

Abstract
More than100 human genetic skin diseases, impacting over 20% of the population, are characterized by disrupted epidermal differentiation. A significant proportion of the 90 genes identified in these disorders to date are concentrated within several functional pathways, suggesting the emergence of organizing themes in epidermal differentiation. Among these are the Notch, TGFβ, IKK, Ras/MAPK, Phosphoinositide 3-kinase, p63, and Wnt signaling pathways as well as core biologic processes mediating calcium homeostasis, tissue integrity, cornification, and lipid biogenesis. Here, we review recent results supporting the central role of these pathways in epidermal differentiation, highlighting the integration of genetic information with functional studies to illuminate the biological actions of these pathways in humans as well as guide development of future therapeutics to correct their dysfunction.
Keywords: epidermis, differentiation, genetics, skin disease, keratinocyte
Epidermal diseases arise from aberrant epidermal differentiation
The epidermis is a self-renewing stratified epithelial tissue that forms the outer barrier of the skin. As such, its function is to protect the organism from outside insults, such as bacterial pathogens, and to prevent water-loss. The epidermis encompasses distinct layers of keratinocytes: the basal layer consists of self-renewing progenitor cells; the spinous layer, lying above the basal layer, consists of upward migrating, differentiating keratinocytes; the granular layer, consists of cells producing the substrates necessary to form the impermeable barrier; and the stratum corneum consists of terminally differentiated enucleated lipid-rich corneocytes that have undergone cornification to form the outer skin surface (Figure 1). Epidermal self-renewal and the mechanisms of stem cell maintenance have been recently reviewed [1]. Here, we focus on genetic alterations underlying epidermal disorders caused by dysfunction of epidermal differentiation, organized by the biological process or signaling pathway most affected (Table 1).
Figure 1. Epidermal differentiation and overview of disorders affecting differentiation.
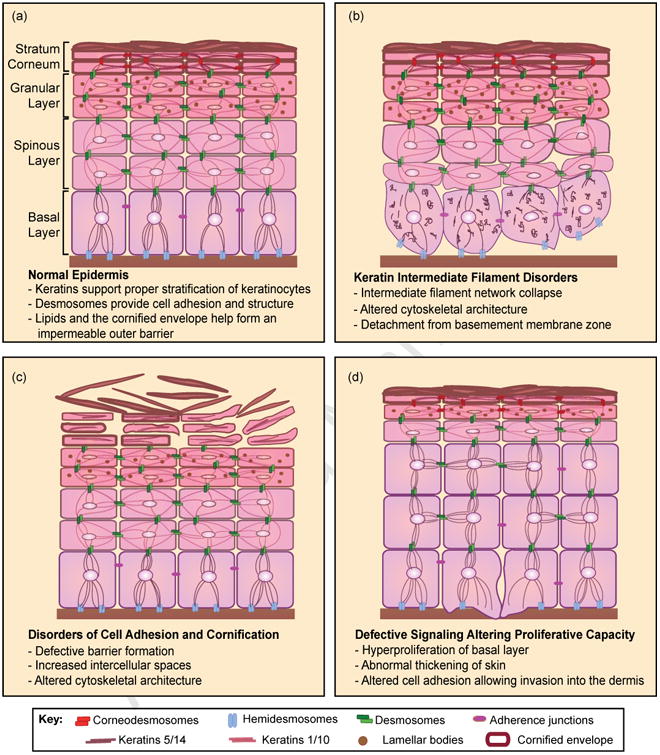
(a) Keratinocytes undergo a process of terminal differentiation involving stratification, which consists of the upward migration of keratinocytes from the basal layer containing progenitor cells into the spinous and granular layers; here, lamellar bodies provide the materials for formation of the impermeable epidermal barrier. (b) Mutations in keratin intermediate filaments 5 and 14 result in detachment from the basement membrane with disruption of the keratin intermediate filament network resulting in altered cytoskeletal architecture. (c) Mutations in components necessary for cell adhesion, such as corneodesmosome mutations exemplified here, result in increased intercellular spaces due to lack of cell-cell contact; mutations affecting this process of cornification result in defective barrier formation and dry, scaly skin. (d) Altered cell signaling can result in defective differentiation with increased proliferative capacity of basal progenitor cells, loss of cell adhesion, and the potential to become invasive squamous cell carcinoma.
Table 1. Epidermal disorders with associated genes and processes affected.
| Disease | Mutated Genes | Process Affecteda | Phenotype |
|---|---|---|---|
| Pachyonychia Congenita | KRT 6a, 16, 17 | IF | Hk, nail dystrophy |
| Epidermolytic Hyperkeratosis | KRT1, KRT10 | IF | Blistering, Hk |
| Epidermolysis Bullosa Simplex | KRT5, KRT14 | IF | Trauma-induced blistering with minimal scarring |
| Icthyosis with Confetti | KRT10 | IF | Blistering, Hk, pale confetti-like spots |
| Epidermolysis Bullosa Simplex-Ogna | PLEC1 | BMZ Adhesion, Hemidesmosomes | Trauma-induced blistering |
| Junctional Epidermolysis Bullosa | LAMA3, LAMB3, LAMC2, COL17A1, ITGB4, ITGA6 | BMZ Adhesion | Generalized blistering and erosions |
| Dystrophic Epidermolysis Bullosa | COL7A1 | BMZ Adhesion | Generalized blistering of skin and erosions with scarring |
| Lethal Congenital Epidermolysis Bullosa | JUP | Desmosomes | Generalized blistering and erosions, alopecia, onycholysis |
| Palmoplantar Keratoderma | JUP, PG, DSP, DSG1, KRT9, KRT16, KRT1 | CA, Desmosomes, IF | Hk affecting palms and soles, curly hair, arrythmia |
| Neonatal Ichthyosis Sclerosing Cholangitis Syndrome | CLDN1 | TJ | Sclerosing cholangitis, ichthyosis, hypotrichosis, dental abnormalities |
| Generalized Peeling Skin Disease | CDSN | Desmosomes | Pruritus, patchy peeling of entire skin |
| Netherton's Syndrome | SPINK5 | Desmosomes | Hk, hair shaft defects |
| Exfoliative Ichthyosis | CSTA | CA | Scaly skin and peeling of non-erythematous skin on the palms and soles |
| Vohwinkle Syndrome | LOR, GJB2 | CE, Gap Junction | Honeycomb Hk on palms and soles |
| Erythrokeratodermia variabilis | GJB3, GJB4 | Gap Junction | Hk, transient patches of erythema |
| Ichthyosis vulgaris | FLG | CE | Dry, scaly, itchy skin covering entire body (Hk) |
| Lamellar Ichthyosis | TGM1, ABCA12, ALOX12B, ALOXE3, ICHTHYIN, CYP4F22 | CE, FA & Chol. metabolism | Hk with scales and erythema covering entire body |
| Recessive X-linked Ichthyosis (RXLI) | STS | FA and Chol. metabolism | Large, brown scales covering limbs and trunk |
| Harlequin's Ichthyosis | ABCA12 | FA and Chol. metabolism | Hk, severe form of congenital ichthyosis with grave prognosis |
| Darier Disease | ATP2A2 | Ca2+ signaling | Hk affecting the seborrheic areas |
| Hailey-Hailey Disease | ATP2C1 | Ca2+ signaling | Hk affecting the flexural areas |
| Ankyloblepharon-Ectodermal Defects-Cleft Lip/Palate | TP63 | Transcription | Fusion of eyelids, epidermal erosion, dystrophic nails, wiry hair, cleft lip/palate |
| Multiple Self-healing Squamous Epithelioma | TGFBR1 | Signal transduction | SCC-like invasive tumors |
| Noonan Syndrome | PTPN11, KRAS | MAPK signaling | Heart, skeletal, hematologic and cognitive defects, distinctive facial dysmorphisms, Hk, lymphedema |
| Cowden's Syndrome | PTEN | PI3K/Akt signaling | Hamartomas of skin and mucosal surfaces |
| Cardio-facio-cutaneous (CFC) syndrome | KRAS, BRAF, MEK1/2 | MAPK signaling | Heart and cognitive defects, distinctive facial dysmorphisms, Hk, sparse hair |
| Costello Syndrome | HRAS | MAPK signaling | Similar to CFC syndrome in addition to loose skin of hands and feet |
| Nevus Sebaceous | HRAS | MAPK signaling | Hairless plaque on scalp with distinct orange color and pebble-like texture |
| Pilomatricoma | CTNNB1 | CA, Wnt signaling | Solitary hard, subcutaneous nodule |
Abbreviations: IF, intermediate filaments; Hk, hyperkeratosis; BMZ, basement membrane zone; CA, cell adhesion; TJ, tight junction; CE, cornified envelope; FA, fatty acid; Chol., cholesterol.
Loss of tissue integrity
Epidermal differentiation is dependent on proper structural architecture. and this begins with the structural integrity of the keratinocyte, which is critical for forming a squamous epithelium. Keratin intermediate filaments (KIF) form a network essential for cellular organization and cytoskeletal structural integrity to withstand mechanical stress. Keratins 5 and 14 are expressed in the basal layer of the skin and are replaced by keratins 1 and 10 in the differentiated suprabasal layers. Mutations in KRT5 and KRT14 are associated with a subset of epidermolysis bullosa (EB), a heterogeneous skin disease characterized by detachment of the epidermis from the dermis and severe blistering (Figure 2) [2,3]. In epidermolysis bullosa simplex (EBS), the severity of disease frequently correlates with the site of mutation in KRT5 and KRT14, with mutations in the region responsible for linking the small acidic type I keratins (KRT1, KRT5) to the larger neutral-basic type II keratins (KRT10, KRT14) giving rise to more severe phenotypes due to the overall disturbance of the KIF network [2]. A recent study investigating the genomic changes occurring in EB versus normal skin confirmed recurrent mutations in KRT5/KRT14 and identified EB gene signatures that point to dysfunction in lipid metabolism as well as epidermal keratinization [4]. EB is also characterized by mutations in various cell adhesion genes, including lamin 332, integrins, and collagen VII recently reviewed in [5]. Among the mutations in EB associated with hemidesmosome, a recent knock-in mouse model of Epidermolysis Bullosa Simplex-Ogna exploits a mutation in the hemidesmosome component, plectin 1a. The EBS-Ogna mutation leads to degradation of plectin 1a and results in disorganization of hemidesmosome assembly leading to skin blistering in response to trauma [6]. Together, these findings provide insight into novel molecular mechanisms of EBS and present a model to test potential therapeutic treatments for this specific type of EB.
Figure 2. Diseases of the epidermis.
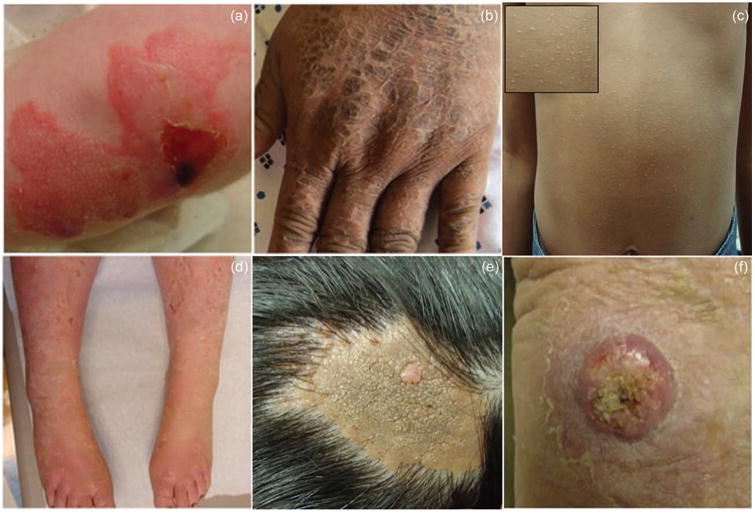
(a) Junctional epidermolysis bullosa features blistering and erosions at birth. (b) Lamellar ichthyosis is characterized by brown, tile-like scales over the entire body. (c) Ichthyosis vulgaris manifests as dry, scaly skin with diffuse general involvment. (d) Epidermolytic hyperkeratosis is characterized by recurrent blisters and hyperkerotic scales. (e) Nevus sebaceous presents as an orange plaqu with pebbly surface. (f) Squamous cell carcinoma appears sharply demarcated and can present with erosions, scaling, and hyperkeratosis.
Mutations in KRT1 and KRT10 are characteristic of epidermolytic hyperkeratosis, a skin disease initially characterized by redness and blistering later developing hyperkeratosis (Figure 2) [2,7]. While deletion of KRT10 in humans results in a severe skin phenotype, KRT1-/-/KRT10-/- mice do not display blistering or hyperkeratosis observed in humans, perhaps due to the abundance of hair follicles on the skin of mice [2,8]. Of note, the mouse model suggests the involvement of desmosomes, components of the cell adhesion machinery, showing that Krt1 and Krt10 are necessary for proper desmosome formation and overall cytoskeletal structure [8]. Interestingly, dominant mutations in KRT10 have recently been identified in ichthyosis with confetti, a rare disease characterized by skin redness, blistering, and skin thickening [9]. The authors observed that, through mitotic recombination, certain areas of the skin reverted back to wild-type KRT10 status. This rare event may be therapeutically targeted to correct the KRT10 mutation in this disease and possibly be employed in other diseases with dominant keratin mutations harboring a wild-type allele.
Disruptions in calcium-signaling
Calcium signaling is recognized for its exquisite control of terminal differentiation in keratinocytes. Hallmarks of epidermal differentiation are dependent on different stages in the calcium-signaling cascade; an intracellular calcium gradient increases from the basal layer to the stratum corneum. Calcium signaling in keratinocytes begins with binding of Ca2+ to plasma membrane Ca2+ receptor (CaR) which stimulates differentiation by inducing E-cadherin-mediated cell-cell adhesion and transcription of differentiation-specific genes [10,11]. Release of intracellular Ca2+ stores stimulates influx of extracellular Ca2+ through transient receptor protein channels (TRPC). Calcium pumps, sarco/endoplasmic reticulum Ca2+ ATPase isoform 2 (SERCA2) and secretory pathway Ca2+/Mn2+ ATPase isoform 1 (SPCA1), replenish intracellular Ca2+ stores in the endoplasmic reticulum (ER) and Golgi, respectively. Depletion of ER Ca2+ induces XBP1-mediated ER stress signaling, which enhances epidermal differentiation, similar to physiological processes that occur during normal barrier wound repair [12].
Calcium homeostasis is pathologically perturbed in Darier Disease (DD) and Hailey-Hailey Disease (HHD), which are characterized by autosomal-dominant mutations in the calcium ATPases ATP2A2 encoding SERCA2 and ATP2C1 encoding SPCA1, respectively [13,14]. Keratinocytes isolated from DD and HHD lesions contained less calcium in the basal layer compared with normal keratinocytes, as well as abnormal expression of keratins 10 and 14 and the ATP receptor P2X7, suggesting an overall defect in keratinocyte differentiation [15]. Compensatory mechanisms to restore intracellular Ca2+ exist in DD keratinocytes, including up-regulation of SPCA1 and the TRPC1 Ca2+ channel [16,17]. Furthermore, differentiation defects caused by SERCA2 inhibition in keratinocytes can be rescued by inhibiting the breakdown of sphingosine-1-phosphate (S1P), thereby representing a novel approach to treat DD [18]. Intracellular SIP has also been demonstrated to enhance expression of Ca2+ dependent differentiation genes [19].
Desmosomal dysfunction in the epidermis
Intercellular junctions in the epidermis are formed by tight junctions, hemidesmosomes, desmosomes and corneodesmosomes, attach to intermediate filaments within the cell, and impart the ability to withstand mechanical stress [20,21]. Three families of proteins make up the desmosome: desmosomal cadherins (desmocollins/desmogleins), plakins, and armadillo proteins (plakoglobin). Mutations in various desmosomal components (DSG1, DSC3, JUP, PKP1, DSP, CDSN) are observed in Palmoplantar Keratoderma (PPK) and Striate PPK [21]. Desmoglein 1 (Dsg1) expression is limited to the suprabasal layer, particularly at the interface of the basal and suprabasal layer, suggesting an important role for Dsg1 in structural morphology linked to terminal differentiation. Mutations in DSG1 are characterized by increased intercellular spaces in the skin and partial detachment of keratinocytes from the suprabasal layer [22]. Depletion of DSG1 in an organotypic model of human epidermis also leads to this phenotype along with concurrent loss of DSC1 expression, which encodes a companion cadherin to Dsg1 [21]. Intriguingly, epidermal differentiation defects due to DSG1 depletion can be rescued in a desmosome-independent manner. Increased EGFR-ERK1/2 signaling resulting from DSG1 depletion was reduced after introduction of a Dsg1 mutant unable to bind desmosomal components, suggesting that Dsg1 itself promotes differentiation by regulating growth signaling [21].
Mutations in the gene JUP, which encodes plakoglobin (PG), a Dsg1 binding partner, have been linked to skin fragility, including a nonsense mutation in EB [21]. Similar to what is observed in humans, a recent study showed that JUP-/- mice have altered epidermal differentiation marked by epidermal hyperproliferation, atypical desmosomes, and increased intercellular spaces [23]. Mutation of desmoplakin (DP), a linker between desmosomes and the KIF network, also results in PPK. Interestingly, DP has been linked to microtubule organization based on the finding that centrosomal proteins Lis1 and Ndel1 re-localize to the cell cortex upon calcium-induced differentiation in a DP-dependent manner [24]. The conclusions drawn from these studies suggest exquisite crosstalk between cell adhesion and epidermal differentiation, much of which remains to be carefully elucidated.
Mutations in additional cell adhesion molecules include a mutation in PVRL4, which encodes nectin-4, a protein involved in forming adherence junctions, identified in patients suffering from ectodermal dysplasia. The mutation prevented its association with nectin-1, and analysis of diseased skin displayed mislocalization of desmosomal components [25]. Furthermore, mutations in CSTA, a protease inhibitor of cystatin A, have recently been described in exfoliative ichthyosis. By means of a mechanical stretch assay and an organotypic model of epidermis in which CSTA was depleted, overall epidermal disorganization distinguished by hyperkeratosis, disturbance of the basal epidermal architecture, and expanded intercellular spaces was observed [26]. Although these findings suggest a role for cystatin A in cell-cell adhesion, no defect in barrier formation was detected.
A nonsense mutation in CDSN, encoding corneodesmosin, a glycoprotein localized to the extracellular part of the corneodesmosome joining the desmosome core to the cornified envelope, has been attributed to generalized peeling skin disease. Affected skin cells deficient in CDSN were reconstituted in an organotypic model and revealed a less organized epidermis with irregular expression of late differentiation markers and severe epidermal barrier defects [21]. Likewise, CDSN-/- mice display detachment of the stratum corneum at sites of friction and trauma and die shortly after birth due to these skin defects [21]. Furthermore, skin grafts of these mice exhibit hair defects and psoriasis-like lesions.
Mutations in cornification and lipid metabolism
In the stratum corneum, keratinocytes undergo terminal differentiation into corneocytes, enucleated cells surrounded by a lipid-rich cornified envelope. The organization of the stratum corneum has been compared to a brick wall, where the corneocytes represent the “bricks” surrounded by extracellular matrix of non-polar lipids, the “mortar” [27]. Granular cells just below the stratum corneum secrete lamellar bodies (LB), which consist of a distinct composition of phospholipids, cholesterol sulfate, glucosylceramides, and catabolic enzymes, into the intercellular spaces. Here, the lipids are enzymatically converted into free fatty acids, cholesterol, and ceramides and then assembled into lamellar membranes in the stratum corneum interstices. Together, the skin forms a robust permeability barrier that prevents trans-epidermal water loss. The formation and maintenance of the permeability barrier is in part dependent on the KLF4 and GRHL3 transcription factors, as well as Wnt-mediated T cell factor (TCF)/lymphoid enhancing-binding factor 1 (Lef1) signaling [28–30].
Impaired permeability barriers are observed in a heterogeneous family of monogenic disorders with underlying ichthyosis. Among common mutations in genes important in epidermal differentiation are those in the FLG gene. FLG encodes the profilaggrin protein, which is critical for formation of keratohyaline granules and whose absence causes ichthyosis vulgaris and contributes strongly to the pathogenesis of atopic dermatitis (Figure 2) [31]. Of note, FLG expression is altered in psoriasis and GWAS studies have identified mutations in late-cornified envelope genes LCE3B and LCE3C [32].
Defects in lipid metabolism or transportation can alter the lamellar membrane leading to barrier abnormalities. Such defects underlie lamellar ichthyosis (LI) (Figure 2), a group of autosomal recessive ichthyoses without systemic involvement, as well as recessive X-linked ichthyosis (RXLI). Lamellar ichthyosis can arise from several genetic aberrations, including mutations in ATP binding cassette 12, (ABCA12), arachidonate lipoxygenase 12B and E3 (ALOX12B, ALOXE3), transglutaminase 1 (TGM1), patatin-like phospholipase 1 (PNPLA1), ichthyin (ichthyin), and cytochrome P450 4F22 (CYP4F22) [33]. While the mechanisms are not well understood, all mutations except TGM1 affect the peroxidated lipid pathway, resulting in metabolite deficiency and substrate accumulation. By contrast, RXLI is caused by mutations in steroid sulfatase (SSase), which hydrolyzes cholesterol sulfate (CSO4) to cholesterol, and is characterized by defects in permeability barrier and desquamation. Excess CSO4 results in lamellar membrane phase separation and inhibition of corneodesmosome proteases, kallikrein 5 and 7 (KLK5 and KLK7), disrupting the permeability barrier and inhibiting desquamation [34].
Mutations in ABCA12 prevent proper delivery of glucosylceramides from the Golgi apparatus into LB. Missense mutation in one allele of ABCA12 is clinically distinct from truncation or deletion mutations in both alleles, resulting in the milder type II LI and the more severe Harlequin's ichthyosis (HI), respectively [35]. Corrective gene transfer of ABCA12 in HI keratinocytes rescued ABCA12 expression and LB formation/secretion, suggesting ABCA12 mutations disrupt LB lipid assembly [35]. Moreover, in HI patients with residual ABCA12, topical treatments with activators of peroxisome proliferator-activated receptor (PPAR) and liver X receptor (LXR) as well as ceramides, glucosylceramide precursors, have been successful in rescuing ABCA12 expression [36,37]. Loss of ABCA12 also leads to abnormal desquamation caused by decrease in expression of KLK5 and cathepsin D proteases, suggesting that ABCA12 regulates both permeability barrier homeostasis and desquamation [35].
Pathways of epidermal homeostasis and dysfunction
Epidermal homeostasis requires a finely tuned balance between basal layer proliferation and commitment to terminal differentiation. Multiple pathways regulate epidermal progenitor exit from the cell cycle and transition into the process of barrier formation, including the TGFβ/SMAD, NF-κB, PI-3K/PTEN, and NOTCH signal transduction pathways [38–42]. At the center of all these processes exists the p53 family member, TP63, essential for both the proliferative capacity and differentiation potential of epidermal progenitors [43,44].
TP63-ZNF750-KLF4 transcriptional circuit in epidermal disorders
TP63 exists in multiple isoforms, but ΔNp63α is the dominant isoform in developmentally mature epidermis. Heterozygous TP63 mutations in the human syndromes Ankyloblepharon-Ectodermal Dysplasia-Clefting (AEC), Ectrodactyl-Ecotdermal Dysplasia (EEC), Limb-Mammary syndrome (LMS), Acro-Dermato-Ungual-Lacrimal-Tooth syndrome (ADULT), Rapp-Hodgkin syndrome (RHS), and Split-Hand/Foot Malformation (SHFM) have been well chronicled [45]. Remarkably, mutations in the C-terminal sterile-alpha-motif (SAM) domain, specific to the ΔNp63α isoform, give rise to AEC, the only TP63 syndrome that presents with severe epidermal erosions. Interestingly, in addition to impaired expression of differentiation markers, including those involved in cell adhesion and barrier formation, AEC patient skin also presents with abnormal suprabasal Ki-67 staining, indicating dysregulated cell cycle exit may contribute to disease features [46]. A recent mouse model of AEC suggests this dysregulation is in part mediated by impaired FGF signaling [47].
Two recent studies have shed light on the pathomechanism of AEC mutant p63. Here, ZNF750, which is associated with familial psoriasis in two extended families, was found to be a direct target of ΔNp63α and is required for the terminal differentiation of human keratinocytes [48–50]. Mechanistic analyses revealed that ZNF750 is required for the induction of KLF4, which is essential for the barrier function of skin [30]. Follow up work further demonstrated that AEC mutant p63 prevented the transcriptional induction of ZNF750 in an organotypic human tissue model [51]. Remarkably, forced expression of ZNF750 rescued impaired epidermal differentiation resulting from AEC mutant p63. Together, these findings provide new insight into the existence of a p63-ZNF750-KLF4 network in human epidermal development and homeostasis.
TGFβ and IKK signaling in inherited epidermal neoplasia
TGFβ signaling participates in a multitude of biological processes including tissue homeostasis and carcinogenesis [52,53]. Although evidence supports a tumor promoting role for TGFβ signaling late in tumorigenesis, the downstream mediators of TGFβ, namely Smads -2, -3 and -4 have anti-proliferative activities in epithelial cells [54,55]. Suppression of Smad2 and Smad4 has been observed in human squamous cell carcinomas (SCCs) (Figure 2) supporting the finding of hyperplasia and spontaneous SCC in mice with conditional ablation of Smad4 in epidermis [39,54] Multiple self-healing squamous epithelioma (MSSE) is an autosomal dominant skin disorder characterized by rapidly growing SCC-like locally invasive tumors. Remarkably, after several weeks of growth, tumors spontaneously regress resulting in deep scars. Though the disease locus was mapped to 9q22.3 almost two decades ago, the lack of recombination events near this genomic region prevented identification of the disease gene. Using the powerful technique of exon capture and deep sequencing, a recent report identified loss-of-function mutations in TGFBR1 in individuals with MSSE [56]. The IκB kinase α (IKK1) is critical for cytoplasmic initiation of non-canonical NF-kB signaling [57]. Independent of its kinase activity, and in a nuclear-dependent manner, IKK1 is also essential for epidermal progenitor cell cycle exit and terminal differentiation [58]. Consistent with these mouse studies, several recent reports have demonstrated IKK1 re-localization, suppression, or deletion in SCC [59–61]. Interestingly, IKK1 physically interacts with Smad-2 and -3 and is required for nuclear accumulation of active Smad-2 and -3 in epidermis [55]. Taken together, these data support a critical role for a TGFβ-Smad2/3-IKK1 axis in promoting epidermal differentiation and suppressing tumorigenesis. Of interest, IKKγ, encoded by NEMO, is mutated in incontinentia pigmenti, a disorder which manifests as epidermal hyperplasia, vesiculation, and resultant pigmentary abnormalities, as well as hypohidrotic ectodermal dysplasia with immune deficiency [62,63]. Unlike IKK1, which displays NF-κB independent actions in epidermal differentiation, NEMO impacts appear largely mediated through IKK2 and the NF-κB pathway, although the precise basis for its phenotypic impacts remain undefined. NEMO interacts physically with the CYLD gene product, a deubiquitinating enzyme that also binds the TRAF2 NF-κB pathway regulator and, when mutated, can cause the benign neoplastic disorder of cutaneous epithelial neoplasms, hereditary cylindromatosis [64].
Cadherin and Wnt signaling defects
In addition to the desmosomal cadherins discussed above, classic cadherins regulate the actin cytoskeleton and also play a role in skin differentiation. Mice with deletion of both E-cadherin and P-cadherin display epidermal barrier defects and hyperthickening of the epidermis with a squamous appearance [65]. Although desmosomal function appeared to be unaltered, a defect in cell structure due to mislocalization of polarity proteins resulted in abnormal differentiation.
Wnts are small, secreted, lipid-modified morphogens involved in developmental signaling and differentiation. Mutations in WNT10A are associated with ectodermal dysplasia [66,67]. Wnts bind to extracellular receptors, effecting an intracellular signaling response, through β-catenin, converging on TCF/LEF transcription factors that act on Wnt target genes. Wnt signaling has been shown to play an important role in hair follicle self-renewal, although recent evidence points to a role for Tcf3 and Tcf4 in maintenance of the progenitor compartment of interfollicular epidermis. Peroxisome proliferator-activated receptor (PPAR) was shown to be a downstream target repressed by overexpression of Tcf3, thus promoting self-renewal by preventing lipid-mediated differentiation [68]. Whereas deletion of Tcf3 has no overt skin phenotype, deletion of both Tcf3 and Tcf4 results in hair follicle deficiency with thin skin due to diminished stem cell potential of interfollicular progenitor cells [69]. These findings appear to be independent of β-catenin-mediated signaling as the Cnntb1-/- mouse displayed hyperproliferative skin rather than tissue collapse [69]. The cytoplasmic tails of classic cadherins bind β-catenin, which can in turn bind to α-catenin to regulate actin-cytoskeletal architecture and mediate cell signaling [70]. Moreover, mutational stabilization of β-catenin is responsible for the common skin neoplasm, pilomatricoma, and increased β-catenin signaling has been linked to cutaneous cancer stem cell maintenance [71,72]. A recent study also described an essential role for β-catenin in mediating proliferation in response to activation of ROCK, a Rho-GTPase responsible for actomyosin contractility [73]. In this study, β-catenin signaling in response to tissue stiffness resulted in a tumor-promoting environment. Together, these finds point to a crucial role for the integrity of cytoskeletal architecture in preventing tumorigenesis.
Notch in epidermal differentiation and tumor suppression
The role of Notch signaling in epidermal differentiation has been well studied. Notch is a transmembrane receptor whose activity is stimulated by extracellular ligand binding resulting in cleavage of its cytoplasmic domain, which functions as a transcriptional co-regulator serving to activate or repress target genes [74]. Notch protein expression is primarily localized to the suprabasal epidermis and has been shown to act in concert with AP-2 factors to regulate terminal differentiation [75]. Importantly, Notch has been implicated in skin tumorigenesis: loss of Notch1 in mice results in skin carcinoma formation as a result of a defective epidermal barrier and crosstalk with the underlying stroma, suggesting loss of Notch1 promotes tumorigenesis [76]. Furthermore, depletion of IRF6, a positively regulated Notch target, leads to decreased differentiation and increased tumor formation [77]. In this study, analysis of a panel of cutaneous SCC indicated a strong correlation between decreased expression of IRF6 and Notch and simultaneous increased expression of EGFR. Strikingly, a recent study using whole-exome and transcriptome sequencing identified mutations in NOTCH1 or NOTCH2 in roughly 75% of cutaneous SCC and SCC cell lines tested [78]. The majority of mutations were due to a G>A transition, typically attributed to UV damage, and resulted in loss of function. These findings not only demonstrate the power of next-generation sequencing in identifying critical mutations in disease, but also point to Notch as a novel biomarker for SCC as well as prioritizing it as a therapeutic target.
The Ras-MAPK pathway in multi-system syndromes
The evolutionarily conserved RAS-ERK/MAP kinase (MAPK) cascade functions to transduce extracellular signals to intracellular responses regulating cell proliferation, differentiation, and survival. Essential for normal tissue homeostasis, this pathway becomes pathologically dysregulated in many types of cancer, including via RAS gene mutations in 10% of human epidermal squamous cell carcinomas and the benign neoplasm nevus sebaceous (Figure 2), as well as in the so-called RASopathies, developmental disorders [79–82]. Attributed to germline mutations within this cascade, RASopathies often present with many interrelated phenotypic characteristics including facial dysmorphism, dermatological manifestations, cardiac abnormalities, and mental retardation (Figure 3) [82]. Thus, there has often been a failure to clinically differentiate each disorder, and recent work in this field has aimed at more distinct classifications to improve diagnostics and therapeutics.
Figure 3. Mutations in the Ras/MAPK and PI3-K/Akt pathway in cutaneous disorders.

“RASopathies” are multi-system disorders characterized by epidermal phenotypes and typically resulting in tumor syndromes. Enumerated circles represent mutations observed in the following disorders: (1) Noonan syndrome exhibits mutations in PTPN11 (SHP-2) and KRAS. (2) Cardio-facio-cutaneous (CFC) syndrome exhibits mutations in KRAS, BRAF, and MEK. (3) Costello syndrome exhibits mutations in HRAS. (4) LEOPARD syndrome exhibits mutations in PTPN11. (5) Neurofibromatosis contains mutations in RasGAP, NF1. (6) Legius syndrome exhibits mutations in SPRED1. (7) Banayan-Zonona and Cowden's syndromes contain mutations in PTEN. Phenotypes associated with these mutations are listed in Table 1.
The RASopathy Cardio-facio-cutaneous (CFC) syndrome is a result of mutations in the RAS-ERK/MAPK cascade including KRAS, BRAF, MEK1 and MEK2 [82]. Recent work has suggested that in spite of a variety of mutations, CFC patients might benefit from small molecular inhibitors of MEK as evidenced by observed responses to multiple inhibitors of MEK in both human cell lines and in developing zebrafish embryos characterized by varied mutant forms [83,84]. Phenotypically similar to CFC syndrome, Costello syndrome (CS) is characterized solely by mutations in HRAS [82,85]. Farnesylation inhibitors, which target posttranslational modifications of RAS, will be employed in a recently characterized mouse model for CS and should yield interesting therapeutic strategies [86].
Phosphoinositide 3-kinase pathway-related disorders
Phosphoinositide 3-kinase (PI3K)/Akt signaling, another important signaling pathway in the epidermis, protects differentiating keratinocytes from caspase-3 dependent apoptosis via engagement of epidermal growth factor receptor (EGFR) and Src families of tyrosine kinases with E-cadherin [87]. Phosphatase and tensin homolog (PTEN) negatively regulates the PI3K/Akt pathway and is mutated or deficient in a spectrum of sporadic cancers and hereditary cancer syndromes characterized by hyperproliferation and resistance to apoptosis [41]. Cowden's and Bannayan-Zonana syndromes are caused by germ line mutations in the tumor suppressor. Sporadic PTEN loss or mutations are associated with skin cancers including melanoma, SCC, and keratoacanthoma (KA). Aggressive SCCs with deletion in Grainyhead-like 3 (GRHL3), coding for a transcription factor required for normal epidermal homeostasis and differentiation, also displays PTEN deficiency and addiction to PI3K/Akt signaling [88]. However, in the context of activated Fos and PTEN loss, an increase in glycogen synthase kinase-3β (GSK3β) inactivation induces differentiation via p53 and/or p21Waf cascade activation, thereby eliciting KA instead of SCC formation [89].
Concluding Remarks
Genetic disruptions in epidermal differentiation cause diseases that impact more than one in five persons. Increasing investigations into the genes affected in the resulting disorders has revealed some emerging organizing etiologic themes. Prominent among these are dominant signaling pathways, including the Notch, TGFβ, IKK, Ras/MAPK, Phosphoinositide 3-kinase, p63, and Wnt signaling pathways as well as central biological processes that regulate calcium homeostasis, tissue integrity, cornification, and lipid biogenesis. The functional non-redundancy of genes impacted within these pathways and processes underscores their potential for therapeutic targeting. As the genetic basis for all human disorders of epidermal differentiation becomes established in the coming years, additional opportunities for effective therapeutic intervention that integrate affected pathways with newly discovered genetic alterations will emerge.
Glossary
- Corneocytes
terminally differentiated keratinocytes in the stratum corneum
- Corneodesmosomes
specialized desmosomes that bridge corneocytes together
- Cornification
transformation of keratinocytes into metabolically inactive corneocytes over a span of 2 weeks. The differentiation process is characterized by loss of organelles and nuclei and accumulation of keratin in terminally differentiated keratinocytes
- Cornified Envelope (CE)
a structural layer of CE proteins in the stratum corneum that is irreversibly crosslinked by calcium-activated transglutaminases and essential in maintaining skin barrier function. CE proteins include loricrin, involucrin, late cornified envelope (LCE), and small proline-rich (SPRRs) proteins
- Desmosomes
intercellular protein complexes composed of cell surface adhesion proteins linked to intracellular intermediate filaments for cell-to-cell adhesion
- Desquamation
natural cycle of shedding and replacing corneocytes
- Hyperkeratosis
thickening of the stratum corneum caused by keratin buildup resulting in abnormal thickening of the skin
- KIF
keratin intermediate filament generated by assembly of keratin monomers form a structural component of the cell maintaining the overall cellular architecture
- Ichthyosis
heterogeneous family of genetically inherited disorders characterized by defective desquamation resulting in the appearance of fishlike scales
- Keratoacanthoma
considered a self-regressing variant of well-differentiated squamous cell carcinoma, appear as a symmetric, dome shaped tumor with a central depression filled with keratin debris
- Lamellar bodies/granules
secretory organelles found in keratinocytes that are filled with lipids, enzymes, and protein and important for normal skin barrier function
- Organotypic model
reconstitution of cell culture in a three-dimensional manner generating tissue resembling the organ of origin
- Squamous cell carcinoma
cancer of epithelial origin frequently arising from sun-exposed regions of the skin
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Blanpain C, Fuchs E. Epidermal homeostasis: a balancing act of stem cells in the skin. Nat Rev Mol Cell Biol. 2009;10:207–17. doi: 10.1038/nrm2636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chamcheu JC, et al. Keratin gene mutations in disorders of human skin and its appendages. Arch Bioch Biophys. 2011;508:123–137. doi: 10.1016/j.abb.2010.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Arin MJ, et al. Identification of novel and known KRT5 and KRT14 mutations in 53 patients with epidermolysis bullosa simplex: correlation between genotype and phenotype. Br J Dermatol. 2010;162:1365–1369. doi: 10.1111/j.1365-2133.2010.09657.x. [DOI] [PubMed] [Google Scholar]
- 4.Bchetnia M, et al. Expression signature of epidermolysis bullosa simplex. Hum Genet. 2012;131:393–406. doi: 10.1007/s00439-011-1077-7. [DOI] [PubMed] [Google Scholar]
- 5.Sawamura D, et al. Overview of epidermolysis bullosa. J Dermatol. 2010;37:214–219. doi: 10.1111/j.1346-8138.2009.00800.x. [DOI] [PubMed] [Google Scholar]
- 6.Walko G, et al. Targeted Proteolysis of Plectin Isoform 1a Accounts for Hemidesmosome Dysfunction in Mice Mimicking the Dominant Skin Blistering Disease EBS-Ogna. PLoS Genet. 2011;7:e1002396. doi: 10.1371/journal.pgen.1002396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Arin Mj, et al. Expanding the keratin mutation database: novel and recurrent mutations and genotype–phenotype correlations in 28 patients with epidermolytic ichthyosis. Br J Dermatol. 2011;164:442–447. doi: 10.1111/j.1365-2133.2010.10096.x. [DOI] [PubMed] [Google Scholar]
- 8.Wallace L, et al. Deletion of K1/K10 Does Not Impair Epidermal Stratification but Affects Desmosomal Structure and Nuclear Integrity. J Cell Sci. 2012;125:1750–1758. doi: 10.1242/jcs.097139. [DOI] [PubMed] [Google Scholar]
- 9.Choate KA, et al. Mitotic Recombination in Patients with Ichthyosis Causes Reversion of Dominant Mutations in KRT10. Science. 2010;330:94–97. doi: 10.1126/science.1192280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tu CL, et al. Inactivation of the Calcium Sensing Receptor Inhibits E-cadherin-mediated Cell-Cell Adhesion and Calcium-induced Differentiation in Human Epidermal Keratinocytes. J Biol Chem. 2008;283:3519–3528. doi: 10.1074/jbc.M708318200. [DOI] [PubMed] [Google Scholar]
- 11.Tu CL, et al. Ablation of the Calcium-Sensing Receptor in Keratinocytes Impairs Epidermal Differentiation and Barrier Function. J Invest Dermatol. 2012;132:2350–2359. doi: 10.1038/jid.2012.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Celli A, et al. Endoplasmic reticulum Ca2+ depletion activates XBP1 and controls terminal differentiation in keratinocytes and epidermis. Br J Dermatol. 2011;164:16–25. doi: 10.1111/j.1365-2133.2010.10046.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sakuntabhai A, et al. Mutations in ATP2A2, encoding a Ca2+ pump, cause Darier disease. Nat Genet. 1999;21:271–277. doi: 10.1038/6784. [DOI] [PubMed] [Google Scholar]
- 14.Hu Z, et al. Mutations in ATP2C1, encoding a calcium pump, cause Hailey-Hailey disease. Nat Genet. 2000;24:61–65. doi: 10.1038/71701. [DOI] [PubMed] [Google Scholar]
- 15.Leinonen PT, et al. Reevaluation of the Normal Epidermal Calcium Gradient, and Analysis of Calcium Levels and ATP Receptors in Hailey–Hailey and Darier Epidermis. J Invest Dermatol. 2009;129:1379–1387. doi: 10.1038/jid.2008.381. [DOI] [PubMed] [Google Scholar]
- 16.Foggia L, et al. Activity of the hSPCA1 Golgi Ca2+ pump is essential for Ca2+-mediated Ca2+ response and cell viability in Darier disease. J Cell Sci. 2006;119:671–679. doi: 10.1242/jcs.02781. [DOI] [PubMed] [Google Scholar]
- 17.Pani B, et al. Up-Regulation of Transient Receptor Potential Canonical 1 (TRPC1) following Sarco(endo)plasmic Reticulum Ca2+ ATPase 2 Gene Silencing Promotes Cell Survival: A Potential Role for TRPC1 in Darier's Disease. Mol Biol Cell. 2006;17:4446–4458. doi: 10.1091/mbc.E06-03-0251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Celli A, et al. SERCA2-Controlled Ca2+-Dependent Keratinocyte Adhesion and Differentiation Is Mediated via the Sphingolipid Pathway: A Therapeutic Target for Darier's Disease. J Invest Dermatol. 2012;132:1188–1195. doi: 10.1038/jid.2011.447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hong JH, et al. K6PC-5, a Direct Activator of Sphingosine Kinase 1, Promotes Epidermal Differentiation Through Intracellular Ca2+ Signaling. J Invest Dermatol. 2008;128:2166–2178. doi: 10.1038/jid.2008.66. [DOI] [PubMed] [Google Scholar]
- 20.Morita K, et al. Tight junctions in epidermis: from barrier to keratinization. Eur J Dermatol. 2011;21:12–17. doi: 10.1684/ejd.2010.1192. [DOI] [PubMed] [Google Scholar]
- 21.Petrof G, et al. Desmosomal genodermatoses. Br J Dermatol. 2012;166:36–45. doi: 10.1111/j.1365-2133.2011.10640.x. [DOI] [PubMed] [Google Scholar]
- 22.Bergman R, et al. Disadhesion of epidermal keratinocytes: A histologic clue to palmoplantar keratodermas caused by DSG1 mutations. J Am Acad Dermatol. 2010;62:107–113. doi: 10.1016/j.jaad.2009.05.016. [DOI] [PubMed] [Google Scholar]
- 23.Li D, et al. Lack of Plakoglobin in Epidermis Leads to Keratoderma. J Biol Chem. 2012;287:10435–10443. doi: 10.1074/jbc.M111.299669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sumigray KD, et al. Lis1 Is Essential for Cortical Microtubule Organization and Desmosome Stability in the Epidermis. J Cell Biol. 2011;194:631–642. doi: 10.1083/jcb.201104009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Brancati F, et al. Mutations in PVRL4, Encoding Cell Adhesion Molecule Nectin-4, Cause Ectodermal Dysplasia-Syndactyly Syndrome. Am J Hum Genet. 2010;87:265–273. doi: 10.1016/j.ajhg.2010.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Blaydon DC, et al. Mutations in CSTA, Encoding Cystatin A, Underlie Exfoliative Ichthyosis and Reveal a Role for This Protease Inhibitor in Cell-Cell Adhesion. Am J Hum Genet. 2011;89:564–571. doi: 10.1016/j.ajhg.2011.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Elias PM, et al. Ichthyoses. Curr Probl Dermatol. 2010;39, 43:1–29. [Google Scholar]
- 28.Fehrenschild D, et al. TCF/Lef1-Mediated Control of Lipid Metabolism Regulates Skin Barrier Function. J Invest Dermatol. 2011;132:337–345. doi: 10.1038/jid.2011.301. [DOI] [PubMed] [Google Scholar]
- 29.Yu Z, et al. The Grainyhead-like epithelial transactivator Get-1/Grhl3 regulates epidermal terminal differentiation and interacts functionally with LMO4. Dev Biol. 2006;299:122–36. doi: 10.1016/j.ydbio.2006.07.015. [DOI] [PubMed] [Google Scholar]
- 30.Segre JA, et al. Klf4 is a transcription factor required for establishing the barrier function of the skin. Nat Genet. 1999;22:356–360. doi: 10.1038/11926. [DOI] [PubMed] [Google Scholar]
- 31.Brown SJ, McLean WHI. One remarkable molecule: filaggrin. J Invest Dermatol. 2012;132:751–762. doi: 10.1038/jid.2011.393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Roberson EDO, Bowcock AM. Psoriasis genetics: breaking the barrier. Trends Genet. 2010;26:415–423. doi: 10.1016/j.tig.2010.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fischer J. Autosomal Recessive Congenital Ichthyosis. J Invest Dermatol. 2009;129:1319–1321. doi: 10.1038/jid.2009.57. [DOI] [PubMed] [Google Scholar]
- 34.Ekholm IE, et al. Stratum Corneum Tryptic Enzyme in Normal Epidermis: a Missing Link in the Desquamation Process? J Invest Dermatol. 2000;114:56–63. doi: 10.1046/j.1523-1747.2000.00820.x. [DOI] [PubMed] [Google Scholar]
- 35.Akiyama M. ABCA12 mutations and autosomal recessive congenital ichthyosis: A review of genotype/phenotype correlations and of pathogenetic conceptsa. Hum Mutat. 2010;31:1090–1096. doi: 10.1002/humu.21326. [DOI] [PubMed] [Google Scholar]
- 36.Jiang YJ, et al. PPAR and LXR Activators Regulate ABCA12 Expression in Human Keratinocytes. J Invest Dermatol. 2007;128:104–109. doi: 10.1038/sj.jid.5700944. [DOI] [PubMed] [Google Scholar]
- 37.Jiang YJ, et al. Ceramide Stimulates ABCA12 Expression Via Peroxisome Proliferator-Activated Receptor Δ in Human Keratinocytes. J Biol Chem. 2009;284:18942–18952. doi: 10.1074/jbc.M109.006973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yang L, et al. Targeted disruption of Smad4 in mouse epidermis results in failure of hair follicle cycling and formation of skin tumors. Cancer Res. 2005;65:8671–8678. doi: 10.1158/0008-5472.CAN-05-0800. [DOI] [PubMed] [Google Scholar]
- 39.Hoot KE, et al. Keratinocyte-specific Smad2 ablation results in increased epithelial-mesenchymal transition during skin cancer formation and progression. J Clin Invest. 2008;118:2722–2732. doi: 10.1172/JCI33713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang JY, et al. NF-kappaB RelA opposes epidermal proliferation driven by TNFR1 and JNK. Genes Dev. 2004;18:17–22. doi: 10.1101/gad.1160904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Suzuki A, et al. Keratinocyte-specific Pten Deficiency Results in Epidermal Hyperplasia, Accelerated Hair Follicle Morphogenesis and Tumor Formation. Cancer Res. 2003;63:674–681. [PubMed] [Google Scholar]
- 42.Demehri S, et al. Epidermal Notch1 Loss Promotes Skin Tumorigenesis by Impacting the Stromal Microenvironment. Cancer Cell. 2009;16:55–66. doi: 10.1016/j.ccr.2009.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Truong AB, et al. p63 regulates proliferation and differentiation of developmentally mature keratinocytes. Genes Dev. 2006;20:3185–3197. doi: 10.1101/gad.1463206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Su X, et al. Rescue of key features of the p63-null epithelial phenotype by inactivation of Ink4a and Arf. EMBO J. 2009;28:1904–1915. doi: 10.1038/emboj.2009.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Koster MI. p63 in Skin Development and Ectodermal Dysplasias. J Invest Dermatol. 2010;130:2352–2358. doi: 10.1038/jid.2010.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Clements Se, et al. Mutations in AEC syndrome skin reveal a role for p63 in basement membrane adhesion, skin barrier integrity and hair follicle biology. Br J Dermatol. 2012;167:134–144. doi: 10.1111/j.1365-2133.2012.10888.x. [DOI] [PubMed] [Google Scholar]
- 47.Ferone G, et al. Mutant p63 causes defective expansion of ectodermal progenitor cells and impaired FGF signalling in AEC syndrome. EMBO Mol Med. 2012;4:192–205. doi: 10.1002/emmm.201100199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Birnbaum RY, et al. Seborrhea-like dermatitis with psoriasiform elements caused by a mutation in ZNF750, encoding a putative C2H2 zinc finger protein. Nat Genet. 2006;38:749–751. doi: 10.1038/ng1813. [DOI] [PubMed] [Google Scholar]
- 49.Yang CF, et al. A promoter sequence variant of ZNF750 is linked with familial psoriasis. J Invest Dermatol. 2008;128:1662–1668. doi: 10.1038/jid.2008.1. [DOI] [PubMed] [Google Scholar]
- 50.Sen GL, et al. ZNF750 is a p63 target gene that induces KLF4 to drive terminal epidermal differentiation. Dev Cell. 2012;22:669–677. doi: 10.1016/j.devcel.2011.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zarnegar BJ, et al. Genomic Profiling of a Human Organotypic Model of AEC Syndrome Reveals ZNF750 as an Essential Downstream Target of Mutant TP63. Am J Hum Genet. 2012;91:435–443. doi: 10.1016/j.ajhg.2012.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wu MY, Hill CS. Tgf-beta superfamily signaling in embryonic development and homeostasis. Dev Cell. 2009;16:329–343. doi: 10.1016/j.devcel.2009.02.012. [DOI] [PubMed] [Google Scholar]
- 53.Fuxe J, et al. Transcriptional crosstalk between TGF-β and stem cell pathways in tumor cell invasion: Role of EMT Promoting Smad Complexes. Cell Cycle. 2010;9:2363–2374. doi: 10.4161/cc.9.12.12050. [DOI] [PubMed] [Google Scholar]
- 54.Qiao W, et al. Hair follicle defects and squamous cell carcinoma formation in Smad4 conditional knockout mouse skin. Oncogene. 2006;25:207–217. doi: 10.1038/sj.onc.1209029. [DOI] [PubMed] [Google Scholar]
- 55.Descargues P, et al. IKKalpha is a critical coregulator of a Smad4-independent TGFbeta-Smad2/3 signaling pathway that controls keratinocyte differentiation. Proc Natl Acad Sci USA. 2008;105:2487–2492. doi: 10.1073/pnas.0712044105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Goudie DR, et al. Multiple self-healing squamous epithelioma is caused by a disease-specific spectrum of mutations in TGFBR1. Nat Genet. 2011;43:365–369. doi: 10.1038/ng.780. [DOI] [PubMed] [Google Scholar]
- 57.Senftleben U, et al. Activation by IKKalpha of a second, evolutionary conserved, NF-kappa B signaling pathway. Science. 2001;293:1495–1499. doi: 10.1126/science.1062677. [DOI] [PubMed] [Google Scholar]
- 58.Hu Y, et al. Abnormal morphogenesis but intact IKK activation in mice lacking the IKKalpha subunit of IkappaB kinase. Science. 1999;284:316–320. doi: 10.1126/science.284.5412.316. [DOI] [PubMed] [Google Scholar]
- 59.Liu B, et al. A critical role for I kappaB kinase alpha in the development of human and mouse squamous cell carcinomas. Proc Natl Acad Sci USA. 2006;103:17202–17207. doi: 10.1073/pnas.0604481103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Maeda G, et al. Epigenetic inactivation of IkappaB Kinase-alpha in oral carcinomas and tumor progression. Clin Cancer Res. 2007;13:5041–5047. doi: 10.1158/1078-0432.CCR-07-0463. [DOI] [PubMed] [Google Scholar]
- 61.Park E, et al. Reduction in IkappaB kinase alpha expression promotes the development of skin papillomas and carcinomas. Cancer Res. 2007;67:9158–9168. doi: 10.1158/0008-5472.CAN-07-0590. [DOI] [PubMed] [Google Scholar]
- 62.Nelson DL. NEMO, NFkappaB signaling and incontinentia pigmenti. Curr Opin Genet Dev. 2006;16:282–288. doi: 10.1016/j.gde.2006.04.013. [DOI] [PubMed] [Google Scholar]
- 63.Keller MD, et al. Hypohidrotic Ectodermal Dysplasia and Immunodeficiency with Coincident NEMO and EDA Mutations. Front Immunol. 2011;2:61. doi: 10.3389/fimmu.2011.00061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Salhi A, et al. Multiple familial trichoepithelioma caused by mutations in the cylindromatosis tumor suppressor gene. Cancer Res. 2004;64:5113–5117. doi: 10.1158/0008-5472.CAN-04-0307. [DOI] [PubMed] [Google Scholar]
- 65.Tinkle CL, et al. New Insights into Cadherin Function in Epidermal Sheet Formation and Maintenance of Tissue Integrity. Proc Natl Acad Sci USA. 2008;105:15405–15410. doi: 10.1073/pnas.0807374105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Adaimy L, et al. Mutation in WNT10A Is Associated with an Autosomal Recessive Ectodermal Dysplasia: The Odonto-onycho-dermal Dysplasia. Am J Hum Genet. 2007;81:821–828. doi: 10.1086/520064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Nawaz S, et al. WNT10A missense mutation associated with a complete Odonto-Onycho-Dermal Dysplasia syndrome. Eur J Hum Genet. 2009;17:1600–1605. doi: 10.1038/ejhg.2009.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Nguyen H, et al. Tcf3 Governs Stem Cell Features and Represses Cell Fate Determination in Skin. Cell. 2006;127:171–183. doi: 10.1016/j.cell.2006.07.036. [DOI] [PubMed] [Google Scholar]
- 69.Nguyen H, et al. Tcf3 and Tcf4 are essential for long-term homeostasis of skin epithelia. Nat Genet. 2009;41:1068–1075. doi: 10.1038/ng.431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Harris TJC, Tepass U. Adherens junctions: from molecules to morphogenesis. Nat Rev Mol Cell Biol. 2010;11:502–514. doi: 10.1038/nrm2927. [DOI] [PubMed] [Google Scholar]
- 71.Chan EF, et al. A common human skin tumour is caused by activating mutations in beta-catenin. Nat Genet. 1999;21:410–413. doi: 10.1038/7747. [DOI] [PubMed] [Google Scholar]
- 72.Malanchi I, et al. Cutaneous cancer stem cell maintenance is dependent on β-catenin signalling. Nature. 2008;452:650–653. doi: 10.1038/nature06835. [DOI] [PubMed] [Google Scholar]
- 73.Samuel MS, et al. Actomyosin-Mediated Cellular Tension Drives Increased Tissue Stiffness and β-Catenin Activation to Induce Epidermal Hyperplasia and Tumor Growth. Cancer Cell. 2011;19:776–791. doi: 10.1016/j.ccr.2011.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Watt FM, et al. Epidermal Notch signalling: differentiation, cancer and adhesion. Curr Opin Cell Biol. 2008;20:171–179. doi: 10.1016/j.ceb.2008.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Wang X, et al. AP-2 factors act in concert with Notch to orchestrate terminal differentiation in skin epidermis. J Cell Biol. 2008;183:37–48. doi: 10.1083/jcb.200804030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Demehri S, et al. Epidermal Notch1 Loss Promotes Skin Tumorigenesis by Impacting the Stromal Microenvironment. Cancer Cell. 2009;16:55–66. doi: 10.1016/j.ccr.2009.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Restivo G, et al. IRF6 is a mediator of Notch pro-differentiation and tumour suppressive function in keratinocytes. EMBO J. 2011;30:4571–4585. doi: 10.1038/emboj.2011.325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Wang NJ, et al. Loss-of-function mutations in Notch receptors in cutaneous and lung squamous cell carcinoma. Proc Natl Acad Sci USA. 2011;108:17761–17766. doi: 10.1073/pnas.1114669108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Scholl FA, et al. Mek1/2 MAPK Kinases Are Essential for Mammalian Development, Homeostasis, and Raf-Induced Hyperplasia. Dev Cell. 2007;12:615–629. doi: 10.1016/j.devcel.2007.03.009. [DOI] [PubMed] [Google Scholar]
- 80.Forbes SA, et al. COSMIC: mining complete cancer genomes in the Catalogue of Somatic Mutations in Cancer. Nucleic Acids Res. 2010;39:D945–D950. doi: 10.1093/nar/gkq929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Groesser L, et al. Postzygotic HRAS and KRAS mutations cause nevus sebaceous and Schimmelpenning syndrome. Nat Genet. 2012;44:783–787. doi: 10.1038/ng.2316. [DOI] [PubMed] [Google Scholar]
- 82.Tidyman W, Rauen K. The RASopathies: developmental syndromes of Ras/MAPK pathway dysregulation. Curr Opin Genet Dev. 2009;19:230–6. doi: 10.1016/j.gde.2009.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Senawong T, et al. Germline mutations of MEK in cardio-facio-cutaneous syndrome are sensitive to MEK and RAF inhibition: implications for therapeutic options. Hum Mol Genet. 2008;17:419–430. doi: 10.1093/hmg/ddm319. [DOI] [PubMed] [Google Scholar]
- 84.Anastasaki C, et al. Kinase-activating and kinase-impaired cardio-facio-cutaneous syndrome alleles have activity during zebrafish development and are sensitive to small molecule inhibitors. Hum Mol Genet. 2009;18:2543–2554. doi: 10.1093/hmg/ddp186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Niihori T, et al. Germline KRAS and BRAF mutations in cardio-facio-cutaneous syndrome. Nat Genet. 2006;38:294–296. doi: 10.1038/ng1749. [DOI] [PubMed] [Google Scholar]
- 86.Schuhmacher AJ, et al. A mouse model for Costello syndrome reveals an Ang II-mediated hypertensive condition. J Clin Invest. 2008 doi: 10.1172/JCI34385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Calautti E, et al. Phosphoinositide 3-Kinase Signaling to Akt Promotes Keratinocyte Differentiation Versus Death. J Biol Chem. 2005;280:32856–32865. doi: 10.1074/jbc.M506119200. [DOI] [PubMed] [Google Scholar]
- 88.Darido C, et al. Targeting of the Tumor Suppressor GRHL3 by a miR-21-Dependent Proto-Oncogenic Network Results in PTEN Loss and Tumorigenesis. Cancer Cell. 2011;20:635–648. doi: 10.1016/j.ccr.2011.10.014. [DOI] [PubMed] [Google Scholar]
- 89.Yao D, et al. Fos cooperation with PTEN loss elicits keratoacanthoma not carcinoma, owing to p53/p21WAF-induced differentiation triggered by GSK3β inactivation and reduced AKT activity. J Cell Sci. 2008;121:1758–1769. doi: 10.1242/jcs.021147. [DOI] [PubMed] [Google Scholar]