

Abstract
The labile nature of phosphoryl groups has presented a long-standing challenge for the characterization of protein phosphorylation via conventional mass spectrometry-based bottom-up proteomics methods. Collision-induced dissociation (CID) causes preferential cleavage of the phospho-ester bond of peptides, particularly under conditions of low proton mobility, and results in the suppression of sequence-informative fragmentation that often prohibits phosphosite determination. In the present study, the fragmentation patterns of phosphopeptides are improved through ion/ion-mediated peptide derivatization with 4-formyl-1,3-benezenedisulfonic acid (FBDSA) anions using a dual spray reactor. This approach exploits the strong electrostatic interactions between the sulfonate moieties of FBDSA and basic sites to facilitate gas-phase bioconjugation and to reduce charge sequestration and increase the yield of phosphate-retaining sequence ions upon CID. Moreover, comparative CID fragmentation analysis between unmodified phosphopeptides and those modified online with FBDSA or in solution via carbamylation and 4-sulfophenyl isothiocyanate (SPITC) provided evidence for sulfonate interference with charge-directed mechanisms that result in preferential phosphate elimination. Our results indicate the prominence of charge-directed neighboring group participation reactions involved in phosphate neutral loss, and the implementation of ion-ion reactions in a dual spray reactor set-up provides a means to disrupt the interactions by competing hydrogen-bonding interactions between sulfonate groups and the side-chains of basic residues.
TOC Image
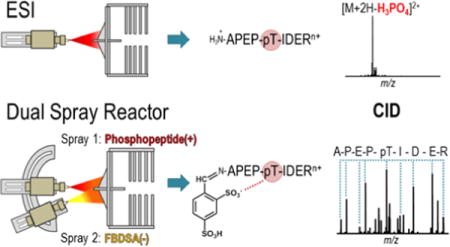
Introduction
Protein phosphorylation is a highly dynamic post-translational modification (PTM) that plays a central role in the signaling and regulatory machinery that mediate nearly all cellular processes including transcription, differentiation, cell cycle progression, and metabolism.1–3 These processes are controlled through the coordinated interplay of protein kinases and phosphatases that modulate the function of target proteins by transiently altering their phosphorylation states at serine, threonine and tyrosine sites.4 Moreover, aberrant phosphorylation arising from the dysregulation of this activity has been linked to the onset and progression of numerous neurodegenerative, oncogenic and metabolic diseases.5–7 Consequently, the molecular-level characterization of protein phosphorylation is essential for the comprehensive understanding of complex mechanisms governing cell health and disease and offers critical insight for the development of new therapeutics.
Mass spectrometry (MS) has emerged as the analytical method of choice for the identification and characterization of phosphorylated proteins on both the individual and global scale.8,9 However, despite exceptional speed and sensitivity, common MS-based approaches suffer from several key impediments arising from the intrinsic biological and chemical properties of phosphoproteins and their peptide constituents. For example, phosphorylation often occurs at substoichiometric levels that are below the sampling depth of most bottom-up data-dependent driven workflows.10,11 Enrichment strategies such as immobilized metal affinity chromatography (IMAC) and metal oxide affinity chromatography (MOAC) have helped to overcome this limitation by selectively increasing the relative abundance of phosphorylated targets within full MS survey scans.9 While this additional step facilitates improved detection, subsequent tandem mass spectrometric (MS/MS) analysis to obtain sequence and phosphosite information by direct fragmentation of selected phosphopeptides is often inhibited by the higher gas phase lability of the phospho-ester bond relative to the polypeptide backone.12 Collision-induced dissociation (CID) remains the most established and widely utilized ion activation method;13 however, the slow heating mechanism that governs ion dissociation promotes cleavage at the most labile sites, thereby inducing preferential neutral loss of the phosphate group and suppression of diagnostic sequence and phosphosite-informative fragmentation.12,14,15 This outcome has proven particularly problematic under conditions of low proton mobility where hydrogen bonding interactions between basic side-chains and the phosphate group facilitate nearly exclusive charge-directed neutral loss of the phosphate.12,16 This shortcoming has prompted the use of alternative activation strategies that provide more informative MS/MS spectra for phosphopeptide characterization, including electron-driven approaches such as electron transfer dissociation and electron capture dissociation (ETD and ECD),17,18 higher-energy collisional activation (HCD),19 ultraviolet photodissociation (UVPD),20–23 and several combinations thereof (i.e., ETcaD,24 EThcD,25 ETUVPD26).
In addition to alternative activation methods, chemical and enzymatic strategies that modify the intrinsic properties of phosphopeptides to make them more suitable for MS/MS interrogation have also been reported, albeit at the cost of more extensive sample preparation and experimental complexity. Approaches based on β-elimination of phosphoryl groups from phosphoserine and phosphothreonine residues followed by Michael addition with a nucleophilic reactant have been used in a diversity of protein phosphorylation studies.27–32 The purpose of this type of strategy is generally two-fold: 1) removal of the CID labile phosphate group to generate more informative MS/MS spectra and 2) incorporation of novel chemistry that can be exploited for streamlined phosphopeptide analysis.28,31–33 Despite these merits, one major drawback to β-elimination-based approaches arises from their lack of selectivity toward phosphotyrosine residues.34 Other strategies aimed at eliminating or minimizing the hydrogen bonding interactions that lead to preferential phosphate cleavage have also shown success for generating more informative CID spectra with the added benefit of being universally applicable to all phosphopeptide types (S/T/Y).35–38 This has been accomplished via selective derivatization of either the phosphate moiety35,36 or basic side-chains of the peptide,37 or alternatively by the complete enzymatic removal of basic residues.38
Recently, we described a method for the high-throughput bioconjugation of peptide cations via pseudo-droplet phase initiated ion/ion reactions using a front-end dual spray reactor.39 This previous work relied on well-characterized gas-phase ion/ion-mediated covalent chemistry using 4-formyl-1,3-benzenedisulfonic acid (FBDSA) anions40–42 to both facilitate rapid derivatization on a timescale compatible with chromatographic separations and to increase the intrinsic photoabsorption cross-section of peptides for enhanced photodissociation at 193 nm.39 Herein, we demonstrate that this same chemistry can also be leveraged to modulate the collisional dissociation behavior of basic phosphopeptides in real time for improved sequence coverage and phosphosite localization relative to their unmodified counterparts. In a manner similar to the removal or selective derivatization of basic sites, this method relies on the preferential formation of noncovalent interactions between the sulfonate moieties of FBDSA and the basic sites within the peptide to overcome or partially disrupt the mechanisms leading to preferential phosphate loss by collisional activation.
Experimental
Materials and Reagents
Phosphopeptides RQpSVELHSPQSLPR, GGGPApTPKKAKKL, and KKALRRQEpTVDAL were purchased from AnaSpec Inc. (Fremont, CA). RRLIEDAEpYAARG-NH2 was purchased from American Peptide Company (Sunnyvale, CA). LHpSQSLPR was custom ordered from United BioSystems (Herndon, VA). Mass spectrometry grade trypsin was purchased from Promega (Madison, WI). 4-formyl-1,3-benzenedisulfonic acid (FBDSA), 4-sulfophenyl isothiocyanate (SPITC), urea and all other solvents and materials were obtained from Sigma-Aldrich (St. Louis, MO). Peptides and reagents were used without further purification.
Solution Phase N-terminal Derivatization
Aliquots of phosphopeptides lacking internal lysine residues were subjected to N-terminal derivatization via 4-sulfophenyl isothiocyanate (SPITC) and carbamylation in the presence of excess urea. SPITC modification was accomplished by reacting 20 μL of stock solution (1 mg of SPITC in 100 μL of 1× PBS, pH 7.4) with 10 nmol of peptide for 30 minutes at 55°C. Carbamylation reactions were carried out via incubation with 8 M urea in 50 mM Tris-HCl (pH 8) at 80°C for 4 h. N-terminally labeled peptides were then desalted on C18 spin columns (Life Technologies, Grand Island, NY), evaporated to dryness and resuspended in 50:50 water/methanol for infusion.
Trypsin Digestion of Phosphopeptides
A 50 μL aliquot of 1 mM RRLIEDAEpYAARG-NH2 stock solution prepared in 25 mM ammonium bicarbonate buffer (pH 8.0) was digested at a 1:50 trypsin-to-peptide ratio for 3 h at 37°C. The digested phosphopeptide was then separated from trypsin using a 10 kDa molecular weight cutoff centrifugal filter that was washed 3x with 200 μL of water. The flow-through from each wash was pooled, evaporated to dryness, and resuspended to 10 μM in 50:50 water/methanol for infusion.
Mass Spectrometry and Front-end Ion/Ion Reactions
All experiments were conducted on a Thermo Scientific Velos Pro dual linear ion trap mass spectrometer (San Jose, CA) equipped with a front-end dual spray reactor as previously described.39 Briefly, the reactor was equipped with two electrospray ionization (ESI) sources (Prosolia Inc., Indianapolis, IN) mounted on a U-shaped railing system that surrounded the front-end of the mass spectrometer. The first source was fully integrated to allow for direct control of spray voltage and polarity in the Thermo Tune Plus control software, whereas the spray voltage of the second source was supplied via an external 5 kV dual polarity high voltage power supply (Stanford Research Systems Inc., Sunnyvale, CA).
For standard infusion experiments, the dual spray reactor was operated in single source mode in a manner comparable to conventional ESI. Phosphopeptide cations were generated by positive mode electrospray ionization of 5 μM working solutions infused at rate of 1.5 μL/min using a spray voltage of 1.5 kV. Alternatively, ion/ion reactions were carried out using dual source mode, during which the second source was simultaneously operated to generate a second population of reagent anions via negative mode ESI of 2 mM FBDSA prepared in 50:50 water/methanol infused at a rate of 3 μL/min. Electrostatic ion/ion complexes were formed at or near atmospheric pressure in the region of spray overlap prior to the inlet of the mass spectrometer. Anion source voltage was varied between −2.0 and −2.75 kV to achieve optimal complex formation and spray stability. Schiff base reactions were performed by collisionally activating the electrostatic phosphopeptide/FBDSA complexes with low normalized collision energy (NCE = 10–18%) to overcome the activation barrier for imine formation. Covalent Schiff base products were then isolated and subjected to MS3 collision-induced dissociation (CID) to generate diagnostic product ions using 20–30% NCE and a q-value of 0.25.
Results and Discussion
Gas-phase ion/ion-mediated Schiff base derivatization with 4-formyl-1,3-benezenedisulfonic acid (FBDSA) is an established method for rapid covalent transformation of peptides within the context of a tandem mass spectrometry-based experiment.39–43 This chemistry, as demonstrated in Figure S1, proceeds via the formation of long-lived electrostatic complexes arising from noncovalent interactions between the sulfonate moieties of FBDSA reagent anions and protonated sites of peptide cations.40 Subsequent collisional activation of these ion/ion intermediates promotes nucleophilic attack on the FBDSA aldehyde by an unprotonated primary amine in the substrate peptide, resulting in concerted dehydration and imine bond formation.40 Although well-defined for unmodified substrates, no studies to date have explored this chemistry with peptides containing labile post-translational modifications. Thus, we sought to evaluate both the feasibility and analytical utility of gas-phase derivatization with FBDSA for a series of phosphorylated peptides containing modified serine, threonine, and tyrosine residues. We hypothesized that this Schiff base reaction could provide a means to modulate hydrogen-bonding interactions in phosphopeptides and minimize phosphate cleavage, and at the same time allow implementation of this method in an on-line fashion via a dual spray reactor set-up.
Peptides that contain multiple basic sites pose a particularly difficult challenge for conventional CID-based characterization owing to the immobilization of charges caused by the presence of multiple basic residues that sequester ionizing protons. Previous mechanistic studies indicate that the protonated basic residues form strong hydrogen bonding interactions with the phosphate group.16 This consequently lowers the energy barrier for charge-directed mechanisms that lead to preferential neutral loss of phosphate and suppression of sequence-informative fragmentation upon collisional activation, thus further exacerbating the phosphate loss problem prevalent for MS/MS analysis of phosphopeptides.16 An illustrative example of this phenomenon is shown in the CID product ion spectrum of doubly charged KKALRRQEpTVDAL (Figure S2). The uninformative loss of H3PO4 from the precursor accounts for approximately 80% of the total product ion signal, while the remaining 20% arises from the b82+ through b122+ ions split between their phosphate retained and neutral loss forms. All diagnostic product ions contain the N-terminus, which is consistent with proton sequestration at basic arginine (R) or lysine (K) side-chains that occur near the N-terminal region of the peptide. As underscored by this example, the impediments associated with charge-immobilization make this a compelling gas-phase environment in which to probe the effects of FBDSA incorporation on phosphopeptide fragmentation.
Dual Spray Reactor-Initiated Schiff Base Bioconjugation of Phosphopeptides
The formation of long-lived phosphopeptide/FBDSA complexes was accomplished in realtime using a front-end dual spray reactor as previously described.39 Briefly, the reactor utilized two oppositely biased ESI sources to simultaneously generate overlapping populations of phosphopeptide cations and FBDSA anions in the high pressure region prior to the inlet of the mass spectrometer. The anionic reagent of interest, FBDSA, is negatively charged and thus has the potential to cause neutralization of peptides during formation of ion-ion complexes. To ensure successful detection of ion/ion complexes in the positive mode, the phosphopeptide substrates each contained at least two positive charge-bearing residues (i.e., arginine or lysine). This process is demonstrated in Figure S3, which compares the mass spectrum of KKALRRQEpTVDAL before (Figure S3a) and after (Figure S3b) interaction with FBDSA anions. Two highly abundant ions consistent with the 1+ and 2+ charge states of the charge-reduced KKALRRQEpTVDAL/FBDSA complex (denoted by the addition of “∆” in the label) are observed exclusively in the post-reaction spectrum at m/z 1874 and 937, respectively. Together the complexed species account for approximately 60% of the total analyte signal, thus demonstrating high ion/ion reaction efficiency in the region of overlap between the dual sprays prior to transmission into the mass spectrometer. The average ion/ion reaction efficiency observed for all peptides in this study was approximately 40%. Collectively, these results suggest that the presence of phosphorylated side-chains do not have a prohibitive effect on FBDSA binding despite possible competition with the phosphate moiety to form stabilizing noncovalent interactions with the protonated basic sites of the peptide.16,44
Once formed, ion/ion complexes were isolated in the linear ion trap and subjected to low energy CID to initiate covalent conversion to products, as demonstrated for the doubly charged complex of KKALRRQEpTVDAL/FBDSA (Figure S3c). Unlike collisional activation of the unreacted phosphopeptide, which is dominated by phosphate neutral loss (Figure S2), the most energetically favored pathway of the electrostatic complex results in dehydration with complete retention of the phosphate group and formation of the covalent Schiff base product. This is reflected by the single product ion observed in the MS2 spectrum which is 18 Da lower in mass from the precursor (Figure S3c), which is consistent with dehydration that occurs upon imine bond formation. The Schiff base product is confirmed via an additional collisional activation step (MS3) (Figure S3d). Collectively, these results demonstrate the ability to covalently modify phosphopeptides in the gas-phase while preserving the integrity of the labile phosphosite.
Collisional Dissociation of Unmodified versus FBDSA-Modified Phosphopeptides
As described above and shown in Figure S2, the product ion spectrum of doubly charged KKALRRQEpTVDAL illustrates the poor performance of CID under proton immobilized conditions; however, a marked improvement in fragmentation is observed following derivatization with FBDSA (Figure S3d). This is reflected in part by a 65% decrease in product ion signal comprised of non-sequence phosphate neutral loss from the precursor. Suppression of this preferential cleavage is accompanied by a concomitant gain in both the number and relative abundance of diagnostic fragment ions, resulting in an increase in sequence coverage from 42% to 92%. A small subset of product ions containing the phosphothreonine residue exhibit phosphate loss; however, in each case the relative abundance of these ions is lower than that of their corresponding phosphate-retaining forms. Furthermore, the emergence of y-ions and singly charged b◆-ions is consistent with greater proton mobility across the peptide backbone.
This dramatic change in fragmentation behavior upon incorporation of FBDSA is proposed to arise from the disruption and displacement of hydrogen bonding interactions between the phosphate group and basic sites of the peptide that lead to selective phosphate cleavage by CID. This hypothesis is supported by the lower pKa of sulfonate moieties in FBDSA relative to the phosphate group,44 as well as previous reports describing the gas-phase stability of acid-base interactions between sulfonate moieties and basic side-chains of peptides.43,45 Consequently, the strength of these interactions, and by effect the degree of change in subsequent fragmentation, should exhibit a dependence on the gas-phase basicity of the interacting side-chains. To explore this further, the relative change in fragmentation following FBDSA derivatization was evaluated for the arginine-containing peptide, RRLIEDAEpYAARG-NH2, and the lysine-containing peptide, GGGPApTPKKAKKL.
A comparison of the CID product ion spectra for the 2+ charge state of RRLIEDAEpYAARG-NH2 before and after gas-phase derivatization is shown in Figure 1. Collisional activation of the underivatized peptide promotes dominant neutral loss of phosphate and ammonia with limited product ion signal arising from cleavage at six of the twelve amide bonds of the peptide backbone (Figure 1a). Resulting low abundance sequence ions are derived from both termini of the peptide and in all cases are singly charged, which indicates protonation of both the N- and C-terminally located arginine residues. This distribution is likely more energetically favorable than protonation of the two adjacent N-terminal arginines due to coulombic repulsion of side-chains. Despite low overall abundance, the b6 and y7 ions originating from cleavage C-terminal to the aspartic acid residue are more abundant than the other b/y fragment ions, an outcome consistent with the aspartic acid effect commonly observed under conditions of low proton mobility.46,47 As demonstrated in Figure 1b, conversion of peptide RRLIEDAEpYAARG-NH2 to its Schiff base analogue profoundly alters its fragmentation. The resulting CID spectrum shows complete suppression of selective phosphate cleavage and instead displays extensive pairwise fragmentation across the peptide backbone, resulting in 92% coverage of the b-ion series and 75% coverage of the y-ion series. Moreover, the collective lack of evidence for charge-directed loss of phosphate or enhanced cleavage C-terminal to aspartic acid is highly indicative of greater proton mobility following FBDSA derivatization.
Figure 1.
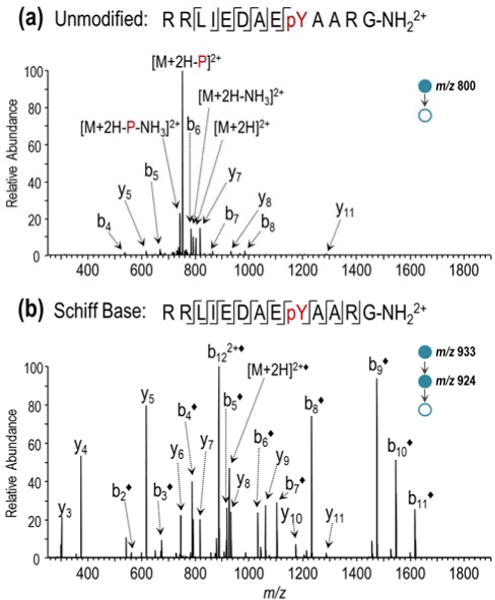
CID product ion mass spectra of RRLIEDAEpYAARG-NH2 (2+) before and after Schiff base modification: (a) MS2 CID mass spectrum of unlabeled peptide and (b) MS3 CID mass spectrum following online dual spray reactor-initiated derivatization. The addition of “◆” to the label indicates covalent FBDSA Schiff base modification and “-P” indicates loss of phosphate.
The effect of FBDSA incorporation on the fragmentation of GGGPApTPKKAKKL is expected to be less pronounced than that of RRLIEDAEpYAARG-NH2 owing to the lower gas-phase basicity of lysine side-chains relative to arginine and thus resultant weaker acid-basic interactions with FBDSA. The CID spectra of the underivatized and FBDSA-derivatized peptide are shown in Figure 2. The magnitude of the overall change in MS/MS patterns before and after derivatization generally agrees with this expectation about the impact of the basic side-chains. Notably, the dominance of phosphate neutral loss from the precursor suggests the unimpeded formation of hydrogen bonding interactions that facilitate charge-directed phosphate cleavage pathways. Differences also arise due to the fact that the ε-amino group of the lysine side-chain serves as a substrate for covalent FBDSA attachment. As a result, the location of the addition of the benzene disulfonic acid group is distributed across multiple reactive sites. This is reflected in the greater spectral complexity of the CID fragmentation spectrum of FBDSA-labeled GGGPApTPKKAKKL 2+, which exhibits contributions of product ions arising from various modified forms of the peptide (isomers) (Figure 2b). Despite these differences, enhanced fragmentation is still observed for the FBDSA modified peptide as demonstrated by more complete coverage of the peptide backbone. Additionally, the enhanced formation of the y7 ion corresponding to N-terminal to proline cleavage is consistent with greater proton mobility following incorporation of FBDSA.47,48
Figure 2.
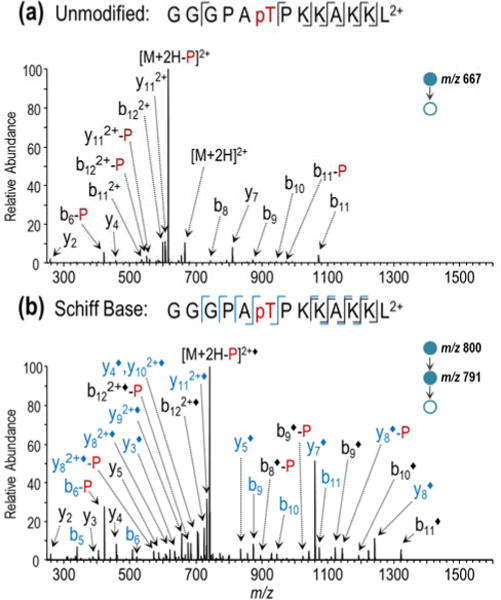
CID product ion mass spectra of GGGPApTPKKAKKL (2+) before and after Schiff base modification: (a) MS2 CID mass spectrum of unlabeled peptide and (b) MS3 CID mass spectrum following online dual spray reactor-initiated derivatization. The addition of “◆” to the label indicates covalent FBDSA Schiff base modification and “-P” indicates loss of phosphate. Ions shown in blue arise from lysine modification, while those shown in black may arise from either lysine or N-terminally labeled species.
The dual spray Schiff base strategy was also evaluated for two tryptic-like peptides, LIEDAEpYAAR and LHpSPQSLPR. Owing to the fact that these are tryptic-like peptides, they are much less basic than the ones described previously, and each possesses a single Arg at the C-terminus. Upon ESI, these peptides are observed in the 1+ and 2+ charge states, and when subjected to the ion-ion mediated workflow with FBDSA yield only singly charged Schiff base products (Figures S4 and S5). The resulting singly charged Schiff base products were subsequently characterized by CID, generating the MS/MS spectra shown in Figures S4C and S5C. The CID spectra for the corresponding unmodified peptides (1+) are shown in Figures S4B and S5B. A diagnostic series of y ions is observed for each of the Schiff base modified peptides, but b ions are not observed because of the location of the sulfonate group at the N-terminus which renders the b products negatively charged or neutral. These results indicate the potential application of the dual spray method for tryptic peptides in conventional bottom-up proteomics applications.
The percent reduction in phosphate neutral loss from the precursor before and after derivatization provides a useful metric by which to evaluate the successful suppression of charge-directed mechanisms that facilitate selective phosphate cleavage. This data is summarized in Figure 3 for representative singly and doubly charged phosphopeptide/FBDSA complexes generated by front-end ion/ion reactions and subsequently converted to their Schiff base modified forms. Substantial improvements in phosphate retention were observed in all cases following FBDSA incorporation; however, this effect was most pronounced for doubly protonated arginine-containing peptides for which the uninformative phosphate-loss pathways plummeted. This result further supports the hypothesis that the strength of the interaction between the sulfonate groups of FBDSA and the basic-side chains of the peptide has a direct impact on the observed change in fragmentation of FBDSA-labeled phosphopeptides.
Figure 3.
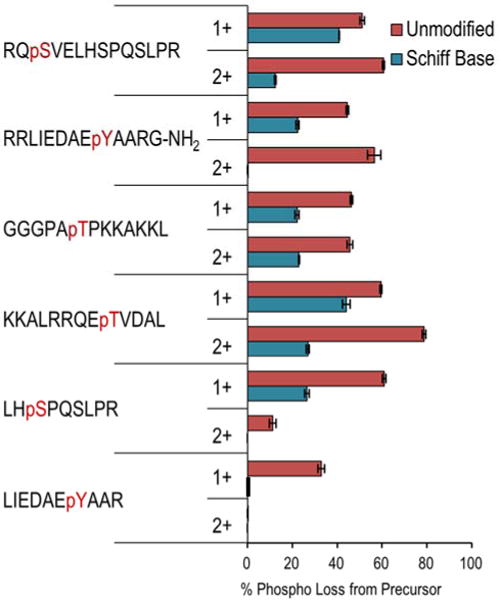
Percent of total product ion abundance arising from neutral loss of phosphate from the precursor ion of unmodified and Schiff based-labeled phosphopeptides subjected to CID.
Exploring the Role of the Sulfonate Moiety
In an effort to gain greater insight into the mechanistic role of the sulfonate moiety during collision-induced dissociation, the fragmentation behavior of unmodified and FBDSA-derivatized RRLIEDAEpYAARG-NH2 and RQpSVELHSPQSLPR was compared to that of their carbamylated and SPITC-modified analogues prepared in solution. Carbamylation converts primary amines of peptides (in this case the N-terminal amine) to less basic carbamate functionalities that are not expected to interact strongly with the basic side-chains of the peptide, thus serving as an experimental control for N-terminal modification (Figure S6a). Alternatively, the SPITC reagent introduces a mono-sulfonated phenyl group at the N-terminus of the peptide that more closely resembles FBDSA (Figure S1), albeit appended via a rather different pathway (Figure S6b). As demonstrated in Figure S7, both reactions are very efficient, and the charge state distributions of the resulting peptides are shifted to lower values (i.e. enhancement of 2+, diminishment of 3+) relative to the unmodified peptide, consistent with removal of the N-terminal protonation site. As expected, this shift is greatest for the SPITC-modified peptide due to the fixed negative charge of the sulfonate moiety.
Energy variable collisional activated dissociation was carried out on the doubly charged precursors for all forms of two representative phosphopeptides, RRLIEDAEpYAARG-NH2 (Figure 4a) and RQpSVELHSPQSLPR (Figure 4b), to evaluate their dissociation thresholds, one measure of stability in the gas phase. In both cases, the survival curves for precursors arising from sulfonate-bearing peptides exhibit a shift toward lower collisional energies relative to unmodified precursors, whereas the carbamylated species show nearly perfect overlap with the dissociation curves of the unmodified peptides. Such variations in fragmentation efficiency curves as a function of collision energy have been purported to reflect the degree of charge sequestration, essentially indicating the energy required for proton mobilization.47 As such, the fragmentation efficiency curves in Figure 4 suggest greater stabilization of the unmodified and carbamylated peptides (lower proton mobility) relative to the sulfonate-bearing peptides. The trends observed from the energy-variable CID curves correlate closely with the resulting fragmentation patterns; the unmodified (Figure 4c) and carbamylated (Figure 4d) peptides exhibit extensive phosphate loss, in contrast to enhanced backbone cleavages and production of ample diagnostic sequence ions with phosphate retention for the FBDSA-modified peptides (Figure 4e). The relative abundances of informative sequence ions were similarly enhanced relative to preferential phosphate loss ions for the SPITC-derivatized peptides; however, selective cleavage of sulfanilic acid from the SPITC label via cleavage of the labile C-N bond of the thiocarbamoyl group biased the resulting fragmentation pattern (Figure S8). These results are consistent with previous reports by Keough and co-workers that demonstrated enhanced structurally informative fragmentation following derivatization with sulfonic acid-incorporating reagents within the context of matrix-assisted laser desorption/ionization (MALDI) mass spectrometry due to the greater mobility of the ionizing proton.49
Figure 4.
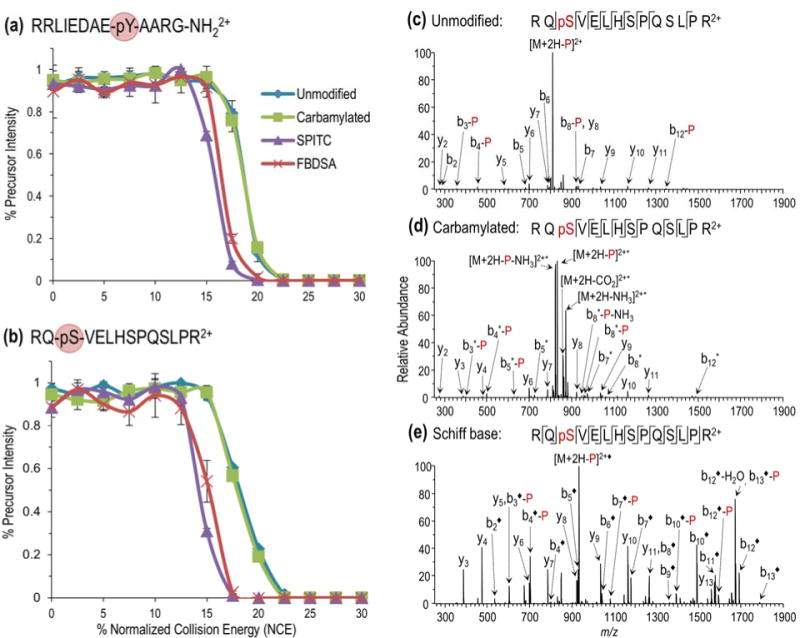
Variable energy CID analysis of unmodified and N-terminally carbamylated, SPITC-and FBDSA-modified (a) RRLIEDAEpYAARG-NH2 (2+) and (b) RQpSVELHSPQSLPR (2+). Normalized precursor abundances are plotted as a function of increasing collision energy. The CID product ion spectra are shown for RQpSVELHSPQSLPR (2+) in the following states: (c) unmodified, (d) carbamylated (*), and (e) FBDSA Schiff base modified (◆). The addition of “-P” to the label indicates loss of phosphate.
Based on the empirical observations made herein, two competing routes for the collision-induced dissociation of FBDSA-derivatized phosphopeptides are proposed as variations of the charge-directed SN2 reaction pathway previously described by Reid and co-workers (Scheme 1a).16 The first pathway gives rise to preferential acid-base interactions between a sulfonate moiety of FBDSA and a protonated basic side-chain in the peptide allowing enhanced formation of sequence-informative product ions (Scheme 1b). However, these acid-base interactions compete with hydrogen-bonding between the phosphate group and basic side-chains that facilitate selective phosphate neutral loss (Scheme 1c). Our preliminary results point to the gas-phase basicity of the side-chains as a primary determinant of the dominant dissociation pathway following FBDSA incorporation.
Scheme 1.
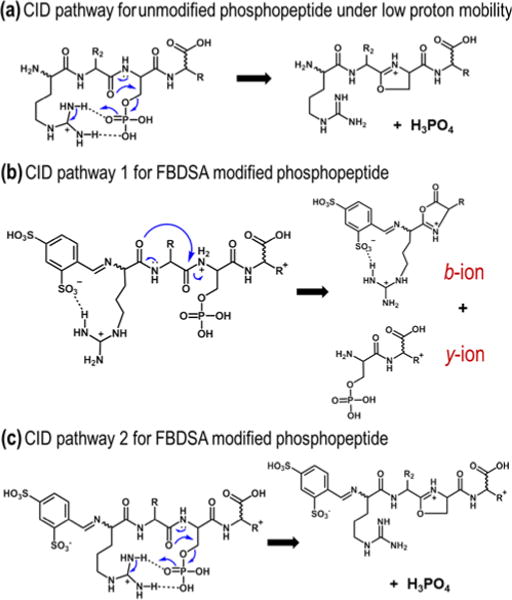
(a) Proposed mechanism for charge-directed neutral loss of phosphate for an unmodified phosphopeptide.16 Proposed competing dissociation pathways for FBDSA-labeled phosphopeptides with (b) and without (c) sulfonate-modulated suppression of charge-directed neutral loss of phosphate.
Conclusions
This work represents the first demonstration of ion/ion-mediated bioconjugation of peptides bearing one of the most common types of post-translational modifications, phosphorylation, in the gas-phase. Electrostatic phosphopeptide/FBDSA complexes were shown to undergo facile collision-induced conversion to covalent products with complete preservation of phosphosite integrity. The resulting FBDSA-derivatized phosphopeptides exhibited suppressed phosphate elimination that is the dominant process of conventional protonated phosphopeptides along with concomitant enhancement in the formation of sequence-informative product ions. The sulfonate moieties introduced upon FBDSA-incorporation are critical to this observed change in fragmentation behavior, as validated through comparative fragmentation analysis with other charge-site mediating modifications (i.e. carbamylation and thiocarbamoylation). Our results provide additional experimental evidence for charge-directed neighboring group participation reactions involved in phosphate neutral loss,16 which appear to be disrupted by competing hydrogen-bonding interactions between sulfonate groups and the side-chains of basic residues. This was further supported by the relative change in phosphate neutral loss observed depending on the gas-phase basicity of the side-chains present, with the greatest changes observed for FBDSA-labeled phosphopeptides containing arginine residues. This suggests that the relative strengths of the hydrogen-bonding interactions play an important role in subsequent fragmentation of the peptides upon CID.
The use of a front-end dual spray reactor to facilitate the ion/ion reaction step allows this method to be widely adaptable to nearly all mass spectrometer platforms, making it a viable option for phosphopeptide analysis when conventional CID-based characterization yields ambiguous sequence and phosphosite information. Moreover, the applicability of this method for tryptic-like peptides, as well as previous demonstrations of reactor compatibility with high-throughput chromatographic workflows39 hint at the potential utility of dual source integration into phosphoproteomic analysis.
Supplementary Material
Acknowledgments
Funding from the NIH (Grant R21GM103553 and R21EB018391) and the Welch Foundation (Grant F1155) is gratefully acknowledged. V.C.C. acknowledges an NSF Graduate Research Fellowship (Grant DGE-110007). W.M.M. acknowledges support from an NIH IRACDA training grant (K12GM10275).
Footnotes
Supporting Information: This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Cohen P. Nat Cell Biol. 2002;4:E127–E130. doi: 10.1038/ncb0502-e127. [DOI] [PubMed] [Google Scholar]
- 2.Ubersax JA, Ferrell JE., Jr Nat Rev Mol Cell Biol. 2007;8:530–541. doi: 10.1038/nrm2203. [DOI] [PubMed] [Google Scholar]
- 3.Johnson LN. Biochem Soc Trans. 2009;37:627–641. doi: 10.1042/BST0370627. [DOI] [PubMed] [Google Scholar]
- 4.Hunter T. Cell. 1995;80:225–236. doi: 10.1016/0092-8674(95)90405-0. [DOI] [PubMed] [Google Scholar]
- 5.Lahiry P, Torkamani A, Schork NJ, Hegele RA. Nat Rev Genet. 2010;11:60–74. doi: 10.1038/nrg2707. [DOI] [PubMed] [Google Scholar]
- 6.Hendriks WJAJ, Pulido R. Biochim Biophys Acta BBA - Mol Basis Dis. 2013;1832:1673–1696. doi: 10.1016/j.bbadis.2013.05.022. [DOI] [PubMed] [Google Scholar]
- 7.Humphrey SJ, James DE, Mann M. Trends Endocrinol Metab. 2015;26:676–687. doi: 10.1016/j.tem.2015.09.013. [DOI] [PubMed] [Google Scholar]
- 8.Mann M, Ong S-E, Grønborg M, Steen H, Jensen ON, Pandey A. Trends Biotechnol. 2002;20:261–268. doi: 10.1016/s0167-7799(02)01944-3. [DOI] [PubMed] [Google Scholar]
- 9.Riley NM, Coon JJ. Anal Chem. 2016;88:74–94. doi: 10.1021/acs.analchem.5b04123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Michalski A, Cox J, Mann M. J Proteome Res. 2011;10:1785–1793. doi: 10.1021/pr101060v. [DOI] [PubMed] [Google Scholar]
- 11.Bauer M, Ahrné E, Baron AP, Glatter T, Fava LL, Santamaria A, Nigg EA, Schmidt A. J Proteome Res. 2014;13:5973–5988. doi: 10.1021/pr500860c. [DOI] [PubMed] [Google Scholar]
- 12.Palumbo AM, Smith SA, Kalcic CL, Dantus M, Stemmer PM, Reid GE. Mass Spectrom Rev. 2011;30:600–625. doi: 10.1002/mas.20310. [DOI] [PubMed] [Google Scholar]
- 13.Brodbelt JS. Anal Chem. 2016;88:30–51. doi: 10.1021/acs.analchem.5b04563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Boersema PJ, Mohammed S, Heck AJR. J Mass Spectrom. 2009;44:861–878. doi: 10.1002/jms.1599. [DOI] [PubMed] [Google Scholar]
- 15.Jones AW, Cooper H. J Analyst. 2011;136:3419–3429. doi: 10.1039/c0an01011a. [DOI] [PubMed] [Google Scholar]
- 16.Palumbo AM, Tepe JJ, Reid GE. J Proteome Res. 2008;7:771–779. doi: 10.1021/pr0705136. [DOI] [PubMed] [Google Scholar]
- 17.Chi A, Huttenhower C, Geer LY, Coon JJ, Syka JEP, Bai DL, Shabanowitz J, Burke DJ, Troyanskaya OG, Hunt DF. Proc Natl Acad Sci. 2007;104:2193–2198. doi: 10.1073/pnas.0607084104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Stensballe A, Jensen ON, Olsen JV, Haselmann KF, Zubarev RA. Rapid Commun Mass Spectrom. 2000;14:1793–1800. doi: 10.1002/1097-0231(20001015)14:19<1793::AID-RCM95>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 19.Nagaraj N, D’Souza RCJ, Cox J, Olsen JV, Mann M. J Proteome Res. 2010;9:6786–6794. doi: 10.1021/pr100637q. [DOI] [PubMed] [Google Scholar]
- 20.Kim T-Y, Reilly JP. J Am Soc Mass Spectrom. 2009;20:2334–2341. doi: 10.1016/j.jasms.2009.08.021. [DOI] [PubMed] [Google Scholar]
- 21.Shin YS, Moon JH, Kim MS. J Am Soc Mass Spectrom. 2010;21:53–59. doi: 10.1016/j.jasms.2009.09.003. [DOI] [PubMed] [Google Scholar]
- 22.Madsen JA, Kaoud TS, Dalby KN, Brodbelt J. S PROTEOMICS. 2011;11:1329–1334. doi: 10.1002/pmic.201000565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fort KL, Dyachenko A, Potel CM, Corradini E, Marino F, Barendregt A, Makarov AA, Scheltema RA, Heck AJR. Anal Chem. 2016;88:2303–2310. doi: 10.1021/acs.analchem.5b04162. [DOI] [PubMed] [Google Scholar]
- 24.Swaney DL, McAlister GC, Wirtala M, Schwartz JC, Syka JEP, Coon JJ. Anal Chem. 2007;79:477–485. doi: 10.1021/ac061457f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Frese CK, Zhou H, Taus T, Altelaar AFM, Mechtler K, Heck AJR, Mohammed S. J Proteome Res. 2013;12:1520–1525. doi: 10.1021/pr301130k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shaffer CJ, Slováková K, Tureček F. Int J Mass Spectrom. 2015;390:71–80. [Google Scholar]
- 27.Meyer HE, Hoffmann-Posorske E, Korte H, Heilmeyer LMG. FEBS Lett. 1986;204:61–66. doi: 10.1016/0014-5793(86)81388-6. [DOI] [PubMed] [Google Scholar]
- 28.Oda Y, Nagasu T, Chait BT. Nat Biotechnol. 2001;19:379–382. doi: 10.1038/86783. [DOI] [PubMed] [Google Scholar]
- 29.Molloy MP, Andrews PC. Anal Chem. 2001;73:5387–5394. doi: 10.1021/ac0104227. [DOI] [PubMed] [Google Scholar]
- 30.Thompson AJ, Hart SR, Franz C, Barnouin K, Ridley A, Cramer R. Anal Chem. 2003;75:3232–3243. doi: 10.1021/ac034134h. [DOI] [PubMed] [Google Scholar]
- 31.Knight ZA, Schilling B, Row RH, Kenski DM, Gibson BW, Shokat KM. Nat Biotechnol. 2003;21:1047–1054. doi: 10.1038/nbt863. [DOI] [PubMed] [Google Scholar]
- 32.Diedrich JK, Julian RR. Anal Chem. 2011;83:6818–6826. doi: 10.1021/ac201647w. [DOI] [PubMed] [Google Scholar]
- 33.Diedrich JK, Julian RR. J Am Chem Soc. 2008;130:12212–12213. doi: 10.1021/ja8023719. [DOI] [PubMed] [Google Scholar]
- 34.McLachlin DT, Chait BT. Anal Chem. 2003;75:6826–6836. doi: 10.1021/ac034989u. [DOI] [PubMed] [Google Scholar]
- 35.Gronert S, Huang R, Li KH. Int J Mass Spectrom. 2004;231:179–187. [Google Scholar]
- 36.Gronert S, Li KH, Horiuchi M. J Am Soc Mass Spectrom. 2005;16:1905–1914. doi: 10.1016/j.jasms.2005.07.018. [DOI] [PubMed] [Google Scholar]
- 37.Leitner A, Foettinger A, Lindner W. J Mass Spectrom. 2007;42:950–959. doi: 10.1002/jms.1233. [DOI] [PubMed] [Google Scholar]
- 38.Lanucara F, Lee DCH, Eyers CE. J Am Soc Mass Spectrom. 2013;25:214–225. doi: 10.1007/s13361-013-0770-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cotham VC, Shaw JB, Brodbelt JS. Anal Chem. 2015;87:9396–9402. doi: 10.1021/acs.analchem.5b02242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Han H, McLuckey SA. J Am Chem Soc. 2009;131:12884–12885. doi: 10.1021/ja904812d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hassell KM, Stutzman JR, McLuckey SA. Anal Chem. 2010;82:1594–1597. doi: 10.1021/ac902732v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Stutzman JR, McLuckey SA. Anal Chem. 2012;84:10679–10685. doi: 10.1021/ac302374p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Stutzman JR, Luongo CA, McLuckey SA. J Mass Spectrom. 2012;47:669–675. doi: 10.1002/jms.2968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schug KA, Lindner W. Chem Rev. 2005;105:67–114. doi: 10.1021/cr040603j. [DOI] [PubMed] [Google Scholar]
- 45.Woods AS, Wang H-YJ, Jackson SN. J Proteome Res. 2007;6:1176–1182. doi: 10.1021/pr060529g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tsaprailis G, Somogyi A, Nikolaev EN, Wysocki VH. Int J Mass Spectrom. 2000:195–196. [Google Scholar]
- 47.Wysocki VH, Tsaprailis G, Smith LL, Breci LA. J Mass Spectrom. 2000;35:1399–1406. doi: 10.1002/1096-9888(200012)35:12<1399::AID-JMS86>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- 48.Bleiholder C, Suhai S, Harrison AG, Paizs B. J Am Soc Mass Spectrom. 2011;22:1032–1039. doi: 10.1007/s13361-011-0092-1. [DOI] [PubMed] [Google Scholar]
- 49.Keough T, Youngquist R, Lacey MP. Anal Chem. 2003;75:156A–165A. doi: 10.1021/ac031274i. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.