

Abstract
As the universal machine that transfers genetic information from RNA to protein, the ribosome synthesizes proteins with remarkably high fidelity and speed. This is a result of the accurate and efficient decoding of mRNA codons via multistep mechanisms during elongation and termination stages of translation. These mechanisms control how the correct sense codon is recognized by a tRNA for peptide elongation, how the next codon is presented to the decoding center without change of frame during translocation, and how the stop codon is discriminated for timely release of the nascent peptide. These processes occur efficiently through coupling of chemical energy expenditure, ligand interactions, and conformational changes. Understanding this coupling in detail required integration of many techniques that were developed in the past two decades. This multidisciplinary approach has revealed the dynamic nature of translational control and uncovered how external cellular factors such as tRNA abundance and mRNA modifications affect the synthesis of the protein product. Insights from these studies will aid synthetic biology and therapeutic approaches to translation.
Keywords: ribosome, translation, elongation, termination, tRNA, mRNA, fidelity
Introduction
Cellular proteins are all synthesized by the ribosome, a conserved macromolecular machine that translates the genetic material encoded in messenger RNA (mRNA). The ribosome consists of multiple protein and RNA components that coordinate among themselves to regulate translation. In addition to the small and large ribosomal subunits bound to the mRNA, the translation machinery consists of transfer RNAs (tRNAs) that recognize specific codons to incorporate the respective amino acids. The whole process is also mediated by protein factors that regulate the four stages of translation: initiation, elongation, termination, and recycling (Fig. 1).
Figure 1.
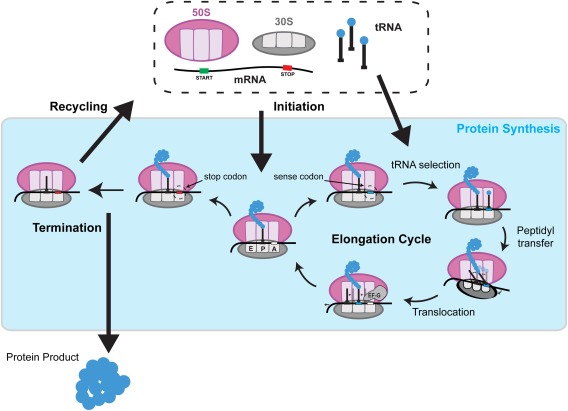
Overview of bacterial translation cycle. The 30S and 50S ribosomal subunits assemble at the start site of mRNA during initiation. The codons in the open reading frame are decoded for nascent peptide extension if a sense codon is presented in the A site during elongation. Elongation involves a sequential cycle: tRNA selection in the A site, peptidyl transfer from the P‐site tRNA to the A‐site tRNA, and translocation of the P‐site and A‐site tRNAs to E site and P site, respectively. If a stop codon is presented in the A site, it is decoded for nascent peptide release during termination. The terminated ribosome complex is disassembled during recycling.
The small (30S in prokaryotes and 40S in eukaryotes) and large ribosomal subunits (50S in prokaryotes and 60S in eukaryotes) assemble at the translation start site of the mRNA during initiation. With the help of the initiator tRNA and initiation factors, the reading frame is established to set the identity of the nascent protein. The sense codons on the mRNA downstream of the start site are subsequently decoded to build the protein during elongation until a stop codon signals release of the completed protein from the ribosome during termination. The ribosome is then disassembled into the subunits via ribosome recycling.
Of these stages of translation, elongation and termination show unique coupling of ligand binding, energy consumption, and conformational changes that produce high fidelity and high rates during protein synthesis. The multistep process of tRNA selection and accommodation into the aminoacyl‐tRNA acceptor site (A site) on the ribosome minimizes the frequency of erroneous amino acids incorporated during elongation. The motor‐like action of elongation factor G (EF‐G) is synchronized with the precise one‐codon shift of tRNAs and mRNA through the ribosome, maintaining the reading frame after each elongation cycle. A protein of correct length is created by accurate recognition of a stop codon, which leads to termination of protein synthesis. These dynamic features of elongation and termination were elucidated through a combination of biochemical, structural, single‐molecule, and computational studies. This review highlights the current models developed from interdisciplinary work, illustrating the molecular choreography that results in the observed high fidelity of bacterial translation.
tRNA Selection and Accommodation: Faithful Decoding of Sense Codons
Translation elongation begins with decoding one mRNA codon, where a cognate aminoacylated tRNA is selected and accommodated into the ribosome for the peptidyl transfer reaction. The translation apparatus has evolved to balance fidelity and efficiency during the last step of genetic information transfer, and structural dynamics and chemical energy expenditures are at the heart of the decoding machinery design. Selection of aminoacylated tRNA (aa‐tRNA, or simply tRNA in this section) and accommodation kinetics are likely key dynamic regulatory steps during translation elongation, supported by recent reports on their importance during nascent protein folding.1, 2 While structural and energetic aspects of the translational decoding have been superbly reviewed previously,3, 4 this section will focus on concepts used in achieving high fidelity and efficiency of decoding via a multistep, dynamic selection process.
Translational decoding is the process to select cognate (for an A‐site codon) tRNA substrates from the pool of different tRNA species with high accuracy (error rate of 10−3 to 10−4)5, 6, 7 and rates (5–20 amino acids per second).8 To achieve this combination of speed and accuracy, the decoding process has two necessary components: rapid sampling and commitment. During sampling, the stability of each interaction, quantified in terms of a free‐energy of codon–anticodon pair binding (ΔG), can be measured as a lifetime of the tRNA bound to the ribosome or a frequency of related structural rearrangement. The intrinsic accuracy of decoding will be determined by the difference of ΔG (ΔΔG) between a cognate substrate and a noncognate (or near‐cognate) substrate for initial ribosome–mRNA interaction. After sampling, an irreversible commitment step prohibits a reverse reaction to ensure the processivity of the reaction. Here, we note an inherent tradeoff among the accuracy, the efficiency, and the energy expenditure: for a single‐step selection process, where ΔG manifests as a kinetic parameter of an exponentially distributed population during sampling, discriminating for a longer bound substrate or more frequent structural rearrangement—thus a less efficient process—would always ensure equal or higher accuracy. This tradeoff between accuracy and efficiency can be improved if multiple selection processes are used in tandem to include a proofreading mechanism, which geometrically increases accuracy while linearly decreasing efficiency, at the expense of added irreversible chemical steps via energy expenditures.9, 10 Within this framework, studies of translational decoding machinery have attempted to answer the following mechanistic questions: how is the stability of codon–anticodon interaction measured on the ribosome, what is the time‐scale of selection process, what are the irreversible chemical steps, and how many of irreversible selection steps are used to ensure highly efficient and accurate decoding? Use of biochemical, single‐molecule, and structural methods has helped answering many of these questions.
The decoding process can be divided into two phases: initial selection and proofreading. The use of structural and single‐molecule methods unveiled a structural aspect of the initial selection mechanism. Initial selection starts with the mRNA‐independent initial binding of each aa‐tRNA molecule to the ribosome as a ternary complex (TC) with the elongation factor Tu (EF‐Tu) and GTP. This binding is mediated by the interaction between EF‐Tu and the ribosomal L7/L12 stalk, and followed by the sampling of the codon–anticodon interaction in the decoding center (DC) of the small ribosomal subunit.4 An induced‐fit mechanism was proposed by which the ribosome discriminates noncognate/near‐cognate codon–anticodon interactions.11, 12 That is, the ribosome uses three universally conserved “monitoring bases” (A1492, A1493, and G530 of 16S rRNA) to stereoselect the cognate codon–anticodon recognition geometry,11, 13 which leads to a domain closure of the small ribosomal subunit.12 Such conformational rearrangement of the ribosomal subunit is postulated to accelerate the forward steps in decoding, namely, the GTPase activation and GTP hydrolysis on EF‐Tu.14
Single‐molecule and bulk biochemical methods have revealed the dynamic nature of initial selection. By comparing the cognate and the near‐cognate aa‐tRNA species sampling characteristics to the ribosome, single‐molecule methods revealed that the cognate tRNA binds longer during its initial selection,15 and leads to a more stable tRNA–ribosome interaction via more frequent bending or movement of the tRNA toward the fully accommodated state.16 Based on these observations, the cognate tRNA selection mechanism may use both the bound lifetime and/or the frequency of the structural rearrangement to measure ΔG of codon–anticodon interaction. Both mechanisms support kinetic discrimination and place the timescale of the selection process on the order of tens of milliseconds. Similar approaches to characterize the difference between the cognate and the near‐cognate species have been taken using in vitro bulk kinetics techniques. Utilizing parametrization of efficiency and accuracy tradeoff by magnesium ions, Johansson et al. elegantly depicted the linear tradeoff between the efficiency of cognate codon reading and the accuracy of tRNA initial selection.17 This work formulated a framework to measure the maximally possible initial selection accuracy of various tRNAs in reading different near‐cognate codons, which revealed the large variation in initial selection accuracy depending on the codon–anticodon identities and suggested error hotspots in initial selection.18
The initial selection accuracy alone is not sufficient to account for the high fidelity of intracellular protein synthesis. After passing through initial selection, the genetic code reading accuracy can be amplified by a proofreading step, where the noncognate (or near‐cognate) substrates can be discarded with a higher probability than the cognate substrates. This additional selection step is thermodynamically driven by the chemical potential drop from the GTP‐hydrolysis reaction on EF‐Tu. Similar to the initial selection, the proofreading step needs to distinguish a cognate tRNA species from near‐cognate tRNA species by sampling and harnessing the free energy of the codon–anticodon interaction, before an irreversible commitment step takes place. On the translation apparatus, the free energy of interaction is amplified through the dynamic rearrangement of structure, which allows stable interaction to be kinetically selected and accommodated further. The dynamic nature of proofreading demanded the use of time‐resolved methods, such as single‐molecule and bulk kinetics, to elucidate its mechanism, while structural methods provided the context of involved molecular movements.
Bulk biochemical measurements have contributed deeply to our understanding of proofreading accuracy and its complimentary nature to the initial selection. Experimentally, proofreading accuracy can be measured as the number of GTP hydrolyzed by EF‐Tu per peptide bond formation, or be calculated by dividing the overall peptide bond formation accuracy by the initial selection accuracy. By comparing three different tRNA isoacceptors in reading their cognate and all near‐cognate codons, Zhang et al. demonstrated that proofreading and initial selection accuracies are positively correlated at high initial selection.19 Interestingly, at low initial selection, proofreading accuracy does not decrease further.19 This provides an explanation for how proofreading neutralizes potential error hotspots in tRNA selection. In conjunction with translation accuracies measured in vivo,20 this study also opened a window to understand how much of the intrinsic accuracy is used to tune the accuracy and efficiency of protein synthesis in living bacterial cells.19
Using reagents to hinder GTP‐hydrolysis after the initial selection, single‐molecule experiments have reported that tRNA can assume accommodated conformation even prior to GTP‐hydrolysis, albeit transiently.21 Based on this observation, it has been speculated that ΔG of the codon–anticodon interaction is measured via frequency of such accommodated conformation during proofreading steps, and a higher frequency of more stable interaction leads to the selection of cognate tRNA.
On the other hand, recent reports have suggested that there are two‐selection steps during the proofreading phase.22 By studying the translation accuracies of tRNA mutants with altered EF‐Tu binding affinities in the presence or absence of EF‐Tu:GTP, Ieong et al. hinted that the EF‐Tu:GDP departure event can be another irreversible step in addition to GTP hydrolysis and peptide bond formation (Fig. 2). Thus, the decoding involves at least three selection steps, each with its own sampling and commitment components, to ensure highly accurate and efficient process.
Figure 2.
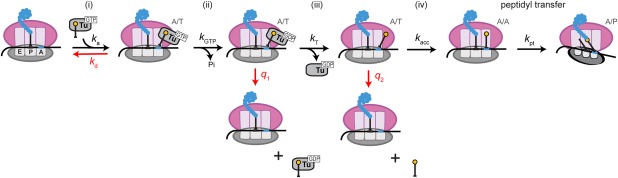
Two‐step kinetic proofreading model of tRNA accommodation. Initial selection of aa‐tRNA:EF‐Tu:GTP ternary complex in the A site (i) presents the first chance of rejection of the complex (k d), followed by GTP hydrolysis by EF‐Tu (ii) as the first commitment step. The second chance of rejection of aa‐tRNA:EF‐Tu:GDP ternary complex (q1) occurs after GTP hydrolysis, followed by the second commitment step of EF‐Tu:GDP dissociation (iii). One last chance of rejection of the aa‐tRNA happens afterwards (q2), followed by accommodation of the aa‐tRNA (iv) and the final commitment step of peptidyl transfer.
The endpoint of proofreading is the peptidyl transfer reaction, in which the α‐N nucleophile of the A‐site aa‐tRNA attacks the ester carbonyl carbon of the peptidyl‐tRNA bound to the peptidyl‐tRNA binding site (P site) to form a peptide bond. This key chemical step in protein synthesis occurs in the peptidyl transferase center (PTC) of the large ribosomal subunit. Structural, biochemical, and computational studies revealed that there were no proteins contributing to the catalysis of peptidyl transfer in the PTC and thus the PTC was regarded as a ribozyme.23, 24, 25, 26 The ribosomal PTC can catalyze this reaction to a >107‐fold faster rate in relation to that of the uncatalyzed reaction, which is the result of at least 13 kcal/mol more favorable activation entropy brought about by the ribosome positioning the substrates in the optimal orientation.27, 28 The ultimate commitment to the incorporated aa‐tRNA is marked by a successful peptidyl transfer reaction, which enables EF‐G to bind and begin a translocation process, where the next codon can be brought into the ribosome for the subsequent round of decoding.
With such a complex decoding process, kinetics of tRNA selection and accommodation can be modulated via multiple ways. tRNA selection inherently depends on the concentration of available cognate aa‐tRNA species, and the strength of the codon–anticodon interaction. Several recent reports have tried disrupting native rhythm of protein synthesis by scrambling codons with less represented synonymous codons without affecting amino acid sequence, which resulted in differently folded structures of nascent proteins.1, 2 In addition, the discovery of numerous chemical modifications within the coding region of mRNA has been suggested to be another way to modulate translation elongation dynamics.29, 30, 31, 32, 33 In particular, the N6‐methylation of adenosine within a codon has been shown to affect initial selection step during decoding.34 Given that the N6‐methyladenosine modification is dynamically regulated by its writer and eraser factors, this revealed a possibility of dynamic regulation of local translation elongation kinetics, which may be used as a cellular signal such as DNA repair.35
On the other hand, the peptidyl transfer reaction has been revealed to be sensitive to the interaction between the nascent peptide chain and its exit tunnel within the ribosome. While the rate of peptidyl transfer reaction depends on the identity of incoming amino acid similar to that of tRNA selection, the sequence of recently incorporated amino acids may serve as an intrinsic memory to affect the reaction rate cooperatively. Using a single‐molecule approach to observe translation elongation over multiple codons, two recent reports have revealed such cooperativity in delaying peptidyl transfer reaction of particular peptide sequence.36, 37 A similar mechanism may be used by a family of antibiotics including macrolides, where a peptide sequence that normally does not inhibit peptidyl transfer reaction may interact with the drug to do so.38, 39, 40 Given such a wide presence of modulators of translation elongation, its dynamic control may be one of the key regulating factors of protein synthesis in health and disease (reviewed by Richter & Coller).41 However, further studies would be needed to see how all these factors come together to regulate the rhythm of protein synthesis.
Translocation: Ribosome as a Molecular Motor
Peptide bond formation sets the stage for translocation catalyzed by EF‐G in bacteria, where tRNAs and mRNA needs to be moved by one codon. Decades of research delineated the main features of the translocation. Following peptidyl transfer, the small ribosomal subunit undergoes a rapid (∼48 s−1)42 counterclockwise rotation of 3–10° with respect to the large subunit [Fig. 3(A)], resulting in a so‐called rotated state conformation.43, 44 The tRNAs also transitions from the classical (P/P, A/A) to the hybrid (P/E, A/E) state [Fig. 3(B)]. EF‐G interacts with ribosome and stabilizes the tRNAs in this hybrid state25, 45, 46, 47 [Fig. 3(D)]. The GTP hydrolysis by EF‐G permits counterclockwise swivel of the head of the small subunit by ∼18–21° [Fig. 3(C)], followed by rapid relaxation back. Head swivel is accompanied by tRNA and mRNA movement placing them in partially translocated states.48, 49 Simultaneously, the small ribosomal subunit rotates back clockwise relative to the large subunit placing the ribosome back in the nonrotated intersubunit conformation.50, 51, 52 The emerging detailed picture of the ribosome in action highlights remaining unanswered questions: What is the conformation of the ribosome before and after translocation? What is the conformational path of translocation? How is the energy from GTP hydrolysis used and what is the role of EF‐G?
Figure 3.
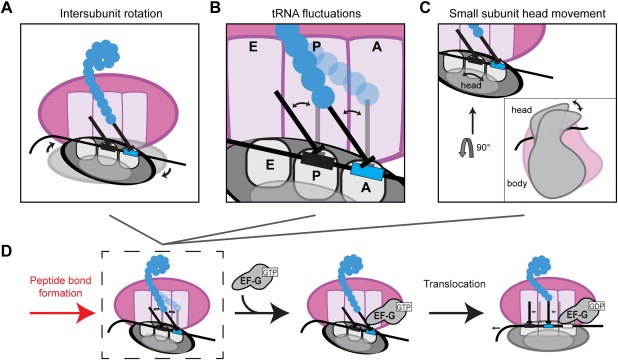
Conformational changes in the ribosome during translocation. Peptide bond formation unlocks the ribosome allowing ribosome intersubunit movements between the nonrotated and rotated states (A), movements of the A‐site and P‐site tRNAs between the classical (A/A, P/P) and hybrid (A/P, P/E) conformers (B), and swiveling of the head domain of the small subunit (C). EF‐G binding stabilizes the hybrid rotated state (D) and translocation via GTP hydrolysis brings the ribosome back to the nonrotated state, with the tRNAs in the classical conformation.
Translocation is the only part of the elongation cycle when the ribosome performs a directional motor function and moves by one codon over mRNA toward the 3′ end. This movement might occur via two principally different mechanisms. The chemical energy of the GTP hydrolysis by EF‐G could be directly converted into mechanical work through a mechanism known as the power stroke. Based on thermodynamic concerns, the alternative Brownian ratchet hypothesis was proposed. In this model, the mRNA and tRNA are moved back and forth by thermal fluctuations and recuperating energy of GTP hydrolysis is used to bias the forward reaction. Distinguishing between these two models is required to understand the mechanism of frame maintenance, recoding, and drug function.
Movement of mRNA and tRNAs occurs in a single mechanical step.53 In principle, the step size allows distinguishing between power stroke and Brownian ratchet models. Power stroke will occur with a step that is equal to the mechanical change corresponding to the transition intermediate, and with the Brownian ratchet the step is expected to be equal to the full extent of the movement. The behavior of the ribosomes under mechanical load shows that step size is close to a triplet length.53, 54 Thus, the ribosome is either moving by power stroke mechanism at a step size equal to triplet length or employs a Brownian ratchet.
The ribosome conformation unlocks upon peptide bond formation, permitting a number of thermally driven movements that were suppressed prior to peptide bond formation. tRNAs are exchanging between classical and hybrid conformations, and ribosomal subunits are possibly spontaneously rotating. Defining movement timescale will allow assigning their significance in the translocation process and might help to distinguish between the two translocation models.
Following actively translating ribosomes demonstrated that prior to A‐site tRNA binding, ribosomal subunits are in the nonrotated conformation. Peptidyl transfer induces the rotated conformation and the ribosome rotates back during translocation.50, 51, 55 The rotational and functional states are linked with lack of the spontaneous conformational exchange at the observed time scales (slower than 10 s−1), implying a high‐energy barrier between the two rotamers.50, 51, 55 Alternatively, ensemble measurements and single‐molecule measurements on stalled complexes showed a very different picture where ribosomal subunits spontaneously rotate at rates between 0.2 and 40 s−1. The ensemble measurements showed rapid reversible intersubunit rotations at 40 s−1 and 27 s−1 for forward and reverse rotations, respectively.42 The static single‐molecule measurements show much slower rotations at the order of 0.1 to 10 s−1.42, 56 In both cases, the presence of spontaneous rotations was viewed as an evidence of the Brownian ratchet mechanism. This created a seeming discrepancy that could be attributed to the methodology rather than to the principal differences in the results. The rapid fluctuations observed in the bulk measurements will be averaged out in real time translation experiments. The static single‐molecule measurements are done at lower temperatures and high magnesium (up to 15 mM), which both slow down RNA rearrangement rates. In agreement, the real‐time translation measurements to achieve efficient protein synthesis are performed at low free Mg (1.5 mM), which might speed up the rotation rates beyond detection limit (10 s−1). Therefore, it is possible that rapid ribosomal rotations indeed exist. This means the energy barrier between rotational states is low, with high‐energy barrier separating the two grand states that have different spontaneous rotation kinetics. In this case, the actively translating ribosomes measurements detect a superposition of rotational states and report on transition between the two grand states. However, additional research will be required to fully reconcile these observations.
Translation occurs fast at ∼20 amino acids per second in vivo,57, 58 equivalent of 50 ms per codon. The translocation step occurs at ∼30–100 µM−1s−1 at 37°C,59, 60, 61 thus at cellular concentrations of EF‐G (in µM range),62, 63 the entire process is expected to take <30 ms. Thus for spontaneous fluctuations to be a part of the Brownian ratchet mechanism they have to occur faster than translocation, which could be the case if rapid rotations are observed in ensemble measurements of actively translating ribosomes. Alternatively, the observed spontaneous modes of the ribosome movements are too slow to occur on their own during translation at in vivo speeds and thus are not evidence for a Brownian ratchet mechanism. They rather demonstrate the decreased energy barriers between these conformations, which represent ribosome in various states along the translocation pathway. In this view, translocation is driven by the power stroke of EF‐G, while the decreased energy barrier between translocated and nontranslocated states permits rapid catalysis of translocation by EF‐G.
The additional case for Brownian ratchet comes from evolution considerations. In the RNA world, the peptidyl transferase evolved prior to decoding.64, 65 Thus, early primordial ribosomes may have been able to conduct peptide elongation in absence of the protein factors and EF‐G and GTP hydrolysis. The protoribosomes would have to rely on the Brownian motor mechanism, where translocation would have to be driven thermally and recuperating energy comes from difference between pre‐ and post‐translocation complexes, or from formation of the next peptide bond. Surprisingly, ΔG of peptidyl transfer27, 66 is similar to the mechanical work of the ribosome, implying no need for EF‐G.53
In the end, the debate on translocation mechanism remains to be settled, as we could not clearly discriminate between two models of translocation. Yet, the most recent report argues that translocation may happen in a hybrid of two mechanisms, where conformational change by EF‐G leads to a partial translocation, then thermal fluctuation completes the translocation.67 Thus, it is possible that translocation is occurring via a combination of both mechanisms.
Termination: Factor‐Mediated Release of Nascent Peptide
The well‐orchestrated movements of the ribosome, tRNAs, and protein factors during elongation ensure synthesis of protein with the correct sequence. Another important mission in translation is to recognize the correct stopping point of protein synthesis. During the last elongation cycle, the ribosome translocates a stop codon (UAG, UGA, or UAA) into the A site, signaling the end of elongation and release of the newly synthesized protein from the ribosome, a process known as termination. During termination, the stop codon recruits two classes of release factors (RFs) to facilitate the release of the nascent peptide. Class I RF, analogous to aa‐tRNA in elongation, recognizes the stop codon and catalyzes the hydrolysis of the ester bond linking the peptide to the P‐site tRNA. The two bacterial class I RFs, RF1 and RF2, specifically recognize UAG and UGA codons, respectively, and both recognize UAA codon. A subset of bacterial species also have a class II RF or RF3 that regulates the process of termination.68 Although RF3 is nonessential for cell viability,69, 70 it was found to accelerate the dissociation of class I RFs after peptide release.71
Misrecognition of sense codons by the class I RFs leads to energy‐costly and potentially toxic truncated proteins, making accurate discrimination of stop codons during termination critical to the fidelity of translation. The in vivo error rate of termination for a sense codon is 10−5 per codon,72 making stop codon recognition by the class I RFs even more accurate than tRNA selection during elongation (10−3 to 10−4 error rate).73 Analogous to the anticodons and the 3′CCA acceptor ends of tRNAs, the class I RFs have tripeptide sequences (PVT in RF1 and SPF in RF2) called the tripeptide anticodon (TA) motifs that discriminate cognate stop codons and the universally conserved GGQ motif responsible for triggering peptide release in the PTC.74, 75 However, the mechanism of stop codon recognition that produces such high fidelity is distinct from the one observed in elongation. Unlike the role of EF‐Tu as a GTPase in the kinetic proofreading mechanism of elongation, the GTPase activity of RF3 in termination is not involved in a proofreading mechanism.76 The accuracy of termination is solely due to the ability of class I RFs to discriminate the stop codons.
Structural studies provided the first hint that the mechanism of stop codon recognition involves structural rearrangements of the release factor. Crystal structures of RF1 and RF2 defined the class I RFs to have four domains, with the TA and GGQ motifs located in domains II and III, respectively [Fig. 4(A)].77, 78 With the distance between them at 23Å, these conserved motifs did not span the distance between the DC and the PTC of the ribosome (75Å). Cryo‐EM structures of ribosome‐bound RF2 showed an open conformer with its two motifs interacting with the DC and PTC.79 This is achieved with disruption of the interactions between domains III and II and rotation of domain III by 75° toward the PTC [Fig. 4(B)]. In contrast, domains II and IV of the RFs form a stable structural core that shows little movement between the closed and open conformers. Crystal structures of class I RFs bound to the ribosome resolving the specific RF‐ribosome interactions led to the proposal that the class I RF initially binds to the ribosome in the low‐affinity closed state [Fig. 4(A)] and a successful conformational change into the high‐affinity open state only occurs upon recognition of the cognate stop codon in the DC.80, 81 Upon initial RF binding, the proposed switch loop of RF connecting domains III and IV docks in the pocket in the DC formed by ribosomal protein S12, A1492 and A1493 of 16S rRNA, and A1913 of 23S rRNA. Structural rearrangements in the DC upon stop codon recognition by RF triggers a conformational change of the switch loop opening domain III to the catalytically active conformer for peptide release.
Figure 4.
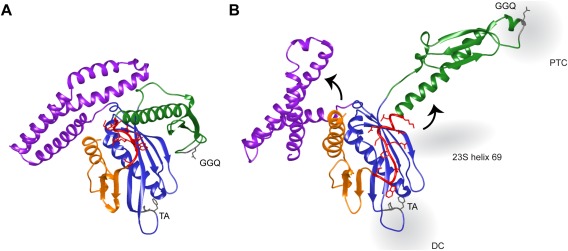
Structures of RF2 in the (A) closed (PDB: 1GQE) and (B) open (PDB: 4V67) conformations. Domains II (blue) and IV (orange) form a structural core that shows little change in the two structures. Domain I (purple) rotates 30° with respect to the static domains in the open RF2 structure. Successful stop codon recognition in the decoding center (DC) by the tripeptide anticodon (TA) motif of domain II results in the closed‐to‐open conformational transition. This is mediated by the interaction of the switch loop (red) with Helix 69 of the 23S rRNA, rotating domain III (green) by 75° placing its GGQ motif into the peptidyl transferase center (PTC) to induce peptide release.
Bulk kinetics studies supported this model by showing that the dissociation rate of RF1 is significantly faster from a ribosome complex with A‐site sense codon.82 This low‐affinity complex with the sense codon still had the class I RF in the closed conformation,83 requiring cognate stop codon for this ribosome‐induced conformational change. This induced‐fit model is depicted in Figure 5. Of the steps shown in this model, two key parameters that determine the likelihood of peptide release upon RF binding to a codon are the rate of RF dissociation from the ribosome (k off) and measured rate of peptide release (k release).76, 82 Changes in the codon identity during termination changed both the measured k off and k release values by up to 3 orders of magnitude, with no significant change in initial association rate of RF (k on). The measured k release consists of three elementary steps: RF conformational change (k conf), hydrolysis of the peptide (k hydr), and dissociation of the peptide from the ribosome (k diss) (Fig. 5). The chance of peptide release upon RF binding is determined by the competition between the forward steps of RF opening and peptide hydrolysis and the reverse step of RF dissociation.
Figure 5.
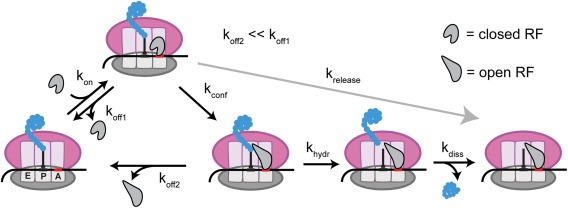
Induced‐fit model of stop codon recognition by class I RFs. Initial binding of closed RF to cognate stop codon in A site (k on) is followed by a ribosome‐induced conformational change (k conf) into the higher affinity open conformation (k off2 ≪ k off1). This active conformation of RF then catalyzes the hydrolysis of the nascent peptide from the P‐site tRNA (k hydr), releasing the peptide from the ribosome (k diss). The experimentally measured peptide release rate (k release) is dependent on k conf, k hydr, and k diss.
The dramatic changes in affinity (k off) of RFs to cognate versus noncognate codons76, 82 are also explained by the network of interactions between the RFs and the respective stop codons. In contrast to the codon–anticodon base‐pairing during elongation, domain 2 of RF interacts with both the Watson–Crick edges and Hoogsteen edges of the stop codon bases. The conserved 16S rRNA bases G530, A1492, and A1493 that are involved in sense codon discrimination during elongation are not directly involved in stop codon recognition, but instead stabilize the open conformation of RF. The specific interactions of the RFs with the stop codons resolved by structural studies80, 81 produced differences in binding free energies to the noncognate codons (ΔΔG) of about 4 kcal/mol, corresponding to a factor of ∼1,000 in affinity.84 These free energy differences agree with the affinity differences measured in the biochemical studies.76, 82 The discriminatory ability of RFs together with their stop codon‐dependent switch into the active conformation provides the mechanistic features that reflect on the accuracy of termination.
After successful stop codon recognition, the RFs position the conserved GGQ motif in the PTC to catalyze the hydrolysis of the ester bond linking the nascent peptide to the P‐site tRNA. To prevent premature peptide hydrolysis during translation, the ribosomal PTC protects the ester bond from nucleophilic attack by a water molecule, as shown by the 10‐fold reduction in nonenzymatic hydrolysis of the peptidyl‐tRNA when bound to the ribosome.85 Interactions with RF induces conformational rearrangements in the PTC that exposes the ester carbon for the nucleophilic attack, accelerating the rate of the reaction by ∼105‐fold,86 which corresponds to lowering of the activation free energy by 7–8 kcal/mol.87 These entropic contributions of the PTC and RFs provide the required specificity for accurate release of the nascent peptide during termination.
Concluding Remarks
Translation of codons in mRNA into a polypeptide sequence is a hallmark of the central dogma made possible by the tight regulation of protein synthesis. Multidisciplinary studies of translation dynamics in the last two decades have illustrated the intricate network of movements and interactions of this multicomponent machinery that result in the observed high fidelity of translation. Many of these discussed regulatory features of bacterial translation are conserved and can be extended to our study of eukaryotic translation. Last, the molecular underpinnings of translation provide the foundation for optimization of ribosomes for expression of proteins and development of new strategies to interfere with the translation machine to treat diseases.
Conflict of Interest
The authors declare no conflict of interest.
References
- 1. Shieh Y‐W, Minguez P, Bork P, Auburger JJ, Guilbride DL, Kramer G, Bukau B (2015) Operon structure and cotranslational subunit association direct protein assembly in bacteria. Science 350:678–680. [DOI] [PubMed] [Google Scholar]
- 2. Buhr F, Jha S, Thommen M, Mittelstaet J, Kutz F, Schwalbe H, Rodnina MV, Komar AA (2016) Synonymous codons direct cotranslational folding toward different protein conformations. Mol Cell 61:341–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Voorhees RM, Ramakrishnan V (2013) Structural basis of the translational elongation cycle. Annu Rev Biochem 82:203–236. [DOI] [PubMed] [Google Scholar]
- 4. Frank J, Gonzalez RL (2010) Structure and dynamics of a processive Brownian motor: the translating ribosome. Annu Rev Biochem 79:381–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Zaher HS, Green R (2009) Quality control by the ribosome following peptide bond formation. Nature 457:161–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Edelmann P, Gallant J (1977) On the translational error theory of aging. Proc Natl Acad Sci USA 74:3396–3398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Precup J, Ulrich AK, Roopnarine O, Parker J (1989) Context specific misreading of phenylalanine codons. Mol Gen Genet 218:397–401. [DOI] [PubMed] [Google Scholar]
- 8. Young R, Bremer H (1976) Polypeptide‐chain‐elongation rate in Escherichia coli B/r as a function of growth rate. Biochem J 160:185–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hopfield JJ (1974) Kinetic proofreading: a new mechanism for reducing errors in biosynthetic processes requiring high specificity. Proc Natl Acad Sci USA 71:4135–4139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ninio J (1975) Kinetic amplification of enzyme discrimination. Biochimie 57:587–595. [DOI] [PubMed] [Google Scholar]
- 11. Ogle JM, Brodersen DE, Clemons WM, Tarry MJ, Carter AP, Ramakrishnan V (2001) Recognition of cognate transfer RNA by the 30S ribosomal subunit. Science 292:897–902. [DOI] [PubMed] [Google Scholar]
- 12. Ogle JM, Murphy FV, Tarry MJ, Ramakrishnan V (2002) Selection of tRNA by the ribosome requires a transition from an open to a closed form. Cell 111:721–732. [DOI] [PubMed] [Google Scholar]
- 13. Yoshizawa S, Fourmy D, Puglisi JD (1999) Recognition of the codon‐anticodon helix by ribosomal RNA. Science 285:1722–1725. [DOI] [PubMed] [Google Scholar]
- 14. Gromadski KB, Rodnina MV (2004) Kinetic determinants of high‐fidelity tRNA discrimination on the ribosome. Mol Cell 13:191–200. [DOI] [PubMed] [Google Scholar]
- 15. Juette MF, Terry DS, Wasserman MR, Altman RB, Zhou Z, Zhao H, Blanchard SC (2016) Single‐molecule imaging of non‐equilibrium molecular ensembles on the millisecond timescale. Nat Methods 13:1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lee T, Blanchard S, Kim H, Puglisi J, Chu S (2007) The role of fluctuations in tRNA selection by the ribosome. Proc Natl Acad Sci USA 104:13661–13665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Johansson M, Zhang J, Ehrenberg M (2012) Genetic code translation displays a linear trade‐off between efficiency and accuracy of tRNA selection. Proc Natl Acad Sci USA 109:131–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zhang J, Ieong K‐W, Johansson M, Ehrenberg M (2015) Accuracy of initial codon selection by aminoacyl‐tRNAs on the mRNA‐programmed bacterial ribosome. Proc Natl Acad Sci USA 112:9602–9607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zhang J, Ieong K‐W, Mellenius H, Ehrenberg M (2016) Proofreading neutralizes potential error hotspots in genetic code translation by transfer RNAs. RNA 055632.115‐. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Manickam N, Nag N, Abbasi A, Patel K, Farabaugh PJ (2014) Studies of translational misreading in vivo show that the ribosome very efficiently discriminates against most potential errors. RNA 20:9–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Blanchard SC Jr RLG, Kim HD, Chu S, Puglisi JD (2004) tRNA selection and kinetic proofreading in translation. Nat Struct Mol Biol 11:1008–1014. [DOI] [PubMed] [Google Scholar]
- 22. Ieong K‐W, Uzun Ü, Selmer M, Ehrenberg M (2016) Two proofreading steps amplify the accuracy of genetic code translation. Proc Natl Acad Sci USA 201610917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Noller HF, Hoffarth V, Zimniak L (1992) Unusual resistance of peptidyl transferase to protein extraction procedures. Science 256:1416–1419. [DOI] [PubMed] [Google Scholar]
- 24. Nissen P, Hansen J, Ban N, Moore PB, Steitz TA (2000) The structural basis of ribosome activity in peptide bond synthesis. Science 289:920–930. [DOI] [PubMed] [Google Scholar]
- 25. Selmer M, Dunham CM, Murphy FV, Weixlbaumer A, Petry S, Kelley AC, Weir JR, Ramakrishnan V (2006) Structure of the 70S ribosome complexed with mRNA and tRNA. Science 313:1935–1942. [DOI] [PubMed] [Google Scholar]
- 26. Trobro S, Aqvist J (2008) Role of ribosomal protein L27 in peptidyl transfer. Biochemistry 47:4898–4906. [DOI] [PubMed] [Google Scholar]
- 27. Sievers A, Beringer M, Rodnina MV, Wolfenden R (2004) The ribosome as an entropy trap. Proc Natl Acad Sci USA 101:7897–7901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Trobro S, Aqvist J (2005) Mechanism of peptide bond synthesis on the ribosome. Proc Natl Acad Sci USA 102:12395–12400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Dominissini D, Nachtergaele S, Moshitch‐Moshkovitz S, Peer E, Kol N, Ben‐Haim MS, Dai Q, Di Segni A, Salmon‐Divon M, Clark WC, Zheng G, Pan T, Solomon O, Eyal E, Hershkovitz V, Han D, Doré LC, Amariglio N, Rechavi G, He C (2016) The dynamic N1‐methyladenosine methylome in eukaryotic messenger RNA. Nature 530:441–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Meyer KD, Saletore Y, Zumbo P, Elemento O, Mason CE, Jaffrey SR (2012) Comprehensive analysis of mRNA methylation reveals enrichment in 3′ UTRs and near stop codons. Cell 149:1635–1646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Schwartz S, Bernstein DA, Mumbach MR, Jovanovic M, Herbst RH, León‐Ricardo BX, Engreitz JM, Guttman M, Satija R, Lander ES, Fink G, Regev A (2014) Transcriptome‐wide mapping reveals widespread dynamic‐regulated pseudouridylation of ncRNA and mRNA. Cell 159:148–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Carlile TM, Rojas‐Duran MF, Zinshteyn B, Shin H, Bartoli KM, Gilbert WV (2014) Pseudouridine profiling reveals regulated mRNA pseudouridylation in yeast and human cells. Nature 515:143–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Dominissini D, Moshitch‐Moshkovitz S, Schwartz S, Salmon‐Divon M, Ungar L, Osenberg S, Cesarkas K, Jacob‐Hirsch J, Amariglio N, Kupiec M, Sorek R, Rechavi G (2012) Topology of the human and mouse m6A RNA methylomes revealed by m6A‐seq. Nature 485:201–206. [DOI] [PubMed] [Google Scholar]
- 34. Choi J, Ieong K‐W, Demirci H, Chen J, Petrov A, Prabhakar A, O'Leary SE, Dominissini D, Rechavi G, Soltis SM, Ehrenberg M, Puglisi JD (2016) N6‐methyladenosine in mRNA disrupts tRNA selection and translation‐elongation dynamics. Nat Struct Mol Biol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Slobodin B, Han R, Calderone V, Vrielink JAFO, Loayza‐puch F, Elkon R, Agami R, Methylation N (2017) Transcription impacts the efficiency of mRNA translation via co‐transcriptional N6‐adenosine article transcription impacts the efficiency of mRNA translation via co‐transcriptional. Cell 169:326–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Navon SP, Kornberg G, Chen J, Schwartzman T, Tsai A, Puglisi EV, Puglisi JD, Adir N (2016) Amino acid sequence repertoire of the bacterial proteome and the occurrence of untranslatable sequences. Proc Natl Acad Sci USA 113:7166–7170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Tsai A, Kornberg G, Johansson M, Chen J, Puglisi J (2014) The dynamics of SecM‐induced translational stalling. Cell Rep 7:1521–1533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Marks J, Kannan K, Roncase EJ, Klepacki D, Kefi A, Orelle C, Vázquez‐Laslop N, Mankin AS (2016) Context‐specific inhibition of translation by ribosomal antibiotics targeting the peptidyl transferase center. Proc Natl Acad Sci USA 113:12150–12155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kannan K, Kanabar P, Schryer D, Florin T, Oh E, Bahroos N, Tenson T, Weissman JS, Mankin AS (2014) The general mode of translation inhibition by macrolide antibiotics. Proc Natl Acad Sci USA 111:15958–15963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Johansson M, Chen J, Tsai A, Kornberg G, Puglisi JD (2014) Sequence‐dependent elongation dynamics on macrolide‐bound ribosomes. Cell Rep 7:1534–1546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Richter JD, Coller J (2015) Pausing on polyribosomes: make way for elongation in translational control. Cell 163:292–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Sharma H, Adio S, Senyushkina T, Belardinelli R, Peske F, Rodnina MV [YEAR] Kinetics of spontaneous and EF‐G‐accelerated rotation of ribosomal subunits. Cell Rep [VOL:PAGE #S]. [DOI] [PubMed] [Google Scholar]
- 43. Frank J, Agrawal RK (2000) A ratchet‐like inter‐subunit reorganization of the ribosome during translocation. Nature 406:318–322. [DOI] [PubMed] [Google Scholar]
- 44. Valle M, Zavialov A, Sengupta J, Rawat U, Ehrenberg M, Frank J (2003) Locking and unlocking of ribosomal motions. Cell 114:123–134. [DOI] [PubMed] [Google Scholar]
- 45. Spiegel PC, Ermolenko DN, Noller HF (2007) Elongation factor G stabilizes the hybrid‐state conformation of the 70S ribosome. RNA 13:1473–1482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Tourigny DS, Fernández IS, Kelley AC, Ramakrishnan V (2013) Elongation factor G bound to the ribosome in an intermediate state of translocation. Science 340:10.1126/science.1235490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Brilot AF, Korostelev AA, Ermolenko DN, Grigorieff N (2013) Structure of the ribosome with elongation factor G trapped in the pretranslocation state. Proc Natl Acad Sci USA 110:20994–20999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Guo Z, Noller HF (2012) Rotation of the head of the 30S ribosomal subunit during mRNA translocation. Proc Natl Acad Sci USA 109:20391–20394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Zhou J, Lancaster L, Donohue JP, Noller HF (2014) How the ribosome hands the A‐site tRNA to the P site during EF‐G–catalyzed translocation. Science 345:1188–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Marshall RA, Dorywalska M, Puglisi JD (2008) Irreversible chemical steps control intersubunit dynamics during translation. Proc Natl Acad Sci USA 105:15364–15369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Chen J, Petrov A, Tsai A, O'Leary SE, Puglisi JD (2013) Coordinated conformational and compositional dynamics drive ribosome translocation. Nat Struct Mol Biol 20:718–727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Ermolenko DN, Majumdar ZK, Hickerson RP, Spiegel PC, Clegg RM, Noller HF (2007) Observation of intersubunit movement of the ribosome in solution using FRET. J Mol Biol 370:530–540. [DOI] [PubMed] [Google Scholar]
- 53. Liu T, Kaplan A, Alexander L, Yan S, Wen J‐D, Lancaster L, Wickersham CE, Fredrick K, Noller H, Tinoco Jr I, Bustamante CJ (2014) Direct measurement of the mechanical work during translocation by the ribosome. Elife 3:e03406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Wen J‐D, Lancaster L, Hodges C, Zeri A‐C, Yoshimura SH, Noller HF, Bustamante C, Tinoco I (2008) Following translation by single ribosomes one codon at a time. Nature 452:598–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Aitken CE, Puglisi JD (2010) Following the intersubunit conformation of the ribosome during translation in real time. Nat Struct Mol Biol 17:793–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Cornish PV, Ermolenko DN, Noller HF, Ha T (2008) Spontaneous intersubunit rotation in single ribosomes. Mol Cell 30:578–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Sørensen MA, Pedersen S (1991) Absolute in vivo translation rates of individual codons in Escherichia coli. J Mol Biol 222:265–280. [DOI] [PubMed] [Google Scholar]
- 58. Zhu M, Dai X, Wang Y‐P (2016) Real time determination of bacterial in vivo ribosome translation elongation speed based on LacZα complementation system. Nucleic Acids Res 44:e155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Rodnina MV, Savelsbergh A, Katunin VI, Wintermeyer W (1997) Hydrolysis of GTP by elongation factor G drives tRNA movement on the ribosome. Nature 385:37–41. [DOI] [PubMed] [Google Scholar]
- 60. Holtkamp W, Cunha CE, Peske F, Konevega AL, Wintermeyer W, Rodnina MV (2014) GTP hydrolysis by EF‐G synchronizes tRNA movement on small and large ribosomal subunits. EMBO J 33: 1073 LP‐1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Cunha CE, Belardinelli R, Peske F, Holtkamp W, Wintermeyer W, Rodnina MV (2013) Dual use of GTP hydrolysis by elongation factor G on the ribosome. Translation 1:e24315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Gordon J (1970) Regulation of the in vivo synthesis of the polypeptide chain elongation factors in Escherichia coli. Biochemistry 9:912–917. [DOI] [PubMed] [Google Scholar]
- 63. Bakshi S, Siryaporn A, Goulian M, Weisshaar JC (2012) Superresolution imaging of ribosomes and RNA polymerase in live Escherichia coli cells. Mol Microbiol 85:21–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Petrov AS, Bernier CR, Hsiao C, Norris AM, Kovacs NA, Waterbury CC, Stepanov VG, Harvey SC, Fox GE, Wartell RM, Hud NV, Williams LD (2014) Evolution of the ribosome at atomic resolution. Proc Natl Acad Sci USA 111:10251–10256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Petrov AS, Gulen B, Norris AM, Kovacs NA, Bernier CR, Lanier KA, Fox GE, Harvey SC, Wartell RM, Hud NV, Williams LD (2015) History of the ribosome and the origin of translation. Proc Natl Acad Sci USA [VOL:PAGE #S]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Rodriguez‐Correa D, Dahlberg AE (2008) Kinetic and thermodynamic studies of peptidyltransferase in ribosomes from the extreme thermophile Thermus thermophilus. RNA 14:2314–2318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Chen C, Cui X, Beausang JF, Zhang H, Farrell I, Cooperman BS, Goldman YE (2016) Elongation factor G initiates translocation through a power stroke. Proc Natl Acad Sci 113:7515–7520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Margus T, Remm M, Tenson T (2007) Phylogenetic distribution of translational GTPases in bacteria. BMC Genomics 8:15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Grentzmann G, Brechemier‐Baey D, Heurgue V, Mora L, Buckingham RH (1994) Localization and characterization of the gene encoding release factor RF3 in Escherichia coli. Proc Natl Acad Sci USA 91:5848–5852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. O'Connor M (2015) Interactions of release factor RF3 with the translation machinery. Mol Genet Genomics 290:1335–1344. [DOI] [PubMed] [Google Scholar]
- 71. Freistroffer DV, Pavlov MY, MacDougall J, Buckingham RH, Ehrenberg M (1997) Release factor RF3 in E.coli accelerates the dissociation of release factors RF1 and RF2 from the ribosome in a GTP‐dependent manner. EMBO J 16:4126–4133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Jørgensen F, Adamski FM, Tate WP, Kurland CG (1993) Release factor‐dependent false stops are infrequent in Escherichia coli. J Mol Biol 230:41–50. [DOI] [PubMed] [Google Scholar]
- 73. Parker J (1989) Errors and alternatives in reading the universal genetic code. Microbiol Rev 53:273–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Ito K, Uno M, Nakamura Y (2000) A tripeptide “anticodon” deciphers stop codons in messenger RNA. Nature 403:680–684. [DOI] [PubMed] [Google Scholar]
- 75. Frolova LY, Tsivkovskii RY, Sivolobova GF, Oparina NY, Serpinsky OI, Blinov VM, Tatkov SI, Kisselev LL (1999) Mutations in the highly conserved GGQ motif of class 1 polypeptide release factors abolish ability of human eRF1 to trigger peptidyl‐tRNA hydrolysis. RNA 5:1014–1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Freistroffer DV, Kwiatkowski M, Buckingham RH, Ehrenberg M (2000) The accuracy of codon recognition by polypeptide release factors. Proc Natl Acad Sci USA 97:2046–2051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Shin DH, Brandsen J, Jancarik J, Yokota H, Kim R, Kim S‐H (2004) Structural analyses of peptide release factor 1 from Thermotoga maritima reveal domain flexibility required for its interaction with the ribosome. J Mol Biol 341:227–239. [DOI] [PubMed] [Google Scholar]
- 78. Vestergaard B, Van LB, Andersen GR, Nyborg J, Buckingham RH, Kjeldgaard M (2001) Bacterial polypeptide release factor RF2 is structurally distinct from eukaryotic eRF1. Mol Cell 8:1375–1382. [DOI] [PubMed] [Google Scholar]
- 79. Klaholz BP, Pape T, Zavialov AV, Myasnikov AG, Orlova EV, Vestergaard B, Ehrenberg M, van Heel M (2003) Structure of the Escherichia coli ribosomal termination complex with release factor 2. Nature 421:90–94. [DOI] [PubMed] [Google Scholar]
- 80. Korostelev A, Asahara H, Lancaster L, Laurberg M, Hirschi A, Zhu J, Trakhanov S, Scott WG, Noller HF (2008) Crystal structure of a translation termination complex formed with release factor RF2. Proc Natl Acad Sci 105:19684–19689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Laurberg M, Asahara H, Korostelev A, Zhu J, Trakhanov S, Noller HF (2008) Structural basis for translation termination on the 70S ribosome. Nature 454:852–857. [DOI] [PubMed] [Google Scholar]
- 82. Hetrick B, Lee K, Joseph S (2009) Kinetics of stop codon recognition by release factor 1. Biochemistry 48:11178–11184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Trappl K, Joseph S (2016) Ribosome induces a closed to open conformational change in release factor 1. J Mol Biol 428:1333–1344. [DOI] [PubMed] [Google Scholar]
- 84. Sund J, Andér M, Åqvist J (2010) Principles of stop‐codon reading on the ribosome. Nature 465:947–950. [DOI] [PubMed] [Google Scholar]
- 85. Pierson WE, Hoffer ED, Keedy HE, Simms CL, Dunham CM, Zaher HS (2016) Uniformity of peptide release is maintained by methylation of release factors. Cell Rep 17:11–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Zavialov AV, Mora L, Buckingham RH, Ehrenberg M (2002) Release of peptide promoted by the GGQ motif of class 1 release factors regulates the GTPase activity of RF3. Mol Cell 10:789–798. [DOI] [PubMed] [Google Scholar]
- 87. Trobro S, Åqvist J (2007) A model for how ribosomal release factors induce peptidyl‐tRNA cleavage in termination of protein synthesis. Mol Cell 27:758–766. [DOI] [PubMed] [Google Scholar]