

Abstract
The complexity of transcriptome-wide protein—RNA interaction networks is incompletely understood. While emerging studies are greatly expanding the known universe of RNA-binding proteins, methods for the discovery and characterization of protein—RNA interactions remain resource intensive and technically challenging. Here we introduce a UV-C crosslinking and immunoprecipitation platform, irCLIP, which provides an ultraefficient, fast, and nonisotopic method for the detection of protein—RNA interactions using far less material than standard protocols.
UV-C crosslinking immunoprecipitation (CLIP) is a powerful technique for interrogating direct protein—RNA interactions1,2. Combined with high-throughput sequencing, CLIP platforms, namely HITS-CLIP3–5, PAR-CLIP6,7, iCLIP8,9, and derivations thereof10,11, have revealed transcriptome-wide protein—RNA interaction networks. Additionally, several recent CLIP mass spectrometry studies have greatly expanded the known mRNA-binding proteome in mammalian cells12–14. Surprisingly, approximately half of the proteins that these studies identified lacked known RNA-binding domains12–14. These findings indicate that the post-transcriptional regulation of RNA is more complex than previous studies have indicated. Therefore, the development of efficient, robust, and easy-to-use tools to study direct RNA-protein interactions is critical to the full characterization of gene regulatory networks.
Current CLIP-based methodologies present considerable technical challenges to widespread implementation15,16. Radioisotopes are required for visualization of UV-crosslinked protein—RNA complexes, and standard radioactive labeling of protein–RNA complexes occurs on the free 5′ ends of crosslinked RNA molecules and therefore does not report successful library adaptor ligation of RNA 3′ ends. Also, radioactive reagents decay, resulting in variable autoradiography signal across experiments. To address these shortcomings, we developed an infrared-dye-conjugated and biotinylated ligation adaptor for rapid and quantitative analysis of in vivo captured protein–RNA interactions (Fig. 1a and Supplementary Fig. 1a–c). The infrared-CLIP (irCLIP) adaptor exhibits the same efficiency in ligation reactions as a standard adaptor, fully recapitulates protein–RNA binding activity as visualized by radioisotope, and reduces the time required for protein–RNA complex visualization >10- to 100-fold (Supplementary Fig. 1d,e; infrared imaging performed with a LI-COR Odyssey CLx Imager). Importantly, the attamole sensitivity of irCLIP adaptor detection also facilitates rapid and quantitative quality controls at critical steps of irCLIP by simple nitrocellulose dot blotting (Supplementary Fig. 1b).
Figure 1.
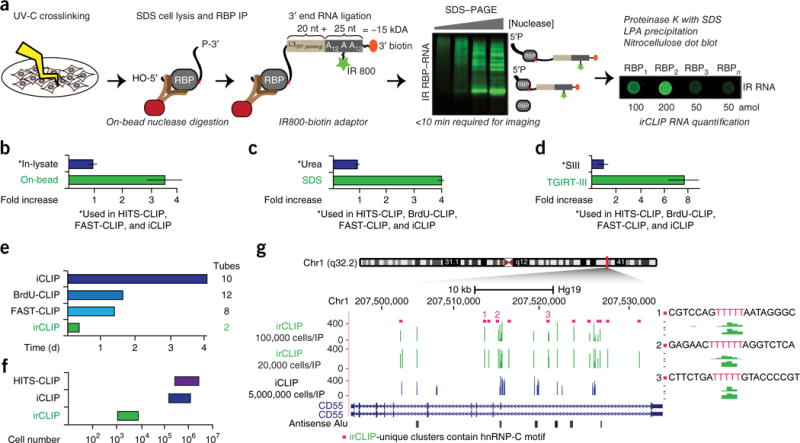
Ultrasensitive, efficient, and fast CLIP platform facilitates low cell number CLIP, (a) Schematic of irCLIP workflow with improved methodologies versus standard CLIP protocols in italics. Green stars denote internal IR800CW dye and orange ovals 3′ biotin modification. IP, immunoprecipitation. (b–d) Average fold increases in (b) target RNA fragments produced by on-bead versus standard in-lysate nuclease digestions across multiple RBPs; (c) purified RNA resulting from irCLIP reformulation of standard CLIP PK digestions and RNA precipitation; and (d) conversion of purified CLIP RNA to cDNA resulting from irCLIP replacement of the standard CLIP RT. (N = 8 (b), 6 (c), 4 (d) per group.) (e) Approximate time requirement for conversion of precipitated RNA to double-stranded DNA (dsDNA). (f) Approximate required input material for the indicated CLIP methodologies versus irCLIP. (g) Comparison of significant (false discovery rate (FDR) < 0.05) hnRNP-C clusters identified with irCLIP at the CD55 locus using 100,000 or 20,000 cells versus published9 iCLIP data using approximately 5,000,000 cells. irCLIP-unique clusters are marked above by magenta squares. Green, irCLIP; blue, iCLIP. 11 of 12 irCLIP-unique clusters contained the strong hnRNP-C consensus-binding motif, UUUUU. Three examples are shown to the right. Data plotted as mean ± s.e.m.
Despite the high protein copy number of many endogenous RNA-binding proteins (RBPs)17, published CLIP studies typically report the use of 5–20 million cells per immunoprecipitation. We suspected that systematic inefficiencies might accumulate across CLIP procedures, resulting in the need for large quantities of input material. We therefore took a stepwise approach to evaluate each biochemical transformation that is critical for the CLIP methodology.
Methodological papers for both HITS-CLIP15 and iCLIP16 recommend in-lysate nuclease digestions to cleave crosslinked RNA flanking the protein–RNA crosslink site. iCLIP, a recently developed version of CLIP, provides base resolution of protein–RNA contacts8 and relies on UV-C crosslinked peptide-RNA interactions to halt reverse transcriptase (RT). Given the high frequency of RT halting (~8O%18), cDNA molecules generated in iCLIP strategies are, on average, shorter than the isolated RNA crosslinked to the RBP of interest. Optimally digested RNA fragments should range from ~40 to 100 nucleotides (nt) in order to produce uniquely mappable cDNA inserts ranging from 20 to 50 nt. We reasoned that the complexity of initial lysates and/or the presence of inhibitory buffer components, which may vary greatly depending on experimental circumstances, might impede optimal digestion of crosslinked RNA. In support of this, we found that on-bead nuclease digestion markedly increased optimal RNA fragments for multiple classes of RNA-binding proteins (Fig. 1b and Supplementary Fig. 2a–h). To determine whether a particular nuclease was better at generating optimal RNA fragments, we compared on-bead nuclease digestions across the same panel of RBPs using RNase A, RNase 1, and S1 nuclease. While on-bead S1 digestions significantly outperformed in-lysate RNase A digestions (Supplementary Fig. 2), on-bead RNase A digestions produced the maximal number of optimal RNA fragments (Supplementary Fig. 3a–h). On-bead digestions may therefore maximize information recovery across CLIP experiments.
Next, we the addressed liberation of protein–RNA complexes from nitrocellulose membranes by proteinase K (PK) digestion. This is commonly done at physiologic temperature (37 °C) in the presence of urea15,16. Initial quantification of urea—PK-released irCLIP-adaptor-ligated RNA revealed >80% loss of material during precipitation. Because urea increases the dielectric constant of the surrounding medium and therefore inhibits precipitation of nucleic acid19, we reasoned that replacement of urea with alternative stimulators of PK activity could increase the efficiency of RNA recovery. We found that replacing urea with SDS and increasing the temperature facilitated liberation of nitrocellulose-bound RNA-peptide complexes. These changes significantly improved precipitation of irCLIP-adaptor-ligated RNA compared to urea-PK digestions and did not impact RNA integrity (Fig. 1c and Supplementary Fig. 4a–c).
Although it requires more than 30 steps and ~4 d, when compared to HITS-CLIP15,16, the iCLIP library construction method requires less time and fewer molecular biology steps. To assess the efficiency of iCLIP library construction, we used irCLIP-adaptor-ligated RNA and a control ‘cDNA’ (which bypassed reverse transcription and cDNA purification) and discovered 90–95% loss of irCLIP-adaptor-ligated RNA in the initial library steps.
Recently, a pair of novel RT enzymes derived from group II introns was described. Compared with previously described enzymes, these enzymes have higher yield, efficiency, fidelity, and processivity, and they operate at higher temperatures for cDNA extension20. We compared one of these enzymes, TGIRT-III, to the commonly used SuperScript III enzyme. TGIRT-III produced four- to eight-fold more cDNA than SuperScript III (as assessed by requiring two or three fewer PCR cycles to generate the same amount of library material; Fig. 1d and Supplementary Fig. 5a). We further hypothesized that loss of cDNA could occur during cleanup reactions preceding PCR. To assess this, we replaced silica-column-based cDNA purification with an AMPure bead-isopropanol mixture (Beckman Coulter), which increased cDNA recovery two- to three-fold (Supplementary Fig. 5b). Lastly, while newer CLIP strategies including BrdU-CLIP10 and FAST-iCLIP11 reduce overall library construction times from ~4 to ~1.5 d, these methods still include a significant number of tube-column transfers that could contribute to systemic loss of material. We therefore leveraged the above use of magnetic-bead-based purification to develop an 8 h, 96-well-based ‘two-tube’ strategy that includes on-bead library size selection, eliminating all precipitations and column purifications present in other protocols (Fig. 1e and Online Methods). The cumulative methodological improvements of irCLIP thus allow a > 100-fold reduction in cellular inputs, approaching a strategy with minimal information loss (Fig. 1f,g).
Incorporating the above improvements into a coherent irCLIP protocol (Online Methods), we performed irCLIP on hnRNP-C (Supplementary Fig. 6a,b), an RBP with multiple published iCLIP data sets available for comparison8,9. Pearson correlations of RT stops from individual biological replicates mapping to human genome (hgl9) were 0.649, 0.534, and 0.553, corresponding to 100,000-cell irCLIP, 20,000-cell irCLIP, and ~5,000,000-cell iCLIP, respectively (Fig. 2a). We confirmed that this reproducibility was stable across other RBPs, as irCLIP of PTBP1 and HuR (Supplementary Fig. 6c,d) from 100,000 cells had similar Pearson correlation values between biological replicates (Supplementary Fig. 7a,b). Importantly, even at these low cell input amounts, irCLIP libraries produced sequencing data with a high unique read fraction as a result of the efficiency gains at the biochemical processing steps (Fig. 2b and Supplementary Fig. 7c).
Figure 2.
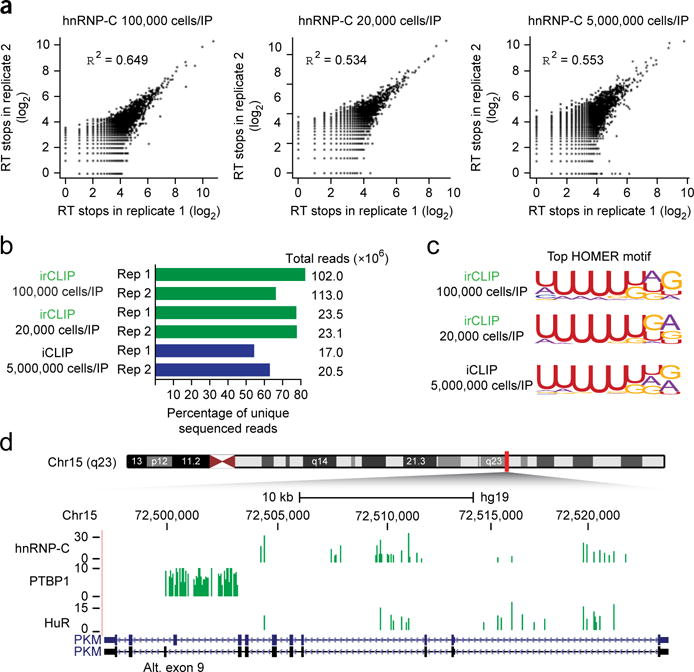
irCLIP produces high-quality data sets with greatly reduced cellular inputs, (a) Pearson correlations of RT stops between biological replicates of hnRNP-C irCLIP of the indicated number of cells per immunoprecipitation. (b) Histogram of the unique read fractions of the hnRNP-C irCLIP and iCLIP biological replicates, (c) Top HOMER motif identified for all significant hnRNP-C clusters from the indicated data set. (d) Comparison of significant (FDR < 0.05) hnRNP-C, PTBP1, and HuR clusters identified with irCLIP at the PKM locus. Alt., alternate. Green, irCLIP; blue, iCLIP.
iCLIP defines the end of each cDNA molecule as the RT stop (and thus one base 3′ of the direct protein–RNA crosslink8). Analytic tools such as FAST-iCLIP use this precise positional information to intersect replicate CLIP data sets at the RT-stop level to obtain high-confidence RBP binding sites for subsequent peak calling and downstream analysis11. Published hnRNP-C iCLIP9 has 6.2% intersection at the RT-stop level between biological replicates (Supplementary Table 1). irCLIP outperforms this; at very low cell input (20,000) we found 7.4% intersection, and at high cell input (100,000) we found 19.6% intersection (Supplementary Table 1). The biological relevance of clusters resulting from irCLIP RT-stop intersections was confirmed by regeneration of the expected top motif, which matched published iCLIP (Fig. 2c). High RT-stop intersection fraction was also evident in the 100,000-cell irCLIP experiments of HuR (19.8%) and PTBP1 (14.5%) (Supplementary Table 1). Additionally, we found minimal correlation of RT stops within significant clusters between hnRNP-C, PTBP1, and HuR (0.087 for HuR versus PTBP1, 0.133 for hnRNP-C versus PTBP1, and 0.176 for hnRNP-C versus HuR), indicating high specificity of high-confidence irCLIP clusters for each RBP (Supplementary Fig. 7d). Previous iCLIP analysis of hnRNP-C identified a major role for hnRNP-C in binding intronic antisense Alu elements9. irCLIP faithfully captured this biology, as 27.7% of hnRNP-C clusters identified by irCLIP (versus 25% identified by iCLIP) bound intronic antisense Alu elements (Supplementary Table 2). Finally, HITS-CLIP analysis of PTBP1 described a direct role for PTBP1 in the control of pyruvate kinase Μ (PKM) alternative exon 9 inclusion5. Visualization of irCLIP RT stops at the PKM locus also identified highly unique PTPB1 clusters surrounding exons 8 and 9 of PKM (Fig. 2d).
The irCLIP methodology at once greatly improves per-cell capture of protein–RNA information and simplifies the biochemical steps involved. These advances provide the basis for irCLIP application on small cell samples. Additionally, the highly sensitive nature of iCLIP will facilitate further expansion of the list of noncanonical RBPs12–14 that may be weakly expressed or may only contextually bind RNA in a limited manner. Finally, simultaneous elimination of radioisotope and preservation of the ability to visualize protein–RNA complexes, as well as procedural simplification, should expand the use of CLIP-based assay in the scientific community.
ONLINE METHODS
irCLIP adaptor construction
The L3-azide-biotin substrate oligonucleotide for irCLIP (5′-OH-AGATCGGAAGAGCGGTTCAGAAAAAAAAAAAA/iAzideN/AAAAAAAAAAAA/3Bio/-3′) is custom synthesized at the 1 μmol synthesis scale (approximate yield of 20 nmol) by Integrated DNA Technologies (IDT). The nonstandard modifications are the internal NHS-azide modification at nucleotide position 32 (/iAzideN/) and the 3′ biotin modification (/3bio/) at the terminal base. In a 1.5-mL Eppendorf tube, 20 nmol of oligonucleotide is phosphorylated with 100 units of OptiKinase (Affymetrix, cat# 78334) in a reaction volume of 0.2 mL containing 1 mM ATP for 1 h at 37 °C. The entire reaction was precipitated at −20 °C overnight by addition of 20 μL 3 Μ NaAcetate, pH 5.5, and 0.55 mL of 100% ethanol. The precipitated phospho-oligonucleotide was pelleted for 30 min at 4 °C in a microcentrifuge at > 13,000 r.p.m., washed one time with ice-cold 80% ethanol, dried, and resuspended in 0.5 mL of nuclease-free water. The entire volume of the phospho-oligonucleotide was next preadenylated using the 5′ DNA Adenylation Kit (NEB, cat# E2610L) by addition of 75 μL each of 1 mM ATP and 10× buffer and 100 μL of Mth RNA ligase. The reaction was incubated for 2 h at 65 °C (it is normal for the reaction to become cloudy) and inactivated at 85 °C for 10 min. Half of the reaction (0.375 mL) was transferred into a second 1.5-mL tube and precipitated overnight at −20 °C by addition of 40 μL of 3 Μ NaAcetate, pH 5.5, and 1 mL of 100% ethanol. The precipitated preA-oligonucleotide was pelleted for 30 min in a microcentrifuge at > 13,000 r.p.m., washed one time with ice-cold 80% ethanol, dried, and resuspended in 0.18 mL of PBS. Click-chemistry conjugation of the infrared dye was done by addition of 20 μL of 10 mM IRdye-800CW-DBCO (LiCor, cat# 929-50000) and incubated for 2 h at 37 °C. The final preA-L3-IR800-biotin DNA adaptor was then column purified using the QIAquick Nucleotide Removal Kit (Qiagen, cat# 28304). The 200 μL click reaction was transferred to a 15-mL Falcon tube and mixed with 4.8 mL of PNI buffer (PNI buffer can also be generated from Qiagen PB buffer by addition of 1.5 volumes of 100% isopropanol). 250 μL aliquots were dispensed across 20 QIAquick nucleotide removal columns (or any purple DNA Qiagen column) and spun at 6,000 r.p.m. for 30 s. Flow-through was decanted and columns washed with 0.75 mL of 80% ethanol. Ethanol washes were decanted and columns spun dry for 2 min at 13,000 r.p.m. Each column was transferred to a new, 1.5-mL Eppendorf tube and eluted by addition of 50 μL of nuclease-free water for 2 min. Columns were then spun for 1 min at 13,000 r.p.m. Eluates were pooled into a single fraction and quantitated. The eluate should appear pale blue with an expected concentration of ~10 μM. The adaptor is stored at −20 °C and is stable for at least 1 year. Working stocks of 1 μM can be freeze—thawed at least 20 times without any detectable loss in activity.
Antibodies used for irCLIP
Anti-hnRNP-C (4F4), anti-HuR (N-16), and anti-PTBPl (N-20) were purchased from Santa Cruz Biotechnology. Anti-ILF3 (cat# 612155) was purchased from BD Biosciences. Anti-DDX5 was purchased from Abeam (cat# ab2l696). Antibodies were preconjugated to Protein G Dynabeads (Thermo Fisher Scientific, cat # 10004D) overnight in PBS. Rabbit anti-mouse IgG, purchased from Thermo Fisher Scientific (cat# 31188), was added to overnight preconjugations of primary mouse antibodies at a 1:1 ratio. For immunoblots, antibodies were used at 1:1,000. For immunoprecipitations, 1 μg of antibody was used per 50 μg of lysate.
irCLIP protein purification and ligation
Adherent HeLa cells grown to ~80% confluence in six-well plates were quickly rinsed with ice-cold PBS, aspirated on-plate and crosslinked on ice with 254 nM UV-C at 0.3 J/cm2. Cells were then incubated with ice-cold PBS/10 mM EDTA for 5 min, collected by cell scraping, and counted. Pelleted cells were lysed in 0.2 mL of SDS lysis buffer (1% SDS; 50 mM Tris, pH 7.5; 1 mM EDTA) and sonicated using a Bioruptor (Diagenode) to remove all viscosity (high setting, six cycles of 30 s on, 45 s rest). Lysates were clarified by centrifugation at maximum speed for 15 min, and soluble fractions were transferred to new tubes. Two volumes of IP Dilution Buffer (1.1% Triton X-100; 50 mM Tris, pH 7.5; 1 mM EDTA; 450 mM NaCl) were added to the clarified SDS lysate and then quantitated in duplicate using the Pierce ΒCA Protein Assay Kit. Lysates were rotated end over end for 1.5 h at 4 °C with antibody preconjugated to Protein G Dynabeads. Immunoprecipitates were washed sequentially for 10 min each at 4 °C with 1 mL of high-stringency buffer (20 mM Tris, pH 7.5; 120 mM NaCl; 25 mM KCl; 5 mM EDTA; 1% Trition-X100; 1% Na-deoxycholate), 1 mL of high-salt buffer (20 mM Tris, pH 7.5; 1 Μ NaCl; 5 mM EDTA; 1% Trition-X100; 1% Na-deoxycholate; 0.001% SDS), 1 mL of low-salt buffer (20 mM Tris, pH 7.5; 5 mM EDTA). Prior to on-bead nuclease digestions, beads were rinsed twice on-magnet with 0.25 mL then with 0.1 mL of NT2 buffer (50 mM Tris, pH 7.5; 150 mM NaCl; 1 mM MgCl2; 0.0005% Igepal). RNAse A and RNAse 1 were diluted in NT2 buffer. RNAse A (Affymetrix #70194Z) was used at a final concentration between 20 and 1.6 ng/mL. RNAse 1 (Thermo Fisher #AM2294) was used at final concentration between 0.4 and 0.0625 u/μL. S1 nuclease (Clontech, cat# 2410) was diluted using the manufacturer-provided 10× buffer, 300 mM NaAcetate pH 4.6,2,800 mM NaCl, and 10 mM ZnSO4, and used at a final concentration ranging from 12–1 u/μL. All nuclease reactions were a total aqueous volume of 30 μL and supplemented with 6 μL of PEG400 (16.7% final). Reactions were chilled on ice before resuspension of immunoprecipitates. All nuclease reactions were incubated at 30 °C for 15 min in an Eppendorf Thermomixer, 15 s 1,400 r.p.m., 90 s rest. Nuclease digestions were stopped by addition of 0.5 mL of ice-cold high-stringency buffer. Immunoprecipitates were then quickly rinsed with 0.25 mL then with 0.05 mL of ice-cold NT2 buffer. RNAse-A- and RNAse-1-digested complexes were then dephosphorylated with T4 PNK (NEB #M0210) for 30 min in an Eppendorf Thermomixer at 37 °C, 15 s 1,400 r.p.m., 90 s rest in a 30-μL reaction (50 mM Tris, pH 7.0; 10 mM MgCl2; 5 mM DTT) containing 10 units of T4 PNK and 0.1 μL SUPERase-IN (Thermo Fisher #AM2694) and supplemented with 6 μL of PEG400. Dephosphorylation reactions were removed (not applicable to S1 digestions) and immunoprecipitates were 3′-end ligated with T4 RNA Ligase 1 (NEB, cat# M0204) overnight in an Eppendorf Thermomixer at 16 °C, 15 s 1,400 r.p.m., 90 s rest in a 30-μL reaction containing 10 units T4 RNA Ligase 1, 1 pmole preA-DNA-adaptor, and 0.1 μL SUPERase-IN. The 30-μL ligation reactions were supplemented with 6 μL of PEG400 (16.7% final). The following day, beads were placed on the magnetic stand, ligation reactions were removed, and beads were resuspended in 10 μL of 1× LDS sample buffer + reducing agent and heated for 15 min at 75 °C. Samples were either stored at −20 °C or immediately resolved by SDS-PAGE using NuPAGE 4–12% Bis-Tris Gels (1.0 mm × 12 well) at 180 V for 45 min. Resolved RNP complexes were wet transferred to nitrocellulose at 400 mA for 60 min at 4 °C.
Proteinase K recovery of RNA-peptide fragments
Nitrocellulose membranes should not be allowed to dry post-transfer. Siliconized tubes were used for all library steps. Membranes were placed on prewetted Whatman filter paper and target region(s) excised with scalpels. Membranes were cut into ~0.5 by 1-mm narrow strips that easily come to rest in the bottom of a siliconized 1.5-mL Eppendorf tube. 0.2 mL of proteinase K reaction buffer (100 mM Tris, pH 7.5; 50 mM NaCl; 1 mM EDTA; 0.2% SDS) containing 10 μL of proteinase K (Thermo Fisher Scientific, cat# AM2546) was added to each tube and incubated for 60 min at 50 °C in an Eppendorf Thermomixer, 15 s 1,000 r.p.m., 30 s rest. After 60 min, tubes were removed from the thermomixer and briefly centrifuged. One microliter of the proteinase K reaction was dot blotted on nitrocellulose and scanned on an Odyssey Clx infrared imager to confirm full release of expected RNA-adaptor-ligated products. 200 μL of saturated-phenol-chloroform, pH, 6.7, was added to each tube and incubated for 10 min at 37 °C in an Eppendorf Thermomixer, 1,400 r.p.m. Tubes were briefly centrifuged and the entire contents were transferred to a 2-mL Heavy Phase Lock Gel tube (5Prime, cat# 2302830). After 2 min centrifugation at > 13,000 r.p.m., the aqueous layer was re-extracted with 1 mL of chloroform (invert tube 10 times to mix; do not vortex, pipet, or shake) in the same 2-mL Heavy Phase Lock Gel tube and centrifuged for 2 min at > 13,000 r.p.m. The aqueous layer was then transferred to a new 2-mL Heavy Phase Lock Gel tube and extracted again with an additional 1 mL of chloroform. After 2 min centrifugation at > 13,000 r.p.m., the aqueous layer was transferred to a siliconized 1.5-mL Eppendorf tube and precipitated overnight at −20 °C by addition of 10 μl 5M NaCl, 3 μL Linear Polyacrylamide (Thermo Fisher Scientific, cat# AM9520), and 0.8 mL of 100% ethanol.
irCLIP library oligonucleotide sequences
cDNA synthesis primers were purchased from IDT with three modifications, 5′ phosphorylation (/5phos/), and two internal carbon spacers (/iSpl8/), which are required for generation of linear PCR product using circularized cDNA as template. The 17 nt of the 5′ end comprised a degenerate barcode using the 6-bp Truseq Illumina barcode sequences. cDNA-barcode1 (6-bp TruSeq barcode in bold):/5phos/WWWNNNATCACGNNNNNTACCCTTCGCTTCACACACAAG/iSp 18/GGATCC/iSp 18/TACTGAACCGC. Additional TruSeq barcodes 2–24: CGATGT, TTAGGC, TGACCA, ACAGTG, GCCAAT, CAGATC, ACTTGA, GATCAG, TAGCTT, GGCTAC, CTTGTA, AGTCAA, AGTTCC, ATGTCA, CCGTCC, GTCCGC, GTGAAA, GTGGCC, GTTTCG, CGTACG, GAGTGG, ACTGAT, ATTCCT. P3short (cDNA elution oligonucleotide): CTGAACCGCTCTTCCGATCT. PCR1 primers, P3tall: GCATTCCTGCTGAACCGCTCTTCCGATCT, P6tall: TTTCCCCTTGTGTGTGAAGCGAAGGGTA. PCR2 primers (PAGE purified). P3solexa: CAAGCAGAAGACGGCATACGAGATCGGTCTCGGCATTCCTGCTGAACCGCTCTTCCGATCT, P6solexa: AATGATACGGCGACCACCGAGATCTACACTCTTTCCCCTTGTGTGTGAAGCGAAGGGTA.
P6 sequencing primer (note: irCLIP uses a custom sequencing oligonucleotide): CACTCTTTCCCCTTGTGTGTGAAGCGAAGGGTA.
irCLIP library construction
RNA fragments were pelleted at > 13,000 r.p.m. for 45 min at 4 °C, washed once with 1 mL of ice-cold 75% ethanol, and air dried. Pellets were resuspended in 15 μL of 10 mM NaCl and heated at 37 °C for 10 min. After brief centrifugation, 1 μL of the dissolved RNA was dot blotted on nitrocellulose and scanned on an Odyssey Clx infrared imager to quantify the recovery of RNA-adaptor-ligated products. 13.75 μL of RNA was mixed with 1 μL of 1 μM cDNA synthesis primer in a 0.2-mL PCR tube and heated to 70 °C for 10 min then rapidly cooled to 4 °C. 4 μL of 5× cDNA synthesis buffer (100 mM Tris, pH 7.5; 50 mM MgCl2,250 mM NaCl; 25 mM DTT, 0.1%; Tween) and 0.2 μL of TIGRT-III reverse transcriptase (Ingex, #TGIRT50) was added to the annealed RNA and incubated at room temperature for 30 min. 1 μL of 25 mM dNTPs was added to the reaction, mixed well, and incubated in a thermocycler for 2 h at 60 °C. cDNA-RNA hybrids were captured by addition of 5 μL of MyOne Streptavidin C1 Dynabeads (Thermo Fisher Scientific, #65001) that had been rinsed and suspended in 100 μL of NT2 buffer and end-over-end rotation for 30 min at 4 °C. Beads were placed on a 96-well magnet and washed twice with 0.1 mL high-stringency buffer and twice with 0.1 mL of PBS. Beads were resuspended in 10 μL of cDNA elution/RNA degradation buffer (8.25 μL water, 1 μL of 1 μM P3short oligonucleotide, and 0.75 μL of 50 mM MnCl2) and placed in a thermocycler with the program: 5 min 95 °C, 1 min 75 °C, ramp 0.1° s−1 to 60 °C forever. After 15 min, tubes were removed and mixed with 5 μL of Circligase-II reaction buffer (3.3 μL water, 1.5 μL 10× Circligase-II buffer, and 0.2 μL of Circligase-II; Epicentre, #CL9021K). cDNA was circularized in a thermocycler for 1.5 h at 60 °C. cDNA was captured by addition of 30 μL of Ampure XP beads (Beckman Coulter, cat# A63880), 75 μL of isopropanol, and 15 min of incubation (the solution was remixed after 7.5 min). Beads were washed once with 80% ethanol, dried for 5 min, and resuspended in 14 μL of water. For maximal elution, tubes were placed in a thermocycler at 95 °C for 2 min and immediately transferred to a 96-well magnet. The 14 μL eluate was transferred to a new 0.2-mL PCR tube containing 15 μL of 2× Phusion HF-PCR Master Mix (NEB, #M0531), 0.5 μL of 30 μM P3/P6 PCR1 oligonucleotide mix, and 0.5 μL of 15× SYBR Green I (Thermo Fisher Scientific, cat# S7563). The tubes were then placed in a Stratagene MX3000P qPCR machine with the following program: 98 °C 2 min and then 15 cycles of 98 °C 15 s, 65 °C 30 s, and 72 °C 30 s, with data acquisition set to the 72 °C extension. PCR1 reactions were removed from the machine after reaching the ‘knee’ of exponential growth. PCR1 reactions were then subjected to one round of magnetic bead size selection by addition of 4.5 μL of isopropanol, 54 μL of Ampure XP beads, and incubation for 10 min. Beads were washed once with 80% ethanol, dried for 5 min and eluted in 10 μL of water. PCR1 products were subjected to a second round of size selection by addition of 1.5 μL of isopropanol, 18 μL of Ampure XP beads, and incubation for 10 min. Beads were washed once with 80% ethanol, dried for 5 min, and eluted in 10 μL 500 nM P3solexa/P6solexa oligo mix. 10 μL of 2× Phusion HF-PCR Master was added to each tube and placed in a thermocycler with the following program: 98 °C 2 min, three cycles of 98 °C 15 s, 65 °C 30 s, and 72 °C 30 s. Final libraries were purified by addition of 36 μL of Ampure XP beads and incubation for 5 min. Beads were washed twice with 70% ethanol, dried for 5 min, and eluted in 20 μL of water. 1–2 μL of libraries were quantitated by HS-DNA bioanalysis.
irCLIP data analysis
irCLIP data was processed using a redesigned FAST-iCLIP pipeline downloaded from GitHub: https://github.com/ChangLab/FAST-iCLIP. Briefly, we made several improvements to the FAST-iCLIP pipeline. We sped up FASTQ preprocessing significantly by doing duplicate removal (with fastx-collapser) before trimming of 5′ barcodes and 3′ adaptors, and we also improved the speed of figure making and annotation of mapped reads. The pipeline now handles ‘zipped’ and ‘unzipped’ FASTQ files to reduce the disk space of source files. We have implemented STAR alignment algorithm to map more accurately to the genome for nonrepeat mapping reads. In addition, we now filter reads by their mapping quality using the −q 30 parameter in samtools view (this value can be set by the user). Finally, to avoid double counting clusters of RT stops, we now remove clusters that overlap.
To increase the types of repetitive transcripts analyzed, we now separately report reads that map to tRNA loci using a tRNA index that contains one copy for each type. For the irCLIP processing we used the following specific parameters:
f 18 (trims 17 nt from the 5′ end of the read).
l 15 (includes all reads longer than 15 nt).
tr (repeat threshold rule) m, n: at least m samples must each have at least n RT stops mapped to repeat RNA molecules. Default is 1,4(1 sample); 2, 3 (2 samples); and x, 2 (x > 2 samples).
tn (nonrepeat threshold rule) m, n: at least m samples must each have at least n RT stops mapped to nonrepeat RNA molecules. Default is 1,4 (1 sample); 2, 3 (2 samples); and x, 2 (x > 2 samples).
sr (STAR Ratio) Maximum mismatches per base allowed for STAR genome mapping (corresponds to outFilterMismatchNo-verLmax). Default is 0.08 (2 mismatches per 25 mapped bases).
bm (Bowtie MAPQ) Minimum MAPQ (Bowtie alignment to repeat/tRNA/retroviral indexes) score allowed. Default is 42.
We designed and implemented a custom script to discover RNA primary sequence motifs (motif Analysis.py, https://github.com/ChangLab/FAST-iCLIP). In the script, we ensure that there are no overlapping RT stop ‘windows’ where we search for motifs by selecting RT stops from highest to lowest enrichment. We have also added capabilities to search for motifs in 5′UTR, 3′UTR, coding sequence, introns, exons, and the whole transcript as well as to count the percentage of RT stops in these regions for each gene. For irCLIP analysis, we masked out any RT stop that occurred ±25 nt surrounding this strongest RT stop. After identifying the highest RT stops within each irCLIP data set, the HOMER tool (http://homer.salk.edu/homer/motif/) findMotifsGenome.pl is used with the following parameters -rna -bg -len 8 -S 10.
Supplementary Material
Acknowledgments
We thank A. Fire, P. Sarnow, and M. Kay for presubmission review. We thank L. Morcom and P. Bernstein for expert administrative assistance and members of the Khavari lab for helpful discussions. This work was supported by the US VA Office of Research and Development, NIH AR49737 and NIH CA142635 (P.A.K.), P50-HG007735 and R01-ES023168 (H.Y.C.), and the Stanford Medical Scientist Program and NIH 1F30CA189514-01 (R.A.F.).
Footnotes
Accession codes. irCLIP data have been deposited in the Gene Expression Omnibus under the accession code GSE78832.
Note: Any Supplementary Information and Source Data files are available in the online version of the paper.
AUTHOR CONTRIBUTIONS
B.J.Z. designed and executed experiments, analyzed the data, and wrote the manuscript with input from the coauthor R.A.F. R.A.F. and B.T.D. designed the FAST-iCLIP pipeline for analysis of ir/iCLIP data. Y.S. executed statistical analysis of ir/iCLIP data replicates. P.A.K, and H.Y.C. designed experiments and wrote the manuscript.
COMPETING FINANCIAL INTERESTS
The authors declare no competing financial interests.
References
- 1.Brimacombe R, Stiege W, Kyriatsoulis A, Maly P. Methods Enzymol. 1988;164:287–309. doi: 10.1016/s0076-6879(88)64050-x. [DOI] [PubMed] [Google Scholar]
- 2.Ule J, Jensen K, Mele A, Darnell RB. Methods. 2005;37:376–386. doi: 10.1016/j.ymeth.2005.07.018. [DOI] [PubMed] [Google Scholar]
- 3.Licatalosi DD, et al. Nature. 2008;456:464–469. doi: 10.1038/nature07488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chi SW, Zang JB, Mele A, Darnell RB. Nature. 2009;460:479–486. doi: 10.1038/nature08170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Xue Y. et at Mol Cell. 2009;36:996–1006. doi: 10.1016/j.molcel.2009.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hafner M, et al. Cell. 2010;141:129–141. doi: 10.1016/j.cell.2010.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lebedeva S, et al. Mol Cell. 2011;43:340–352. doi: 10.1016/j.molcel.2011.06.008. [DOI] [PubMed] [Google Scholar]
- 8.König J, et al. Nat Struct Mol Biol. 2010;17:909–915. doi: 10.1038/nsmb.1838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zarnack K, et al. Cell. 2013;152:453–466. doi: 10.1016/j.cell.2012.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Weyn-Vanhentenryck SM, et al. Cell Rep. 2014;6:1139–1152. doi: 10.1016/j.celrep.2014.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Flynn RA, et al. RNA. 2015;21:135–143. doi: 10.1261/rna.047803.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Castello A, et al. Cell. 2012;149:1393–1406. doi: 10.1016/j.cell.2012.04.031. [DOI] [PubMed] [Google Scholar]
- 13.Baltz AG, et al. Mol Cell. 2012;46:674–690. doi: 10.1016/j.molcel.2012.05.021. [DOI] [PubMed] [Google Scholar]
- 14.Kwon SC, et al. Nat Struct Mol Biol. 2013;20:1122–1130. doi: 10.1038/nsmb.2638. [DOI] [PubMed] [Google Scholar]
- 15.Moore MJ, et al. Nat Protoc. 2014;9:263–293. doi: 10.1038/nprot.2014.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Huppertz I, et al. Methods. 2014;65:274–287. doi: 10.1016/j.ymeth.2013.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kulak NA, Pichler G, Paron I, Nagaraj N, Mann M. Nat Methods. 2014;11:319–324. doi: 10.1038/nmeth.2834. [DOI] [PubMed] [Google Scholar]
- 18.Urlaub H, Hartmuth K, Lührmann R. Methods. 2002;26:170–181. doi: 10.1016/S1046-2023(02)00020-8. [DOI] [PubMed] [Google Scholar]
- 19.Choe WS, Middelberg AP. Biotechnol Prog. 2001;17:1107–1113. doi: 10.1021/bp010110p. [DOI] [PubMed] [Google Scholar]
- 20.Mohr S, et al. RNA. 2013;19:958–970. doi: 10.1261/rna.039743.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.