

Abstract
RNA plays a fundamental, ubiquitous role as either substrate or functional component of many large cellular complexes—“molecular machines”—used to maintain and control the readout of genetic information, a functional landscape that we are only beginning to understand. The cellular mechanisms for the spatiotemporal organization of the plethora of RNAs involved in gene expression are particularly poorly understood. Intracellular single‐molecule fluorescence microscopy provides a powerful emerging tool for probing the pertinent mechanistic parameters that govern cellular RNA functions, including those of protein coding messenger RNAs (mRNAs). Progress has been hampered, however, by the scarcity of efficient high‐yield methods to fluorescently label RNA molecules without the need to drastically increase their molecular weight through artificial appendages that may result in altered behavior. Herein, we employ T7 RNA polymerase to body label an RNA with a cyanine dye, as well as yeast poly(A) polymerase to strategically place multiple 2′‐azido‐modifications for subsequent fluorophore labeling either between the body and tail or randomly throughout the tail. Using a combination of biochemical and single‐molecule fluorescence microscopy approaches, we demonstrate that both yeast poly(A) polymerase labeling strategies result in fully functional mRNA, whereas protein coding is severely diminished in the case of body labeling.
Keywords: single‐molecule fluorescence imaging, noncoding RNA, fluorophore labeling, single particle tracking
Introduction
Nature has long perfected the use of large assemblies of biomolecules that convert chemical energy into quasi‐mechanical movements to perform life's essential functions. For example, cells utilize the translation machinery—the ribosome—to manufacture proteins from coding messenger RNA (mRNA) templates. Cellular nanomachines also modulate the extent of translation by employing short (∼22‐nucleotide), single‐stranded noncoding RNAs termed microRNAs (miRNAs) to guide Argonaute proteins assembled into an RNA‐induced silencing complex (RISC) to the 3′ untranslated region (UTR) of their target mRNAs.1, 2 RISC will inhibit translation and then recruit destabilizing machinery to the target,3 designed to decap, deadenylate, and degrade the target.3, 4, 5, 6 These nanomachines, and innumerous others involved in RNA metabolism, synergistically drive the cellular enterprise, thus meriting initiatives to probe their components to learn how they naturally function, and identify when and how they become dysfunctional to cause disease.
Similar to any machine, ribonucleoprotein complexes (RNPs) are modular in nature and thus require the proper assembly of the correct number and type of molecule for their function. Malformation of any of these components—through misfolding or genetic mutation—can lead to loss of or aberrant function, resulting in disease. Owing to their size, complexity, and difficulty to purify, there are still too few strategies available to study these macromolecular complexes either inside or outside the cell. As an added layer of complexity, some of these cellular nanomachines are too few in number to sufficiently purify from cells via traditional biochemistry. Thus, electron and fluorescence microscopy have emerged as powerful approaches to probe the structure and activities of individual machine components within their cellular context.7, 8, 9, 10
Intracellular (“in cellulo”) single‐molecule fluorescence microscopy (SMFM) imaging is a powerful tool for measuring expression levels, kinetics of interaction between two molecules, cellular localization, intramolecular dynamics, and 3D architecture of assembled structures in their native environment.7, 11, 12, 13, 14 Given the stochastic nature of biological processes in the cellular environment, single‐molecule assays are pivotal not only for probing biochemical mechanisms but also for understanding how biology is controlled spatiotemporally, ultimately deconvolving what separates disease from archetypical. Some examples of intracellular SMFM techniques used to measure these phenomena are fixed‐cell particle counting through single‐molecule fluorescence in situ hybridization (smFISH) to assess expression levels,15 single‐particle tracking for interaction kinetics and cellular localization,16 fixed‐cell stepwise photobleaching counting of subunit stoichiometry,17 and super‐resolution microscopy for 3D architecture,18 among others.7, 11 Despite the value that SMFM approaches provide, there is still an insufficient number of tools available to label RNA components of RNP nanomachines by comparison to those that are protein based.7 This discrepancy exists in part because RNA research dramatically accelerated only over the past two decades with the discovery of a plethora of noncoding RNAs, and thus still trails protein research. Accordingly, the most common labeling techniques take advantage of mega‐Dalton RNPs appended to an mRNA's 3′ end in the form of an RNA recognition sequence that associates with a specific RNA binding protein fused to a relatively dim fluorescent protein, whose signal is amplified by massive repetition of the RNA recognition sequence.7, 11, 12, 13, 14, 19, 20 In some cases, aberrant localization and diffusion properties have been noted for these large and compositionally ill‐defined constructs.21
An eukaryotic mRNA is canonically viewed as divided into four functional segments: the 5′ UTR (including the m7G cap),22, 23 the coding sequence,24 the 3′ UTR,25 and the poly(A) tail.26 Incorporating a fluorescent probe into any one of these segments may be deleterious to the mRNA's function, potentially resulting in spurious findings and incorrect conclusions. For this reason, there is a great need for labeling strategies that can precisely incorporate a controllable number of fluorophores at exact locations in an RNA. Ideally, these methods should require few steps; are of high efficiency and yield; and use gentle, commercially available, and noncytotoxic materials. Diverse fluorescent labeling methods of endogenous RNAs are available to meet these goals.27, 28, 29, 30, 31, 32 Many of these methods are plagued by issues relating to limited dye permeability through the cell membrane, nonspecific dye binding, cytotoxicity, necessary modifications to the genome, high‐molecular‐weight RNA extensions, and high intrinsic background; hence, they are often impractical for monitoring low‐abundance and short transcripts.33, 34 Many of these problems are overcome by covalently labeling an exogenous RNA in vitro where the number of dyes can be controlled and the RNA be purified away from unbound dye. However, endogenous RNAs invoke challenges associated with their method of delivery.33, 34, 35, 36 We have previously surmounted this challenge for 3′ fluorophore‐labeled miRNAs through microinjection and proper control design.3, 37
Herein, we exploited T7 RNA polymerase and yeast poly(A) polymerase35, 38 to integrate modified nucleotides into one of the three segments of an mRNA: randomly into the body (which includes the 5′ UTR, coding region and 3′ UTR), randomly into the poly(A) tail, or specifically between the body and tail (BBT). Coupling transcription and tailing with modification and labeling methods has the potential to save time and cost, as well as to increase the yield. In the case of body labeling, the modified nucleotide was precoupled to the fluorophore, while in the other two cases, yeast poly(A) polymerase required a separate dye coupling step. Following modification and labeling, each strategy was tested for its ability to produce an mRNA that remains a substrate of the translation, RISC, and degradation machineries. We discovered through ensemble and intracellular single‐molecule probing that the tail and BBT strategies preserve these functional activities of the mRNA. By contrast, body labeling drastically impairs the coding function, although the mRNA's 3′ UTR remains accessible to RISC.
Results
T7 RNA polymerase and yeast poly(A) polymerase efficiently body and tail label mRNAs, respectively
Aside from the m7G cap,22 functional eukaryotic mRNAs require a poly(A) tail comprising a successive string of 100–250 adenosines, which play an intricate role in mRNA stability and translation.39 For labeling firefly luciferase (FLuc) or Renilla luciferase (RLuc) mRNA, two types of chemically modified nucleoside triphosphates (CNTPs) were integrated into the RNA chain using enzymatic labeling methods: co‐transcriptional incorporation of cyanine 5‐aminoallyluridine‐5′‐triphosphate [Cy5‐UTP, Fig. 1(A)] using T7 RNA polymerase and incorporation of 2′‐azido‐2′‐deoxyadenosine‐5′‐triphosphate [azido‐ATP, Fig. 1(B)] using yeast poly(A) polymerase after transcription. Three labeling strategies [Fig. 1(C)] were implemented to either randomly incorporate Cy5‐UTP throughout the body of the RNA [5′ UTR, coding sequence and 3′ UTR, Fig. 1(C), top], add consecutive azido‐ATP moieties between the body and poly(A) tail [Fig. 1(C), middle], or randomly incorporate azido‐ATP throughout the poly(A) tail [Fig. 1(C), bottom]. Each of these strategies offers the unique capacity to place the fluorophore distally from a region of interest. As an example, body or BBT labeling strategies may best suited for studying deadenylation, as the modifications are integrated distally from the tail.
Figure 1.
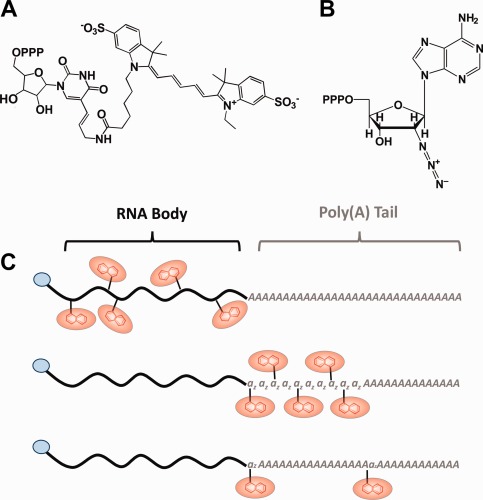
Labeling of mRNA. Modified CNTPs (A, B) are enzymatically incorporated into the RNA chain either cotranscriptionally (C, Top) or using yeast poly(A) polymerase, yPAP (C, middle and bottom). First, the body (black) of the mRNA is transcribed with (C, top) or without cyanine 5‐aminoallyluridine‐5′‐triphosphate (C, middle and bottom) using T7 RNA polymerase and an m7G cap (blue). In strategy C, middle, the mRNA is modified with a series of consecutive 2′‐azido‐2′‐deoxyadenosine‐5′‐triphosphates (B, az) and subsequently poly(A) tailed (grey), both using yPAP. In strategy C, bottom, the capped mRNA is simultaneously, and randomly, modified with 2′‐azido‐2′‐deoxyadenosine‐5′‐triphosphate (az) during the poly(A) tailing procedure with yPAP. In both strategies (C, middle and bottom), the 2′‐azido‐2′‐deoxyadenosine‐5′‐triphosphate (az) modifications are coupled with either DIBO Alexa Fluor® 647 or Alkyne Alexa Fluor® 647 (both red) via click reactions.
It has long been known that T7 RNA polymerase tolerates pyrimidine‐based CNTPs if they are altered at the 5‐position.38, 40, 41, 42 Thus, supplementing a transcription reaction with Cy5‐UTP will permit the co‐transcriptional body labeling of a Firefly Luciferase mRNA [FLuc, Fig. 1(C), top]. To test the extent of tolerability and control of the labeling process, we substituted increasing fractions of the UTP pool with Cy5‐UTP (0–10% of total UTP), incubated each reaction using standard conditions, and tested for the extent of incorporation. In agreement with previous findings,38, 40, 41 we found that the CNTP is a well‐tolerated substrate as indicated by the consistency in RNA yield (Table 1) and lack of an increase in aborted side‐products beyond those observed for the 0% Cy5‐UTP control transcription [Fig. 2(A)]. Furthermore, quantification revealed a linear correlation between the number of fluorophores per purified RNA molecule (Table 1) and the fraction of Cy5‐UTP during transcription [Fig. 2(B)]. This suggests that predicting and controlling the extent of labeling is easily achieved by adjusting the starting concentration of Cy5‐UTP in the transcription mixture. The RNA was subsequently 5′ capped and polyadenylated for use in translation assays.
Table 1.
Calculated Parameters for Each Labeling Strategy
| mRNA labeling strategy | |||
|---|---|---|---|
| Body labeled | BBT labeled | Tail labeled | |
| 5 labels/RNA | 0.79% Cy5‐UTP | 13.2 min | 10.9% Azide‐ATP |
| Normalized yields | 107% | 100% | 87% |
| Normalized in vitro translation output | 28% | 112% | 97% |
| Fast population diffusion coefficient | NA | 0.917 µm2/s (0.664 µm2/s) | 1.03 µm2/s (1.32 µm2/s) |
| Fast population AUC | NA | 0.075 (0.078) | 0.062 (0.038) |
| Slow population diffusion coefficient | NA | 0.108 µm2/s (0.0787 µm2/s) | 0.124 µm2/s (0.126 µm2/s) |
| Slow population AUC | NA | 0.17 (0.17) | 0.18 (0.21) |
Adjusting only CNTP input or incubation time, the estimated quantities or times required to achieve 5 labels per mRNA using standard incubation conditions. Normalized yields are calculated based on final concentrations of each mRNA following CNTP modification and purification, relative to unmodified control. In vitro translation output is calculated as the FLuc protein band intensity for the labeled, from rabbit reticulocyte extract, normalized to unlabeled control. Average MSD calculated diffusion coefficients for the slow and fast populations, and the areas under the curve (AUC) for each distribution, with (brackets) or without co‐injected miR‐7.
Figure 2.
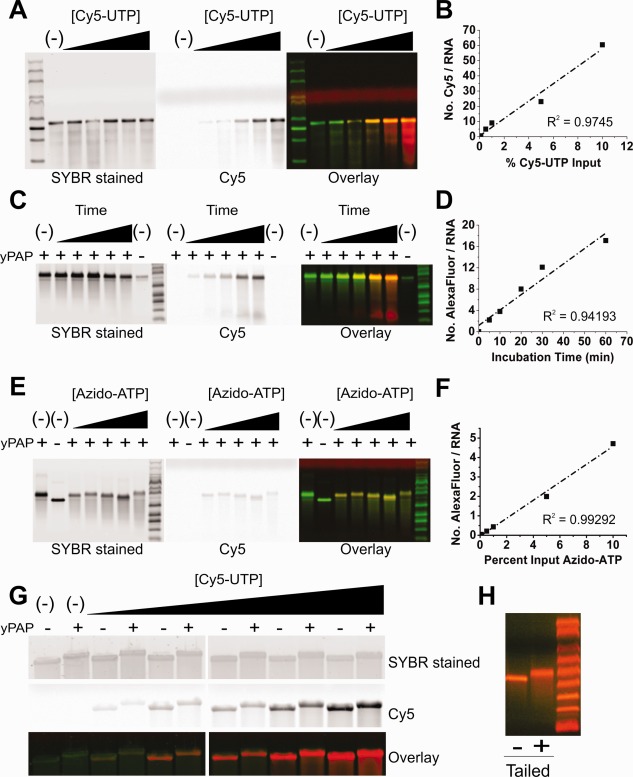
Extent of labeling and post‐transcriptional mRNA modification for all three labeling strategies. (A, C, E) Gel images of denaturing (1M urea) 1.2% agarose gels stained for total RNA (left panel, green in overlay), Cy5 or Alexa647‐labeled RNA (right panel, red in overlay), indicate the extent of RNA labeling. Graphs of the Cy5 or Alexa647 dye‐to‐RNA ratio, derived using absorbance measurements and Beer's law, are plotted against yPAP incubation time or CNTP % input, demonstrating a strong positive‐linear correlation for our body (B), BBT (D), and tail (F) labeling strategies. (A) Cotranscriptional body labeling of FLuc mRNA demonstrates a clear trend of increasing Cy5 intensity with percent Cy5‐UTP (0, 0.1, 0.5, 1.0, 5.0, and 10%) input. (C) Increasing the yPAP incubation time (0, 5, 10, 20, 30, and 60 min) of capped RLuc mRNA in the presence of 2′ azide‐modified ATP increases the extent of BBT labeling by comparison to untailed and tailed (ATP only) controls. (E) Fluorescence increases modestly with increasing percent azide‐ATP input (0, 0.1, 0.5, 1.0, 5.0, and 10%) in the tail labeling strategy. (G) SYBR Gold‐stained FLuc mRNA (top panel, green in overlay) was body labeled with increasing concentrations of Cy5 CNTP (bottom panel, red in overlay) and, regardless of the extent of labeling, remains capable of being yPAP polyadenylated. (H) Denaturing 1.2% agarose gel analysis of heavily BBT‐labeled FLuc mRNA either with or without yPAP treatment. In all cases, M signifies lanes with RNA markers.
Labeling an RNA between its body and tail [BBT, Fig. 1(B)] ensures that the CNTP is incorporated outside a functionally relevant sequence. For this strategy, the RNA was first transcribed, 5′ capped, then 3′ modified with a successive chain of azido‐ATP moieties using yeast poly(A) polymerase (yPAP). The extent of labeling was controlled kinetically by incubating the mRNA for 0, 5, 10, 20, 30, and 60 min. Following further polyadenylation (without modification) and purification, the RNA was fluorescently labeled using the Click‐IT® Alexa Fluor® 647DIBO Alkyne (Alexa647) reagent. We chose to label the RNA using a strained‐promoted alkyne‐azide cycloaddition (SPAAC) click approach because the reagent is commercially available, uses gentle reaction conditions, and leads to a high yield. Copper‐based click approaches resulted in excessive RNA degradation [Supporting Information, Fig. S1(A)]. BBT modification and SPAAC labeling of the RNA was extensive and rapid, as an average of 2.2 Alexa647 molecules per RNA were incorporated in as little as 5 min (Table 1). Gel analysis of the purified RNA demonstrated an increase in Alexa647 band intensity with time of incubation with yPAP and azido‐ATP [Fig. 2(C)], leading to a linear correlation—particularly in early time points—once quantified [Fig. 2(D)].
Our third strategy involved the random incorporation of azide‐ATP during the poly(A) tailing step [Fig. 1(C), bottom]. This strategy is designed to save time and number of necessary steps, which may be important when RNA yields are low. To test the extent and controllability of RNA labeling, the transcribed and capped RLuc message was yPAP tailed with varying concentrations of azide‐ATP (0, 0.1, 0.5, 1, 5, and 10% of total ATP). Subsequently, the purified tail‐modified RNA was Alexa647 labeled as before and gel analyzed [Fig. 2(E)]. As expected, calculating the molar ratio of Alexa647 to RNA and plotting it against the azido‐ATP fraction in the ATP pool revealed a linear correlation [Fig. 2(F)]. While a modest average 4.7 labels per RNA were observed for the 10% azido‐ATP condition, this strategy appears best when the extent of labeling needs to be tightly controlled. While this tail labeling strategy inherently installed the poly(A) tail, body and BBT labeling required an additional step. To assess the impact of the incorporated CNTPs on poly(A) tailing, body‐ and BBT‐labeled mRNAs were assessed after their yPAP‐mediated tailing reaction by denaturing 1.2% agarose gel electrophoresis. Regardless of the extent of labeling, the body‐labeled mRNAs were easily tailed, as indicated by the change in molecular weight following yPAP treatment [Fig. 2(G)]. Similarly, the azido‐ATP modifications in the BBT labeling strategy did not impede yPAP from further modifying the mRNA with a typical poly(A) tail [Fig. 2(H)]. Next, the resulting 5′‐capped and 3′‐tailed mRNAs were tested for protein coding capacity.
BBT and tail labeling of an mRNA are compatible with translation
To test the compatibility of our fluorescent mRNAs with full‐length translation, we heavily labeled FLuc mRNA with each one of our three approaches (Body = 60 labels/RNA, BBT = 17 labels/RNA, and Tail = 4.7 labels/RNA), and incubated the resulting mRNAs in translation‐active rabbit reticulocyte extract. To detect all protein products, the reaction was supplemented with BIODIPY® labeled, lysine‐charged tRNA (FluoroTect™), then quenched and analyzed for fluorescently labeled proteins of all molecular weights on a gradient, 4–20% Tris‐Glycine SDS gel. In all cases, a protein product was observed at the proper molecular weight, identified by translation of the unlabeled mRNA, with few aborted side products beyond those observed from the unlabeled control [Fig. 3(A,B)]. As may be expected, the body‐labeled mRNA produced 72% less FLuc protein relative to control [Fig. 3(A)], suggesting that this labeling strategy impedes the ribosome. By contrast, both our BBT and tail labeling strategies produced protein bands with intensities within 10% of the control [Fig. 3(B)]. Considering these data, the body labeling strategy was not assessed further for its coding functionality.
Figure 3.
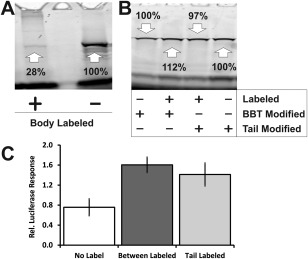
BBT and tail‐labeled mRNAs produce translated protein. (A) SDS‐PAGE gel analysis of translated FluoroTect® labeled FLuc protein from 1 µg of either body labeled or unlabeled mRNA incubated in rabbit reticulocyte extracts. (B) SDS‐PAGE gel analysis of translated FluoroTect®‐labeled FLuc protein from 1 µg of either unlabeled or BBT‐labeled FLuc mRNA, and either unlabeled or tail‐labeled FLuc mRNA, incubated in rabbit reticulocyte extract. (C) Relative luciferase responses of BBT‐ or tail‐labeled FLuc mRNA, normalized with RLuc mRNA, after transfection into HeLa cells.
Next, to assess their protein coding capabilities in cellulo, HeLa cells were transfected with heavily labeled BBT (17 labels/RNA) and tail (4.7 labels/RNA) modified FLuc mRNAs and measured for their relative gene expression [Fig. 3(C)]. Strikingly, we observed an approximately twofold increase in FLuc expression for labeled mRNA versus unmodified. We attribute this finding to a stabilizing effect either directly from the fluorophores or the longer poly(A) tail43 as found by gel analysis [Fig. 2(E)] to result from the labeling procedure. As a further test of translation in cellulo, we microinjected labeled and unlabeled mRNAs into both U2OS and HeLa cells and detected FLuc by immunofluorescence. A 10‐kDa Cascade Blue® Dextran injection marker was co‐microinjected to identify injected cells. Similar to the unlabeled version, BBT‐ and tail‐labeled RNA produced significant quantities of FLuc protein, by comparison to noninjected cells and RNase‐treated tail‐labeled mRNA [Fig. 4(A,B)]. Collectively, these data suggest that both these strategies are viable options for labeling an mRNA and maintaining its coding functionality.
Figure 4.
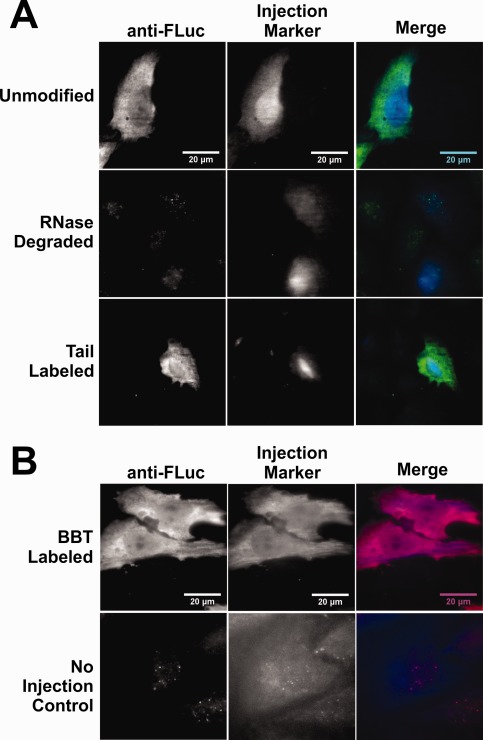
Microinjected BBT‐ and tail‐labeled mRNAs selectively express FLuc protein. U2OS cells (A) and HeLa cells (B) were microinjected with either 10 nM unmodified BBT‐ or tail‐labeled mRNA in 1× PBS and 0.025% 10 kDa Cascade Blue® Dextran. Twelve hours after microinjection, cells were fixed in 4% formaldehyde solution, ethanol permeabilized, and stained with primary and secondary antibodies labeled with either Cy3 (A) or DyLight 650 (B) fluorophores. (A) Microinjected U2OS cells selectively express protein that colocalize exclusively with the Cascade Blue® injection marker for both fluorescent tail‐labeled and unlabeled FLuc mRNA. As a control, microinjected RNase A‐treated fluorescent tail‐labeled FLuc mRNA did not express protein. (B) Microinjected fluorescent BBT‐labeled FLuc RNA into HeLa cells selectively expressed protein that colocalize with the cell injection marker, in contrast to noninjected cells.
The 3′ UTRs of BBT‐ and tail‐labeled mRNAs are accessible to miRNA regulation
As miRNAs serve to inhibit translation initiation, mRNA targets regulated by miRNA‐loaded RISC should have a repressed protein expression signature relative to a counterpart without a corresponding target site in their 3′ UTR. We therefore designed a set of dual luciferase reporter plasmids (RLuc and FLuc) containing an FLuc gene engineered with 0, 1, 2, 3, 4, 5, 6, or 11 miR‐7 miRNA recognition element (MRE) sites to test the functional accessibility of the 3′ UTR of our labeled mRNAs [Supporting Information, Fig. S2(A,B)]. Transfection experiments with the unlabeled mRNAs revealed an increase in miRNA‐dependent gene repression with increasing MRE number in both HeLa and DCP1a‐EGFP stably transfected U2OS cells [Supporting Information, Fig. S2(C,D)]. We found this observation to be strictly dependent on co‐transfection with miR‐7, consistent with the notion that both HeLa and U2OS cells contain negligible quantities of endogenous miR‐7.44 Importantly, transfecting labeled FLuc BBT (∼17 labels/RNA) and tail (∼5 labels/RNA) modified mRNAs bearing 0, 1, 3, 6, or 11 miR‐7 MRE sites into HeLa cells demonstrated the same increasing repression signature as their unlabeled and unmodified control [Fig. 5(A)]. Thus, the strategic placement of these fluorophores appears to be compatible with the miRNA‐dependent translation initiation inhibition by RISC.
Figure 5.
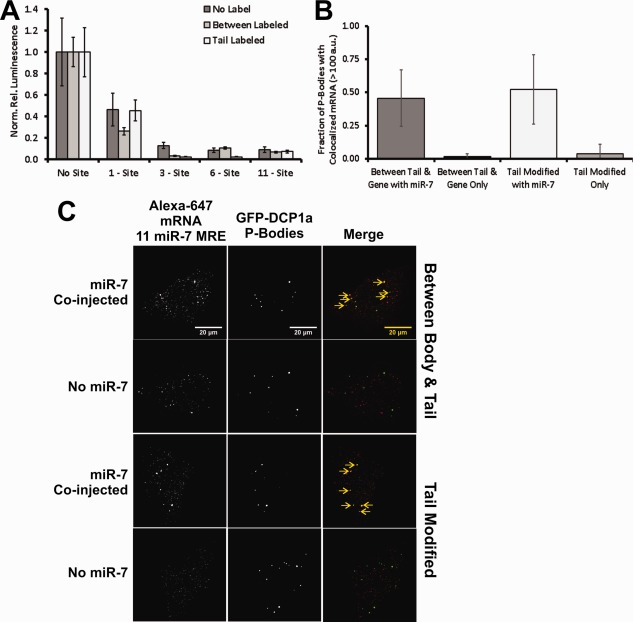
Fluorescent BBT‐ and tail‐labeled FLuc mRNAs are repressed and degraded by miRNA. (A) RLuc normalized luminescence from transfected unlabeled and fluorescent BBT‐ or tail‐labeled FLuc mRNA, containing either 0, 1, 3, 6, or 11 miR‐7 MRE sites, in HeLa cells. (B) Live cell imaging of DCP1a‐EGFP stably transfected U2OS cells, microinjected with either fluorescent BBT‐ or tail‐labeled FLuc mRNA with or without miR‐7, analyzed 2 h after microinjection. Yellow arrows indicate colocalized fluorescent mRNA with cytoplasmic P‐Body granules. (C) Quantification of extent of P‐Body colocalization with fluorescent mRNA in DCP1a‐EGFP stably transfected U2OS cells. Fluorescent P‐Body granules possessing Alexa647 fluorescence exceeding a threshold of 100 arbitrary units within a 4‐pixel radius of the P‐Body centroid and persisting for over 9 frames are counted as a colocalization event.
Another aspect of miRNA regulation of an mRNA target is its eventual degradation.3, 45 mRNA destabilization involves the recruitment of deadenylase complexes (CCR4‐Not1 and PAN 2/3), for the shortening of the poly(A) tail,46 the DCP1/2 decapping complex,47 and XRN1‐dependent 5′‐to‐3′ mRNA digestion.48 These destabilization processes are thought to partially occur within large cytoplasmic aggregates, termed processing bodies (P‐Bodies).49 Thus, the recruitment of a target mRNA to a P‐Body granule is typically viewed as an indication of degradation. To assess whether our labeled mRNAs are capable of miRNA‐dependent destabilization, we created a stably transfected U2OS cell line that expresses DCP1a protein chimerically linked to EGFP, as described elsewhere,50 and measured the extent of colocalization of our labeled mRNAs with P‐Bodies in cellulo. More specifically, microinjected labeled mRNAs were assessed for the extent of colocalization with EGFP‐DCP1a, 2 h after injection [Fig. 5(B)]. Microinjection solutions contained 1 nM Alexa647‐labeled BBT or tail‐modified mRNA—all heavily labeled—with 11 miR‐7 MRE sites in 1× PBS, with or without 1 µM mature miR‐7 duplex, and with 0.025% Cascade Blue®‐labeled 10 kDa Dextran for identifying injected cells. After 2 h, live cells were imaged for Alexa647 foci within a 4‐pixel radius of the P‐Body centroid that persisted for >9 imaging frames (0.9 s). At this threshold, in the presence of miR‐7, ∼50% of P‐Bodies were found colocalized with Alexa647‐labeled mRNA, regardless of the labeling strategy [Fig. 5(B,C)]. In fact, even the body‐labeled mRNA, incapable of efficient protein coding, colocalized with P‐Body foci (Supporting Information, Fig. S3). By contrast, in the absence of miR‐7 on average <10% of the P‐Bodies were found colocalized with the mRNA [Fig. 5(B,C)]. Taken together, our results show that an mRNA's 3′ UTR remains accessible to miRNA‐dependent RNA silencing with all three labeling strategies.
Single‐particle tracking of microinjected fluorescent BBT‐ and tail‐labeled mRNAs
Our analysis so far suggests that our mRNA labeling strategies for BBT and tail are compatible with both protein translation and RNA silencing. The power of extracellular labeling of a fully functional mRNA lies in the ability to capture its entire spatiotemporal distribution at the single‐molecule level in cellulo with low background, as any free fluorophores have been removed from a labeled RNA that is minimally altered relative to its unlabeled counterpart. We therefore microinjected 1 nM of both versions of labeled FLuc mRNA, bearing 11 miR‐7 MRE sites, in the presence and absence of miR‐7 duplex, into U2OS cells stably transfected with DCP1a‐EGFP, and monitored their diffusion characteristics 30 min after injection. Of note, nondenaturing gel analysis of the microinjection solutions indicated that assembly between the duplexed miRNA and mRNA prior to injection was negligible [Supporting Information, Fig. S4(A)]. We observed a wide range of diffusion coefficients among the mRNA population as calculated from their mean‐square displacement (MSD), obtained from single‐particle tracking trajectories [Fig. 6(A), Movie 1], similar to our prior observations using miRNAs binding to endogenous mRNAs.3, 37 Histograms of diffusion coefficients (n = 4–6 cells, >1,000 total trajectories) could be broadly fitted with two populations, slow (red dashes, BBT = 0.108 µm2/s; tail = 0.124 µm2/s) and fast (green dashes, BBT = 0.917 µm2/s; tail = 1.03 µm2/s); the averages of these distributions closely resemble those observed for an FLuc gene in literature.51 Comparing these values with the similarly calculated diffusion coefficients of the EGFP‐labeled P‐Bodies [Fig. 6(B)] suggests that the slow‐diffusing mRNAs may be associated with a (faster) subset of these granules. Interestingly, the diffusion coefficient distributions of each mRNA in the presence and absence of miR‐7 were largely indistinguishable (Table 1), suggesting that diffusion constants are not a good indicator of regulation 30 min after microinjection. However, P‐Bodies were found to be extensively colocalized with fluorescent FLuc mRNA in the presence, but not absence, of miR‐7 [Fig. 6(C)]. On an individual molecule level, we observed that, in the absence of miR‐7, fluorescent mRNAs occasionally encroached upon a P‐Body, but upon encountering a physical barrier rapidly diffused away [Fig. 6(C)]. By contrast, in the presence of miR‐7, we observed colocalized mRNAs persist at the P‐Body core for >9 imaging frames. Furthermore, we can elucidate details about the interactions between the two particles. As an example, a fluorescent FLuc mRNA, sequestered by a P‐Body in the presence of miR‐7 for over 0.4 s, escaped it to potentially re‐enter the translating mRNA pool [top panels of Fig. 6(E), Movie 2].
Figure 6.
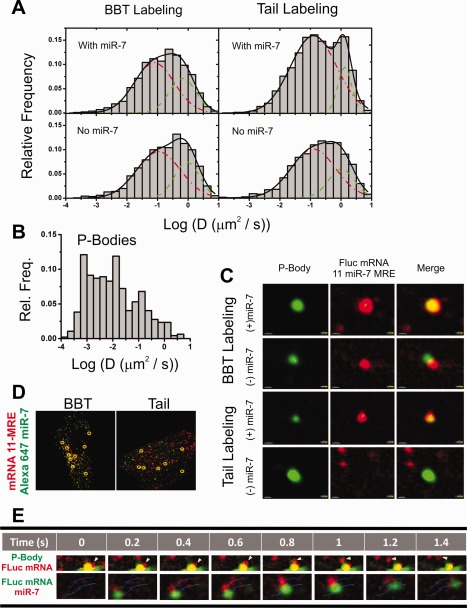
Single‐molecule analysis of fluorescent mRNAs in cellulo. (A) MSD‐derived diffusion coefficients of Alexa647 BBT (left two panels) and tail‐labeled (right two panels) FLuc mRNAs comicroinjected with (top two panels) and without (bottom two panels) duplexed miR‐7. Particle diffusion coefficients were calculated f 0.5 h after microinjection into DCP1a‐EGFP stably transfecting U2OS cells. Distributions were generated from >1,000 particle tracks from 5 cells and were fit to two Gaussians, the slower distribution is designated by a red dashed line and the faster population by a green dashed line. (B) MSD‐derived diffusion coefficients of P‐Bodies. Distributions were generated from ∼500 particle tracks from 16 cells. (C) Pseudocolored images of Alexa647‐labeled mRNA (red) and DCP1a‐EGFP fluorescent P‐Bodies (green), cotracked with and without coinjected miR‐7. (D) Live cell analysis of Alexa555 BBT‐ (left pane) and tail‐labeled (right pane) FLuc mRNA (green) coinjected with Alexa647‐labeled miR‐7 (red) HeLa cells. Yellow circles represent instances of colocalization observed for periods exceeding 9 frames. (E) Pseudocolored images of a dissociation event between a P‐Body (green) and a fluorescent mRNA (red, top panels) and a co‐tracking event between a fast‐diffusing fluorescent mRNA (green) and an miRNA (red, bottom panels) observed in live‐cell single‐particle tracking experiments. Thin blue lines represent the tracks in which the particles traverse during the course of the event.
Next, to co‐track mRNA with a regulating miRNA, we comicroinjected 1 nM Alexa555‐labeled BBT‐ or tail‐modified mRNA with 100 nM 3′ Alexa647‐labeled miR‐7 [Supporting Information, Fig. S4(B)]. Of note, while testing the functionality of 3′ fluorophore‐labeled miR‐7, we observed derepression in a dual luciferase reporter gene assay in which the FLuc mRNA contained 11 MRE sites, when increasing the fraction of labeled versus unlabeled guide strand [Supporting Information, Fig. S4(C)]. We hypothesized that the 3′ fluorophore disfavors use of the labeled strand as guide, which was supported by the observation that additionally modifying the 5′ end of the passenger strand with an Iowa Black® RQ label reverted this loss of repression. As Ago preferentially loads the RNA strand with the weakest thermodynamic interaction with its duplexed counterpart at its 5′ end,52 we conclude that 3′ labeling of the miRNA weakens or in other ways interferes with these interactions. Presumably, labeling the 5′ end of the passenger strand with Iowa Black® RQ hinders Ago's ability to load the passenger strand, moving its preference back to the 3′ labeled miR‐7.
Following a 1 h incubation after microinjection, we performed live‐cell single‐particle tracking of the injected cells and co‐tracked the fluorescent miRNA with the mRNA. While the majority of the observable interactions between the miRNA and targeted mRNA were relatively stationary [Fig. 6(D)], we were also able to capture highly mobile interacting particles [bottom panels of Fig. 6(E), Movie 3]. Similarly, microinjecting Cy5‐body‐labeled FLuc with 11 miR‐7 MRE sites with Cy3‐labeled miR‐7 also revealed extensive colocalization in fixed U2OS cells [Supporting Information, Fig. S3(B)]. Collectively, all three labeling strategies are adequate for in cellulo single‐molecule fluorescence microscopy assays, with the caveat that the body‐labeled approach is viable only for noncoding RNAs.
Discussion
Herein, we systematically tested and validated two enzymatic methods to strategically incorporate chemically modified nucleic acids (CNTP) in vitro into one of the three selected regions of an mRNA molecule: the body, between the body and tail (BBT), and throughout the tail (Fig. 1). Each strategy has the unique capacity to place the fluorophores within distinct regions of the RNA molecule depending on the experimental need. To this end, we employed two enzymes: T7 RNA polymerase and yeast poly(A) polymerase (yPAP). For body labeling, T7 RNA polymerase will incorporate 5‐position‐modified pyrimidines, such as Cy5‐labeled UTP [Fig. 1(A)]. For BBT‐ and tail‐modifying approaches, the yPAP enzyme can efficiently incorporate small modifications at the 2′ position of ATP, such as azido‐ATP [Fig. 1(B)]. Fluorophore labeling of the azido‐moiety was rapidly and gently accomplished by incubating the modified RNA with a strained‐promoted alkyne‐azide cycloaddition (SPAAC) fluorescent click reagent, which avoids the background degradation observed when using the more common Cu+‐mediated click chemistry [Supporting Information, Fig. S1(B)].
Co‐transcriptional body (5′ through 3′ UTR) labeling of an RNA molecule using T7 RNA polymerase was efficient and high‐yielding (Fig. 2 and Table 1), yet rendered an mRNA noncoding [Fig. 3(A)]. For the proper translation of a viable protein, the highly processive components of the translation machinery must traverse the coding sequence without obstruction.53, 54 The fact that the average mammalian coding sequence is ∼1,200 nt, while the 3′ UTR is ∼1,000 nt55 and the 5′ UTR is ∼150 nt,56 suggests that there is a high probability a fluorophore will be randomly incorporated into the coding region. Thus, this strategy turned out to be impractical for labeling intracellularly functional mRNAs, albeit was compatible with their P‐Body association (Supporting Information, Fig. S3). As ∼80% of the transcribed genome is predicted to be noncoding,57, 58, 59 long noncoding RNAs (lncRNAs) of >200 nucleotides (nt) length might still be amenable to a body‐labeling approach. By keeping the fluorophore‐to‐RNA ratio as low as possible, one can in principle minimize the impact of these labels on RNA function, but would need to assess this impact.
The other two strategies, BBT and tail labeling, were both efficient and high‐yielding (Fig. 2 and Table 1) and rendered mRNAs indistinguishable from their unmodified counterparts in terms of both protein coding and miRNA‐mediated regulation (Figs. 3, 4, 5, 6). Owing to the nature of the labeling strategy and the relative ease with which yeast poly(A) polymerase (yPAP) incorporates azido‐ATP,27, 35 the BBT strategy accrues extensive labeling over short time intervals. Thus, BBT labeling is best suited in instances where numerous fluorophores and a bright mRNA are desired, while containing the labels between, but outside of, functionally important regions. By contrast, the tail‐modification strategy importantly dilutes the azido‐ATP with unmodified ATP, thereby more easily controlling the extent of incorporation into the RNA molecule. However, this strategy randomly incorporates the CNTP within a functional region of the RNA molecule, even though sufficient stretches of poly(A) sequence may remain sufficiently unmodified to retain functional activity such as binding of poly(A) binding protein and protection from exonucleolytic degradation. Consequently, the coding functionality of both BBT‐ and tail‐labeled mRNAs proved unhindered; as judged by in vitro rabbit reticulocyte translation [Fig. 3(B)], in vivo luciferase assays [Fig. 3(C)], and immunofluorescence (Fig. 4), both types of labeled mRNAs translated luciferase protein comparably to an unmodified control. Furthermore, BBT‐ and tail‐labeled FLuc mRNAs containing 0, 1, 3, 6, or 11 miR‐7 MRE sites in their 3′ UTR demonstrated an MRE‐dependent decrease in luciferase expression similar to that of control [Fig. 5(A)]. Finally, the miR‐7 MRE‐containing fluorescent FLuc mRNAs colocalized with cytoplasmic P‐Bodies in U2OS cells in miRNA‐dependent fashion and co‐tracked with miRNAs (Figs. 5 and 6). Taken together, both yPAP approaches yield mRNAs with coding function and accessible 3′ UTRs that can provide real‐time, mechanistic insights into the RNA silencing pathway.
Our study sought to probe the merits of three labeling strategies to best guide the user on which technique is most appropriate for their experimental needs. So far, the MCP‐EGFP and similar protein‐based approaches are the most widely used labeling systems for studying single mRNA molecules intracellularly in real time.7, 11, 12, 13, 14, 19, 20 By genetically appending the RNA with a repeating ∼21‐nucleotide MS2 coat protein (MCP) binding sequence, recombinant overexpressed fluorescent‐protein‐tagged MCP will reversibly bind and thus label the RNA molecule. To visualize the RNA molecule over the diffuse background of unbound fluorescent MCP, 24 MCP binding sites are genetically encoded into the mRNA, typically between the body and poly(A) tail. This system presents the advantages that the mRNA gene is transcribed and the RNA is labeled within the cell and that it has been amply validated. However, the large background contributed by the overexpression of fluorescent MCP necessitates image processing to sufficiently visualize and study the RNA of interest. Furthermore, the expression levels of both the MCP and mRNA are controlled by DNA promoters, thus making it challenging to precisely control the number of molecules being visible. Last, the insertion of 504 nt of unnatural sequence into the RNA is required to yield 24 MCP binding sequences, which are bound by, on average, ∼13 MCP dimers at any given time. The addition of mass and sequence increase the risk of impaired biological function. The direct labeling methods described here, by comparison, drastically reduce background by removal of unbound fluorophores in vitro prior to cell exposure, do not require the incorporation of non‐native RNA sequences, add only small chemical modifications to endogenous RNA nucleotides, require no additional cloning or transfection of challenging repetitive sequences, and easily control the number of labels per RNA molecule for the desired brightness. They thus represent a complementary new tool for the study of intracellular mRNAs, including their interactions with pathway components such as P‐Bodies and miRNAs by two‐color single‐particle imaging, with the promise to reveal the stochasticity of these nanomachines and how they adapt under various cellular conditions. Ultimately, modeling of these observables using systems biology approaches will help predict gene regulatory outcomes.
Materials and Methods
Preparation of fluorescent miRNA duplexes
All labeled and unlabeled miRNA guide and passenger strands were ordered from Integrated DNA Technologies (IDT, see oligonucleotide list in Supporting Information). For all combinations of guide and unlabeled passenger strand, each RNA molecule was modified to contain a 5′ phosphate and, in the case of the labeled guide strand, a 3′ NHS‐ester‐linked Alexa Fluor 647 or Cy3 fluorophore. The synthesis of a 5′ Iowa Black® RQ‐labeled passenger strand precluded a 5′ phosphate. Guide and passenger strands were HPLC purified by IDT, and their size and purity verified by denaturing, 8M urea, 20% polyacrylamide gel electrophoresis. For the Alexa Fluor 647‐ and Cy3‐labeled guide strands, >90% of the RNA was found to be singly labeled as determined by quantifying the molar ratio of fluorophore to RNA through UV–visible absorbance measurements. Duplex formation between guide and passenger strand were performed at a 1:1.5 ratio in 1× phosphate‐buffered solution (PBS, Gibco, Cat# 70011) to a final concentration of 10 µM. Duplex formation was assessed by electrophoretic mobility shift assay on a nondenaturing 20% polyacrylamide gel in 1× Tris/Borate/EDTA buffer.
Plasmid reporter design
Dual luciferase reporter plasmids are variants of the pmiRGLO Dual‐Luciferase miRNA Target Expression Vector (Promega, Cat# E1330). Depending on the number of miR‐7 MRE sites cloned into the FLuc gene, pools of 5′ phosphate containing primers (Supporting Information) were heat‐annealed followed by T4 DNA ligase treatment. Both the annealed primers and the pmiRGLO vector were restriction enzyme digested using Xba1 and Sbf1 and purified using the QIAquick PCR Purification Kit (Qiagen, Cat# 28104). Restriction enzyme digested pmiRGLO vector were Antarctic Phosphatase treated and purified for the second time using the QIAquick PCR Purification Kit. Purified primer pool and pmiRGLO vector were pooled at a ratio of 7:1 and ligated using T4 DNA Ligase. The reaction was QIAquick PCR purified and transformed into XL10‐Gold® Ultracompetent Cells (Agilent, Cat# 200314). Transformation mixture was smeared onto LB agar plates with 100 µg/mL ampicillin to achieve single colonies. Colonies were PCR screened for the proper insertion of the miR‐7 MRE into the FLuc 3′ UTR. Selected colonies were grown in LB medium with 100 µg/mL ampicillin to a cell density of approximately 3–4 × 109 cells/mL and the plasmid was purified using the QIAGEN® Plasmid Maxi Prep Kit (Qiagen, Cat# 12162). Purified plasmid sequences were further characterized by the University of Michigan Sequencing Core via Sanger Sequencing.
To transcribe FLuc‐modified mRNA, we restriction cut the FLuc gene, with the appropriate number of miR‐7 MRE sites, from the pmiRGLO plasmid and cloned it into the pcDNA™ 3.1 (‐) Mammalian Expression Vector (Invitrogen, Cat# V795‐20). To do this, both plasmids were treated with PspOMI and Xba1 restriction enzymes, whereas pmiRGLO vector was additionally Antarctic Phosphatase treated, and both were purified via QIAquick purified. Purified pmiRGLO and pcDNA restriction digested mixtures were pooled in a ratio of 2:1 and T4 DNA ligase treated and QIAquick purified. The ligated mixture was then transformed into XL10‐Gold® Ultracompetent Cells, smeared onto LB agar plates containing 100 µg/mL ampicillin, and single colonies containing the proper plasmid were PCR identified. Selected colonies were grown in LB medium with 100 µg/mL ampicillin to a cell density of approximately 3–4 × 109 cells/mL and the plasmid was extracted and purified using the QIAGEN® Plasmid Maxi Prep Kit (Qiagen, Cat# 12162). Purified plasmid sequences were further characterized by the University of Michigan Sequencing Core via Sanger Sequencing.
In vitro RNA preparation
Plasmid templates, from which RNA was prepared, were first linearized using restriction enzymes. Renilla Luciferase RNA was transcribed from the Not1 restriction enzyme cut pRL‐CMV Vector (Promega, Cat# E2261) plasmid. FLuc RNA, bearing 0, 1, 3, 6, or 11 miR‐7 MRE sites, was transcribed from AFlIII restriction enzyme cut pcDNA™ 3.1 (‐) Mammalian Expression Vectors. Linearized plasmids were further purified using the QIAquick PCR Purification Kit.
Unlabeled RLuc and FLuc RNA were first transcribed from 1 µg (per reaction) of linearized plasmid, using MEGAscript® T7 Transcription Kit (ThermoFisher Scientific, Cat# AM1333), incubated at 37°C for 6 h, and purified using MEGAclear™ Kit (ThermoFisher Scientific, Cat# AM1980). RNA was capped using ScriptCap™ m7G Capping System (CellScript™, Cat# C‐SCCE0610) and yeast poly(A) polymerase (yPAP) tailed using USB® Poly(A)Polymerase, Yeast kit (Affymetrix, Cat# 74225Y/Z) per the manufacturer's instructions, with a MEGAclear™ purification step following each treatment. For body labeling, fractions of the total UTP pool were replaced with 0.1, 0.5, 1.0, 5.0, or 10% cyanine 5‐aminoallyluridine‐5′‐triphosphate (Trilink Biotechnologies, Cat# N‐5108). For modifying the RNA between body and tail (BBT), an additional yPAP tailing and purification step was inserted between cap and tail procedures. In this instance, the RNA was yPAP treated with the total pool of ATP replaced with 2′‐azido‐2′‐deoxyadenosine‐5′‐triphosphate (Trilink Biotechnologies, Cat# N‐1045), using standard kit conditions, but incubating only for 10, 20, 30, or 60 min. The tail‐modified RNA procedure hijacked the yPAP poly(A) tailing step to cointegrate 2′‐azido‐2′‐deoxyadenosine‐5′‐triphosphate into the poly(A) tail. To do this, portions of the total ATP pool were replaced with 0.1, 0.5, 1.0, 5.0, or 10% of 2′‐azido‐2′‐deoxyadenosine‐5′‐triphosphate nucleic acid.
2′‐Azido‐2′‐deoxyadenosine‐modified mRNAs were fluorescently labeled using either Click‐IT® Protein Reaction Buffer Kit (Life Technologies, Cat# C10276) with Alexa Fluor® Alkyne (Life Technologies, Cat# A10278) or Click‐IT® Alexa Fluor® 647/555 DIBO Alkyne (Invitrogen, Cat# C10408/C10406). Alkyne Alexa Fluor® 647, Click‐IT® Alexa Fluor® 647 DIBO Alkyne, and Click‐IT® Alexa Fluor® 647 were all dissolved in anhydrous DMSO to a stock concentration of 1 mM. Cu‐based click chemistry using the Click‐IT® Protein Reaction Buffer Kit and Alkyne Alexa Fluor® 647 was used as described per the manufacturer's instructions, using a final RNA concentration of 1 µM. RNA labeled using the strained‐promoted alkyne‐azide cycloaddition (SPAAC) click reactions, 1 µM of azide‐ATP‐modified RNA is mixed with 100 µM Click‐IT® Alexa Fluor® 647/555 DIBO alkyne in 1× PBS for 2 h at 37°C. Following all labeling procedures, the RNA was MEGAclear™ purified and EtOH precipitated overnight. Precipitated RNA was dissolved in miliQ H2O to a final concentration of approximately 1 µM. RNA integrity was analyzed via either 4% denaturing, 8M urea, polyacrylamide or 1.2% denaturing, 1M urea, agarose gel electrophoresis. RNA was detected via SYBR® Gold Nucleic Acid Gel Stain (ThermoFisher Scientific, Cat# S11494) or GelRed™ (Biotium, Cat# 41003).
Cell culture and transfection
HeLa (CCL‐2, ATCC) and DCP1a‐EGFP stably transfected U2OS cells were grown in an incubator, held at 37°C and in an atmosphere with 5% CO2 and 95% relative humidity. HeLa cells were maintained in DMEM medium (GIBCO, Cat# 11995) and U2OS cells in phenol red free McCoy's 5A medium with l‐glutamine (GE Health, Cat# SH30270.01). For both cell lines, the medium was additionally supplemented with 10% (v/v) fetal bovine serum (FBS) and 100 U/mL penicillin–streptomycin (ThermoFisher Scientific, Cat# 15140122).
DCP1a‐EGFP stably transfected U2OS cell line generation
U2OS cells were stably transfected with our DCP1a‐EGFP plasmid using a protocol described elsewhere.50 In short, U2OS cells were seeded to a density of 5 × 105 cells in a 6‐well plate and allowed to adhere overnight. Into each well, a mixture of 2 µg of linearized DCP1a‐EGFP containing plasmid DNA, 4 µL of FuGENE® HD (Promega, Cat# E2311) to a total 50 µL with Optimem minimal medium was added. After 6 h, fresh cell medium was placed into each well and cells were allowed to continue to incubate for an additional 18 h. Following a 24 h period, cell medium was supplemented with 0.5 mg/mL G418 selection marker. Cell medium was replaced every 2–3 days to ensure a high concentration of selection marker. After cell colonies become large enough, cells were split, the cell density was counted, and cells were seeded onto a 96‐well plate at a density of approximately 1 cell per well. After cells reached ∼80% confluency, they were tested for phenotypic EGFP expression. Furthermore, EGFP‐DCP1a expression was tested further by Western blot analysis using an Anti‐DCP1a (C‐terminal) antibody (Sigma, Cat# D5444).
Repression assays
To validate that miR‐7 increasingly regulate mRNA in an MRE number‐dependent process, dual luciferase reporter plasmids bearing 0, 1, 2, 3, 4, 5, 6, or 11 miR‐7 MRE sites in the FLuc 3′ UTR were co‐transfected with either unlabeled miR‐7 duplex or control miRNA (Life Technologies, Cat# 4390843) into HeLa and DCP1a‐EGFP stably transfected U2OS cells. To do this, a 96‐well plate was seeded with 15,000 cells per well and allowed to adhere for 24 h. Next, half of the cell medium was replaced by an equivalent volume of transfection mixture. For each well, the transfection mixture comprised 60 ng of the dual luciferase reporter, containing the appropriate number of miR‐7 MRE sites in the FLuc 3′ UTR, 100 nM of either duplexed miR‐7 or control miRNA, 0.4 µL of Lipofectamine 2000, and diluted to a total of 10 µL with Optimem medium (Gibco, Cat# 31985070). Upon the addition of the transfection mixture, samples were allowed to incubate for a period of 6 h, when the cells were given fresh medium. After a 24 h period, the cells were washed thrice with 1× PBS, lysed and analyzed for total RLuc and FLuc luminescence per the instructions of the Dual‐Luciferase Reporter Assay System (Promega, Cat# E1910), and analyzed on a luminometer (Lmax Molecular Devices Luminometer Reader, BERTHOLD Luminescence). For transfected mRNA, the same protocol was used, but substituting plasmid DNA with 100 ng of RLuc mRNA and 100 ng of either fluorescent BBT‐ or tail‐labeled FLuc mRNA, or unmodified control FLuc, cloned with 0, 1, 3, 6, or 11 miR‐7 MRE sites.
Validating fluorescent miRNA functionality
HeLa cells were split and seeded into a 96‐well culture plate to a density of 15,000 cells/well. Cells were allowed to adhere for 24 h, after which half of their medium was replaced with transfection mixture. Transfection mixture, per well, contained 0.4 µL of lipofectamine 2000, 60 ng of the dual luciferase plasmid reporter containing the FLuc gene with 11 miR‐7 MRE sites, and 100 nM of duplexed miR‐7 into a total of 10 µL in Optimem medium (Gibco, Cat# 31985070). While the concentration of the duplexed pool of miR‐7 remained constant, proportions of total comprised a mixture of fluorescently labeled miR‐7 guide strand heat‐annealed to either an unlabeled or a 5′‐Iowa Black®‐labeled passenger strand. After 6 h, cell medium was replaced with fresh medium and incubated for an additional 18 h. Cells were then washed thrice with 1× PBS, lysed and analyzed for total RLuc and FLuc luminescence per the instructions of the Dual‐Luciferase Reporter Assay System (Promega, Cat# E1910), and analyzed on a luminometer (Lmax Molecular Devices Luminometer Reader, BERTHOLD Luminescence).
In vitro translation assay
The ability of labeled and unlabeled transcribed RNA to translate protein was tested using the Rabbit Reticulocyte Lysate System (Promega, TM232). All components were thawed and stored on ice prior to mixing. Next, 35 µL of rabbit reticulocyte lysate was mixed with 0.5 µL of each amino acid mixture, 2 µg of the mRNA, and 2 µL FluoroTect™ GreenLys in vitro Translation Labeling System (Promega, Cat# L5001), and diluted to final volume of 50 µL with water. Mixtures were incubated at 30°C for 90 min. Then, 5 µL of sample was mixed with 5 µL of milliQ water and 10 µL of 2× SDS gel‐loading buffer, and heated to 70°C for 3 min. Sample was then loaded onto an Novex™ Value™ 4–20% Tris–glycine Mini Gel (ThermoFisher Scientific, Cat# XV04200PK20) and imaged using a Typhoon 9410 Variable Mode Imager (GE Healthcare Life Sciences).
Immunofluorescence of firefly luciferase
HeLa cells (1.5 × 105 cells) were seeded onto Delta T Culture Dishes (Bioptechs, Cat# 04200417C) 24 h before microinjection. Just before microinjection, cells were washed once and immersed in minimal HEPES‐buffered saline (HBS) solution, containing 20 mM HEPES‐KOH (pH 7.4), 135 mM NaCl, 5 mM KCl, 1 mM MgCl2, 1.8 mM CaCl2, and 5.6 mM glucose. Microinjection solutions comprised 10 nM of labeled or unlabeled mRNA, 1 mg/mL Cascade Blue® 10 kDa Dextran (Life Technologies, Cat# D‐1976), and 1× PBS to a total volume of 30 µL. Samples were spin filtered in a 0.22 µm pore sized Ultrafree‐MC GV Centrifugal Filter (EMD Millipore, Cat# UFC30GV00) to remove any large particulates from clogging the microinjection needle, and kept on ice until further use. A Femtotip micropipette (Eppendorf, Cat# 930000035) was loaded with 4 µL of injection solution and cells were injected using a Femtojet pump and Injectman NI2 micromanipulator (Eppendorf) for 0.5 s at 100 hPa with 20 hPa compensation pressure. Once completed, microinjected cells were returned to complete medium and incubated at 37°C and in an atmosphere with 5% CO2 and 95% relative humidity. After 12 h, cells were washed thrice with warm 1× PBS solution, fixed in 4% (w/v) paraformaldehyde solution in PBS for 20 min, washed twice with PBS, and stored overnight in 70% ethanol. Next, cells were washed twice with 1× PBS and blocked in a solution of 5% rabbit serum (Sigma, Cat# R9133), 0.02% sodium azide, and 1× PBS for 45 min at 25°C. Blocked cells were then immersed in a 1:50 diluted Anti‐Luciferase pAb (Promega, Cat# G7451) in blocking solution for 1 h at 25°C. Subsequently, cells were washed twice with 1× PBS (5–10 min/wash) and soaked in 1:200 diluted rabbit anti‐goat IgG (H + L) Cross Adsorbed Secondary Antibody, DyLight 650 conjugated, (Pierce, Cat# SA5‐10081) in blocking solution for 1 h at 25°C. Cells were washed thrice with 1× PBS and imaged for DyLight 650 fluorescence at either 20× or 60× magnification.
Single‐molecule fluorescence microscopy
Microscopy imaging was conducted similar to before,3, 37 on a home‐built IX‐81 Olympus microscope with a 60×, 1.49 NA oil immersion objective (Olympus), 2× magnification wheel, P‐545.3C7 capacitive piezoelectric x–y–z stage (Physik Instrumente), IXon 897 (Andor) EMCCD camera, and a Cell‐TIRF module (Olympus). Cells were illuminated using solid‐state lasers with wavelengths of 405 nm (0.8 mW at the objective) and 640 nm (8 mW at the objective). Highly inclined laminar optical sheet (HILO) microscopy was used to achieve sufficient illumination depth while minimizing background. A quadband dichroic (Chroma) 405/488/532/647 was used to detect miR‐7 fluorescent particles and cell boundaries. Cells were maintained at 37°C on the Delta T open dish system (Bioptechs). All videos were acquired at 100 ms camera exposure time for 50–200 frames.
Live‐ and fixed‐cell imaging of RNA
HeLa or DCP1a‐EGFP stably transfected U2OS were seeded (1–1.5 × 105 cells) and allowed to adhere for 1 day. Before microinjection, cells were immersed in 1× HBS (see above). Microinjection solutions comprised 1 nM mRNA, 1 mg/mL Cascade Blue® 10 kDa dextran, with and without 1 µM unlabeled miR‐7 duplex, or 100 nM Cy3‐ or Alexa 647‐labeled miR‐7 duplexed with 5′ – Iowa Black®‐labeled passenger strand, and 1× PBS to a total volume of 30 µL. Injection solutions were spin filtered (0.22 µm pore size) and stored on ice until injection. Femtotip micropipette is filled with 2–4 µL of injection solution and cells were injected using a Femtojet pump and Injectman NI2 micromanipulator (Eppendorf) for 0.5 s at 100 hPa with 20 hPa compensation pressure. Once completed, microinjected cells were returned to complete medium and incubated at 37°C and in an atmosphere with 5% CO2 and 95% relative humidity. Cells were allowed to incubate for the delegated time (0.5, 1, or 2 h postinjection) where they were subsequently washed thrice in HBS medium and live cells were either immediately imaged or 4% formaldehyde fixed (20 min) and subsequently imaged in Oxygen Scavenger System (5 mM protocatechuic acid (PCA), protocatechuate‐3,4‐dioxygenase (PCD), and 2 mM Trolox (6‐hydroxyl‐2,5,7,8‐tetramethylchroman‐2‐carboxylic acid)).
All images were background subtracted using a rolling‐ball radius of 5 pixels using the ImageJ Software (NIH) to remove the diffuse and nonuniform background in each image. Each video was further subtracted from a binary mask, created by outlining the cell boundary discovered in the Cascade Blue Dextran images, to eliminate any particles located outside of the cell of interest. Videos were then tracked using the tracking module of Imaris (Bitplane) software. Particles persisting for more than 9 frames were considered for mean square displacement (MSD) analysis, as the accuracy of an MSD calculated diffusion coefficients begin to exceed 50% error.60 In‐house MATLAB routines were then used to calculate the MSD and diffusion coefficients for all particle tracks, as described elsewhere.3, 37 Colocalization between any two fluorescent particles was identified as those that persist within a 4‐pixel radius of one another for more than 9 frames and exceed a threshold of >100 arbitrary units (a.u.) using a custom in‐house ImageJ routine.
miRNA:mRNA preassembly gel shift assay
Cy3‐miR‐7 guide strand was diluted to 10 µM with and without 15 µM miR‐7 unlabeled passenger strand and heat‐annealed in 1× PBS. Duplexed Cy3‐miR‐7 was diluted to 0.1 µM in PBS with 1 mg/mL Cascade Blue 10 kDa dextran, and either 0, 0.001, 0.01, or 0.1 µM Cy5 body‐labeled mRNA with 11 miR‐7 MRE sites. As a positive control, 0.1 µM single‐stranded Cy3‐miR‐7 was mixed with dextran and 0.01 µM Cy5 body‐labeled mRNA with 11 miR‐7 MRE sites in 1× PBS. As negative controls, the dextran injection marker was incubated with either 0.1 µM of duplexed miR‐7 in 1× PBS or with 0.1 µM Cy5 body‐labeled mRNA containing no miR‐7 MRE. All samples were allowed to incubate for 2 h at 25°C. At the end of each incubation, 10 µL of each sample was diluted with 10 µL of 2× nondenaturing loading dye and loaded onto a nondenaturing 15% polyacrylamide gel, run at a constant 13 W for 2.5 h at 4°C. The gel was scanned for both Cy5 mRNA and Cy3 miRNA and analyzed for gel shifting of the Cy3 miRNA to slower migrating products.
Supporting information
Supporting Information
Acknowledgment
The authors thank the Single Molecule Analysis in Real‐Time (SMART) Center of the University of Michigan, seeded by NSF MRI‐R2‐ID award DBI‐0959823 to NGW, as well as Dr Damon Hoff for training and technical advice on the Single Particle Tracker microscope.
References
- 1. Bartel DP (2009) MicroRNAs: target recognition and regulatory functions. Cell 136:215–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Czech B, Hannon GJ (2011) Small RNA sorting: matchmaking for Argonautes. Nat Rev Genet 12:19–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Pitchiaya S, Androsavich JR, Walter NG (2012) Intracellular single molecule microscopy reveals two kinetically distinct pathways for microRNA assembly. EMBO Rep 13:709–715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Huntzinger E, Izaurralde E (2011) Gene silencing by microRNAs: contributions of translational repression and mRNA decay. Nat Rev Genet 12:99–110. [DOI] [PubMed] [Google Scholar]
- 5. Fabian MR, Sonenberg N (2012) The mechanics of miRNA‐mediated gene silencing: a look under the hood of miRISC. Nat Struct Mol Biol 19:586–593. [DOI] [PubMed] [Google Scholar]
- 6. Fabian MR, Sonenberg N, Filipowicz W (2010) Regulation of mRNA translation and stability by microRNAs. Annu Rev Biochem 79:351–379. [DOI] [PubMed] [Google Scholar]
- 7. Pitchiaya S, Heinicke LA, Custer TC, Walter NG (2014) Single molecule fluorescence approaches shed light on intracellular RNAs. Chem Rev 114:3224–3265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Stoddart JF (2001) Molecular machines. ACC Chem Res 34:410–411. [DOI] [PubMed] [Google Scholar]
- 9. Nogales E, Grigorieff N (2001) Molecular Machines: putting the pieces together. J Cell Biol 152:F1–F10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. McEwen BF, Frank J (2001) Electron tomographic and other approaches for imaging molecular machines. Curr Opin Neurobiol 11:594–600. [DOI] [PubMed] [Google Scholar]
- 11. Yu J (2016) Single‐molecule studies in live cells. Annu Rev Phys Chem 67:565–585. [DOI] [PubMed] [Google Scholar]
- 12. Wu B, Eliscovich C, Yoon YJ, Singer RH (2016) Translation dynamics of single mRNAs in live cells and neurons. Science 352:1430–1435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Coleman RA, Liu Z, Darzacq X, Tjian R, Singer RH, Lionnet T (2015) Imaging transcription: past, present, and future. Cold Spring Harb Symp Quant Biol 80:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Buxbaum AR, Yoon YJ, Singer RH, Park HY (2015) Single‐molecule insights into mRNA dynamics in neurons. Trends Cell Biol 25:468–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Itzkovitz S, van Oudenaarden A (2011) Validating transcripts with probes and imaging technology. Nat Methods 8:S12–S19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ruthardt N, Lamb DC, Brauchle C (2011) Single‐particle tracking as a quantitative microscopy‐based approach to unravel cell entry mechanisms of viruses and pharmaceutical nanoparticles. Mol Ther 19:1199–1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Schoen I (2014) Localization precision in stepwise photobleaching experiments. Biophys J 107:2122–2129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sydor AM, Czymmek KJ, Puchner EM, Mennella V (2015) Super‐resolution microscopy: from single molecules to supramolecular assemblies. Trends Cell Biol 25:730–748. [DOI] [PubMed] [Google Scholar]
- 19. Katz ZB, English BP, Lionnet T, Yoon YJ, Monnier N, Ovryn B, Bathe M, Singer RH (2016) Mapping translation ‘hot‐spots’ in live cells by tracking single molecules of mRNA and ribosomes. Elife 5:[ARTICLE #]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hocine S, Raymond P, Zenklusen D, Chao JA, Singer RH (2013) Single‐molecule analysis of gene expression using two‐color RNA labeling in live yeast. Nat Methods 10:119–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Garcia JF, Parker R (2015) MS2 coat proteins bound to yeast mRNAs block 5′ to 3′ degradation and trap mRNA decay products: implications for the localization of mRNAs by MS2‐MCP system. RNA 21:1393–1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Topisirovic I, Svitkin YV, Sonenberg N, Shatkin AJ (2011) Cap and cap‐binding proteins in the control of gene expression. Wiley Interdiscip Rev RNA 2:277–298. [DOI] [PubMed] [Google Scholar]
- 23. Hinnebusch AG, Ivanov IP, Sonenberg N (2016) Translational control by 5′‐untranslated regions of eukaryotic mRNAs. Science 352:1413–1416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Liu SR, Hu CG, Zhang JZ (2016) Regulatory effects of cotranscriptional RNA structure formation and transitions. Wiley Interdiscip Rev RNA 7:562–574. [DOI] [PubMed] [Google Scholar]
- 25. Yeh HS, Yong J (2016) Alternative polyadenylation of mRNAs: 3′‐untranslated region matters in gene expression. Mol Cells 39:281–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Curinha A, Braz SO, Pereira‐Castro I, Cruz A, Moreira A (2014) Implications of polyadenylation in health and disease. Nucleus 5:508–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Winz ML, Samanta A, Benzinger D, Jaschke A (2012) Site‐specific terminal and internal labeling of RNA by poly(A) polymerase tailing and copper‐catalyzed or copper‐free strain‐promoted click chemistry. Nucleic Acids Res 40:e78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Someya T, Ando A, Kimoto M, Hirao I (2015) Site‐specific labeling of RNA by combining genetic alphabet expansion transcription and copper‐free click chemistry. Nucleic Acids Res 43:6665–6676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Rinaldi AJ, Suddala KC, Walter NG (2015) Native purification and labeling of RNA for single molecule fluorescence studies. Methods Mol Biol 1240:63–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Winz ML, Linder EC, Andre T, Becker J, Jaschke A (2015) Nucleotidyl transferase assisted DNA labeling with different click chemistries. Nucleic Acids Res 43:e110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Langereis MA, Feng Q, Nelissen FH, Virgen‐Slane R, van der Heden van Noort GJ, Maciejewski S, Filippov DV, Semler BL, van Delft FL, van Kuppeveld FJ (2014) Modification of picornavirus genomic RNA using ‘click’ chemistry shows that unlinking of the VPg peptide is dispensable for translation and replication of the incoming viral RNA. Nucleic Acids Res 42:2473–2482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Gerowska M, Hall L, Richardson J, Shelbourne M, Brown T (2012) Efficient reverse click labeling of azide oligonucleotides with multiple alkynyl Cy‐Dyes applied to the synthesis of HyBeacon probes for genetic analysis. Tetrahedron 68:857–864. [Google Scholar]
- 33. Holstein JM, Rentmeister A (2016) Current covalent modification methods for detecting RNA in fixed and living cells. Methods 98:18–25. [DOI] [PubMed] [Google Scholar]
- 34. Schulz D, Rentmeister A (2014) Current approaches for RNA labeling in vitro and in cells based on click reactions. Chembiochem 15:2342–2347. [DOI] [PubMed] [Google Scholar]
- 35. Fauster K, Hartl M, Santner T, Aigner M, Kreutz C, Bister K, Ennifar E, Micura R (2012) 2′‐Azido RNA, a versatile tool for chemical biology: synthesis, X‐ray structure, siRNA applications, click labeling. ACS Chem Biol 7:581–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Sawant AA, Mukherjee PP, Jangid RK, Galande S, Srivatsan SG (2016) A clickable UTP analog for the posttranscriptional chemical labeling and imaging of RNA. Org Biomol Chem 14:5832–5842. [DOI] [PubMed] [Google Scholar]
- 37. Pitchiaya S, Krishnan V, Custer TC, Walter NG (2013) Dissecting non‐coding RNA mechanisms in cellulo by single‐molecule high‐resolution localization and counting. Methods 63:188–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Sawant AA, Tanpure AA, Mukherjee PP, Athavale S, Kelkar A, Galande S, Srivatsan SG (2016) A versatile toolbox for posttranscriptional chemical labeling and imaging of RNA. Nucleic Acids Res 44:e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Proudfoot NJ (2011) Ending the message: poly(A) signals then and now. Genes Dev 25:1770–1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Tanpure AA, Srivatsan SG (2011) A microenvironment‐sensitive fluorescent pyrimidine ribonucleoside analogue: synthesis, enzymatic incorporation, and fluorescence detection of a DNA abasic site. Chem Eur J 17:12820–12827. [DOI] [PubMed] [Google Scholar]
- 41. Srivatsan SG, Tor Y (2007) Fluorescent pyrimidine ribonucleotide: synthesis, enzymatic incorporation, and utilization. J Am Chem Soc 129:2044–2053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Walter N, Steiner C (1993) Fast chemiluminescent measurement of RNA polymerase activity based on photon counting technology. Biotechniques 15:926–931. [PubMed] [Google Scholar]
- 43. Moretti F, Kaiser C, Zdanowicz‐Specht A, Hentze MW (2012) PABP and the poly(A) tail augment microRNA repression by facilitated miRISC binding. Nat Struct Mol Biol 19:603–608. [DOI] [PubMed] [Google Scholar]
- 44. Mayr C, Bartel DP (2009) Widespread shortening of 3′ UTRs by alternative cleavage and polyadenylation activates oncogenes in cancer cells. Cell 138:673–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Djuranovic S, Nahvi A, Green R (2011) A parsimonious model for gene regulation by miRNAs. Science 331:550–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Eulalio A, Huntzinger E, Nishihara T, Rehwinkel J, Fauser M, Izaurralde E (2009) Deadenylation is a widespread effect of miRNA regulation. RNA 15:21–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Nishihara T, Zekri L, Braun JE, Izaurralde E (2013) miRISC recruits decapping factors to miRNA targets to enhance their degradation. Nucleic Acids Res 41:8692–8705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Chang JH, Xiang S, Xiang KH, Manley JL, Tong LA (2011) Structural and biochemical studies of the 5′‐> 3′ exoribonuclease Xrn1. Nat Struct Mol Biol 18:270–U249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Anderson P, Kedersha N (2009) RNA granules: post‐transcriptional and epigenetic modulators of gene expression. Nat Rev Mol Cell Biol 10:430–436. [DOI] [PubMed] [Google Scholar]
- 50. Kedersha N, Tisdale S, Hickman T, Anderson P (2008) Real‐time and quantitative imaging of mammalian stress granules and processing bodies. RNA 448:521–552. [DOI] [PubMed] [Google Scholar]
- 51. Ma J, Liu Z, Michelotti N, Pitchiaya S, Veerapaneni R, Androsavich JR, Walter NG, Yang WD (2013) High‐resolution three‐dimensional mapping of mRNA export through the nuclear pore. Nat Commun 4:2414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Noland CL, Doudna JA (2013) Multiple sensors ensure guide strand selection in human RNAi pathways. RNA 19:639–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Bonderoff JM, Lloyd RE (2010) Time‐dependent increase in ribosome processivity. Nucleic Acids Res 38:7054–7067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Alonzo DA, Schmeing TM (2016) Ribosomes make sweeping arrests. Nat Chem Biol 12:127–128. [DOI] [PubMed] [Google Scholar]
- 55. Hendrickson DG, Hogan DJ, McCullough HL, Myers JW, Herschlag D, Ferrell JE, Brown PO (2009) Concordant regulation of translation and mRNA abundance for hundreds of targets of a human microRNA. PLoS Biol 7:e1000238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Mignone F, Gissi C, Liuni S, Pesole G (2002) Untranslated regions of mRNAs. Genome Biol 3:REVIEWS0004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Djebali S, Davis CA, Merkel A, Dobin A, Lassmann T, Mortazavi A, Tanzer A, Lagarde J, Lin W, Schlesinger F, Xue C, Marinov GK, Khatun J, Williams BA, Zaleski C, Rozowsky J, Roder M, Kokocinski F, Abdelhamid RF, Alioto T, Antoshechkin I, Baer MT, Bar NS, Batut P, Bell K, Bell I, Chakrabortty S, Chen X, Chrast J, Curado J, Derrien T, Drenkow J, Dumais E, Dumais J, Duttagupta R, Falconnet E, Fastuca M, Fejes‐Toth K, Ferreira P, Foissac S, Fullwood MJ, Gao H, Gonzalez D, Gordon A, Gunawardena H, Howald C, Jha S, Johnson R, Kapranov P, King B, Kingswood C, Luo OJ, Park E, Persaud K, Preall JB, Ribeca P, Risk B, Robyr D, Sammeth M, Schaffer L, See LH, Shahab A, Skancke J, Suzuki AM, Takahashi H, Tilgner H, Trout D, Walters N, Wang H, Wrobel J, Yu Y, Ruan X, Hayashizaki Y, Harrow J, Gerstein M, Hubbard T, Reymond A, Antonarakis SE, Hannon G, Giddings MC, Ruan Y, Wold B, Carninci P, Guigo R, Gingeras TR (2012) Landscape of transcription in human cells. Nature 489:101–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Guttman M, Amit I, Garber M, French C, Lin MF, Feldser D, Huarte M, Zuk O, Carey BW, Cassady JP, Cabili MN, Jaenisch R, Mikkelsen TS, Jacks T, Hacohen N, Bernstein BE, Kellis M, Regev A, Rinn JL, Lander ES (2009) Chromatin signature reveals over a thousand highly conserved large non‐coding RNAs in mammals. Nature 458:223–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Tuck AC, Tollervey D (2011) RNA in pieces. Trends Genet 27:422–432. [DOI] [PubMed] [Google Scholar]
- 60. Pinaud F, Michalet X, Iyer G, Margeat E, Moore HP, Weiss S (2009) Dynamic partitioning of a glycosyl‐phosphatidylinositol‐anchored protein in glycosphingolipid‐rich microdomains imaged by single‐quantum dot tracking. Traffic 10:691–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information