma: European Journal of Endocrinology Prize Lecture. Eur J Endocrinol. 2006;155:645–53. doi: 10.1530/eje.1.02289. [DOI] [PubMed] [Google Scholar]
- 6.Ju YS, Lee WC, Shin JY, Lee S, Bleazard T, Won JK, et al. A transforming KIF5B and RET gene fusion in lung adenocarcinoma revealed from whole-genome and transcriptome sequencing. Genome Res. 2012;22:436–45. doi: 10.1101/gr.133645.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kohno T, Ichikawa H, Totoki Y, Yasuda K, Hiramoto M, Nammo T, et al. KIF5B-RET fusions in lung adenocarcinoma. Nature medicine. 2012;18:375–7. doi: 10.1038/nm.2644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Takeuchi K, Soda M, Togashi Y, Suzuki R, Sakata S, Hatano S, et al. RET, ROS1 and ALK fusions in lung cancer. Nat Med. 2012;18:378–81. doi: 10.1038/nm.2658. [DOI] [PubMed] [Google Scholar]
- 9.Hechtman JF, Zehir A, Yaeger R, Wang L, Middha S, Zheng T, et al. Identification of targetable kinase alterations in patients with colorectal carcinoma that are preferentially associated with wild-type RAS/RAF. Mol Cancer Res. 2016;14:296–301. doi: 10.1158/1541-7786.MCR-15-0392-T. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Le Rolle AF, Klempner SJ, Garrett CR, Seery T, Sanford EM, Balasubramanian S, et al. Identification and characterization of RET fusions in advanced colorectal cancer. Oncotarget. 2015;6:28929–37. doi: 10.18632/oncotarget.4325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bos M, Gardizi M, Schildhaus HU, Buettner R, Wolf J. Activated RET and ROS: two new driver mutations in lung adenocarcinoma. Transl Lung Cancer Res. 2013;2:112–21. doi: 10.3978/j.issn.2218-6751.2013.03.08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang R, Hu H, Pan Y, Li Y, Ye T, Li C, et al. RET fusions define a unique molecular and clinicopathologic subtype of non-small-cell lung cancer. J Clin Oncol. 2012;30:4352–9. doi: 10.1200/JCO.2012.44.1477. [DOI] [PubMed] [Google Scholar]
- 13.Saito M, Ishigame T, Tsuta K, Kumamoto K, Imai T, Kohno T. A mouse model of KIF5B-RET fusion-dependent lung tumorigenesis. Carcinogenesis. 2014;35:2452–6. doi: 10.1093/carcin/bgu158. [DOI] [PubMed] [Google Scholar]
- 14.Huang Q, Schneeberger VE, Luetteke N, Jin C, Afzal R, Budzevich MM, et al. Preclinical modeling of KIF5B-RET fusion lung adenocarcinoma. Mol Cancer Ther. 2016;15:2521–9. doi: 10.1158/1535-7163.MCT-16-0258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gautschi O, Wolf J, Milia J, Filleron T, Carbone DP, Camidge DR, et al. Targeting RET in patients with RET-rearranged lung cancers: Results from a global registry. J Clin Oncol. 2016;34(suppl) doi: 10.1200/JCO.2016.70.9352. abstr 9014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hart CD, De Boer RH. Profile of cabozantinib and its potential in the treatment of advanced medullary thyroid cancer. Onco Targets Ther. 2013;6:1–7. doi: 10.2147/OTT.S27671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Drilon A, Rekhtman N, Arcila M, Wang L, Ni A, Albano M, et al. Cabozantinib in patients with advanced RET-rearranged non-small-cell lung cancer: an open-label, single-centre, phase 2, single-arm trial. Lancet Oncol. 2016;17:1653–60. doi: 10.1016/S1470-2045(16)30562-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Drilon A, Wang L, Hasanovic A, Suehara Y, Lipson D, Stephens P, et al. Response to cabozantinib in patients with RET fusion-positive lung adenocarcinomas. Cancer Discov. 2013;3:630–5. doi: 10.1158/2159-8290.CD-13-0035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yoh K, Seto T, Satouchi M, Nishio M, Yamamoto N, Murakami H, et al. Vandetanib in patients with previously treated RET-rearranged advanced non-small-cell lung cancer (LURET): an open-label, multicentre phase 2 trial. Lancet Resp Med. 2017;5:42–55. doi: 10.1016/S2213-2600(16)30322-8. [DOI] [PubMed] [Google Scholar]
- 20.Velcheti V, Hida T, Reckamp KL, Yang JC, Nokihara H, Sachdev P, et al. Phase 2 study of lenvatinib (LN) in patients (Pts) with RET fusion-positive adenocarcinoma of the lung. Ann Oncol. 2016;27 doi: 10.1093/annonc/mdw383.05. [DOI] [PubMed] [Google Scholar]
- 21.Drilon A, Fakih M, Fu S, Patel M, Olszanski A, Lockhart C, et al., editors. A phase 1/1b study of RXDX-105, an oral RET and BRAF inhibitor, in patients with advanced solid tumors [Abstract]. 28th EORTC-NCI-AACR Symposium on Molecular Targets and Cancer Therapeutics; 2016; Munich, Germany. [Google Scholar]
- 22.James J, Ruggeri B, Armstrong RC, Rowbottom MW, Jones-Bolin S, Gunawardane RN, et al. CEP-32496: a novel orally active BRAF(V600E) inhibitor with selective cellular and in vivo antitumor activity. Mol Cancer Ther. 2012;11:930–41. doi: 10.1158/1535-7163.MCT-11-0645. [DOI] [PubMed] [Google Scholar]
- 23.Patel M, Fakih M, Olszanski A, Lockhart Drilon A, Fu S, et al. A phase 1 dose escalation study of RXDX-105, an oral RET and BRAF inhibitor, in patients with advanced solid tumors. J Clin Oncol. 2016;34(suppl) 2016 ASCO Annual Meeting. abstr 2574. [Google Scholar]
- 24.Zhang X, Shao Y, Wang K. Incidence and risk of hypertension associated with cabozantinib in cancer patients: a systematic review and meta-analysis. Expert Rev Clin Pharmacol. 2016;9:1109–15. doi: 10.1080/17512433.2016.1190269. [DOI] [PubMed] [Google Scholar]
- 25.Sasai K, Sukezane T, Yanagita E, Nakagawa H, Hotta A, Itoh T, et al. Oncogene-mediated human lung epithelial cell transformation produces adenocarcinoma phenotypes in vivo. Cancer Res. 2011;71:2541–9. doi: 10.1158/0008-5472.CAN-10-2221. [DOI] [PubMed] [Google Scholar]
- 26.Sato M, Vaughan MB, Girard L, Peyton M, Lee W, Shames DS, et al. Multiple oncogenic changes (K-RAS(V12), p53 knockdown, mutant EGFRs, p16 bypass, telomerase) are not sufficient to confer a full malignant phenotype on human bronchial epithelial cells. Cancer Res. 2006;66:2116–28. doi: 10.1158/0008-5472.CAN-05-2521. [DOI] [PubMed] [Google Scholar]
- 27.Holladay MW, Campbell BT, Rowbottom MW, Chao Q, Sprankle KG, Lai AG, et al. 4-Quinazolinyloxy-diaryl ureas as novel BRAFV600E inhibitors. Bioorg Med Chem Lett. 2011;21:5342–6. doi: 10.1016/j.bmcl.2011.07.019. [DOI] [PubMed] [Google Scholar]
- 28.Rowbottom MW, Faraoni R, Chao Q, Campbell BT, Lai AG, Setti E, et al. Identification of 1-(3-(6,7-dimethoxyquinazolin-4-yloxy)phenyl)-3-(5-(1,1,1-trifluoro-2-methylpropa n-2-yl)isoxazol-3-yl)urea hydrochloride (CEP-32496), a highly potent and orally efficacious inhibitor of V-RAF murine sarcoma viral oncogene homologue B1 (BRAF) V600E. J Med Chem. 2012;55:1082–105. doi: 10.1021/jm2009925. [DOI] [PubMed] [Google Scholar]
- 29.Sequist LV, Heist RS, Shaw AT, Fidias P, Rosovsky R, Temel JS, et al. Implementing multiplexed genotyping of non–small-cell lung cancers into routine clinical practice. Ann Oncol. 2011;22:2616–24. doi: 10.1093/annonc/mdr489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lipson D, Capelletti M, Yelensky R, Otto G, Parker A, Jarosz M, et al. Identification of new ALK and RET gene fusions from colorectal and lung cancer biopsies. Nat Med. 2012;18:382–4. doi: 10.1038/nm.2673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pao W, Hutchinson KE. Chipping away at the lung cancer genome. Nat Med. 2012;18:349–51. doi: 10.1038/nm.2697. [DOI] [PubMed] [Google Scholar]
- 32.Stransky N, Cerami E, Schalm S, Kim JL, Lengauer C. The landscape of kinase fusions in cancer. Nat Commun. 2014;5:4846. doi: 10.1038/ncomms5846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wiesner T, He J, Yelensky R, Esteve-Puig R, Botton T, Yeh I, et al. Kinase fusions are frequent in Spitz tumours and spitzoid melanomas. Nat Commun. 2014;5:3116. doi: 10.1038/ncomms4116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ballerini P, Struski S, Cresson C, Prade N, Toujani S, Deswarte C, et al. RET fusion genes are associated with chronic myelomonocytic leukemia and enhance monocytic differentiation. Leukemia. 2012;26:2384–9. doi: 10.1038/leu.2012.109. [DOI] [PubMed] [Google Scholar]
- 35.Gujral TS, Singh VK, Jia Z, Mulligan LM. Molecular mechanisms of RET receptor-mediated oncogenesis in multiple endocrine neoplasia 2B. Cancer Res. 2006;66:10741–9. doi: 10.1158/0008-5472.CAN-06-3329. [DOI] [PubMed] [Google Scholar]
- 36.Wang D PM, Fakih M, Lockhart AC, Olszanski AJ, Patel R, Brown PD, et al., editors. A phase 1 study of RXDX-105, an oral RET, BRAF and EGFR tyrosine kinase inhibitor, in patients with advanced or metastatic cancers [abstract]. AACR-NCI-EORTC International Conference on Molecular Targets and Cancer Therapeutics; 2015; Boston. [Google Scholar]
- 37.Planchard D, Mazieres J, Riely GJ, Rudin CM, Barlesi F, Quoix EA, et al. Interim results of phase II study BRF113928 of dabrafenib in BRAF V600E mutation–positive non-small cell lung cancer (NSCLC) patients. J Clin Oncol. 2013;(suppl):31. abstr 8009. [Google Scholar]
- 38.Mok TS, Wu YL, Thongprasert S, Yang CH, Chu DT, Saijo N, et al. Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. N Engl J Med. 2009;361:947–57. doi: 10.1056/NEJMoa0810699. [DOI] [PubMed] [Google Scholar]
- 39.Shaw AT, Kim D-W, Nakagawa K, Seto T, Crinó L, Ahn M-J, et al. Crizotinib versus chemotherapy in advanced ALK-positive lung cancer. N Engl J Med. 2013;368:2385–94. doi: 10.1056/NEJMoa1214886. [DOI] [PubMed] [Google Scholar]
- 40.Seto T, Yoh K, Satouchi M, Nishio M, Yamamoto N, Murakami H, et al. A phase II open-label single-arm study of vandetanib in patients with advanced RET-rearranged non-small cell lung cancer (NSCLC): Luret study. J Clin Oncol. 2016;34(suppl) abstr 9012. [Google Scholar]
- 41.Lee S, Lee J, Ahn M, Kim D, Sun J, Keam B, et al. A phase II study of vandetanib in patients with non-small cell lung cancer harboring RET rearrangement. J Clin Oncol. 2016;34(suppl) doi: 10.1093/annonc/mdw559. abstr 9013. [DOI] [PubMed] [Google Scholar]
- 42.Drilon A, S CS, S R, S R, G MS, R GJ, et al. Phase II study of cabozantinib for patients with advanced RET-rearranged lung cancers. J Clin Oncol. 2015;33(suppl) abstr 8007. [Google Scholar]
- 43.Patel MR, Fakih M, Olszanski AJ, Lockhart AC, Drilon AE, Fu S, et al. A phase 1 dose escalation study of RXDX-105, an oral RET and BRAF inhibitor, in patients with advanced solid tumors. J Clin Oncol. 2016;34(suppl) abstr 2574. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.